
Turnbaugh, P.J., Hamady, M., Yatsunenko, T., Cantarel, B.L., Duncan, A., Ley, R.E., Sogin, M.L., Jones, W.J., Roe, B.A., Affourtit, J.P., et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484.
Vehik, K., and Dabelea, D. (2011). The changing epidemiology of type 1 diabetes: why is it going through the roof? Diabetes Metab. Res. Rev. 27, 3–13.
Virgin, H.W., Wherry, E.J., and Ahmed, R. (2009). Redefining chronic viral infection. Cell 138, 30–50.
Wen, L., Ley, R.E., Volchkov, P.Y., Stranges, P.B., Avanesyan, L., Stonebraker, A.C., Hu, C., Wong, F.S., Szot, G.L., Bluestone, J.A., et al. (2008). Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455, 1109–1113.
White, D.W., Keppel, C.R., Schneider, S.E., Reese, T.A., Coder, J., Payton, J.E., Ley, T.J., Virgin, H.W., and Fehniger, T.A. (2010). Latent herpesvirus infection arms NK cells. Blood 115, 4377–4383.
Wu, H.J., Ivanov, I.I., Darce, J., Hattori, K., Shima, T., Umesaki, Y., Littman, D.R., Benoist, C., and Mathis, D. (2010). Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 32, 815–827.
Yeung, W.C., Rawlinson, W.D., and Craig, M.E. (2011). Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ 342, d35.
FROM GENETICS OF INFLAMMATORY BOWEL DISEASE TOWARDS MECHANISTIC INSIGHTS90
Daniel B. Graham91,92and Ramnik J. Xavier91,93
Advancements in human genetics now poise the field to illuminate the pathophysiology of complex genetic disease. In particular, genome-wide association studies (GWAS) have generated insights into the mechanisms driving inflammatory bowel disease (IBD) and implicated genes shared by multiple autoimmune and autoinflammatory diseases. Thus, emerging evidence suggests a central role for the mucosal immune system in mediating immune homeostasis and highlights the complexity of genetic and environmental interactions that collectively modulate the risk of disease. Nevertheless, the challenge remains to determine how genetic variation can precipitate and sustain the inappropriate inflammatory response to commensals that is observed in IBD. Here, we highlight recent advancements in immunogenetics
________________
90 Reprinted from Trends in Immunology 34(8), Graham, D. B. and R. J. Xavier. 2013. From genetics of inflammatory bowel disease toward mechanistic insights, pages 371-378, Copyright 2013, with permission from Elsevier.
91 Broad Institute of MIT and Harvard, Cambridge, MA, USA.
92 Department of Medicine, Massachusetts General Hospital, Boston, MA, USA.
93 Gastrointestinal Unit, Center for the Study of Inflammatory Bowel Diseases, and Center for Computational and Integrative Biology, Massachusetts General Hospital, Boston, MA, USA.
and provide a forward-looking view of the innovations that will deliver mechanistic insights from human genetics.
Genetics of IBD
Genetic predisposition to autoimmune and autoinflammatory diseases is well established, yet the interrelationship between multiple genetic components and environmental triggers remains to be elucidated. The observation that some individuals develop IBD at childhood and others during adulthood, suggests distinct environmental contributions to disease initiation versus disease progression. Although the genetic elements predisposing to IBD are present from birth, disease onset occurs later in life, and there is further need to understand how host genetic and epigenetic factors interact with environmental triggers at disease onset and how genetics influence immune regulatory networks that sustain disease. The most recent advancement in the field came from a meta-analysis of 15 prior GWASs to aid in the design of the Immunochip (Jostens et al., 2012). This custom-designed genotyping array contains high-density genomic coverage of single nucleotide polymorphisms (SNPs) implicated by GWASs to fine map disease loci and implicate new genes. In combination with imputed GWAS data, Immunochip validation studies on over 25 000 IBD cases identified 71 new loci for a total of 163 loci associated with IBD (Jostens et al., 2012). As a result, these studies substantially increase the estimated disease heritability for both Crohn’s disease (CD) and ulcerative colitis (UC). Furthermore, they highlight 110 shared loci for both disease subtypes, while 30 are classified as specific for CD and 23 for UC (Jostens et al., 2012). IBD shares the largest number of loci with type 1 diabetes and shows substantial enrichment of overlap with ankylosing spondylitis, psoriasis, and susceptibility to mycobacterial infection (Jostens et al., 2012). Such overlap in the genetics of autoimmune/autoinflammatory diseases points to several common immune processes, such as regulation of mucosal immunity, in mediating inflammatory pathology. Together, the immediate impact of implicating 163 loci in IBD resulted in the identification of new candidate genes and pathways that may drive disease. However, taking human genetic studies a step further requires building new experimental systems to define the biological functions of these genes. For example, genetic studies in psoriasis patients implicated Act1 (Traf3ip2), an adaptor functioning downstream of the interleukin-17 receptor (IL-17R) (Ellinghaus et al., 2010; Huffmeier et al., 2010; Strange et al., 2010). The generation of a knockout mouse that modeled TRAF3IP2 loss of function variants in humans revealed hyperactive TH17 responses driving IL-22-dependent inflammation (Wang et al., 2013). Thus, GWASs led to the development of a novel mouse model that provided key insight into disease mechanisms. Here, we discuss how advancements in genomics and functional genetics have contributed to our current understanding of IBD, and how these approaches can be applied to provide new mechanistic insights and therapeutic opportunities (Figure A20-1).
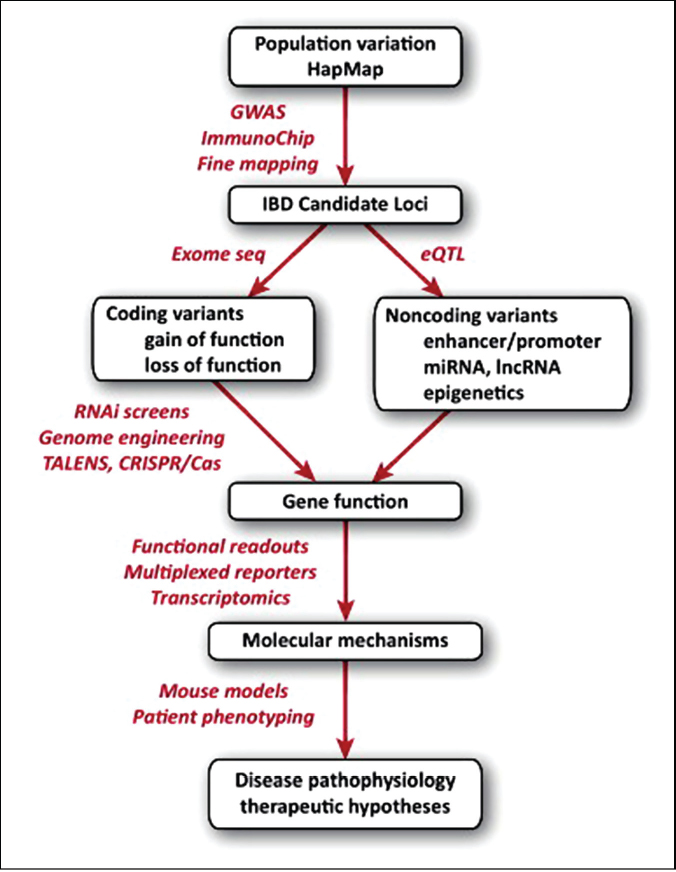
FIGURE A20-1 From genetics to disease mechanisms. Advancements in genomics technology have facilitated the discovery of 163 loci associated with inflammatory bowel disease (IBD). Identifying causal mutations within coding and noncoding regions of these loci has begun to reveal gene function and shed new light on disease mechanisms. Progressing from genetics to mechanistic insight is accelerating as a result of new technology in the field of functional genetics.
Genetic Variation and Functional Repercussions
Many SNPs implicated by GWASs are not directly causal with respect to phenotype, rather they exist in linkage disequilibrium with yet to be discovered variants that are functional. This point highlights the fact that GWASs cover relatively common genetic variants including SNPs with >1% frequency within the population and fail to capture rare or undiscovered variants. Such germlineencoded DNA variation can lead to nonsynonymous coding variants that change protein function and/or posttranslational regulation. However, the majority of the SNPs implicated by GWASs represent variation in noncoding regions of the genome. Thus, alterations in gene expression are likely to be important factors in immune dysregulation, and highlight the need to comprehensively characterize DNA and RNA regulatory elements.
Genetic variation impacts gene expression at the level of transcription, RNA stability, splicing, and epigenetic modification. Accordingly, new genomics tools have been developed to capture genetic regulation at multiple levels. Studies merging proteomics with genomics identified differential binding of transcription factors to SNPs in the IL2RA promoter that regulate gene expression (Butter et al., 2012). As an additional mechanism of transcriptional regulation, long noncoding (lnc) RNAs within the genome have been shown to play key roles in modulating gene expression (Derrien et al., 2012; Guttman et al., 2009). Recent studies have identified a critical requirement for lncRNA in maintaining pluripotency and enforcing lineage-specific gene expression profiles (Guttman et al., 2011). It is now clear that expression of lncRNAs is regulated in a cell type-specific manner. Thus, analysis of the human beta cell transcriptome identified tissue-specific lncRNAs that were transcriptionally dysregulated in type 2 diabetes (T2D) or that mapped to loci previously associated with T2D (Moran et al., 2012). In the context of host defense, the lncRNA NeST has been identified as an epigenetic regulator of interferon-gamma (IFN-γ) expression in CD8 T cells and determines susceptibility to viral and bacterial infection in mice (Gomez et al., 2013). However, the potential role for lncRNAs in regulating human inflammatory diseases remains largely unexplored.
Following transcription, mRNA splicing and stability are tightly regulated. In this context, inflammatory stimuli and microbial components induce a coordinated program of miRNA expression in monocytes that regulates inflammatory responses (Hasler et al., 2012). Consequently, variants in the gene encoding IL23R that associate with IBD are resistant to downmodulation by miRNA, which results in upregulation of IL23R expression (Zwiers et al., 2012). Similarly, variants in the 3′ UTR of genes encoding cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and IL-10 alter recognition by miRNAs, resulting in dysregulated expression (deJong et al., 2012). In addition to regulation at the level of mRNA stability, RNA splicing impacts gene function. SNPs in the 3′ untranslated region of Ctla4 associate with type 1 diabetes (T1D) and correlate with reduced expression of a soluble CTLA-4 splice variant (Ueda et al., 2003). Accordingly,
transgenic mice with germline integration of a cassette encoding short hairpin RNA (shRNA) specifically targeting soluble CTLA-4 impaired T regulatory cell (Treg) function and resulted in accelerated onset of diabetes in the nonobese diabetic (NOD) system and severe colitis in the CD45RBhi transfer model (Gerold et al., 2011).
Determining the impact of genetic variation on expression profiles has been a major focus in the field, as highlighted in studies of expression quantitative trait loci (eQTL) that aim to correlate SNPs with transcriptome data (Knight, 2012). Recent studies have begun to identify SNPs that alter transcription of neighboring genes (cis eQTLs) and distant genes (trans eQTLs). The analysis of 92 strains of inbred mice identified several thousand eQTLs associated with macrophage responses to lipopolysaccharide (LPS) (Orozco et al., 2012). Similarly, eQTL studies in human dendritic cells (DCs) identified 198 eQTLs associated with Mycobacterium tuberculosis infection (Barreiro et al., 2012). These findings are notable given the genetic overlap between susceptibility to M. tuberculosis infection and IBD and given the enrichment of IBD candidate genes in DCs (Jostens et al., 2012). In a direct example of eQTLs in IBD, SNPs associated with CD correlate with increased expression of caspase recruitment domain family member 9 (Card9), which has been suggested with potentiate inflammatory signaling cascades (Jostens et al., 2012). Similarly, SNPs associated with T1D have been shown to act in trans on an antiviral inflammatory network driven by interferon regulatory factor 7 (IRF7) in monocytes (Heinig et al., 2010). Although many of the eQTLs highlighted above are cell type-specific and the mechanistic basis of this specificity is not entirely clear, it is likely to be regulated by differential pathway activity and epigenetic effects. In this context, the Encyclopedia of DNA Elements (ENCODE) project continues to deposit rich datasets of genome-wide epigenetic modifications across multiple cell types. Recent chromatin mapping studies highlight SNPs associated with autoimmunity that are enriched within enhancer elements containing epigenetic modifications in specific cell types (Ernst et al., 2011; Maurano et al., 2012).
As the number of noncoding variants and SNPs associated with autoimmune disease has expanded, so has identification of coding variants. Exome sequencing at IBD loci has identified novel coding variants and helped pinpoint specific genes within these loci that are likely to impact disease (Rivas et al., 2011). For example, many IBD loci are gene-dense, and SNP signals are not able to precisely pinpoint the causative gene. A locus on chromosome 12 implicated by GWAS is comprised of the LRRK2 gene and MUC19. Although a great deal of attention has focused on study of the kinase leucine-rich repeat kinase 2 (LRRK2), the discovery of new coding variants in MUC19 indicates the need for more detailed analysis of this gene in IBD pathogenesis (Rivas et al., 2011). MUC19 is a gelforming apomucin, thus genetic variants of MUC19 may contribute to mucosal barrier dysfunction by causing quantitative changes in its production or structural changes in the glycoprotein core. The barrier function of the mucous layer
acts in concert with its ability to retain antimicrobial effector molecules such as defensins and secreted IgA. Thus, B cell IgA production is a central pathway in mucosal immunity, which is consistent with a newly discovered role for the IBD candidate gene BACH2 in class switch recombination (Muto et al., 2010). In fact, exome sequencing recently defined coding variants in BACH2 and identified a distinct profile in severe UC characterized by mutations in both BACH2 and IL10 (Christodoulou et al., 2012). Although B cell function was not directly tested in this study, BACH2 and IL-10 can induce immunoglobulin class switching (Briere et al., 1994; Muto et al., 2010), thus highlighting the potential of gene–gene interactions to impact cell type-specific phenotypes. Collectively, these studies demonstrate the diverse mechanisms by which genetic variation impacts gene function and highlight the importance of genetic interactions.
Addressing Genetic Epistasis
The diversity of genetic backgrounds in human subjects complicates phenotyping endeavors, but highlights the notion that phenotypes derive from the aggregate effects of multiple genetic factors. Furthermore, IBD genes interact, as highlighted by the discovery of a microtubule-associated serine/threonine-protein kinase 3-regulated transcriptional program that broadly controls expression of inflammatory genes associated with nuclear factor-kappa-B (NFkB) activity, such as those induced by NOD2 and Toll-like receptors (TLRs) (Labbe et al., 2008, 2012). It is now possible to discern the impact of genetic perturbation on transcriptional programs by employing RNAi-mediated knockdown of candidate genes followed by RNAseq. Accordingly, this approach may provide a deeper understanding of how IBD genes interact with one another at the genetic level. Although genetic epistasis is thought to be an important component of disease, it is not captured well by GWASs (Zuk et al., 2012). Nevertheless, studies have shown that T1D risk alleles for human leukocyte antigen (HLA) class II and Ctla4 statistically interact and support a role for gene–gene interactions (Howson et al., 2012). Epistasis is difficult to study systematically, because of the sheer number of possible genetic contributors. Furthermore, perturbing multiple genes simultaneously has practical limits, and addressing the issue of genetic epistasis in complex disease will ultimately require an unbiased discovery-based approach invoking GWASs on a much larger scale.
Exploring Gene-Environment Interactions
Following the logic that host genetic epistasis contributes to disease, emerging evidence indicates that host genetic interactions with microbes also shapes immunologic phenotypes. Twin studies suggest that infant and early childhood infections may be associated with IBD (Ng et al., 2012). In addition, stable interactions of commensal communities with the host shape immune development,
as demonstrated by the observation that segmented filamentous bacteria (SFB) promote development of Th17 cells in mice (Ivanov et al., 2009). Similarly, innate lymphoid cell (ILC) development is shaped by the microbiome (Sonnenberg and Artis, 2012), and ILCs accumulate in inflamed mucosal tissues (Bernink et al., 2013). Dietary factors also promote ILC development, as aryl hydrocarbon receptor (AHR) ligands induce lymphoid follicle development in the intestine by acting on ILCs (Kiss et al., 2011). Conversely, ILCs shape the composition of the microbiome by limiting dissemination of lymphoid resident commensals (Sonnenberg et al., 2012). These key observations begin to provide insight into the interactions between the host immune system, microbiome, and dietary metabolites (Fukuda et al., 2011).
In addition to stable host–commensal interactions, transient interactions between host and pathogens irrevocably change the immune system. Prior pathogen exposure elicits more robust responses to subsequent infection. While memory within the adaptive immune system provides antigen-specific protection upon rechallenge, the innate immune system can be “trained” by prior infection and poised to respond more robustly to a variety of pathogens (Cooper et al., 2009; O’Leary et al., 2006; Quintin et al., 2012; Sun et al., 2009). Thus, previous infection may condition the immune system to mount a pathologic response, which is not to suggest that IBD is caused by a particular pathogen. Rather, emerging evidence suggests a “multiple hit” model for conferring susceptibility to IBD (Cadwell et al., 2010). Similar to IBD patients bearing ATG16L1 risk alleles, Atg16l1 hypomorphic mice exhibit Paneth cell defects associated with abnormalities in granule packaging. Importantly, pathology associated with Paneth cell function depended on prior exposure to virus (Cadwell et al., 2010). Given the interconnection between host and microbes, the future challenge is to catalogue the metagenome and correlate host genotype to alterations in the microbiome. Still more challenging is the prospect of determining past history of pathogen infection in patients and identifying the immunologic consequences of prior infection. Notably, clues to prior pathogen exposure may be encoded in the specificity of gut IgA (Lindner et al., 2012).
Extrapolating Pathways from Human Genetic Data
To date, GWASs have identified 163 loci associated with IBD (Jostens et al., 2012). Using pathway analysis tools to examine genes within these loci immediately reveals patterns related to immunity. For example, cytokines and their respective receptors are abundantly represented. Upon closer examination, additional immunoregulatory pathways can be distilled and implicate genetic programs associated with Treg function, Th17 development, negative regulation of T cell receptor (TCR) signaling, innate pathogen sensing pathways, antigen presentation, and apoptosis (Figure A20-2). Although the function of caspases in eliciting apoptosis of T cells during activation-induced cell death is well
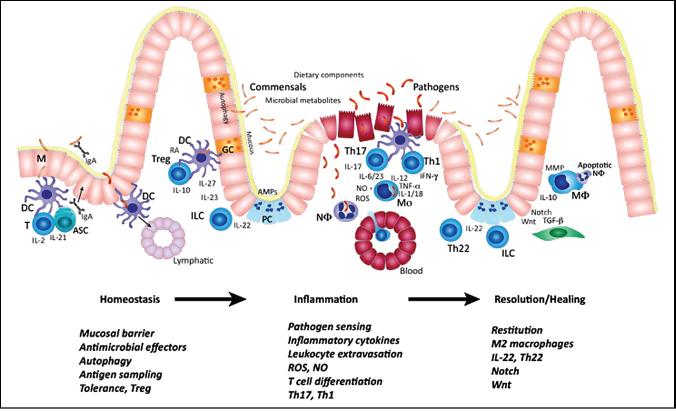
FIGURE A20-2 Inflammatory bowel disease (IBD) pathways and key cell types. Genetic studies have helped to identify critical biological processes and cell types that regulate intestinal inflammation. Genetic variation that perturbs any stage of immune homeostasis, inflammation, or resolution can result in the inappropriate inflammatory response to commensals that is observed in IBD. M cell (M), antibody secreting cells (ASC), dendritic cell (DC), retinoic acid (RA), T cell (T), T regulatory cell (Treg), innate lymphoid cell (ILC), goblet cell (GC), Paneth cell (PC), antimicrobial proteins (AMPs), neutrophil (Nø), inflammatory monocyte (Mo), macrophage (Mø), reactive oxygen species (ROS), nitric oxide (NO), and matrix metalloproteinase (MMP).
characterized, caspases have additional functions in inflammasome signaling and processing of IL-1 family cytokines. Given that dysregulated caspase activity and inflammasome function have been associated with IBD, study of additional caspase-dependent pathways and caspase substrates may reveal novel insights into IBD pathophysiology (Becker et al., 2013).
Gene annotation and functional associations have also highlighted key pathways that surpass the traditional boundaries of immunology and would not have been easily predicted by previous studies (Box A20-1). Paramount among these are cellular processes related to autophagy, redox signaling, and endoplasmic reticulum (ER) stress. Emerging evidence points to multiple pathway interactions; for example, ER stress and autophagy mutually regulate one another to maintain metabolic homeostasis during inflammation and to promote antimicrobial effector responses 45 and 46. In addition, macrophage exposure to bacterial lipopolysaccharide induces a metabolic switch to glycolysis resulting in accumulation of succinate that promotes IL-1β production by stabilizing hypoxia-inducible
BOX A20-1
Key Genes Implicated in IBD
Advancements in genomics have identified specific loci and rare coding variants associated with risk or protection from IBD. The genes identified by these studies highlight specific cellular pathways that may contribute to disease onset and/or progression (reviewed in Khor et al., 2011). Here, we highlight candidate IBD genes that implicate additional pathways that collectively suggest connections between cellular metabolism, inflammation, and mucosal microbial communities (Table A20-1).
TABLE A20-1 Candidate IBD Genes
| Select IBD genes identified by ImmunoChip (Jostens et al., 2012) | ||
| Gene | Locus | Putative function |
| RNF186 | 1p36.13 | Highly expressed in the intestine and contains a RING-type zinc finger that may function as a ubiquitin ligase. Association with IBD has been validated in several populations (Juyal et al., 2011; Yang et al., 2013). Evidence suggests genetic interaction with another IBD gene, HNF4A (Garrison et al., 2006). |
| SP110 | 2q37.1 | Associated with primary immunodeficiency. Expressed in hematopoietic cells and contains a bromodomain with potential involvement in epigenetic regulation. Loss of function mutations can decrease IL-10 production by B cells (Bloch et al., 2012). |
| SP140 | 2q37.1 | Expressed in hematopoietic cells and contains a bromodomain with potential involvement in epigenetic regulation. |
| MST1 | 3p21 | Hepatocyte growth factor-like protein produced in the liver. Activates the receptor tyrosine kinase MST1R on epithelial cells (and some subsets of macrophages). Gain of function variants enhance macrophage motility (Hauser et al., 2012). |
| FUT2 | 19q13.3 | Golgi protein expressed in gastrointestinal tract. Enzymatic activity generates a secreted oligosaccharide that functions as a substrate for synthesis of A and B blood group antigens. Loss of function mutations (nonsecretor phenotype) lack expression of blood group antigens in mucosal surfaces. Secretor status correlates with alterations in the microbiome (Rausch et al., 2011) and risk of IBD (Miyoshi et al., 2011) and T1D (Smyth et al., 2011). |
| SLC22A4 | 5q31.1 | Ergothioneine transporter expressed in intestine and subsets of myeloid cells. May regulate cellular redox state, potentially linking metabolism with inflammatory responses (Kato et al., 2010). |
| GSDMB | 17q12 | May be involved in regulation of epithelial cell apoptosis (Saeki et al., 2009). It is also highly expressed in CD8 T cells. |
| Gene | Locus | Putative function |
| ORMDL3 | 17q12 | Regulates the ER stress response associated with inflammation (McGovern et al., 2010). |
| TNFSF15 | 9q32 | Expressed on endothelial cells and activated APCs. One of its receptors (TNFRSF25) promotes Treg expansion in a ligand-dependent manner (Khan et al., 2013). |
| TNFAIP3 | 6q23 | Ubiquitin modifying enzyme expressed in myeloid cells. Negatively regulates NFkB signaling and inflammatory cytokines (Hammer et al., 2011). |
| SLC6A7 | 5q32 | Proline transporter that may regulate cellular metabolic state and inflammation. |
| IL10RA | 11q23 | Receptor for IL-10 broadly expressed on hematopoietic cells. Transduces immunosuppressive signal through STAT3 and TYK2. Associated with early onset IBD (Moran et al., 2013). |
| Select IBD genes with coding variants identified by exome sequencing (Rivas et al., 2011) | ||
| Gene | Locus | Putative function |
| IL23R | 1p31.3 | Receptor for IL-23 expressed predominantly in T cells. Promotes differentiation of pathogenic Th17 cells (Ghoreschi et al., 2010). |
| CARD9 | 9q34.3 | Expressed in myeloid cells where it promotes activation of NFkB and inflammatory cytokines downstream of pattern recognition receptors (PRRs) that are associated with immunoreceptor tyrosine-based activation motifs (ITAMs) or hemi-ITAMs (Hara et al., 2007). Promotes cytokine environment conducive to Th17 differentiation. |
| NOD2 | 16q21 | Intracellular PRR specific for bacterial peptidoglycans and is expressed in myeloid cells. Activates NFkB and promotes inflammatory cytokines. Can induce bacterial killing in an autophagy-dependent manner (Homer et al., 2010). |
| IL18RAP | 2q12 | Accessory protein for IL-18 receptor expressed on NK and T cells. Promotes stimulatory effect of IL-18 on T cell IFN-γ3 production (Cheung et al., 2005). |
| MUC19 | 12q12 | Gel-forming mucin expressed in epithelial tissues. Potential role in barrier function and interaction with microbial communities. |
| CUL2 | 10p11.21 | Component of E3 ubiquitin-protein ligase complex potentially linking proteosomal system with autophagy. |
| PTPN22 | 1p13.2 | Protein tyrosine phosphatase that regulates T and B cell responses at the level of antigen receptor signaling (Rhee and Veillette, 2012). |
| C1orf106 | 1q32.1 | Expressed in epithelial cells of the gastrointestinal tract. May promote epithelial integrity and barrier function. |
factor 1 α subunit (HIF1-α) (Tannahill et al., 2013). Conversely, metabolism impinges upon the immune system by regulating tolerance. Tryptophan catabolism mediated by indoleamine 2,3-dioxygenase (IDO) generates kynurenine, which promotes IL-10 production by DCs and facilitates Treg differentiation, thus inhibiting Th17 responses (Nguyen et al., 2010). Th17 differentiation is also regulated by environmental factors such as NaCl derived from dietary sources. In this context, elevated levels of salt stimulate expression of serum/glucocorticoid regulated kinase (SGK1), which in turn induces expression of IL-23R to enhance Th17 differentiation (Wu et al., 2013). Further work elucidating Th17 cytokine networks has uncovered previously unrecognized connections between IL-17 and maintenance of epithelial barrier function (Reynolds et al., 2012), thus indicating an important role for the immune system in maintaining epithelial homeostasis and restitution. In particular, neutrophil influx into inflamed tissues initially amplifies inflammation, and later promotes an anti-inflammatory healing response through macrophage-dependent clearance of apoptotic neutrophils (Stark et al., 2005). In this context, mice deficient in the NADPH oxidase subunit P40phox exhibit an impairment in the healing response following acute colonic inflammation (Conway et al., 2012), thus revealing an additional role for reactive oxygen species (ROS) in epithelial restitution in addition to its known role as an antimicrobial agent. The importance of ROS in mucosal immunity is further supported by the discovery of rare variants of NCF4 and NCF2 that associate with IBD (Matute et al., 2009; Muise et al., 2012). These genetic studies implicating IBD genes in the ROS pathway suggest that neutrophils are key cell types in pathology, and further identify new functions for these cells that influence clinical manifestations of disease (Figure A20-3).
Pathway interactions identified by GWASs implicate several cell types in inflammatory pathology. By cross-referencing candidate disease genes and pathways with gene expression patterns in immune cell subsets, DCs and memory T cells feature prominently as central cell types contributing to IBD (Jostens et al., 2012). Consistent with this notion, Ly6Chi monocytes in the inflamed colon generate inflammatory DCs and antigen presenting DCs that drive autoinflammatory T cell responses (Zigmond et al., 2012). Additional evidence implicates ILCs as key mediators of host defense and inflammatory pathology (Sonnenberg and Artis, 2012; Tait Wojno and Artis, 2012). Furthermore, expression of IBD genes identified by GWASs highlight the gut epithelium and innate immune mechanisms including barrier function, goblet cell secretion of mucins, and Paneth cell secretion of antimicrobial mediators (Bevins and Salzman, 2011). With several functional pathways implicated in IBD, the next challenge will be to place unannotated genes in pathways that drive disease and to elucidate regulatory networks.
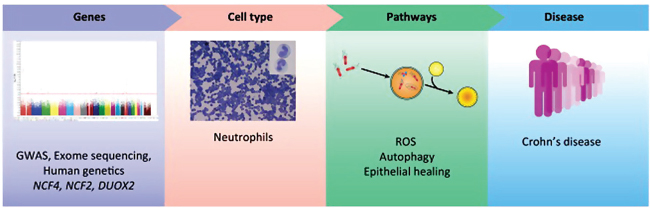
FIGURE A20-3 Molecular pathogenesis of inflammatory bowel disease (IBD): assembling the evidence. Identification of genes in the reactive oxygen species (ROS) pathway point to neutrophils as key cellular mediators of IBD, while functional studies implicated ROS in antibacterial defense, autophagy, and epithelial healing. Thus, genetics can be used to define a mechanism-based subset of IBD patients and guide treatment.
New Approaches in Assigning Function to Genes
Although many of the candidate IBD genes implicated by GWASs can be assembled into known pathways, a substantial fraction of these genes (>40%) are poorly characterized at the functional level. A significant challenge is to identify functions for candidate IBD genes and to determine how signaling pathways work together to regulate mucosal immune effector mechanisms. One can posit that candidate IBD genes with unknown function may control key immune functions that drive disease, and that assigning function to these genes will expand our understanding of immunoregulatory mechanisms. To meet the challenge of assigning functions to candidate IBD genes in relevant immune responses, high throughput RNAi screening approaches have been developed. Implementing RNAi screens has shown initial signs of success, but has yet to be systematically applied to assign IBD candidate genes with immunologic function. As a recent example, RNAi screening was used to identify mediators of innate pathogen recognition through the NOD2 complex. Here, screening approaches identified NOD2-dependent regulators of NFkB and further demonstrated a novel mechanism for the spatial assembly of the NOD2 signaling complex (Lipinski et al., 2012). In another recent study, genome-wide RNAi screening approaches identified 190 cofactors required for mediating endosomal pathogen sensing pathways mediated by TLR7 and TLR9 (Chiang et al., 2012).
Elucidating Mechanisms
Most functional genetic strategies aimed at assigning function to genes involve knockdown or overexpression of candidates. While this approach can establish the requirement, or define a regulatory role for a specific gene in a
defined biological process, it does not effectively capture how genetic variation in human populations impacts immune regulation. For example, coding variants may result in gain of function, loss of function, or exist on a functional continuum somewhere in between. These are important distinctions when attempting to infer disease mechanisms or design new therapeutics. By the same token, determining how genetic variation in risk alleles versus protective alleles impacts a cellular response requires mechanistic insight. The next challenge will be to develop reliable approaches to demonstrate the causal effect of a genetic variant on disease progression and to determine the underlying mechanism of action.
With accessibility of exome sequencing technology on the rise, genetic diversity can be quantified and increasing numbers of coding variants have been identified 60 and 61. It has been estimated that human genomes typically harbor up to 100 loss of function variants and approximately 20 genes that are nonfunctional (MacArthur et al., 2012). Due to limitations in the ability to predict which variants cause a given phenotype and which are benign, rigorous mechanistic studies must necessarily follow. IBD GWASs led to the identification of the autophagy gene ATG16L1 and a putative loss of function coding variant thereof (T300A) (Hampe et al., 2007; Rioux et al., 2007). Knockdown of endogenous ATG16L1 in epithelial cells and overexpression of RNAi-resistant ATG16L1 T300A resulted in impaired antibacterial autophagy and formally demonstrated a role for this coding variant in a relevant biological readout (Kuballa et al., 2008). Although overexpression of coding variant cDNAs is ideally suited for high throughput analyses of many candidate mutations, overexpression can also mask subtle effects or exaggerate phenotypes. In gene replacement or exon replacement approaches, a coding variant is introduced into the endogenous locus so as to retain regulation at the level of chromatin remodeling, transcription, and splicing. Knockin mice have proven successful in this regard and can be cross-bred to introduce mutations at multiple loci. More recent innovations in genome engineering now enable efficient generation of isogenic human cells. Approaches that employ transcription activator like effectors (TALEs) allow for targeting endogenous genes to introduce coding variants (Bedell et al., 2012; Hockemeyer et al., 2011; Mouscou and Bogdanove, 2009; Reyon et al., 2012), and the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR/Cas) system has been recently adapted to target multiple genes simultaneously (Cong et al., 2013; Garneau et al., 2010; Mali et al., 2013).
Assessing Disease Relevance
In progressing from the identification of genetic loci by GWASs to defining functions for disease genes and variants, the next step towards determining the role for a candidate gene in disease pathology requires study in vivo. Here, multiple cell types and signaling pathways coordinately interact to mediate loss of immune tolerance and persistent inflammation. In this context, murine models have
earned a prominent position as a mainstay for determining if a candidate gene is necessary or sufficient for disease. Mouse models have been used to demonstrate that the ubiquitin modifying enzyme A20 limits inflammatory responses, because conditional deletion in DCs resulted in spontaneous colitis and spondyloarthritis (Hammer et al., 2011). Furthermore, the role of DCs in maintaining tolerance is highlighted by the observation that regulatory DCs pulsed with bacterial antigen can mitigate experimental colitis in mice (Yamanishi et al., 2012).
An emerging concept from mouse models and human genetic studies is that IBD cases are likely to be comprised of distinct subsets of mechanism-driven disorders (Virgin and Todd, 2011). In this context, systematic immunophenotyping of genotyped human cohorts will be required to define mechanism-based disease subsets. In disease-free individuals bearing a T1D risk allele at the IL2R locus, IL-2R expression was reduced and Treg function was impaired (Garg et al., 2012). Here, a rigorous approach with large sample sizes combined with rigorous quality control was implemented to address the effects of different genetic backgrounds. In addition, inclusion of healthy controls with disease risk genotypes can help to exclude complicating effects of chronic disease that may alter immune phenotypes.
Concluding Remarks
Human genetics has provided key insights into complex disease. Significant overlap in genes implicated across several autoimmune/autoinflammatory diseases indicates common immunologic mechanisms as well as unique disease-specific pathways that must be tightly regulated to balance host defense against the risk for pathological inflammation. Only with a deeper understanding of the mechanisms driving disease and their underlying genetic components will the goal of interpreting patient genotypes become feasible. Towards this end, the diagnostic power of genetics bears potential to stratify patient subsets based on disease mechanisms and treat them accordingly. Moreover, progress towards deciphering the genetic components of IBD pathophysiology will identify new points of entry for mechanism-based therapeutics. Although it remains a challenge to mitigate pathological inflammation without compromising host defense, advancements in genetics offer the opportunity to treat the underlying mechanisms that incite IBD rather than broadly suppressing immune function.
Acknowledgements
The authors are funded by the Center for the Study of Inflammatory Bowel Diseases (Massachusetts General Hospital), the Helmsley Trust, and NIH R01 DK092405.
References
Abecasis, G.R. et al. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65
Barreiro, L.B. et al. (2012) Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. U.S.A. 109, 1204–1209
Becker, C. et al. (2013) Complex roles of caspases in the pathogenesis of inflammatory bowel disease. Gastroenterology 144, 283–293
Bedell, V.M. et al. (2012) In vivo genome editing using a high-efficiency TALEN system. Nature 491, 114–118
Bernink, J.H. et al. (2013) Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 14, 221–229
Bevins, C.L. and Salzman, N.H. (2011) Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 9, 356–368
Bloch, D.B. et al. (2012) Decreased IL-10 production by EBV-transformed B cells from patients with VODI: implications for the pathogenesis of Crohn disease. J. Allergy Clin. Immunol. 129, 1678– 1680
Briere, F. et al. (1994) Interleukin 10 induces B lymphocytes from IgA-deficient patients to secrete IgA. J. Clin. Invest. 94, 97–104
Butter, F. et al. (2012) Proteome-wide analysis of disease-associated SNPs that show allele-specific transcription factor binding. PLoS Genet. 8, e1002982 Cadwell, K. et al. (2010) Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145
Chang, J.S. et al. (2012) Endoplasmic reticulum stress response promotes cytotoxic phenotype of CD8alphabeta+ intraepithelial lymphocytes in a mouse model for Crohn’s disease-like ileitis. J. Immunol. 189, 1510–1520
Cheung, H. et al. (2005) Accessory protein-like is essential for IL-18- mediated signaling. J. Immunol. 174, 5351–5357
Chiang, C.Y. et al. (2012) Cofactors required for TLR7- and TLR9- dependent innate immune responses. Cell Host Microbe 11, 306–318
Christodoulou, K. et al. (2012) Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes. Gut http://dx.doi.org/10.1136/gutjnl-2011-301833
Cong, L. et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823
Conway, K.L. et al. (2012) p40phox expression regulates neutrophil recruitment and function during the resolution phase of intestinal inflammation. J. Immunol. 189, 3631–3640
Cooper, M.A. et al. (2009) Cytokine-induced memory-like natural killer cells. Proc. Natl. Acad. Sci. U.S.A. 106, 1915–1919
de Jong, V.M. et al. (2012) Post-transcriptional control of candidate risk genes for type 1 diabetes by rare genetic variants. Genes Immun. 14, 58–61
Derrien, T. et al. (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789
Ellinghaus, E. et al. (2010) Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat. Genet. 42, 991–995
Ernst, J. et al. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49
Fukuda, S. et al. (2011) Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469, 543–547
Garg, G. et al. (2012) Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J. Immunol. 188, 4644–4653
Garneau, J.E. et al. (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71
Garrison, W.D. et al. (2006) Hepatocyte nuclear factor 4alpha is essential for embryonic development of the mouse colon. Gastroenterology 130, 1207–1220
Gerold, K.D. et al. (2011) The soluble CTLA-4 splice variant protects from type 1 diabetes and potentiates regulatory T-cell function. Diabetes 60, 1955–1963
Ghoreschi, K. et al. (2010) Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467, 967–971
Gomez, J.A. et al. (2013) The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell 152, 743–754
Guttman, M. et al. (2009) Chromatin signature reveals over a thousand highly conserved large noncoding RNAs in mammals. Nature 458, 223–227
Guttman, M. et al. (2011) lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477, 295–300
Hammer, G.E. et al. (2011) Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat. Immunol. 12, 1184–1193
Hampe, J. et al. (2007) A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 39, 207–211
Hara, H. et al. (2007) The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol. 8, 619–629
Hasler, R. et al. (2012) Microbial pattern recognition causes distinct functional micro-RNA signatures in primary human monocytes. PLoS ONE 7, e31151
Hauser, F. et al. (2012) Macrophage-stimulating protein polymorphism rs3197999 is associated with a gain of function: implications for inflammatory bowel disease. Genes Immun. 13, 321–327
Heinig, M. et al. (2010) A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature 467, 460–464
Hockemeyer, D. et al. (2011) Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 29, 731–734
Homer, C.R. et al. (2010) ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 139, 1630–1641 1641.e1–2
Howson, J.M. et al. (2012) Evidence of gene-gene interaction and age-at-diagnosis effects in type 1 diabetes. Diabetes 61, 3012–3017
Huffmeier, U. et al. (2010) Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat. Genet. 42, 996–999
Ivanov, I.I. et al. (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498
Jostins, L. et al. (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124
Juyal, G. et al. (2011) An investigation of genome-wide studies reported susceptibility loci for ulcerative colitis shows limited replication in north Indians. PLoS ONE 6, e16565
Kato, Y. et al. (2010) Gene knockout and metabolome analysis of carnitine/organic cation transporter OCTN1. Pharm. Res. 27, 832–840
Khan, S.Q. et al. (2013) Cloning, expression, and functional characterization of TL1A-Ig. J. Immunol. 190, 1540–1550
Khor, B. et al. (2011) Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317
Kiss, E.A. et al. (2011) Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565
Knight, J.C. (2012) Resolving the variable genome and epigenome in human disease. J. Intern. Med. 271, 379–391
Kuballa, P. et al. (2008) Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS ONE 3, e3391
Labbe, C. et al. (2008) MAST3: a novel IBD risk factor that modulates TLR4 signaling. Genes Immun. 9, 602–612
Labbe, C. et al. (2012) Genome-wide expression profiling implicates a MAST3-regulated gene set in colonic mucosal inflammation of ulcerative colitis patients. Inflamm. Bowel Dis. 18, 1072–1080
Lindner, C. et al. (2012) Age, microbiota, and T cells shape diverse individual IgA repertoires in the intestine. J. Exp. Med. 209, 365–377
Lipinski, S. et al. (2012) RNAi screening identifies mediators of NOD2 signaling: implications for spatial specificity of MDP recognition. Proc. Natl. Acad. Sci. U.S.A. 109, 21426–21431
MacArthur, D.G. et al. (2012) A systematic survey of loss-of-function variants in human proteincoding genes. Science 335, 823–828
Mali, P. et al. (2013) RNA-guided human genome engineering via Cas9. Science 339, 823–826
Matute, J.D. et al. (2009) A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood 114, 3309–3315
Maurano, M.T. et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195
McGovern, D.P. et al. (2010) Genome-wide association identifies multiple ulcerative colitis susceptibility loci. Nat. Genet. 42, 332–337
Miyoshi, J. et al. (2011) Ectopic expression of blood type antigens in inflamed mucosa with higher incidence of FUT2 secretor status in colonic Crohn’s disease. J. Gastroenterol. 46, 1056–1063
Moran, C.J. et al. (2013) IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm. Bowel Dis. 19, 115–123
Moran, I. et al. (2012) Human beta cell transcriptome analysis uncovers lncRNAs that are tissuespecific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 16, 435–448
Moscou, M.J. and Bogdanove, A.J. (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326, 1501
Muise, A.M. et al. (2012) NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut 61, 1028–1035
Muto, A. et al. (2010) Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. EMBO J. 29, 4048–4061
Ng, S.C. et al. (2012) Role of genetic and environmental factors in British twins with inflammatory bowel disease. Inflamm. Bowel Dis. 18, 725–736
Nguyen, N.T. et al. (2010) Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. U.S.A. 107, 19961–19966
O’Leary, J.G. et al. (2006) T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat. Immunol. 7, 507–516
Orozco, L.D. et al. (2012) Unraveling inflammatory responses using systems genetics and geneenvironment interactions in macrophages. Cell 151, 658–670
Quintin, J. et al. (2012) Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12, 223–232
Rath, E. and Haller, D. (2012) Unfolded protein responses in the intestinal epithelium: sensors for the microbial and metabolic environment. J. Clin. Gastroenterol. 46 (Suppl.), S3–S5
Rausch, P. et al. (2011) Colonic mucosa-associated microbiota is influenced by an interaction of Crohn disease and FUT2 (Secretor) genotype. Proc. Natl. Acad. Sci. U.S.A. 108, 19030–19035
Reynolds, J.M. et al. (2012) Cutting edge: regulation of intestinal inflammation and barrier function by IL-17C. J. Immunol. 189, 4226–4230
Reyon, D. et al. (2012) FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 30, 460–465
Rhee, I. and Veillette, A. (2012) Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat. Immunol. 13, 439–447 Review
Rioux, J.D. et al. (2007) Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604
Rivas, M.A. et al. (2011) Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 43, 1066–1073
Saeki, N. et al. (2009) Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer 48, 261–271
Sanjana, N.E. et al. (2012) A transcription activator-like effector toolbox for genome engineering. Nat. Protoc. 7, 171–192
Smyth, D.J. et al. (2011) FUT2 nonsecretor status links type 1 diabetes susceptibility and resistance to infection. Diabetes 60, 3081–3084
Sonnenberg, G.F. and Artis, D. (2012) Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity 37, 601–610
Sonnenberg, G.F. et al. (2012) Innate lymphoid cells promote anatomical containment of lymphoidresident commensal bacteria. Science 336, 1321–1325
Stark, M.A. et al. (2005) Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 22, 285–294
Strange, A. et al. (2010) A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 42, 985–990
Sun, J.C. et al. (2009) Adaptive immune features of natural killer cells. Nature 457, 557–561
Tait Wojno, E.D. and Artis, D. (2012) Innate lymphoid cells: balancing immunity, inflammation, and tissue repair in the intestine. Cell Host Microbe 12, 445–457
Tannahill, G.M. et al. (2013) Succinate is an inflammatory signal that induces IL-1b through HIF-1a. Nature 496, 238–242
Ueda, H. et al. (2003) Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature 423, 506–511
Virgin, H.W. and Todd, J.A. (2011) Metagenomics and personalized medicine. Cell 147, 44–56
Wang, C. et al. (2013) The psoriasis-associated D10N variant of the adaptor Act1 with impaired regulation by the molecular chaperone hsp90. Nat. Immunol. 14, 72–81
Wu, C. et al. (2013) Induction of pathogenic T17 cells by inducible salt-sensing kinase SGK1. Nature http://dx.doi.org/10.1038/nature11984 Yamanishi, H. et al. (2012) Regulatory dendritic cells pulsed with carbonic anhydrase I protect mice from colitis induced by CD4+CD25- T cells. J. Immunol. 188, 2164–2172
Yang, S.K. et al. (2013) Genome-wide association study of ulcerative colitis in Koreans suggests extensive overlapping of genetic susceptibility with Caucasians. Inflamm. Bowel Dis. 19, 954–966
Zigmond, E. et al. (2012) Ly6C(hi) monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen- presenting cells. Immunity 37, 1076–1090
Zuk, O. et al. (2012) The mystery of missing heritability: genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. U.S.A. 109, 1193–1198
Zwiers, A. et al. (2012) Cutting edge: a variant of the IL-23R gene associated with inflammatory bowel disease induces loss of microRNA regulation and enhanced protein production. J. Immunol. 188, 1573–1577


















