
Pflughoeft, K. J. & Versalovic, J. Human microbiome in health and disease. Annu. Rev. Pathol. 7, 99–122 (2012).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Roth, R. R. & James, W. D. Microbial ecology of the skin. Annu. Rev. Microbiol. 42,441–464 (1988).
Saunders, C. W., Scheynius, A. & Heitman, J. Malassezia fungi are specialized to live on skin and associated with dandruff, eczema, and other skin diseases. PLoS Pathog. 8,e1002701 (2012).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Schoch, C. L. et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl Acad. Sci. USA 109, 6241–6246 (2012).
St-Germain, G. & Summerbell, R. Identifying Fungi: A Clinical Laboratory Handbook (Star Publishing Company, 2011).
Wang, Q., Garrity, G. M., Tiedje, J. M. & Cole, J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol.73, 5261–5267 (2007).
DISTINCT MICROBIAL COMMUNITIES WITHIN THE ENDOSPHERE AND RHIZOSPHERE OF POPULUS DELT OIDES ROOTS ACROSS CONTRASTING SOIL TYPES72
Neil R. Gottel,73Hector F. Castro,73,74Marilyn Kerley,73Zamin Yang,73Dale A. Pelletier,73Mircea Podar,73,75Tatiana Karpinets,73,76Ed Uberbacher, 73Gerald A. Tuskan,73,76Rytas Vilgalys,77Mitchel J. Doktycz,73and Christopher W. Schadt73,75,*
Abstract
The root-rhizosphere interface of Populus is the nexus of a variety of associations between bacteria, fungi, and the host plant and an ideal model
________________
72 Reprinted with permission from Gerald A. Tuskan. Originally printed as Gottel, N. R. et al. 2011. Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Applied and Environmental Microbiology 77(17):5934-5944.
73 Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831.
74 Department of Ecology and Evolutionary Biology.
75 Department of Microbiology.
76 Department of Plant Sciences, University of Tennessee, Knoxville, Tennessee 37796.
77 Department of Biology, Duke University, Durham, North Carolina 27708.
* Corresponding author. Mailing address: Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831-6038. Phone: (865) 576-3982. Fax: (865) 576-8646. E-mail: schadtcw@ornl.gov.
for studying interactions between plants and microorganisms. However, such studies have generally been confined to greenhouse and plantation systems. Here we analyze microbial communities from the root endophytic and rhizospheric habitats of Populus deltoides in mature natural trees from both upland and bottomland sites in central Tennessee. Community profiling utilized 454 pyrosequencing with separate primers targeting the V4 region for bacterial 16S rRNA and the D1/D2 region for fungal28S rRNA genes. Rhizosphere bacteria were dominated by Acidobacteria (31%) and Alphaproteobacteria (30%), whereas most endophytes were from the Gammaproteobacteria (54%) as well as Alphaproteobacteria (23%). A single Pseudomonas-like operational taxonomic unit (OTU) accounted for 34% of endophytic bacterial sequences. Endophytic bacterial richness was also highly variable and 10-fold lower than in rhizosphere samples originating from the same roots. Fungal rhizosphere and endophyte samples had approximately equal amounts of the Pezizomycotina (40%), while the Agaricomycotina were more abundant in the rhizosphere (34%) than endosphere (17%). Both fungal and bacterial rhizosphere samples were highly clustered compared to the more variable endophyte samples in a UniFrac principal coordinates analysis, regardless of upland or bottomland site origin. Hierarchical clustering of OTU relative abundance patterns also showed that the most abundant bacterial and fungal OTUs tended to be dominant in either the endophyte or rhizosphere samples but not both. Together, these findings demonstrate that root endophytic communities are distinct assemblages rather than opportunistic subsets of the rhizosphere.
Introduction
Populus is considered the model organism for the study of woody perennials (Tuskan et al., 2004) and represents the first tree genome to be fully sequenced (Tuskan et al., 2006). Populus has also received attention in bioenergy research for the production of cellulose-derived biofuels (Tuskan et al., 2006). Populus can be grown on land not suitable for food production and increase carbon sequestration, thus minimizing the competition between food and fuel production and reducing the carbon debt incurred through land use changes (Sannigrahi et al., 2010). Populus may also provide an ideal model for understanding a variety of plant-microbe interactions (Taghavi et al., 2009). Populus and other members of the Salicaceae are capable of establishing associations with both arbuscular mycorrhizal (AM) and ectomycorrhizal (ECM) fungi (Gehring et al., 2006) that may result in unique interactions between these fungi, as well as other endophytic and rhizospheric organisms and the host. Populus bacterial rhizosphere and endophytic constituents have received some attention due to their potential role in phytoremediation of industrial chemicals (Moore et al., 2006) and heavy metals (Biro and Takacs, 2007), as well as plant growth-promoting bacteria (PGPB), which benefit plants by providing fixed nitrogen and/or aiding resistance
to infection by pathogens (Jones and Dangl, 2006). However, most studies of such relationships have been greenhouse or plantation based, and the rhizosphere and endophyte communities of Populus from natural systems have not been studied comprehensively by molecular ecology approaches. Newly developed, high-throughput sequencing approaches of bacterial and fungal rRNA gene markers should enable an expanded understanding of such plant-microbe relationships and comprehensive descriptions of the full diversity of associations within the Populus microbiome.
Roots are the primary sites of plant nutrient import and organic molecule export, which provide carbon and energy sources to nearby microorganisms and result in a “rhizosphere” that supports higher bacterial numbers than do bulk soils (Hinsinger et al., 2009). In many cases these relationships seem to have evolved to the point where certain microorganisms appear to live nonpathogenically as endophytes within the roots or, in the case of some fungi, establish mutually beneficial mycorrhizal relationships (Gehring et al., 2006). In the case of bacterial root endophytes, it is unclear whether these microorganisms represent a specialized community or merely opportunistic rhizosphere microorganisms (Hardoim et al., 2008). Due to the complexity of the plant root habitat and the nearby rhizosphere, as in other terrestrial ecosystems, a great fraction of the microorganisms present likely remain unknown and uncultured (Amann et al., 1995).
This study focuses on the root endophyte and the directly associated rhizosphere communities of two populations of Populus deltoides located in upland and bottomland sites near the Caney Fork River in central Tennessee. Both fungi
TABLE A17-1 Selected Tree Dendrometric Measurements and Soil Physicochemical Characteristics for Bottomland and Upland Sitesa
| Tree sample ID no. | DBH* (cm)b | Crown ht* (m) | Crown width (radius, m) | Soil texture | Moisture* (%) | pH* |
| B1 | 58.4 | 25.8 | 7.6 (1.5)d | Sandy loam | 22.2 (0.1)c | 6.6 |
| B2 | 89.6 | 38.4 | 8.2 (2.2) | Sandy loam | 25.3 (0.6) | 6.7 |
| B3 | 91.1 | 32.7 | 7.6 (1.5) | Sandy loam | 29.6 (0.3) | 6.8 |
| B4 | 77.9 | 31.0 | 7.8 (0.4) | Sandy loam | 28.4 (0.2) | 6.6 |
| U1 | 37.0 | 21.5 | 4.3 (0.2) | Clay loam | 40.0 (1.7) | 7.9 |
| U2 | 38.1 | 19.9 | 7.2 (3.1) | Clay | 39.8 (0.4) | 7.7 |
| U3 | 38.4 | 18.9 | 5.1 (1.3) | Clay | 35.0 (0.7) | 7.8 |
a A characteristic is followed by an asterisk when the means were significantly different (P ≤ 0.01) for bottomland (B) versus upland (U) sites.
b DBH, diameter at breast height.
c OM, organic matter.
d Values in parentheses represent standard deviations of 3 measurement replicates.
and bacteria were characterized from the same samples by using bar-coded pyrosequencing of rRNA genes, enabling direct comparisons between these cooccurring communities. By utilizing a nested sampling design, we focused on three main questions: (i) Do endophytic communities reflect a specialized group of organisms or merely an opportunistic subset of the associated rhizosphere community? (ii) To what extent do site and soil conditions modulate endophyte and rhizosphere community composition? (iii) How variable are microbial communities among trees, sites, and the ecological niches of the root endosphere and rhizosphere?
Materials and Methods
Sites Description and Sample Collection
Native P. deltoides samples were collected in the basin of the Caney Fork River in the Buffalo Valley Recreation Area, downstream of the Center Hill Dam (bottomland site) and within Edgar Evans State Park (upland site), both in DeKalb County, TN, during the first week of October 2009. Three and four mature Populus trees were selected from the upland (36°4′N, 85°50′W) and bottomland (36°6′N, 85°50′W) sites, respectively. The soil characteristics of each tree and surrounding soil are presented in Table A17-1. Three soil cores were taken adjacent to each tree and kept on ice and refrigerated until soil characterizations. Soil moisture was determined gravimetrically after sieving to 4 mm. Other soil characterizations were performed by the Agricultural and Environmental Services
| Total C* (%) | Total N* (%) | OM* (%)c | K* (ppm) | NH4-N (ppm) | NO3-N (ppm) | P* (ppm) |
| 1.7 | 0.14 | 2.5 | 38.6 | 1.7 | 5.0 | 47.1 |
| 1.9 | 0.17 | 2.7 | 55.2 | 2.2 | 10.1 | 74.2 |
| 2.5 | 0.20 | 3.7 | 56.9 | 2.5 | 16.0 | 68.6 |
| 2.2 | 0.17 | 3.1 | 52.3 | 2.6 | 11.7 | 78.4 |
| 6.2 | 0.38 | 8.7 | 123.2 | 2.8 | 9.6 | 202.1 |
| 5.6 | 0.41 | 8.2 | 134.6 | 2.9 | 16.9 | 249.5 |
| 5.1 | 0.35 | 7.6 | 121.3 | 2.7 | 5.16 | 233.7 |
Laboratory (AESL) at the University of Georgia (http://aesl.ces.uga.edu/) on these same sieved, composited samples. Statistical comparisons of upland and bottomland site and soil characteristics were conducted using Fisher’s t test as implemented in StatPlus:mac LE (AnalystSoft, Inc.). Three primary lateral roots near each tree were carefully excavated and traced from the originating tree to ensure identity. Tertiary fine roots were removed, shaken over a sieve to remove loose soil, and washed with 100 ml of 10 mM NaCl solution to remove the adhering rhizosphere soil. This wash solution was collected into 50-ml tubes and constituted the rhizosphere samples. Multiple root samples were collected from different lateral roots of each tree, with nine total samples at the upland site (labeled the U site) and 11 root samples at the bottomland site (labeled B site). For a detailed breakdown of the nested sampling schema, refer to Table S1 in the supplemental material. Washed root and rhizosphere samples were transported on ice and stored at 4°C for up to 2 days prior to surface sterilization of the roots and then stored at -80°C thereafter.
Root surface sterilization was performed 2 days after collection by rinsing roots 5 times with sterile distilled water (dH2O) and then transferring roots with a diameter of 2 mm or less to 50-ml centrifuge tubes. The surface sterilization protocol used 3 washes with sterile dH2O before and after the following sterilization steps: 3% H2O2 for 30 s, 100% ethanol for 30 s, 6.15% NaOCl with 2 to 3 drops of Tween 20 per 100 ml for 3 min, and finally again with 3% H2O2 for 30 s. Surface sterility was confirmed for all samples by touching a subsampled root from each collection onto LB plates and incubating overnight at 30°C.
DNA Extraction
Surface-sterilized roots were chopped into 1 mm sections by using a sterilized razor blade in a petri dish. Each root sample was split into five 50 mg subsamples, and total DNA was extracted using the PowerPlant DNA isolation kit (MoBio, Carlsbad, CA), with the following modification to the manufacturer’s instructions: 50 μl of 10% cetyltrimethylammonium bromide was added to each lysis tube containing the lysis solution and roots to enhance plant cell lysis, followed by three freeze-thaw cycles (–80°C/65°C; 10 min each) and homogenization in a mixer mill for 20 min at 30 Hz (model MM400; Retsch Inc., Newtown, PA). Each set of 5 subsampled extractions was then concentrated into a single 50-μl DNA sample. DNA was quantified using an ND-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE). For rhizosphere samples, 2.0 ml of soil slurry and associated cells was pelleted via low-speed centrifugation, and the extractions were carried out using the standard MoBio protocol and PowerSoil DNA extraction kit (MoBio, Carlsbad, CA) and the Retsch mixer mill as described above.
Bacterial and Fungal Ribosomal PCR Amplification and Sequencing
Fifty microliters of 454 sequencing template per sample was produced using the following PCR reagent concentrations: 1× High Fidelity PCR buffer (Invitrogen, Carlsbad, CA), 0.2 mM (each) deoxynucleoside triphosphates (dNTPs), 2 mM MgSO4, 0.6 μM forward and reverse primers, 1.0 mg/ml bovine serum albumin (BSA), and 2 units of Platinum Taq High Fidelity (Invitrogen, Carlsbad, CA). To each 49-μl reaction mixture, 1 μl of template DNA (diluted 1:10 in 1× Tris) was added. Thermocycler settings for rhizosphere samples were 2 min at 95°C, then 30 cycles of 95°C for 15 s, 55°C for 45 s, and 68°C for 45 s, with a final extension for 7 min at 68°C. For endophyte samples, 35 cycles were used instead of 30. Bacterial primers targeted the V4 region of the 16S rRNA gene, as described by Vishnivetskaya et al. (2011), and fungal primers targeted the first ~700 bp of the 28S LSU rRNA gene by using the primers LROR and LR3 (Castro et al., 2010), which had been modified to contain the 454 A and B primer, and the A primer was further modified to contain one of 20 8-bp DNA bar codes downloaded from the Ribosomal Database Project (RDP) (Cole et al., 2009). Unincorporated primers, primer dimers, and dNTPs were removed by using the Agencourt AMPure purification system (Beckman Coulter, Danvers, MA). Product purity and concentration were checked with an Agilent 2100 Bioanalyzer (Santa Clara, CA). Emulsion reactions were performed in paired samples containing 2 sample PCR amplicons that were matched for template quantity and quality (Vishnivetskaya et al., 2011). Sequencing was performed on a Roche genome sequencer FLX system (Indianapolis, IN) using the manufacturer’s recommended conditions. Twenty samples originating from the rhizosphere and 20 samples originating from the endosphere were loaded onto the A and B regions of the sequencing plates, and separate pyrosequencing runs were performed for bacterial and fungal amplicons.
Sequence Analysis
Raw bacterial sequence outputs from the FLX system were uploaded to the Ribosomal Database Project Pyrosequencing Pipeline (http://pyro.cme.msu.edu/index.jsp) (Cole et al., 2009) for primer removal and quality control and then restricted to the desired length (see Results for further details). Populus plastid and mitochondrial 16S rRNA sequences were identified using BLAST search similarity against the entire data set, and those with >95% similarity to the plant sequence were removed from the data set. Bacterial taxonomy was assigned using the RDP Classifier (Cole et al., 2009). Raw fungal sequences were trimmed and quality checked using mothur (Schloss et al., 2009). Fungal sequences were classified by their top BLAST hit compared against the SILVA LSU database in MG-RAST (Meyer et al., 2008) for representative operational taxonomic units (OTUs). The mothur program was further used for alignment of bacterial and fungal sequences based on the full alignments of the rRNA SSU and LSU,
respectively, of the SILVA database, release 104, as a template (Pruesse et al., 2007). The mothur program was also used for preclustering at 2% (20), rarefaction curves, distance calculations, clustering, and further analysis based on OTUs.
Bacterial and fungal data were analyzed separately using the Fast UniFrac program (Meyer et al., 2008), which provides a suite of tools to compare microbial communities under a phylogenetic framework. The bacterial sequences were mapped to relatives contained in the Greengenes database core set. The fungal data set was mapped to a version of the eukaryotic SILVA LSU reference database that was parsed locally to include 565 representative sequences. Phylogenetic trees were built using RAxML v7.0.4 (Vishnivetskaya et al., 2011) using maximum likelihood and an optimized GAMMA model of rate heterogeneity and alpha shape parameter. Trees were analyzed using the unweighted options within Fast UniFrac, and samples were categorized according to sample source (endophyte or rhizosphere) and soil type (bottomland or upland). Both UniFrac and P-test significances were used to compare the microbial communities, as these two methods test different hypotheses and can result in different P values (Meyer et al., 2008). The UniFrac metric tests for differences among treatments by using the branch length, which is unique to each treatment. A P-test uses a phylogenetic tree to test whether two environments are significantly different by using parsimony-based scoring (Lozupone et al., 2006). UniFrac tests were performed using 1,000 permutations and calculated with the Fast UniFrac web application (http://bmf2.colorado.edu/fastunifrac/) (Hamady et al., 2010). UniFrac and P-test significance values were corrected using the Bonferroni correction for multiple comparisons. Principal coordinate analysis (PCoA) was further performed using the Fast UniFrac metric and visualized by origin from either endophyte or rhizosphere in both upland and bottomland sites by using the 3D Java KiNG image program (http://kinemage.biochem.duke.edu/software/).
Hierarchical Clustering
Before clustering, low-abundance and rare OTUs were filtered out by using a two-step procedure. OTUs were first sorted by the sums of their relative abundances across all samples in the data set, and those OTUs accounting for 50% of the total relative abundance in the data set were retained (85 of 8,686 bacterial and 78 of 12,532 fungal OTUs). Second, we removed rare OTUs by retaining only those that occurred in more than 5 samples. The final data set thus included 76 bacterial and 72 fungal/eukaryote OTUs, representing the most abundant and common taxa in our data set. The hierarchical clustering of the percent abundance of the selected OTUs in each sample was implemented using Cluster 3.0 (Eisen et al., 1998) and visualized in TreeView (Schloss et al., 2009) with the Spearman rank correlation coefficients as the similarity metric and a complete linkage clustering criteria.
Results
Tree and Soil Characteristics
A total of 9 upland site samples (3 each from 3 trees) were collected for endophyte, rhizosphere, and bulk soil analysis. The bulk soil samples were pooled from around each tree for soil physical and biochemical characterization. Eleven bottomland site samples, 3 each from 3 trees, and a set of 2 from a fourth tree, were similarly analyzed. Upon examination, the tree ring core sample from the bottomland tree (B3) was found to have significant brown rot in the center of the core. However, other plant phenotypic, soil, and microbial community characteristics from the two samples originating from this tree were not found to substantially differ from the remainder of the samples, and data associated with these samples were thus retained. The upland and bottomland sites differed significantly in both the characteristics of the Populus trees sampled (diameter, height, etc.) and most soil characteristics (texture, percent C, percent N, etc.) with the exception of the concentrations of ammonium and nitrate ions, which were highly variable within each site (Table A17-1).
Sequencing Quality Control and Results
Multiple levels of quality control during sequence processing and data analysis were employed. Sequences with a Phred score of <25 were removed, ensuring that the lowest-quality sequences had only ~0.3% probability of an incorrectly called base (Schloss et al., 2009). Sequences with ambiguous bases and homopolymers (>10 bases) and sequences of <150 bp or >300 bp were removed during the initial tag and primer checks through the RDP. Aligned sequences were then trimmed to a common region. Further quality control was achieved by then clustering the entire data set at 97% similarity (bacteria) or 95% similarity (fungi) and eliminating all singletons as, globally, single-sequence clusters are likely sequencing errors (Kunin et al., 2010). However, this may eliminate some extremely rare actual community members.
After initial processing, a total of 177,291 reads for bacterial endophytic samples and 100,311 reads for bacterial rhizosphere samples were generated in our pyrosequencing survey, with an average length read of 205 bp. In the fungal data set, after trimming and processing using mothur, 169,771 endophytic reads and 152,329 rhizosphere reads remained, with an average read length of 244 bp. After singleton removal, there were 116,685 bacterial sequences and 316,360 fungal sequences remaining, out of the original 120,052 bacterial sequences and 322,100 fungal sequences. The number of bacterial sequences was much lower than the fungal sequences, owing to the removal of 67,000 mitochondrial and 65,266 plastid sequences from the endophytic bacterial sample processing that were identified via BLAST similarity against the genomes of each organelle. A detailed breakdown of recovered sequence numbers sample by sample is
presented in Table S1 of the supplemental material. Representative sequences of all OTUs and the entire quality control data set are available from our project website (http://pmi.ornl.gov) or by contacting the corresponding author.
Bacterial endophytic read counts were distributed unevenly among samples, with an average number of 1,117 per sample and a standard deviation of 1,204. Bacterial rhizosphere sequences were more evenly distributed, with an average of 5,015 sequences per sample and a standard deviation of 617. Fungal libraries had a fairly small number of nontarget sequences. On average our LSU rRNA gene fungal endophyte libraries contained ~78% sequences that were classifiable as fungi, ~11% as Viridiplantae, and ~10% as metazoan, with the small remainder coming from Alveolates, Cercozoa, and other eukaryotes. Rhizosphere fungal libraries were on average ~87% fungi, 3.1% Viridiplantae, 9.7% metazoans, and a small number from other eukaryotes. The fungal primers used in the study were designed for fungal specificity as well as breadth, so the overall abundance of these nonfungal eukaryotic groups should not be considered to have ecological meaning in these studies. Thus, we excluded nonfungal sequences from the overall phylogenetic classifications (see Figure A17-4, below). However, nonfungal sequences were not excluded from OTU-level comparisons, as individual OTUs that amplified are unlikely to be affected by primer biases; thus, Figures A17-2, A17-5, and A17-6, below, contain nonfungal, eukaryotic OTUs.
Characterization of Endophyte and Rhizosphere Bacterial and Fungal Communities
Rarefaction curves were generated via mothur using a 97% identity cutoff for bacterial samples (Figure A17-1) and a 95% identity cutoff for fungal samples (Figure A17-2). As expected, endophytic microbial communities were less diverse than the rhizosphere community for both bacteria and fungi. However, bacterial endophytes exhibited a high degree of variation in the shape of their rarefaction curves compared to the other sample types. The majority of the bacterial endophyte samples saturated below 50 OTUs, although several samples continued to increase past 200 OTUs. No differences were apparent for rarefaction curves for microbial communities originating from upland versus bottomland sites. The fungal rarefaction curves displayed similar trends as the bacterial curves, with greater richness in the rhizosphere; however, endophytic samples were not as variable as the bacterial communities and on average contained more OTUs at equal sequencing depths (Figure A17-2).
Rhizospheric and endophytic bacterial communities exhibited different overall patterns of relative abundance of the major groups at the phylum level (Figure A17-3). No major differences in the phyla relative abundance patterns were observed between the upland and bottomland environments, and sample-to-sample variation was also low (data not shown). Rhizosphere bacterial communities were similar to previously reported soil communities (Kend and Triplett,
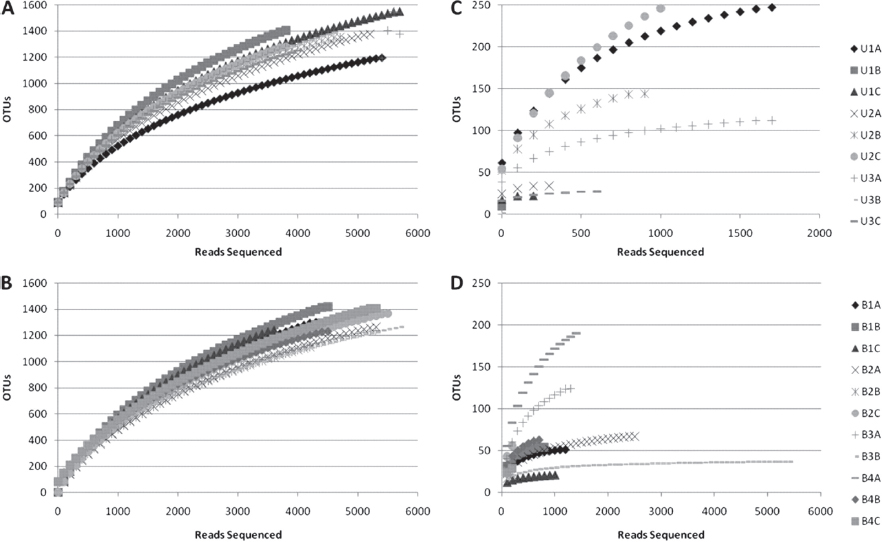
FIGURE A17-1 Rarefaction curves for bacterial OTUs, clustering at 97% rRNA sequence similarity. Curves represent sequences for multiple samples of rhizospheric(A and B) or endophytic (C and D) communities originating from samples of either upland(A and C) or bottomland(B and D) trees. See Table S1 in the supplemental material for a more detailed breakdown of properties on a sample-by-sample basis.
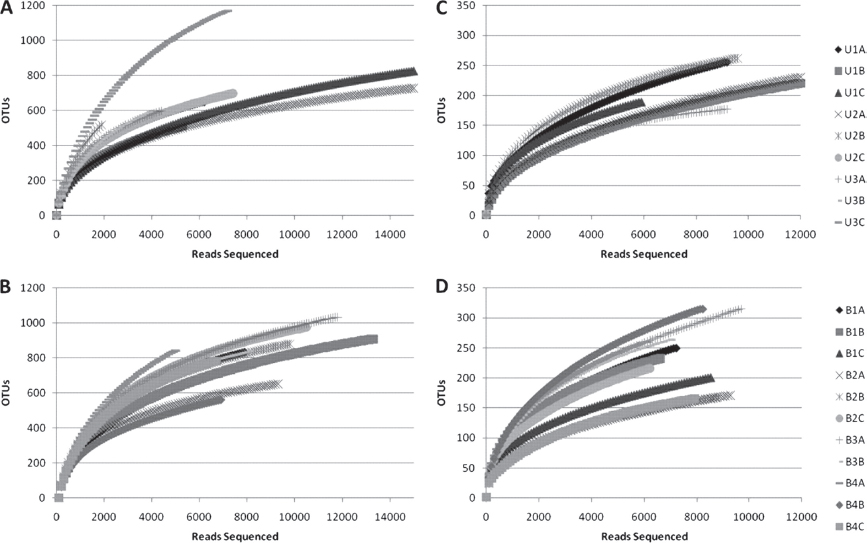
FIGURE A17-2 Rarefaction curves for fungal OTUs, clustering at 95% rRNA sequence similarity. Curves represent sequences for multiple samples of rhizospheric(A and B) and endophytic(C and D) communities originating from samples of either upland (A and C) or bottomland (B and D) trees. See Table S1 in the supplemental material for a more detailed breakdown of properties on a sample-by-sample basis.
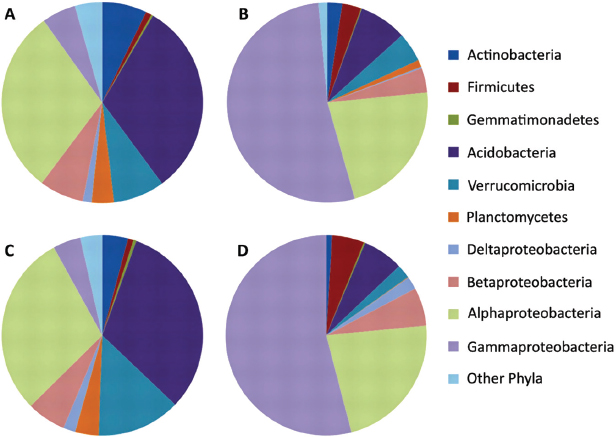
FIGURE A17-3 Bacterial classifications using the RDP classifier at 80% identity as implemented in mothur, shown at the phylum level except for Proteobacteria, which are classified by class. The charts represent average results for rhizospheric (A and B) and endophytic (C and D) or communities originating from samples of either bottomland (B and D) or upland (A and C) trees. To aid in distinguishing the colors, phylogenetic groups are presented in the same order in the pie charts (clockwise) as in the legend (top to bottom) in each subchart.
2002), where Proteobacteria were dominant (~40%), followed by Acidobacteria (33%), and Verrucomicrobia (~10%) in the samples. The Proteobacteria within the rhizosphere were primarily composed of the Alphaproteobacteria subclass (60%), with lower levels of Betaproteobacteria (15%) and Gammaproteobacteria (10%). The majority of Acidobacteria were group 6 (60%), with 15% each from groups 17 and 4, a typical distribution for soil communities (Barns et al., 2007). Endophytic bacterial communities were heavily dominated by Proteobacteria at the phylum level, at >80% of the sequences in each sample, and Acidobacteria comprised only 6%. Within the Proteobacteria, endophyte communities were heavily dominated by Gammaproteobacteria, followed by Alphaproteobacteria.
Fungal classification results indicated that fungal communities within both the rhizosphere and endosphere were dominated by the Ascomycetes within the Pezizomycotina (~40%) and Basidiomycetes within the Agaricomycotina (~25%). Similar to the bacterial patterns, relative abundance patterns of these broad fungal phylogenetic groups showed little difference between upland and
bottomland but major changes between rhizosphere and endosphere habitat (Figure A17-4). Approximately one-quarter of the LSU sequences were unidentified even to a phylum level with the BLAST-based classifications employed. An additional difference was in the bottomland endophytic community, of which 17% of sequences were attributable to Pucciniomycotina, which were at very low abundance levels in upland endophyte samples and both rhizospheres. The majority of these sequences originated from 9 of 11 endophyte samples within the bottomland data set, but Pucciniomycotina were also present in low abundance in rhizosphere samples.
Fast UniFrac and OTU-Based Hierarchical Clustering Analyses
Principal coordinate analysis generated by Fast UniFrac showed that the rhizosphere and endophyte bacterial and fungal communities form distinct clusters; within each of these clusters, the bottomland and upland communities exhibit considerable overlap (Figure A17-5). These results are similar to the trends in
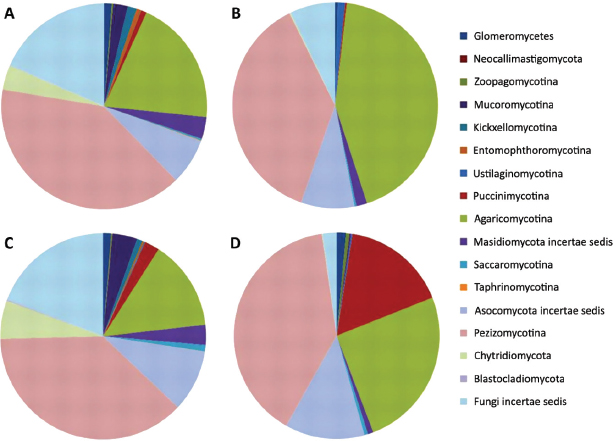
FIGURE A17-4 Fungal sequence classifications as identified from a consensus among the top BLAST scores against the SILVA LSU database. The charts represent average results for rhizospheric (A and B) or endophytic (C and D) or communities originating from samples of either bottomland (B and D) or upland (A and C) trees. To aid in distinguishing the colors, phylogenetic groups are presented in the same order in the pie charts (clockwise) as in the legend (top to bottom) in each subchart.
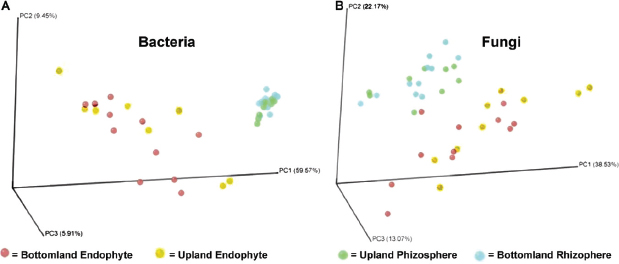
FIGURE A17-5 Principle coordinate analysis of bacterial (A) and fungal (B) communities, based on Fast UniFrac analysis. Circles are color coded by sample type: rhizosphere bottomland, green; rhizosphere upland, blue; endophyte bottomland, red; endophyte upland, yellow.
the phylogenetic classification and rarefaction results, where there were marked differences between the rhizosphere and endophytes but no consistent changes between the upland and bottomland sites. Correcting for multiple comparisons, UniFrac significance values for the bacterial data set significantly differed for the major treatments: endophyte versus rhizosphere and bottomland versus upland sites (see Table S2 in the supplemental material). Using the P-test, there was a highly significant difference between the bottomland endophyte sample and both of the rhizosphere data sets (P < 0.001), but none of the other comparisons resulted in significant differences. For the fungal data set and using the UniFrac test, significant differences were detected according to sample source (endophyte versus rhizosphere) but not according to site (bottomland versus upland) (see Table S2). When corrected for multiple comparisons, the P-test did not reveal any significant differences among fungal samples. We also used the UniFrac and P-test significance tests to check for differences between roots from the same trees and between trees from the same sites. Results from these tests were highly variable (see Table S3 in the supplemental material). While the majority of P-tests showed nonsignificant differences from samples within trees and between trees, many of the UniFrac tests did show significant differences, especially in diverse rhizosphere samples. These differences between the two unique tests (UniFrac and P-test) were likely due to the fact that the P-test takes into account only the tree topology while the UniFrac test takes into account both the tree topology and the branch length (evolutionary distance) between members (Lozupone et al., 2006). Additionally, UniFrac significance tests also have been noted by its developers to overestimate significance from diverse samples using next-generation sequencing, and it has been recommended that these tests be replaced
by PCoA and hierarchical clustering (Hinsinger et al., 2003), as presented in Figure A17-5 and Figure A17-6.
Hierarchical cluster analysis of the relative abundance of the most abundant OTUs in the fungal and bacterial data sets with high occurrence rates revealed two major linkage groups that separated completely by rhizosphere or endosphere origin in both bacterial (Figure A17-6A) and fungal (Figure A17-6B) samples. Soil type also often separated within subgroups, with numerous exceptions. Information on the identity of the major OTUs identified that are numbered and shown in Figure A17-6 can be also be found in Table S4 of the supplemental material.
Discussion
Very little is known about Populus interactions with the microbial community in mature, natural ecosystems, as most studies have originated from either
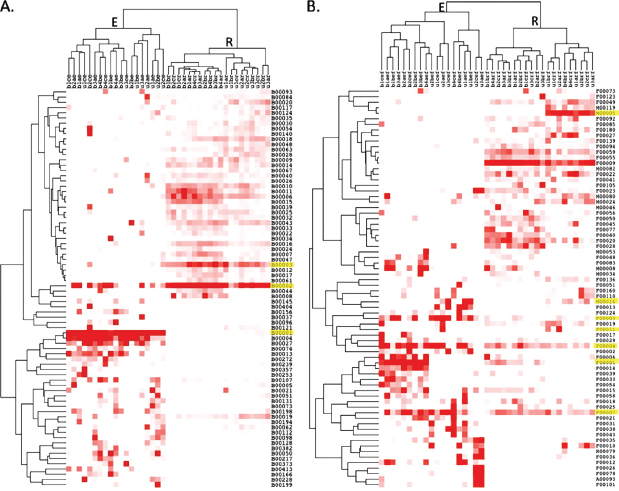
FIGURE A17-6 Heat map and hierarchical cluster analysis based on the relative abundances of the top OTUs identified in >5 samples in the bacterial (A) and fungal (B) data sets. Cluster analysis completely separated OTU abundance by endophyte or rhizosphere origin. OTUs highlighted in yellow are discussed in the text. For classification details of the OTUs depicted here, see Table S4 in the supplemental material.
greenhouse cuttings (Moore et al., 2006; van der Lelie et al., 2009) or shortrotation, plantation-grown trees (Stefani et al., 2009; van der Lelie et al., 2009). To our knowledge no studies have comprehensively examined the bacterial and fungal communities simultaneously in the same sample sets for any part of the Populus microbiome, and few have employed culture-independent methods. In an attempt to disentangle the root endophytic and rhizospheric Populus-associated microbial communities, we used 454 pyrosequencing surveys to characterize the bacterial and fungal communities of P. deltoides trees in upland and bottomland sites along the Caney Fork River in Tennessee. These sites differed significantly in both soil and stand characteristics (Table A17-1). In this study, both the fungal and bacterial communities were described from the same DNA extractions: the paired assessments of rhizospheric (consisting of adhering soils washed from the roots) and endophytic populations (extracted from surface-sterilized roots) were obtained from the same root systems and samples. Our results suggest that the diversity and composition of the associated microbial communities are largely consistent regardless of the differences in host trees and soil physicochemical characteristics associated with the two upland and bottomland sites studied and that the rhizosphere and endophyte communities are largely independent with little overlap in the dominant phyla or OTUs. These findings contradict previous reports that indicated soil properties are a major driver of differences in the distributions and compositions of microbial communities (Buyer et al., 2002; Jesus et al., 2010).
Variability in Rhizosphere and Endosphere Populations of P. deltoides
The number of OTUs in the bacterial endophytic samples (83 ± 78) was much lower than in the rhizosphere (1,319 ± 99) and more variable from sample to sample. The bacterial endophytic samples with the most diversity were an order of magnitude less diverse than the most diverse rhizosphere samples at similar sequencing depths (Figure A17-1). It is possible that this was caused by sporadic and nonuniform colonization of Populus roots by rhizosphere bacteria, which could contribute to the observed high variability. However, this variation may also have arisen partially from our failure to have sequenced the bacterial endophyte community deeply and uniformly. As seen in the rarefaction curves, many endophytic samples had an order of magnitude fewer reads than their counterpart rhizosphere samples. This was due to the high levels of plastid and mitochondrial 16S sequences within the amplified community DNA that were removed from the analysis, including ~67,000 mitochondrial and ~65,266 plastid rRNA gene sequences. The fungal data set was not impacted by this problem (Figure A17-2), as we only found low numbers of fungal reads that were identified as originating from the Populus host tree, or of other soil eukaryotes, such as nematodes. In contrast to the bacterial endophyte rarefaction curves, the fungal endophyte curves exhibited many more OTUs at comparable levels
of sampling effort/sequencing depth and much less variability from sample to sample. However, high-throughput sequencing of fungal rRNA genes currently has far less support than bacterial and archaeal rRNA pyrosequencing based on previous studies, databases, and robust alignments that account for secondary structure. Previous fungal studies typically sequenced the ITS regions (Buee et al., 2009; Jumpponen et al., 2010) or the 18S region (Rousk et al., 2010) of the rRNA genes. The ITS regions are hypervariable, which prevents sequence alignment across the breadth of the fungi impossible, and therefore the identity of many uncultured and underdocumented fungi cannot be determined accurately. Additionally, many community comparison methods based on alignments and phylogenetic methods (e.g., UniFrac) are not possible without alignments. Conversely, 18S regions are often highly conserved and prevent identification past the family level. The D1/D2 region of the 28S LSU rRNA gene was targeted in this study in order to identify known and unknown fungal organisms, even at deep phylogenic branch points (Porter et al., 2008; Schadt et al., 2003), while also enabling alignment and phylogeny-based community analysis methods.
Higher-Order Phylogenetic Composition of Endophyte Versus Rhizosphere Populations of P. deltoides in Contrasting Soils
Rhizosphere bacterial samples were dominated by Acidobacteria and Proteobacteria in both soil types, both of which are common phyla recovered from soil sequencing surveys (Castro et al., 2010; Janssen, 2006; Jesus et al., 2010) (Figure A17-3). The ratio of Proteobacteria to Acidobacteria has been suggested to be an indicator of the trophic level of soils (Castro et al., 2010; Smit et al., 2001), favoring Proteobacteria in nutrient-rich soils and Acidobacteria in nutrient-poor soils. Selection for Proteobacteria in the rhizosphere of Trifolium repens and Lolium perenne has been attributed to the nutrient-rich conditions of the rhizosphere (Marilley and Aragno, 1999), and similar results have been obtained in other plant systems, such as maize (Sanguin et al., 2006), soybean (Xu et al., 2009), and grasslands (Singh et al., 2007). However, in other woody species, such as chestnut trees (Lee et al., 2008) and black spruce (Filion et al., 2004), Acidobacteria have been shown to dominate rhizosphere systems. In our study, Proteobacteria were only slightly more prevalent (43% of sequences) than the Acidobacteria (38%), which may indicate a level intermediate between the copiotrophic and oligotrophic conditions thought to characterize the preferred habitats of these groups. However, numerous other factors may be driving these ratios. Acidobacterial relative abundance in soils has also been shown to correlate with soil pH (Lauber et al., 2009) and relative moisture (Castro et al., 2010). The pH of the bulk soils in the upland site was a full unit higher than the bottomland (Table A17-1), yet on average the relative abundance of Acidobacteria was slightly greater in the upland samples (41%) than in the bottomland (35%). However, we did not measure the pH of the rhizosphere directly to determine
whether it was different than that of the bulk soil, and it is well-established that some plants modify the rhizosphere pH (Hinsinger et al., 2003).
Populus is unusual among most higher plants because it can associate with both endomycorrhizal Glomeromycete fungi as well as ectomycorrhizal fungi in the Ascomycotina and Basidiomycotina (Luckac et al., 2003; Vozzo and Hackayl, 1974). The higher-level fungal communities observed in our study clearly differ between the rhizosphere and endosphere, even at broad taxonomic levels, and in some cases between the upland and bottomland locations (Figure A17-4). The differences between upland and bottomland communities are primarily due to large numbers of an individual OTU (F00001), similar to basidiomycetous yeasts from the Pucciniomycotina, which dominated most bottomland endophyte samples, comprising on average 10% (Figure A17-6). Using traditional ITS cloning techniques targeting the ECM fungi in transgenic plantation-grown Populus, Stefani et al. (Stefani et al., 2009) did not find any differences among the fungal communities of transformed and untransformed Populus deltoides × P. trichocarpa hybrids. They found a high frequency of ECM fungi in the root tips but only recovered about 42 OTUs from the root tips and 58 OTUs from soil cloning (at a sequence similarity of 98%), of which 39 and 26, respectively, were ECM fungi (Stefani et al., 2009). These OTU values are surprisingly low compared to other fungal surveys and to our study of mature natural stands. However, it is likely that the Stefani et al. study also undersampled OTUs, as they used a much lower sequencing depth, which is inherent in clone-based analyses. Jumpponen et al., using ITS pyrosequencing, recovered 1,077 OTUs (at 95% sequence similarity) from Quercus spp. roots (Jumpponen et al., 2010). The number of OTUs recovered from Populus roots in our study was 298 ± 90 (95% sequence similarity). Buée et al. observed a comparable number of fungal OTUs (600 to 1,000) in a survey of forest soils (Buee et al., 2009) as we found in our survey of rhizosphere soils (1,036 ± 278), and they observed some differences in OTU number and composition based on soil type. Our OTU numbers were also lower than estimates from the clone-based study of Fierer et al. of rainforest soils (1,000 to 2,000 OTUs) (Fierer et al., 2007). Many of the difficulties in comparing such OTU levels stem not only from different sampling/sequencing depths but also the inherent differences in variability between the ITS, SSU, and LSU rRNA gene regions employed and differing opinions on OTU cutoff similarities. Our analysis used a conservative 95% similarity cutoff for the D1 region of the LSU, which may have led to lower OTU numbers.
Several authors have proposed that soil type is one of the major drivers of rhizosphere microbial communities (Bachmann and Kinzel 1992; Hinsinger et al., 2009), while other authors have suggested that the plant growth stage can shape the rhizosphere microbial communities (Chiarini et al., 1998; Hinsinger et al., 2009). Our limited study of two contrasting soil types showed that both bacterial and fungal communities did not differ significantly in higher-order composition in either the rhizosphere or endosphere environment. In addition to
divergent soil properties, the upland and bottomland sites had statistically different tree sizes and age classes (Table A17-1), but we did not see major differences in community composition between these sites. Taken together our results suggest that the presence of Populus trees has a dominant effect over other factors in determining overall microbial community patterns in the rhizosphere as well as the endosphere. However, additional studies that incorporate diverse Populus genotypes and development stages as well as broader sets of soil conditions and cooccurring tree species would be required to fully understand and enumerate the effects of such factors.
Is the Root Endosphere Community a Subset of the Rhizosphere Community?
It has been speculated that endophytic root bacterial communities comprise a subset of colonists originating from the surrounding rhizosphere soil (Cocking 2003; Hallmann et al., 1997), and the resulting community composition is affected by the surrounding soil and environmental properties. Therefore, if endophytes are mostly facultative rhizosphere organisms and/or accidental passengers within the root, then the rhizosphere and endosphere will have similar overall patterns of dominant phylogenetic groups and OTU abundance patterns. However, this pattern was not observed in our data. The higher-order classifications of both communities differed in the abundance of major phyla (Figure A17-3 and Biro and Takacs, 2007), and perhaps more tellingly, the phylogeny-based UniFrac PCoA (Figure A17-5) and the OTU-based cluster analysis (Figure A17-6) differed dramatically between these two plant-associated environments. Considering that each of the endosphere and rhizosphere sample pairs were derived from the same root collections, this pattern is especially striking.
Previous reports have shown that endophytic communities are dominated by Gammaproteobacteria, followed by Betaproteobacteria and Alphaproteobacteria (van der Lelie et al., 2009). These studies were based primarily on culture-dependent isolation techniques (Moore et al., 2006; Taghavi et al., 2009). Our pyrosequencing surveys had a similarly high abundance of Gammaproteobacteria, but with a greater number of Alphaproteobacteria than Betaproteobacteria (Figure A17-3). These discrepancies could be due to differences between culture-dependent and -independent methods or because previous knowledge of endosphere and rhizosphere communities has often originated from entirely separate samples or studies. Perhaps we were able to clearly observe differences between these communities because the endosphere and rhizosphere populations originated from the same root samples or because of the more comprehensive analyses enabled by pyrosequencing. With regard to the dominant endophytes of Populus, many studies have shown that the Gammaproteobacteria dominates (49). In our study of natural populations and previous culture-dependent studies, dominant bacterial endophytes were often Pseudomonas spp. One OTU (B00001) attributable to a P. fluorescens-like organism dominated both upland (33.8%) and
bottomland (34.6%) endophyte samples but accounted for small fractions in the rhizosphere (0.2 and 0.08% of upland and bottomland, respectively). A similar disparity between endosphere and rhizosphere abundances can be seen across many of the OTUs in Figure A17-6. Another study in the presence of trichloroethylene (TCE) showed the dominant Gammaproteobacteria to be Serratia spp. in culture-based assessments (Xu et al., 2009). Serratia-like OTUs were exceedingly uncommon in our study and comprised <0.001% of the data set. Ulrich et al. reported differing endophytic phyllosphere communities across various genotypes of plantation-grown Populus in Europe (Ulrich et al., 2008) but did not examine root or rhizosphere communities.
The number of OTUs attributable to ECM and other mycorrhizal fungi was lower than expected in our samples, and some ongoing studies suggest P. deltoides is only weakly ectomycorrhizal compared to other Populus species and hybrids or compared to other tree species, such as oak and pine (F. Martin and R. Vilgalys, unpublished data). A study by Stefani et al. (2009) of plantation-grown Populus hybrids found differences in the rank abundance of different OTUs identified within roots and in bulk soils but also observed that over half of the ECM OTUs cooccurred in both habitats, even at the low sequencing depths achievable with clone library examinations. However, it is unclear how the Stephani et al. sampling strategy would compare with the surface-sterilized endophyte and rhizosphere sampling methods employed in our study. ECM OTUs were also observed in both the rhizosphere and endophyte samples in our study. For example, OTU F00003 is attributable to a Cortinarius-like organism (Basidiomycotina) and occurred at an average frequency of 4.7% in endosphere samples but only at 0.2% in the rhizosphere (Figure A17-6). Conversely, OTU F00011 was dominant in the rhizosphere and endosphere of several samples and attributable to a Tuber-like ECM organism (Ascomycotina).
The uniqueness of rhizosphere root endophyte communities is illustrated in the hierarchical clustering-based analysis of the dominant OTUs from each environment (Figure A17-6). With the exception of a few OTUs, most are abundant in either the endophyte or rhizosphere samples, but not both. Of the OTUs that are prominent in both environments, most are fungal. For example, OTUs F00007 and F00004 were attributable to a Neonectria-like species and a Veronia-like species, present in high abundance in both the endophyte and rhizosphere samples. These Ascomycete genera are known as plant-associated pathogens; however, many species have unknown effects on plant growth (Rossman et al., 1999). The most dominant bacterial rhizosphere OTU (B00002) was a Bradyrhizobium-like organism that was also sporadically present in the endosphere. OTU B0003 is the second most dominant across rhizospheric environments and classified as Chloroflexi-like. Chloroflexi are common soil organisms but have not been reported as plant endophytes. Some nonfungal metazoan taxa (e.g., nematodes) present in the fungal data set have distinct OTU distribution patterns. For example, OTU M00005 represents a Steinernema-like nematode that occurs in the
rhizosphere, whereas M00016 represents a Hoploimus-like nematode prominent in the endosphere. While such patterns of species partitioning in rhizosphere and endosphere habitats are preliminary, considering the small number of environments sampled, they begin to suggest that segregating of the myriad of soil niches may be possible with further dissection and application of deep pyrosequencing techniques.
Conclusions
At broad taxonomic levels as well as the individual OTU level, rhizosphere and endophyte communities of both bacteria and fungi associated with native P. deltoides are clearly distinct, suggesting that the tissues within naturally occurring Populus roots represent a unique niche for microbial communities. There appears to be little variation in dominant phyla within rhizosphere and endophyte habitats between the two soils and ecotypes of Populus examined thus far. Future work that includes more diverse soil types and the analysis of the specific effects of host genotype and chemical phenotypes should further elucidate the relative effects of environment and host factors in microbial associations with Populus.
Acknowledgments
We thank S. Retterer, A. Gorin, D. Weston, C. Pan, and L. Gunter for their assistance in conducting field and lab work. We also thank the Army Corps of Engineers and the Tennessee Parks Department for access to sampling sites.
This research was sponsored by the Genomic Science Program, U.S. Department of Energy, Office of Science, Biological and Environmental Research, as part of the Plant Microbe Interfaces Scientific Focus Area (http://pmi.ornl.gov). Oak Ridge National Laboratory is managed by UT-Battelle LLC, for the U.S. Department of Energy under contract DE-AC05-00OR22725.
The submitted manuscript has been authored by a contractor of the U.S. Government under contract DE-AC05-00OR22725.
References
Amann, R. I., W. Ludwig, and K. H. Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143–169.
Bachmann, G., and H. Kinzel. 1992. Physiological and ecological aspects of the interactions between plant-roots and rhizosphere soil. Soil Biol. Biochem. 24:543–552.
Barns, S. M., E. C. Cain, L. Sommerville, and C. R. Kuske. 2007. Acidobactetia phylum sequences in uranium-contaminated subsurface sediments greatly expand the known diversity within the phylum. Appl. Environ. Microbiol. 73:3113–3116.
Biro, I., and T. Takacs. 2007. Study of heavy metal uptake of Populus nigra in relation to phytoremediation. Cereal Res. Commun. 35:265–268.
Buee, M., et al. 2009. 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol. 184:449–456.
Buyer, J. S., D. P. Roberts, and E. Russek-Cohen. 2002. Soil and plant effects on microbial community structure. Can. J. Microbiol. 48:955–964.
Castro, H. F., A. T. Classen, E. E. Austin, R. J. Norby, and C. W. Schadt. 2010. Soil microbial community responses to multiple experimental climate change drivers. Appl. Environ. Microbiol. 76:999–1007.
Chiarini, L., A. Bevivino, C. Dalmastri, C. Nacamulli, and S. Tabacchioni. 1998. Influence of plant development, cultivar and soil type on microbial colonization of maize roots. Appl. Soil Ecol. 8:11–18.
Cocking, E. C. 2003. Endophytic colonization of plant roots by nitrogenfixing bacteria. Plant Soil 252:169–175.
Cole, J. R., et al. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–D145.
Eisen, M. B., P. T. Spellman, P. O. Brown, and D. Botstein. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U. S. A. 95:14863–14868.
Fierer, N., et al. 2007. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microbiol. 73:7059–7066.
Filion, M., R. C. Hamelin, L. Bernier, and M. St. Arnaud. 2004. Molecular profiling of rhizosphere microbial communities associated with healthy and diseased black spruce (Picea mariana) seedlings grown in a nursery. Appl. Environ. Microbiol. 70:3541–3551.
Gehring, C. A., R. C. Mueller, and T. G. Whitham. 2006. Environmental and genetic effects on the formation of ectomycorrhizal and arbuscular mycorrhizal associations in cottonwoods. Oecologia 149:158–164.
Hallmann, J., A. Quadt-Hallmann, W. F. Mahaffee, and J. W. Kloepper. 1997. Bacterial endophytes in agricultural crops. Can. J. Microbiol. 43:895–914.
Hamady, M., C. Lozupone, and R. Knight. 2010. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 4:17–27.
Hardoim, P. R., L. S. van Overbeek, and J. D. van Elsas. 2008. Properties of bacterial endophytes and their proposed role in plant growth. Trends Microbiol. 16:463–471.
Hinsinger, P., A. G. Bengough, D. Vetterlein, and I. M. Young. 2009. Rhizosphere: biophysics, biogeochemistry and ecological relevance. Plant Soil. 321:117–152.
Hinsinger, P., C. Plassard, C. X. Tang, and B. Jaillard. 2003. Origins of root-mediated pH changes in the rhizosphere and their responses to environmental constraints: a review. Plant Soil 248:43–59.
Huse, S. M., D. M. Welch, H. G. Morrison, and M. L. Sogin. 2010. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 12:1889–1898.
Janssen, P. H. 2006. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 72:1719–1728.
Jesus, E. D., et al. 2010. Bacterial communities in the rhizosphere of biofuel crops grown on marginal lands as evaluated by 16S rRNA gene pyrosequences. Bioenerg. Res. 3:20–27.
Jones, J. D. G., and J. L. Dangl. 2006. The plant immune system. Nature 444:323–329.
Jumpponen, A., K. L. Jones, D. Mattox, and C. Yaege. 2010. Massively parallel 454-sequencing of fungal communities in Quercus spp. ectomycorrhizas indicates seasonal dynamics in urban and rural sites. Mol. Ecol. 19: 41–53.
Kent, A. D., and E. W. Triplett. 2002. Microbial communities and their interactions in soil and rhizosphere ecosystems. Annu. Rev. Microbiol. 56: 211–236.
Kunin, V., A. Engelbrektson, H. Ochman, and P. Hugenholtz. 2010. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12:118–123.
Lauber, C. L., M. Hamady, R. Knight, and N. Fierer. 2009. Pyrosequencingbased assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 75:5111–5120.
Lee, S. H., J. O. Ka, and J. C. Cho. 2008. Members of the phylum Acidobacteria are dominant and metabolically active in rhizosphere soil. FEMS Microbiol. Lett. 285:263–269.
Lozupone, C., M. Hamady, and R. Knight. 2006. UniFrac: an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7:371.
Lukac, M., C. Calfapietra, and D. L. Godbold. 2003. Production, turnover and mycorrhizal colonization of root systems of three Populus species grown under elevated CO2 (POPFACE). Global Change Biol. 9:838–848.
Marilley, L., and M. Aragno. 1999. Phylogenetic diversity of bacterial communities differing in degree of proximity of Lolium perenne and Trifolium repens roots. Appl. Soil Ecol. 13:127–136.
Meyer, F., et al. 2008. The metagenomics RAST server: a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9:386.
Moore, F. P., et al. 2006. Endophytic bacterial diversity in poplar trees growing on a BTEX-contaminated site: the characterisation of isolates with potential to enhance phytoreniediation. Syst. Appl. Microbiol. 29:539–556.
Porter, T. M., et al. 2008. Widespread occurrence and phylogenetic placement of a soil clone group adds a prominent new branch to the fungal tree of life. Mol. Phylogenet. Evol. 46:635–644.
Pruesse, E., et al. 2007. SILVA: a comprehensive online resource for quality checked and aligned rRNA sequence data compatible with ARB. Nucleic Acids Res. 35:7188–7196.
Rossman, A. Y., G. J. Samuels, C. T. Rogerson, and R. Lowen. 1999. Genera of Bionectriaceae, Hypocreaceae and Nectriaceae (Hypocreales, Ascomycetes). Stud. Mycol. 42:1–248.
Rousk, J., et al. 2010. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4:1340–1351.
Sanguin, H., et al. 2006. Potential of a 16S rRNA-based taxonomic microarray for analyzing the rhizosphere effects of maize on Agrobacterium spp. and bacterial communities. Appl. Environ. Microbiol. 72:4302–4312.
Sannigrahi, P., A. J. Ragauskas, and G. A. Tuskan. 2010. Poplar as a feedstock for biofuels: a review of compositional characteristics. Biofuels Bioprod. Bior. 4:209–226.
Schadt, C. W., A. P. Martin, D. A. Lipson, and S. K. Schmidt. 2003. Seasonal dynamics of previously unknown fungal lineages in tundra soils. Science 301:1359–1361.
Schloss, P. D., et al. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541.
Singh, B. K., S. Munro, J. M. Potts, and P. Millard. 2007. Influence of grass species and soil type on rhizosphere microbial community structure in grassland soils. Appl. Soil Ecol. 36:147–155.
Smit, E., et al. 2001. Diversity and seasonal fluctuations of the dominant members of the bacterial soil community in a wheat field as determined by cultivation and molecular methods. Appl. Environ. Microbiol. 67:2284–2291.
Stefani, F. O. P., J. M. Moncalvo, A. Seguin, J. A. Berube, and R. C. Hamelin. 2009. Impact of an 8-year-old transgenic poplar plantation on the ectomycorrhizal fungal community. Appl. Environ. Microbiol. 75:7527–7536.
Taghavi, S., et al. 2009. Genome survey and characterization of endophytic bacteria exhibiting a beneficial effect on growth and development of poplar trees. Appl. Environ. Microbiol. 75:748–757.
Tuskan, G., et al. 2004. The Populus genome: are there discernable differences between the genomes of perennial woody plants and herbaceous annuals? Acta Physiol. Plant 26:292–293.
Tuskan, G. A., et al. 2006. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science 313:1596–1604.
Ulrich, K., A. Ulrich, and D. Ewald. 2008. Diversity of endophytic bacterial communities in poplar grown under field conditions. FEMS Microbiol. Ecol. 63:169–180.























