
van der Lelie, D., et al. 2009. Poplar and its bacterial endophytes: coexistence and harmony. Crit. Rev. Plant Sci. 28:346–358.
Vishnivetskaya, T. A., et al. 2011. Mercury and other heavy metals influence bacterial community structure in low-order Tennessee streams. Appl. Environ. Microbiol. 77:302–311.
Vozzo, J. A., and E. Hacskayl. 1974. Endomycorrhizal and ectomycorrhizal associations in 5 Populus species. B Torrey Bot. Club 101:182–186.
Xu, Y. X., et al. 2009. Bacterial communities in soybean rhizosphere in response to soil type, soybean genotype, and their growth stage. Soil Biol. Biochem. 41:919–925.
INTERACTIONS BETWEEN COMMENSAL FUNGI AND THE C-TYPE LECTIN RECEPTOR DECTIN-1 INFLUENCE COLITIS78
Iliyan D. Iliev,79Vincent A. Funari,80,81Kent D. Taylor,80Quoclinh Nguyen,80Christopher N. Reyes,79Samuel P. Strom,80Jordan Brown,80Courtney A. Becker,79Phillip R. Fleshner,82Marla Dubinsky,79,83Jerome I. Rotter,80Hanlin L. Wang,84Dermot P. B. McGovern,79,80Gordon D. Brown,85and David M. Underhill 79,84,86,*
Abstract
The intestinal microflora, typically equated with bacteria, influences diseases such as obesity and inflammatory bowel disease (IBD). Here we show that the mammalian gut contains a rich fungal community that interacts with the immune system through the innate immune receptor Dectin-1. Mice lacking
________________
78 From Iliev, I. D., et al. 2012. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336(6086):1314-1317. Reprinted with permission from AAAS.
79 Inflammatory Bowel and Immunobiology Research Institute, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA.
80 Medical Genetics Institute, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA.
81 Department of Pediatrics, David Geffen School of Medicine, University of California, Los Angeles, CA 90095, USA.
82 Department of Surgery, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA.
83 Department of Pediatrics, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA.
84 Department of Pathology and Laboratory Medicine, David Geffen School of Medicine, University of California, Los Angeles, CA 90095, USA.
85 Section of Immunology and Infection, Division of Applied Medicine, Institute of Medical Sciences & The Aberdeen Fungal Group, University of Aberdeen, Aberdeen, UK.
86 Research Division of Immunology, Cedars-Sinai Medical Center, Los Angeles, CA 90048, USA.
* Correspondence to: David M. Underhill, Inflammatory Bowel & Immunobiology Research Institute, Cedars-Sinai Medical Center, 8700 Beverly Blvd, Los Angeles, CA 90048, Email: David. Underhill@csmc.edu.
Dectin-1 exhibited increased susceptibility to chemically-induced colitis, which was the result of altered responses to indigenous fungi. In humans we identified a polymorphism in the gene for Dectin-1 (CLEC7A) that is strongly linked to a severe form of ulcerative colitis. Together our findings reveal a novel eukaryotic fungal community in the gut (the “mycobiome”) that coexists with bacteria and substantially expands the repertoire of organisms interacting with the intestinal immune system to influence health and disease.
Interactions between the commensal microflora and the gut immune system are critical for establishing a balance between immunity and tissue health. Changes in gut bacteria described as “dysbiosis” have been associated with intestinal inflammation (Elinav et al., 2011; Lupp et al., 2007; Willing et al., 2010) and metabolic syndrome (Arumugam et al., 2011; Henao-Mejia et al., 2012; Vijay-Kumar, et al., 2010). The vast majority of studies on commensal microbiota have focused on gut bacteria, and the terms “intestinal microbiota” and “intestinal bacteria” are often used interchangeably. Recent studies have begun to note, however, that a fraction of gut microorganisms are not bacterial (Qin et al., 2010). Although a few studies have suggested the presence of commensal fungi in the gut (Ott et al., 2008; Qin et al., 2010), whether they interact with the mucosal immune system or influence diseases is unknown.
Fungi are recognized by a number of immune receptors among which Dectin-1 has emerged as key for phagocytosis and killing by myeloid phagocytes. Dectin-1 is a C-type lectin receptor that recognizes β-1,3-glucans found in the cell walls of nearly all fungi. Dectin-1 activates intracellular signals through caspase recruitment domain–containing protein 9 (CARD9), which leads to inflammatory cytokine production and induction of T helper 17 (TH17) immune responses (Cheng et al., 2011; Conti et al., 2009; Gringhuis et al., 2012; LeibundGut-Ladmann et al., 2007). Deficiencies in either Dectin-1 or CARD9 result in enhanced susceptibility to pathogenic fungal infections in humans and mice (Ferwada, et al., 2009; Glocker et al., 2009; Taylor et al, 2007). Polymorphic variants in the gene for CARD9 are strongly associated with Crohn’s disease and ulcerative colitis in humans (Franke et al., 2010; McGovern et al., 2010). Furthermore, anti–Saccharomyces cerevisiae antibodies (ASCA) against yeast mannan have been strongly associated with Crohn’s disease (Joossens et al., 2002; Seow et al., 2009). Together, these last-named findings suggest a possible link between immune responses to commensal fungi and intestinal disease.
We examined fungal distribution and detected fungal ribosomal DNA (rDNA) throughout the murine gastrointestinal tract with highest densities in the terminal colon of C57BL/6 (Figure A18-1A) and 129S2/Sv (fig. S1A) mice. We stained colonic tissue sections and observed that fungi are abundant and in close proximity with commensal bacteria (Figure A18-1B and figs. S1B and S2 to S4). Furthermore, we found that a soluble Dectin-1 probe (Ganter et al., 2005) binds to 5 to 7% of the fecal material, consisting of fungal cells with various
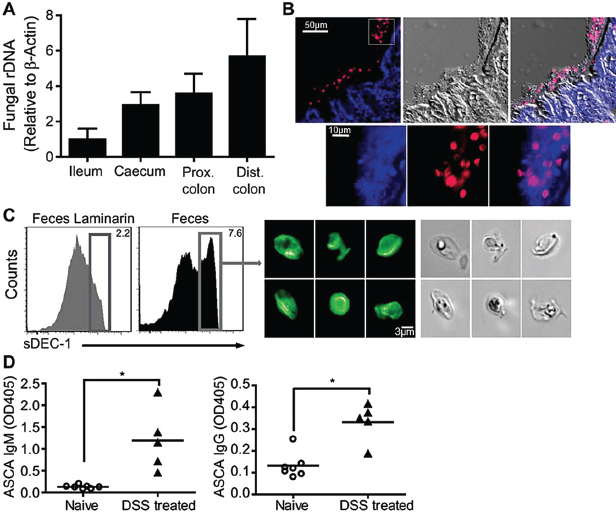
FIGURE A18-1 Commensal fungi are present in the intestine and are recognized by Dectin-1. (A) Prevalence of fungi in mucosa isolated from ileum, cecum, proximal (prox) and distal (dist) colon of C57BL/6J mice. ITS1-2 rDNA level was analyzed by quantitative polymerase chain reaction and normalized to β-actin DNA. (B) Visualization of commensal fungi in the intestine. Colon sections were stained with a soluble Dectin-1 probe (sDEC-1) and counterstained with 4′, 6′-diamidino-2-phenylindole (DAPI). The DAPI signal has been amplified in (B, bottom) to show that DAPI-stained bacteria and fungi are in close proximity to each other. (C) Intestinal fungi are recognized by Dectin-1. Fecal pellets were homogenized and labeled with sDEC-1 in the presence (gray histogram) or absence (black histogram) of laminarin (a soluble β-glucan) to block specific binding. Binding was assessed by flow cytometry (left panels). Dectin-1–binding fungi were sorted (right) and visualized by confocal microscopy. (D) ASCA generation after DSS colitis. Mice were exposed twice to 2.5% DSS-supplemented water for 7 days each separated by 2 weeks of recovery. Serum samples were collected before DSS treatment (day 0) and 2 weeks after the last DSS cycle (42 days total), and ASCA IgM and IgG were measured by enzyme-linked immunosorbent assay (ELISA). Each symbol represents a mouse, all error bars indicate the SD; unpaired t test, *P < 0.05. All data are representative of at least two independent experiments with similar results.
morphologies (Figure A18-1C and fig. S5). Fungi were also present in rat, guinea pig, rabbit, pig, dog, and human feces (fig. S1C). Together, the data demonstrate that commensal fungi contribute to the intestinal microbial community in many species.
We next examined whether gut fungi can be detected by the immune system upon intestinal insult. We utilized a mouse model of dextran sodium sulfate (DSS)–induced colitis extended to allow antibody responses to develop. We found that DSS-induced intestinal inflammation led to the development of circulating immunoglobulin G (IgG) and IgM antibodies against fungi (ASCA) (Figure A18-1D), which suggested that fungal antigens indigenous to the gut might be responsible for the induction of ASCA during colitis.
Because gut commensal fungi are recognized by Dectin-1, we tested whether Dectin-1–deficient mice (Clec7a−⁄−) are susceptible to DSS-induced colitis. Clec7a−⁄− mice experienced increased weight loss (Figure A18-2A) and displayed altered histology characterized by increased mucosal erosion, crypt destruction, inflammatory cell infiltration, and tumor necrosis factor–α (TNF-α) production in the colon (Figure A18-2, B to D) as compared with their wild-type (WT) littermate controls. We further detected augmented production of interferon-γ (IFN-γ) and interleukin-17 (IL-17) in intestines from Clec7a−⁄− mice (fig. S6). Similar results were obtained comparing cohoused animals (fig. S7). These results indicate that Dectin-1 deficiency leads to increased susceptibility to colitis.
Many studies have documented the importance of bacteria in intestinal inflammation, so we examined whether bacteria could contribute to the susceptible phenotype. We observed no significant differences in major phyla of commensal bacteria between WT and Clec7a−⁄− mice (fig. S8). To directly determine whether microflora can transfer disease, we depleted intestinal bacteria and fungi with antibiotics, transplanted fecal microflora from WT or Clec7a−⁄− mice, and exposed mice to DSS. Microflora from Clec7a−⁄− mice did not transfer susceptibility to disease (Figure A18-2, E and F, and figs. S9 and S10). The data demonstrate that the disease phenotype in the Clec7a−⁄− mice is affected by the genotype of the mouse, not by initial differences in microflora.
We know very little about what commensal fungi populate the murine gut or how they contribute to colitis in Dectin-1 deficiency. To define the mouse intestinal fungal microbiome, we isolated DNA from murine feces, amplified the internal transcribed spacer region (ITS1-2) of fungal rDNA, and performed high-throughput sequencing (Ghannoum et al., 2010). Combining data from 23 mice, we obtained >30 Mb of raw data from 454 pyrosequencing and >2.2 Gb of raw data from Illumina genome analyzer (GA) sequencing, together containing >1.3 million individual sequences that passed quality control. Detailed analysis identified >100 different well-annotated fungal species representing at least 50 genera, which illustrated the fungal diversity. In addition, we identified >100 novel and/or unannotated fungi representing the large uncharacterized nature of the mycobiome in the gut (figs. S11 and S12). Note that 97.3% of all the fungal
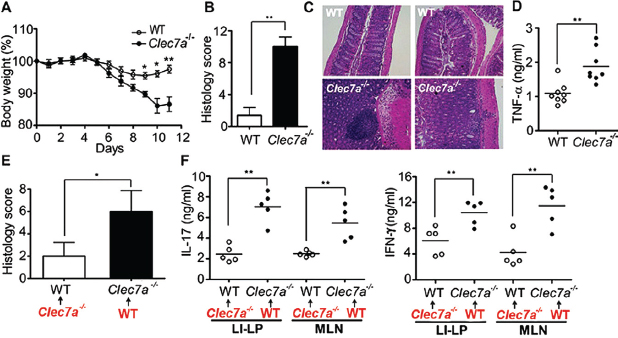
FIGURE A18-2 Dectin-1 regulates the severity of colitis. WT and Clec7a−⁄− littermates were treated with 2.5% DSS for 7 days and kept on water for 4 additional days. Colitis progress and severity were assessed by measuring body weight during treatment (A), histology (B and C), and TNF-α production in the colon (D) on day 11. (E and F) WT and Clec7a−⁄− mice were given an antibiotic cocktail including fluconazole for 3 weeks, given a transplant as indicated (red) with fecal microflora from WT or Clec7a−⁄− mice, and treated with DSS as in (A). Disease severity was accessed by histology score (E) and by cytokine production stimulated by antibodies against CD3 and CD28 in large-intestine lamina propria (LI-LP) and MLN T cells (F). Each symbol represents a different mouse. One of four independent experiments is shown. Error bars, SD; *P < 0.05, **P < 0.01.
sequences identified belonged to 10 fungal species, with 65.2% of the sequences belonging to a single fungus: Candida tropicalis (Figure A18-3A and fig. S13). We found 7 of the 20 most common gut fungi also in mouse food (fig. S13 and S14). These accounted, however, for only 1.5% of total fungi in the intestines, which suggests that highly represented fungal species are indigenous to the gut.
Many studies have shown that intestinal inflammation can lead to changes in commensal bacteria that affect the host (Garret et al., 2010; Lupp et al., 2007; Willing et al., 2010). Whether colitis affects the makeup of the commensal mycobiome is unknown. One study has reported increased fungal burden in intestines of Crohn’s disease patients (Ott et al., 2008), and another has shown increased colonization with exogenously added C. albicans during DSS colitis in mice (Jawhara et al., 2008). Notably, we found that, during colitis in Clec7a−⁄− mice the proportion of opportunistic pathogenic fungi including Candida and Trichosporon increases, whereas nonpathogenic Saccharomyces decreases (Figure A18-3B and fig. S15). Examination of colons revealed that fungi invade inflamed tissues in DSS-treated Clec7a−⁄− mice but remain in the lumen of DSS-treated WT mice
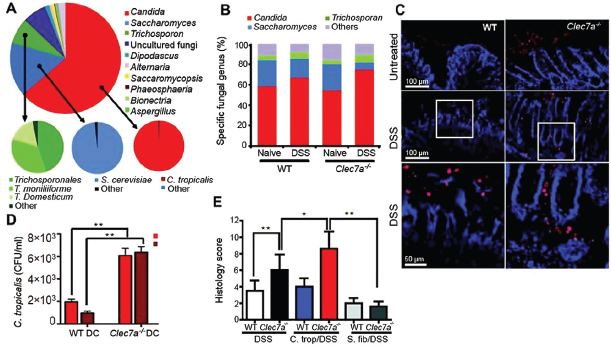
FIGURE A18-3 Defining the fungal microbiome and characterizing the specific role of Dectin-1–mediated host defense during colitis. (A) DNA was isolated from murine feces, and mycobiome analysis was performed using Roche 454 and Illumina GA sequencing of ITS1-2 rDNA. The taxonomic distribution of the most abundant fungal genera is shown (large pie chart), and the species breakdowns for major groups are provided (small pie charts). (B) Quantitative analysis of the major intestinal fungal genera in WT and Clec7a−⁄− mice before and after treatment with DSS. Illumina GA data were analyzed and presented as relative percentage of dominant fungal genera (n =16 mice). (C) Fungal invasion of colonic tissue in Clec7a−⁄− mice during colitis. Colon sections from WT and Clec7a−⁄− mice before and after colitis were stained with the sDEC-1 probe and counterstained with DAPI. (D) Intestinally conditioned dendritic cells were incubated with live C. tropicalis, and killing was assessed after 6 and 18 hours. (E) Histology score of WT and Clec7a−⁄− mice supplemented or not with four doses of C. tropicalis or S. fibuligera every other day, and then treated with 2.5% DSS for 7 days and kept on water for 4 additional days. Data are representative of at least two independent experiments with similar results. Error bars, SD; *P < 0.05, **P < 0.01.
(Figure A18-3C and fig. S16). These data are consistent with the observation that intestinally conditioned Clec7a−⁄− dendritic cells are less capable of killing C. tropicalis in vitro (Figure A18-3D). Together, the data suggest that Dectin-1 deficiency leads to altered immunity to commensal fungi in the gut.
Given that C. tropicalis is an opportunistic pathogen, we further analyzed its role during colitis. We supplemented mice with C. tropicalis and subjected them to DSS. (See fig. S17A for dosing schedule.) For comparison, another group of mice was supplemented with S. fibuligera, a nonpathogenic fungus that, like C. tropicalis, grows in yeast and filamentous forms and is recognized by Dectin-1 (fig. S18). Colitis symptoms including weight loss, crypt loss, and
inflammatory cell infiltration were more severe in Clec7a−⁄− mice supplemented with C. tropicalis compared with the Clec7a−⁄− controls (Figure A18-3E and fig. S17, B and C). In contrast, C. tropicalis supplementation did not aggravate colitis in WT mice. Consistent with the pathology, we detected increased IL-17 and IFN-γ production by T cells from the mesenteric lymph nodes (MLNs) and colons of Clec7a−⁄− mice supplemented with C. tropicalis (fig. S17, D and E), as well as increased message for TNF-α, IL-23p19, IL-17a, Cxcl2, and defensins in colons (fig. S19). This correlated with higher loads of C. tropicalis in the intestines of DSS-treated Clec7a−⁄− mice (fig. S20B). In contrast, S. fibuligera supplementation did not contribute to colitis pathology (Figure A18-3E and figs. S17 and S19), and fungal loads were unchanged (fig. S20C). The data suggest that an inability of Clec7a−⁄− mice to mount effective immune responses to specific intestinal fungi creates conditions that promote inflammation.
To determine whether the altered fungal burden during colitis contributes to disease severity in the absence of Dectin-1, we suppressed fungal growth with fluconazole, a specific antifungal drug (fig. S3). Fluconazole treatment during colitis led to reduced weight loss (Figure A18-4A), and milder histological disease characteristics (Figure A18-4B), specifically in Clec7a−⁄− mice. We similarly observed decreased TH1 and TH17 responses (Figure A18-4, C and D, and fig. S21, A and B) and decreased production of inflammatory cytokines (fig. S21, C and D). Taken together, these results further support the conclusion that an inability to control fungi in the gut leads to more severe colitis in Dectin-1 knockout mice.
Having established a role for Dectin-1 in fungal control during colitis in mice, we next explored whether there is an association between inflammatory bowel disease (IBD) and genetic variation of the human Dectin-1 gene (CLEC7A). Because the mouse model suggested that Dectin-1 is involved in the severity of colonic disease, we focused our human studies on ulcerative colitis (UC), a disease of the colon, and in particular on severe UC. Up to 30% of patients with UC require colectomy, usually for severe disease that will not respond to medical therapy, including systemic corticosteroids, cyclosporine, and biological therapies [that is, medically refractory UC (MRUC)]. We compared CLEC7A alleles in an MRUC group with those from a group of patients with UC who had not required colectomy (non-MRUC) (Haritunians et al., 2010). We identified an association of CLEC7A single-nucleotide polymorphism rs2078178 in patients with MRUC (logistic regression, P = 0.007). Notably, a two-marker haplotype, rs2078178 to rs16910631, was more strongly associated with MRUC (AG haplotype; logistic regression, P = 0.00013; and Fisher’s test, P = 0.0005) (Figure A18-4E and fig. S22 and table S1), a shorter time to surgery, and thus with a more severe UC (Figure A18-4F). Compared with healthy controls, the haplotype is strongly associated with MRUC and not with non-MRUC, further consistent with the idea that the haplotype is associated with severe disease (table S2). CLEC7A has not been identified in any genome-wide association study yet as an IBD susceptibility
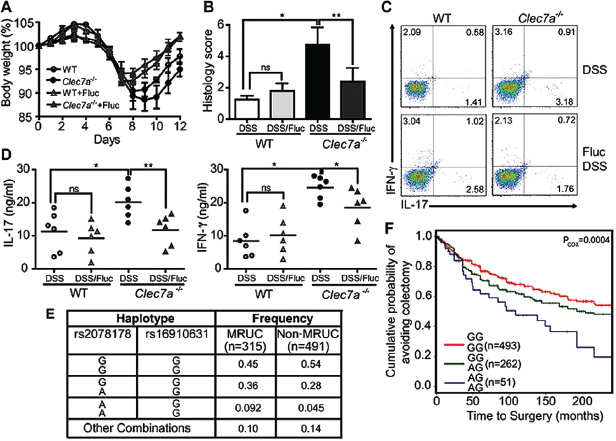
FIGURE A18-4 Anti-fungal therapy ameliorates colitis in Clec7a−⁄− mice and CLEC7A associates with ulcerative colitis severity in humans. (A) WT and Clec7a−⁄− mice were given fluconazole in their drinking water for total of 14 days (starting 2 days prior the induction of DSS colitis), and body weight was measured. Weight loss is shown in (A) (p<0.05). Histology score (B), the percentage of IL-17 and IFN-γ producing CD4+ T cells in LI-LP (C), and IL-17 and IFN-γ production in MLNs (D) were determined 4 days after the 7 days of DSS treatment. Each symbol represents a different mouse. One of three independent experiments with similar results is shown. Error bars, s.d., * P < 0.05, ** P < 0.01. (E) Specific CLEC7A haplotypes associate with medically refractory ulcerative colitis (MRUC). Haplotypes were formed from rs2078178 and rs16910631 using PHASE v2.3. Haplotypes listed as “Other Combinations” were those that could not be reliably determined (posterior p<0.95). (F) The CLEC7A “AG/AG” haplotype associates with severity of disease as indicated by earlier progression to colectomy. Haplotypes were tested for association with time to surgery by fitting the MRUC/non-MRUC and time to surgery with a Cox proportional hazards model.
gene. Unlike susceptibility genes that predispose to disease, severity gene variants aggravate disease that is initially established through other mechanisms. The CLEC7A risk haplotype we report here fits the latter situation and agrees with our observation that Clec7a−⁄− mice do not develop spontaneous colitis. These findings suggest more in-depth studies on the role of the CLEC7A gene and pathway on the natural history of UC and will require further validation in independent cohorts.
A deeper understanding of the mechanisms by which fungi stimulate inflammatory immune responses in the gut may lead to better therapies for IBD and may be especially beneficial to patients with particularly severe forms of UC carrying the risk haplotype of the gene for Dectin-1. Overall, the idea that fungi are present in the gut and that they interact strongly with the immune system will fundamentally alter how we think about the gut microflora and inflammatory bowel diseases.
Acknowledgments
This study was supported in part by the National Institute of Allergy and Infectious Diseases, NIH (AI071116 to D.M.U.) and the Janis and William Wetsman Family Chair in Inflammatory Bowel Disease Research (D.M.U.). I.D.I. held a Research Fellowship Award (3064) from the Crohn’s and Colitis Foundation of America. Further support came from National Institute of Diabetes and Digestive and Kidney Diseases, NIH, grant P01-DK046763; UCLA Clinical and Translational Science Institute grant UL1RR033176; Cedars-Sinai Medical Center Inflammatory Bowel and Immunobiology Research Institute Funds; The Feintech Family Chair in IBD (S. R. Targan); The Cedars-Sinai Board of Governors’ Chair in Medical Genetics (J.I.R.); The Abe and Claire Levine Chair in Pediatric IBD (M.D.); and The Joshua L. and Lisa Z. Greer Chair in IBD Genetics (D.M.). G.D.B. is supported by the Wellcome Trust. Clec7a−⁄− mice are available through a Material Transfer Agreement with the University of Aberdeen. The data presented in this paper are tabulated in the main paper and in the supplementary materials. All sequences generated in this study have been deposited in the National Center for Biotechnology Information, NIH, Short Read Archive (www.ncbi.nlm.nih.gov/Traces/sra, accession no. SRA051853.1). G.D.B. is an advisory board member of MiniVax and Fiberbiotics. M.D. is a consultant for Prometheus Labs.
Supplementary Materials
Visit www.sciencemag.org/cgi/content/full/336/6086/1314/DC1.
References
Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174.
Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335.
Cheng SC, et al. The dectin-1/inflammasome pathway is responsible for the induction of protective T-helper 17 responses that discriminate between yeasts and hyphae of Candida albicans. J Leukoc Biol.2011;90:357.
Chenna R, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res.2003;31:3497.
Conti HR, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299.
Elinav E, et al. NLRP6 Inflammasome Regulates Colonic Microbial Ecology and Risk for Colitis. Cell.2011
Ferwerda B, et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med.2009;361:1760.
Franke A, et al. Genome-wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL) Nat Genet. 2010;42:292.
Fried LP, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263.
Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005;24:1277.
Garrett WS, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292.
Ghannoum MA, et al. Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog. 2010;6:e1000713.
Glocker EO, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727.
Gringhuis SI, et al. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol. 2012
Haritunians T, et al. Genetic predictors of medically refractory ulcerative colitis. Inflamm Bowel Dis.2010;16:1830.
Henao-Mejia J, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179.
Iliev ID, Mileti E, Matteoli G, Chieppa M, Rescigno M. Intestinal epithelial cells promote colitis-protective regulatory T-cell differentiation through dendritic cell conditioning. Mucosal Immunol.2009;2:340.
Jawhara S, et al. Colonization of mice by Candida albicans is promoted by chemically induced colitis and augments inflammatory responses through galectin-3. J Infect Dis. 2008;197:972.
Joossens S, et al. The value of serologic markers in indeterminate colitis: a prospective follow-up study. Gastroenterology. 2002;122:1242.
Kelsall B. Recent progress in understanding the phenotype and function of intestinal dendritic cells and macrophages. Mucosal Immunol. 2008;1:460.
Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656.
Landers CJ, et al. Selected loss of tolerance evidenced by Crohn’s disease-associated immune responses to auto- and microbial antigens. Gastroenterology. 2002;123:689.
LeibundGut-Landmann S, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630.
Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078.
Liu Z, DeSantis TZ, Andersen GL, Knight R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res. 2008;36:e120.
Lupp C, et al. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2:204.
Matteoli G, et al. Gut CD103+ dendritic cells express indoleamine 2,3-dioxygenase which influences T regulatory/T effector cell balance and oral tolerance induction. Gut. 2010;59:595.
McGovern DP, et al. Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat Genet. 2010;42:332.
Nilsson RH, Bok G, Ryberg M, Kristiansson E, Hallenberg N. A software pipeline for processing and identification of fungal ITS sequences. Source Code Biol Med. 2009;4:1.
Ott SJ, et al. Fungi and inflammatory bowel diseases: Alterations of composition and diversity. Scand J Gastroenterol. 2008;43:831.










