
Discussion Framework for Clinical Trial Data Sharing
Clinical trials are crucial to determining the safety of medical interventions and their ability to achieve particular health outcomes. Clinical trials are required by regulatory authorities around the world before a new medical product can be brought to market, or before a new indication, formulation, or target population can be approved for an intervention already on the market (ICH, 1995). After a product’s introduction, additional clinical trials are commonly conducted by industry, government, and academia to further define the relative safety and efficacy (or effectiveness) of the product. Clinical trials are also used to study interventions that do not involve regulated medical products, for example, surgical techniques, behavioral interventions, or studies designed to improve disease management practice (Califf, 2013).
Vast amounts of data are generated over the course of a clinical trial. These data are held by the sponsors conducting the clinical trial, and in some instances, by participants or their advocates (Drazen, 2002; Terry and Terry, 2011). Depending on the regulatory jurisdiction, data might or might not be shared or made available to the public for secondary uses. Shared data might include both summary data and individual patient data. In the United States, if a sponsor is seeking regulatory approval, data are shared in confidence with regulators.1 Select study data might also be made available to individual researchers on a case by case basis upon request, or could be made publicly available, usually at the
____________________
1 However, in the European Union, the European Medicines Agency (EMA) has undertaken regulatory action to share anonymized clinical trial data with external requestors. The EMA’s data sharing initiative is described on p. 32.
summary level, for example, through publication in a peer-reviewed journal or through publicly accessible clinical trial registration sites (e.g., ClinicalTrials.gov). Other data, however, remain largely unavailable to outside researchers and the public, including data found in analyzable data sets, clinical study reports (CSRs), and individual participant data (IPD) that, if accessible, could facilitate new analyses and a deeper understanding of a particular therapy or condition (Doshi et al., 2013; Goldacre, 2012; Gordon et al., 2013; Rawlins, 2012). Increased sharing of IPD, in particular, could facilitate activities such as independent reanalysis of trial results, addressing concerns about publication bias,2 characterizing trial outcomes by subgroups, considering additional questions beyond the original trial hypotheses, carrying out meta-analysis for systematic reviews, and facilitating hypothesis generation and additional research to develop new therapies (Doshi et al., 2012; IOM, 2013; McGauran et al., 2010; Ross et al., 2012).
The data sharing movement has gained substantial momentum during the last decade, in both the clinical trial and larger scientific communities (Boulton et al., 2011; Royal Society, 2012). A cultural change has occurred in which the conversation around data sharing has moved from whether it should happen to how it can be carried out (IOM, 2013). To that end, a large number of people and organizations involved in clinical trials have endorsed principles promoting, in their view, responsible sharing of clinical trial data (Loder, 2013). Prominent examples include the joint statement from funders of health research data, the AllTrials campaign, and the Pharmaceutical Research and Manufacturers of America (PhRMA) and European Federation of Pharmaceutical Industries and Associations (EFPIA) principles (AllTrials, 2013; PhRMA and EFPIA, 2013; Wellcome Trust, 2011). Some organizations have gone beyond general statements and principles and begun to adopt data sharing policies as well. The U.S. National Institutes of Health (NIH), as well as international groups of researchers and nonprofit funders, has been involved in data sharing activities for years (e.g., BioLINCC, MalariaGen). More recently, European regulators and some pharmaceutical and device companies (e.g., GlaxoSmithKline [GSK], Medtronic) have begun to plan and implement their own data sharing policies (EMA, 2013; Nisen and Rockhold, 2013; YODA, 2013). In addition, medical journals have begun to require authors to include data sharing statements
____________________
2 The tendency for positive results of trials of health care interventions to be reported or published, with corresponding underreporting/non-publication of negative or inconclusive results (Cochrane Collaboration, 2002).
and, in the case of the British Medical Journal (BMJ), to agree to make de-identified patient-level data available upon “reasonable request” as a condition for publication (Godlee and Groves, 2012, p. 1).
In October 2012, the Institute of Medicine (IOM) convened individuals with a broad range of expertise and perspectives to discuss sharing of clinical research data in a public workshop. As highlighted by workshop participants, data sharing can provide important potential benefits to industry, nonprofit and government funders of research, academic investigators, patient advocacy groups, and, ultimately, patients and the public, including, for example, speeding medical innovation by reducing redundancies, facilitating the identification and validation of new drug targets, identifying new indications for use, and improving the understanding of the safety and efficacy of therapies (IOM, 2013). In followup to the 2012 IOM workshop, the IOM was asked by a group of federal, industry, and U.S. and international foundation sponsors3 to conduct a consensus study to recommend guiding principles and a framework for the responsible sharing of clinical trial data.
Charge to the Committee and Scope of the Study
As described in the committee’s charge from the sponsors (see Appendix A), over a 17-month period of deliberations, the committee will release two documents:
1. This document, which is a framework for discussion (“framework”), to be released in January 2014 for public comment. The framework will summarize the committee’s initial thoughts on guiding principles that underpin responsible sharing of clinical trial data, define key elements of clinical trial data and data sharing, and describe a selected set of clinical trial data sharing activities. The charge to the committee excludes evidence-based findings and conclusions and recommendations from this document.
2. A final report with findings and recommendations related to the committee’s full charge.
____________________
3 U.S. National Institutes of Health, U.S. Food and Drug Administration, AbbVie Inc., Amgen Inc., AstraZeneca Pharmaceuticals, Bayer, Biogen Idec, Bristol-Myers Squibb, Burroughs Wellcome Fund, Doris Duke Charitable Foundation, Eli Lilly and Company, EMD Serono, Genentech, GlaxoSmithKline, Johnson & Johnson, Medical Research Council (UK), Merck & Co., Inc., Novartis Pharmaceuticals Corporation, Novo Nordisk, Pfizer Inc., Sanofi-Aventis, Takeda, and Wellcome Trust.
To respond to the charge, the IOM convened a committee composed of persons with expertise in key scientific and research-related areas, including academia, industry, funding bodies, regulatory activities, scientific publications, clinicians, and patients. Individual committee member expertise spans academic clinical trial design, performance and dissemination; pharmaceutical product development; statistics, informatics, and data security; ethics of human subjects research; and law and regulatory requirements (including privacy, security, and intellectual property). Committee members also have insight into the global context of data sharing; the concerns of research participants, patients, and their families; and other relevant issues.
As charged in its statement of task, in the next phase of the study the committee will identify the key benefits, challenges, and risks of sharing, as well as key risks of not sharing. This analysis will take into consideration the full range of perspectives of research sponsors and investigators, study participants, regulatory agencies, patient groups, and the public. The committee is also charged, in the final report, with suggesting strategies and practical approaches for responsible data sharing. As part of its recommendations, the committee will offer guiding principles for and characteristics of the optimal infrastructure and governance for data sharing. In particular, the committee has been called to consider, among other issues, resource constraints, implementation, disincentives in the academic research model, changing norms, protection of human subjects and patient privacy, intellectual property and other legal issues, and preservation of scientific standards and data quality.
As outlined in the statement of task (Appendix A), many terms are defined for the purposes of this study. “Data sharing” is defined as the responsible entity (“data generator”) making the data available via open or restricted access, or exchanged among parties. For the purposes of this study, data generator may include industry sponsors, data repositories, and researchers conducting clinical trials. The committee has adopted a working definition of “data holder” to mean the entity or entities that have access to data, including regulatory agencies, journals to which manuscripts are submitted, and other repositories such as ClinicalTrials.gov in the United States. Data holders—for example, persons who collect source data, develop an analyzable data set, or carry out statistical analyses—may or may not have legal authority to provide third-party access to the data. The scope of the study is limited to interventional clinical trials. For the purposes of this study, “interventional clinical trials” are defined as “research in which participants are assigned to receive one
or more interventions (or no intervention) so that the effects of the interventions on biomedical or health-related outcomes can be evaluated. Assignments to [intervention]4 groups are determined by the study protocol” (ClinicalTrials.gov, 2012). Assignments could be at the individual or group level. An intervention is a “process or action that is the focus of a clinical trial. This can include giving participants drugs, medical devices, procedures, vaccines, and other products that are either investigational or already available” (ClinicalTrials.gov, 2012). For the purposes of this study, intervention types are limited to drugs, devices, biologics, other treatments or therapies (such as radiation), surgical procedures, behavioral interventions, and changes in the administration or delivery of clinical care.
A FRAMEWORK FOR DISCUSSION AND CALL FOR COMMENTS
This framework for discussion articulates the committee’s preliminary thoughts on guiding principles that underpin the responsible sharing of clinical trial data, defines key elements of data and data sharing activities, and describes a selected set of data sharing activities. One goal of this framework is to set the stage for identification of the numerous complicated issues that recommended strategies and practical approaches to sharing of clinical trial data might need to take into account. As with any complex policy problem, there are advantages and disadvantages to be weighed and potential for competing interests and incentives among and within various stakeholder groups. The guiding principles as defined below potentially could be in conflict or might be interpreted or prioritized differently, depending on one’s perspective.
As a first step in fulfilling the committee’s charge, the framework identifies key issues so that the public can point out omissions and begin to suggest benefits, interests, risks, and burdens of options that should be considered. The framework also identifies issues about which the committee will gather additional information through public meetings and the submission of written materials by interested parties (see Box 1 and the section below). Comparing the various public responses to the framework will help clarify the countervailing interests and values the commit-
____________________
4 To maintain consistency in its terminology the committee has substituted the term “intervention” here for the term “treatment,” which is used in the ClinicalTrials.gov definition.
tee will take into account in making its recommendations to enhance responsible sharing of clinical trial data.
BOX 1
Specific Topics for Public Feedback
Global Implementation and Practical Consideration
• Because most large clinical trials are global in nature, how can clinical trial data be shared in that global context? How can different national regulations for research participants’ privacy protections, approval of drugs and devices, data exclusivity and intellectual property laws, resources, and health priorities be taken into account?
• How might strategies and approaches regarding data sharing take into account clinical trials conducted in resource-poor settings; trials designed by citizen-scientists using data they contribute directly; and trials designed through participatory research?
Timing and Prioritization
• How might different types of clinical trial data, and different uses of shared data, be prioritized for sharing? What would be the rationale for placing a higher priority on certain types of data or analyses?
• What might be the advantages and disadvantages of distinguishing highest priority sharing of clinical trial data from other sharing activities?
• What might be the advantages and disadvantages to various stakeholders of sharing different types of datasets, at different points in time after the completion of a clinical trial?
• Should programs or approaches calling for or requiring new data sharing apply only to new trials undertaken from the date of a new program forward, or retroactively apply to clinical trials started before the data sharing program was initiated?
Mitigating Risks
• What might be done to minimize the risks to patients and to public health from the dissemination of findings from invalid analyses of shared clinical trial data?
• What measures should be deployed to minimize the privacy and confidentiality risks to trial participants? For example, are current anonymization or de-identification methodologies sufficient?
• Under what circumstances are identifiable data needed to fulfill articulated purposes of a data sharing activity? Under what circumstances might re-identification of trial participants be beneficial (for the participants or the public)? Have there been there examples of instances of re-identification of trial participants (e.g., for safety reasons to warn a patient of a potential risk, or for questionable and potentially unethical reasons) and what were the impacts?
Enhancing Incentives
• What incentives and protections might be established to encourage clinical trial sponsors and clinical investigators to continue to conduct clinical trials in the future, without unduly restricting the sharing of certain types of data? How do we protect or provide incentives for researchers to share data?
• What is the appropriate responsibility of the primary investigator(s) or research institution(s) to support secondary users in their interpretation of shared data, and what infrastructure or resources are needed to enable such ongoing support? For those with experience in data sharing, what is the burden of providing such support to help others understand and use the provided information?
Measuring Impact
• What would be appropriate outcome measures to assess the usefulness of different models of clinical trial data sharing, and how can they be used to guide improvements in data sharing practices?
Invitation for Public Comments
The issues identified and the options and observations described in this framework are preliminary and do not represent a comprehensive review of the subject. This framework does not assess the benefits and risks of different options and, consistent with its charge, does not contain conclusions or recommendations. Instead, this framework serves to identify areas of interest and concern that will be pursued in greater detail during the second phase of the project and addressed in the final report. The final report will also analyze the risks and benefits of options and make conclusions and recommendations. As required in the charge to the committee, the framework is being released for public comment. The committee welcomes comments from interested parties to help ensure that major concerns and issues are not overlooked, and particularly invites comments on the difficult or complex issues outlined in Box 1 below. Comments may be submitted to the committee at either of two forthcoming public workshops, or via the committee’s project website, http://www8.nationalacademies.org/cp/projectview.aspx?key=49578.
During the course of the study, members of the public are encouraged to provide comments on this framework, as well as general comments on issues within the scope of the charge to the committee. The committee is also interested in receiving testimony and suggestions on
topics that are likely to be particularly complex, and where differing perspectives are likely to reside. At future meetings, the public will be invited to discuss these issues or to submit written statements.
GUIDING PRINCIPLES FOR RESPONSIBLE SHARING OF CLINICAL TRIAL DATA
In this framework for discussion, the committee proposes four high-level principles as a starting point for developing a framework for responsible sharing of clinical trial data (see Box 2; the principles are discussed individually below). By explicitly articulating these guiding principles and the rationale behind them, the committee hopes to bring into sharper relief the values of concern for different stakeholders that need to be acknowledged and balanced in data sharing policies and procedures.
In developing this provisional set of guiding principles, the committee drew on recent proposals for principles of data sharing in clinical trials from scholars and working groups (AllTrials, 2013; EMA, 2013; FDA, 2013; Godlee and Groves, 2012; Mello, 2013; PhRMA and EFPIA, 2013; Wellcome Trust, 2011; YODA, 2013), as well as widely accepted guiding principles articulated in official statements of research ethics and international standards, such as the Declaration of Helsinki, the Belmont Report (which presents the ethical rationale for current U.S. regulations for human subjects research), and others (Childress et al., 2005; CIOMS, 2002; ICH, 1996; National Commission, 1979; WMA, 2013). Policies regarding clinical trial data sharing will have a stronger intellectual foundation and practical applicability if they take into account policies on related topics. A framework for sharing clinical trial
BOX 2
Provisional Guiding Principles for Responsible Sharing of Clinical Trial Data
• Respect the individual participants whose data are shared.
• Maximize benefits to participants in clinical trials and to society, while minimizing harms.
• Increase public trust in clinical trials.
• Carry out sharing of clinical trial data in a manner that enhances fairness.
data should therefore be consistent with the principles guiding related issues, including the protection of human research participants, regulation of drugs and medical devices, scientific publications, and intellectual property protections.
In this section the committee presents potential consequences of clinical trial data sharing, which may or may not occur in any particular data sharing activity. In its further deliberations and final report, the committee will assess these potential consequences of clinical trials data sharing.
Respect the Individual Participants Whose Data Are Shared
The committee’s first provisional guiding principle stems from the broadly articulated concept that respect for research participants is a fundamental principle of research ethics (ICH, 1996; National Commission, 1979).
Respect Through Research Participant Protections
Respect for research participants requires protecting their dignity, integrity, and right to self-determination; this includes, at a minimum, compliance with applicable regulations and ethical standards for the conduct of clinical trials and handling of the resulting data. Respect for research participants has historically been understood to require specific informed consent from participants (including consent for how their data will be used) before they enroll in a clinical trial in which the intervention is carried out at the individual participant level (Childress et al., 2005; CIOMS, 2002; WMA, 2013).5 For existing trials, data sharing (particularly sharing beyond other investigators in the trial) might not have been explicitly discussed with participants during the consent process. Sharing of data without specific participant consent might be ethically acceptable and legally permitted in certain instances. For example, if the shared data are de-identified, current U.S. federal regulations on human research protections and U.S. health information privacy regulations (e.g., the Health Insurance Portability and Accountability Act
____________________
5 Specific informed consent is not necessarily required for trials at the group level, such as in certain cluster-randomized trials (Weijer and Emanuel, 2000) or for certain comparative effectiveness trials (Faden et al., 2013).
[HIPAA] of 1996)6 allow other researchers to use the data for research under certain conditions without consent from the original participants.7
Respect also suggests a need to protect the confidentiality and privacy of trial participants when data are shared. Questions have been raised about the sufficiency of commonly used de-identification methodologies (Benitez and Malin, 2010; El Emam, 2013; McGraw, 2012); consequently, additional protections may be needed.
Respect can also be demonstrated and advanced through efforts to engage participants and their representatives in the development of processes for sharing of clinical trial data (CTSA, 2011). For example, new policies and procedures regarding data sharing and subsequent additional analyses (particularly for specific trials or classes of trials as relevant) could be developed with input and feedback from representatives of research participants, disease advocacy groups, community advisory boards, and the public (Jiang et al., 2013). Such an approach would also include dissemination of information and calls for input about data sharing policies and procedures and a rationale for data sharing that is accessible and understandable to the public. The act of seeking and obtaining such input would not in itself constitute surrogate consent or authorization for data sharing. Rather, it would respect participants by actively seeking to identify concerns about and potential unappreciated benefits of data sharing that were not previously taken into account, and allow participants or their advocates to suggest how the process of data sharing might be improved (Stiles and Petrila, 2011).
Maximize Benefits to Participants in Clinical Trials and to Society, While Minimizing Harm
Understanding and balancing the potential benefits and harms of health interventions is a significant component of health care and of health intervention research and development. Similarly, there are poten-
____________________
6 Public Law 104-191, 104th Cong. (August 21, 1996).
7 The U.S. example has been described here for illustrative purposes. Privacy protections with respect to sharing anonymized data without reconsent vary across jurisdictions. For example, the European Union has strong data privacy protections that need to be observed when clinical trial data are shared by its member states (Article 29 Data Protection Working Party, 2013).
tial benefits and harms associated with the sharing of clinical trial data. Strategies for data sharing should maximize benefits to those who give of themselves to participate—and to society as a whole—while minimizing potential harms for various stakeholders. This provisional guiding principle for responsible sharing of clinical trial data is derived from the ethical concept of beneficence.
International ethical standards identify beneficence as a basic ethical principle and obligation of research involving human subjects. The International Conference on Harmonisation (ICH) Guideline for Good Clinical Practice declares: “Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks” and “the rights, safety, and well-being of the trial subjects are the most important considerations and should prevail over interests of science and society” (ICH, 1996). Benefits include both the immediate knowledge gained from answering the hypothesis of a particular clinical trial and the broader utility of the study data in informing development of effective new interventions. As discussed in the Belmont Report, practitioners are faced with deciding “when it is justifiable to seek certain benefits despite the risks involved” (National Commission, 1979, p. 5). The potential utility of data is a component in the balance of potential benefits and risks when making the decision to expose individual participants to risk in order to seek benefits to society as a whole.
Clinical trials are designed and carried out to address research questions about the safety and efficacy (or effectiveness) of one or more health interventions. The interventions that participants receive are determined by the study protocol, not by what their personal physicians consider best for them as individuals. In consenting to participate, clinical trial participants also accept that complying with the study protocol potentially entails inconvenience and risks (Lidz et al., 2004). Although participation in clinical trials, on the whole, might not be significantly riskier than ordinary clinical care or receiving the same intervention outside the trial (Gross et al., 2006), in a specific trial the benefits and risks of the study arms are not known at the outset. In some instances the intervention arm of a trial will be shown to have significantly worse out-
comes than the control arm, a finding that cannot be predicted at the time of enrollment.
From the perspective of clinical trial participants, data sharing increases their contributions to generalizable human health knowledge by potentially facilitating additional findings beyond the original, predefined clinical trial outcomes. Thus, if data are not shared, opportunities are missed to generate additional knowledge from their contributions (Califf, 2013; IOM, 2013; Mello et al., 2013).
From the perspective of society as a whole, sharing data from clinical trials, if it were to yield additional results that contribute to the scientific knowledge base, could provide a more accurate, less biased, and more comprehensive picture of the benefits and risks of an intervention. Sharing clinical trial data could potentially lead to enhanced efficiency and safety of the clinical research process by, for example,
• reducing duplication of efforts and costs of future studies;
• reducing exposure of participants in future trials to avoidable harms that can be identified through data sharing;
• providing a deeper knowledge base for regulatory decisions;
• supporting the development of clinical practice guidelines; and
• allowing health care professionals and patients to make more informed decisions about clinical care.
The usefulness of clinical trial data would be enhanced if, in addition to sharing the protocol, manual of operations, statistical analysis plan, a copy of the case report forms, and metadata about the analyzable data file, the data user could ask the data generator questions regarding logistical and practical issues such as the conduct of the clinical trial or data analyses. The usefulness of shared clinical trial data might also be enhanced if data were collected in a standardized manner, where scientifically indicated (Califf, 2013; IOM, 2013; Mello et al., 2013).
In the long run, proponents have suggested that sharing clinical trial data could improve public health and patient outcomes, reduce the incidence of adverse effects from therapies, and decrease expenditures for medical interventions that are ineffective or less effective than alternatives. In addition, data sharing could open opportunities for exploratory research that might lead to hypotheses about the mechanisms of disease, more effective therapies, or alternative uses of existing or abandoned therapies that could be tested in additional research (Califf, 2013; IOM, 2013; Mello, 2013).
These potential benefits of clinical trial data sharing need to be balanced against any potential harms. Data sharing could place clinical trial participants at increased risk for invasions of privacy or breaches of confidentiality, which could then lead to social or economic harms to the participants (IOM, 2013; Mello et al., 2013).
Data sharing could also result in potential harms to society. For example, shared clinical trial data might be analyzed in a manner that leads to biased effect estimates or invalid conclusions (although this might also occur with the original analyses) (Krumholz and Ross, 2011). Incorrect conclusions or treatment recommendations for either whole-patient populations or subgroups could produce suboptimal care and unnecessary anxiety, and result in possible discrimination (IOM, 2013; Spertus, 2012). Concerns about such future uses of their clinical trial data might also deter some individuals and/or communities from participating in clinical trials.
Clinical trialists and sponsors of clinical trials could also suffer potential harms due to the administrative and financial burden of data sharing, which they might regard as an unfunded mandate. The manner in which data are shared might undermine the incentives of clinical trial sponsors, clinical investigators, coordinating centers, researchers, and other essential stakeholders to invest time and resources in the development and clinical testing of potential new treatment practices (Dickersin, 2013; Rathi et al., 2012). For example, data sharing might allow confidential commercial information (CCI) to be discerned from the data (Teden, 2013). Competitors might use shared data to seek regulatory approval of competing products in countries that do not recognize data exclusivity periods or that do not grant patents for certain types of research. The manner in which clinical trial data are shared might also harm the intellectual capital and professional recognition of academic clinical investigators, who devote considerable effort and time to designing a clinical trial, recruiting and retaining participants, and collecting the primary data. If subsequent independent analyses fail to give appropriate recognition to the original investigators, there will not be incentives for investigators to share clinical trial data.
Increase Public Trust in Clinical Trials
Public trust is an intrinsic value undergirding the biomedical science and health research enterprise, which is fundamentally aimed at improving human health. Trust is also essential for ensuring continued public support for clinical research and for fostering participation in clinical trials. The concept of public trust in clinical trials encompasses both trust in the scientific process of generating data (i.e., that there is accountability for how the trial is carried out) and trust in the validity of the reported trial findings (i.e., that the reported findings are an accurate representation of the underlying data) (IOM, 2013). The sharing of clinical trial data could be carried out in ways that enhance or undermine public trust in clinical research. The process of data sharing should therefore be undertaken in a manner that enhances, rather than undermines, public trust in both the clinical trial process and the data sharing process.
By increasing the transparency of trial design and conduct and the pathway to trial conclusions, sharing clinical trial data might increase public trust in the outcomes of specific trials and of clinical trials generally (Loder, 2013). Data sharing might also increase the usefulness and trustworthiness of clinical trial data and analyses because clinical researchers who know that others will be using their data might be more thorough and more careful in their methodology and its documentation. Such additional attention to detail might also help to reduce bias in the data and findings (Mello et al., 2013).
Sharing clinical trial data could enhance public trust by facilitating confirmatory analyses that could determine whether the final conclusions and summaries of clinical trials are robust and valid inferences from the original evidence, although this must be done in a credible and fair manner (Laine et al., 2007). Whether the inferences from a particular trial are strong or weak, activities seeking to demonstrate widespread applicability of the findings could enhance overall trust in the scientific process and in the resulting evidence-based recommendations for clinical care.
Trust in clinical research could be enhanced further if sharing clinical trial data were accompanied by outreach and engagement to help the public understand that numerous judgments are needed to transform source data into analyzable data (CTSA, 2011), and that highly trained researchers might take different approaches to answering a research
question or to analyzing a given dataset. Different analytical approaches and interpretations of data by researchers are an expected part of the scientific process and discussions. Such outreach could also help the public better understand that findings from individual clinical trials are often not definitive, and that attempts to replicate original analyses or the conduct of meta-analyses using pooled data from multiple clinical trials can strengthen, modify, refute, or extend the original reports from a trial. Moreover, trust would be enhanced if additional analyses using data from a clinical trial were easily connected with the publications from the original trial.
Trust in the Data Sharing Process
Sharing clinical trial data could carry the risk of undermining public trust in clinical trials under certain circumstances, for example, if multiple analyses yield conflicting or invalid conclusions (Califf, 2013). Public trust in clinical trials where data are shared might be undermined unless there are clear, transparent, and accountable processes for the data sharing, including established criteria for clinical trial data sharing, procedures for fairly adjudicating requests for data against those criteria, mechanisms for ensuring or confirming the integrity of data when they are collected and analyzed by different parties, and accountability for both data holders and requesters in adhering to those standards. Clear, transparent, and accountable processes for data sharing would also include protection of patient privacy and respectful handling of individual patient data. Further, public trust could be increased if the public sees evidence of the incorporation of its perspectives (whether through the mechanisms described above or by addressing specific community concerns). If analyses of shared data use inappropriate methods and statistics and lead to biased conclusions, they can undermine patient trust in valid conclusions about the study intervention. Such mistrust might ultimately lead to seriously flawed clinical care decisions, unwarranted patient concerns about the quality of care, and avoidable patient anxiety.
Carry Out Sharing of Clinical Trial Data in a Manner That Enhances Fairness
Fairness, broadly articulated, is a core ethical principle that is applicable in several ways to sharing clinical trial data. In very general terms, fairness entails persons receiving what is due to them or what they de-
serve (Beauchamp and Childress, 2009). Fairness requires similar treatment of all people (as individuals or as part of groups, entities, processes, etc.) unless there are justifiable reasons to treat certain people differently. Disagreements about fairness arise in specifying what an individual or group is due or deserves, what are sufficient reasons for different treatment under what might be perceived by some as similar circumstances, and whether inequity, unfairness, or injustice has occurred. All individuals and organizations involved in the clinical trial enterprise have a stake in the fairness of data sharing. In the context of this discussion framework, fairness addresses the distribution of the risks and potential benefits of clinical trial data sharing, whereas beneficence generally refers to the aggregate risks and potential benefits.
Fairness in Exposure to Potential Risks of Data Sharing
Clinical trial data sharing should be carried out in a fair manner so that it does not repeat, in the data sharing context, well-documented historical examples of placing disproportionate risks of clinical research on vulnerable groups (Bioethics Commission, 2011; Emanuel et al., 2008; Jones, 2008; Wertheimer, 2008). For example, data sharing should include protections for participant subgroups that are particularly vulnerable to breaches of confidentiality or other adverse consequences of data sharing. Clinical trial participants could be particularly vulnerable to harm if they have conditions, or are members of groups, that are often stigmatized. In this regard, there might be justifiable and ethical reasons for handling some types of clinical trial data sharing differently to reduce the potential for unfair treatment of participants. For example, whole-genome sequencing data could be identifiable (Gymrek et al., 2013), which might be considered to put participants at heightened risk, and additional safeguards or protections for participants whose genomic data could be shared might be warranted.
Fairness in the Distribution of the Potential Benefits of Research
Clinical trial data sharing could increase fairness in clinical research by helping to distribute the benefits of clinical research more equitably across different groups of participants and communities. Pooling shared data from several clinical trials could, for example, benefit groups who have been enrolled in clinical trials in such small numbers that the statistical power in any single trial to draw valid inferences about risks and
benefits is limited. Among the underserved groups for whom data sharing might accelerate research are individuals with rare conditions, rare subtypes of common conditions, and members of certain ethnic groups that historically have low enrollment in clinical trials. Underrepresentation of these groups in clinical trials can lead to a weaker evidence base for clinical care decisions, as well as health disparities and discrimination (IOM, 2002).
Clinical trial sponsors, clinical investigators, and coordinating centers that design and carry out clinical trials might consider fairness to include appropriate recognition, protection, and reward. Investigators who make substantial investments of intellectual capital, time, and resources have an interest in carrying out additional analyses of the data they have collected and in receiving due credit when others take advantage of these data. In addition, there are administrative burdens when sharing data with others that need to be fairly distributed among those who collect and control clinical trial data, those who seek access to shared data for additional research, and society. Appropriate protection of these interests could help provide incentives (or reduce disincentives) to share data and to conduct future clinical trials.
ADDITIONAL OBSERVATIONS RELATING TO PRINCIPLES FOR DATA SHARING
Operational Strategies Following from Guiding Principles
The principles described in this framework suggest several possible operational strategies that approaches for responsible data sharing might take into account. The committee will consider such operational strategies in its further deliberations and in formulating recommendations in the final report and welcomes comments about operational strategies. Implementation of these principles might take into account operational considerations, such as
• timing of when data are shared;
• proportional consideration of benefits, burdens, and risks to various parties in adopting a data sharing program or activity;
• opportunities to embed “learning” in a clinical trial data sharing system, permitting tracking of the beneficial outcomes of different approaches to data sharing, identification of unanticipated
adverse consequences of approaches, dissemination of best practices for data sharing, and promotion of ongoing quality improvement in data sharing systems and approaches; and
• need for data sharing systems and approaches to be globally applicable and practically achievable.
Framing Rights and Responsibilities: To Whom Do the Benefits of Clinical Trial Data Belong?
Foundational to any discussion of possibilities and needs for clinical trial data sharing is a fundamental philosophical question: To whom do the benefits of the data belong? Are data primarily for the benefit of the public or for private parties? This is a separate question from a technical or legal analysis of which party owns the data or which parties have certain property interests in the data, which includes control over access to and use of the data.
With respect to ownership of clinical trial data, academic institutions that receive research grants might claim ownership of the data collected during the research in order to comply with regulatory requirements (Drazen, 2002). Private funders of clinical trials might claim that they own the research data, particularly if the data will form part of a submission to the U.S. Food and Drug Administration (FDA) for regulatory approval. The language of research grants and contracts and the wording of informed consent forms that participants sign in clinical trials could also delineate what party owns or has rights of disposition of data. For example, the NIH includes data sharing requirements in the terms and conditions of its research grants with direct costs greater than $500,000 (NIH, 2003). A property owner does not always have absolute dominion over the property in question; others may have legal access to it under certain conditions for certain purposes.8 Ultimately, the question of who owns the data is less important than asking what are the rights and responsibilities of data holders. The committee intends to engage the question of to whom ought the benefits of clinical trial data belong as a thought experiment to help inform its analysis.
On one hand, the benefit of clinical trial data might be regarded as primarily belonging to the public—in order to benefit patients and society through the advancement of science and clinical knowledge that leads to improved patient care. From this perspective, some might argue that
____________________
8 Moreover, property may be taken for public use without consent of the owner, subject to Constitutional requirements for due process and fair compensation (Evans, 2011).
sharing clinical trial data ought to be a prima facie obligation. That is, the default policy—the presumption—should be to share data, with justification needed to restrict sharing or recognize an exception to sharing.
On the other hand, the benefit of clinical trial data might be regarded as primarily belonging to organizations and individuals who invested resources and time to plan and carry out the clinical trial and analyze the data. The rationale for this perspective might be the provision of fair rewards for investment and work in carrying out the trial, or it might be the risk that organizations and individuals might otherwise lack appropriate incentives to develop new tests and therapies. From this perspective, the policy presumption might be that clinical trial data sharing should only be undertaken if the interests and rights of those who carried out the trial are appropriately protected and incentivized. Some might argue that clinical trial data sharing should be optional and voluntary, at the discretion of the organization and individuals who invested resources and time to conduct the clinical trial.
While we have framed these positions as a dichotomy for the sake of discussion, another approach would be to balance both perspectives in a policy of clinical trial data sharing, in addition to adhering to the other guiding principles discussed above, as well as applicable existing, modified, or new legal requirements. Any attempt to balance competing interests would require careful attention to the conditions of sharing, responsibilities, limitations, and exceptions.
DATA SHARING ELEMENTS AND ACTIVITIES
The following sections define key elements of data and data sharing activities, such as the type(s) of data to be shared; provider(s) and recipient(s) of shared data; and whether and when data are disclosed publicly, with or without restrictions, or exchanged privately among parties. This section then describes a selected set of data sharing activities. The purpose of outlining potential data sharing activities is to provide an heuristic approach to organizing the work of the committee throughout the course of the study, including information gathering and discussions in public sessions. In its final report, the committee will, with respect to each of these data sharing activities, present findings relating to benefits, risks, and burdens associated with these data sharing activities and suggest strategies and practical approaches to facilitate responsible data sharing. The data sharing activities noted in this discussion should not be
interpreted as a conceptual framework that would necessarily lead to particular findings, conclusions, or recommendations.
What Types of Data Could Be Shared?
Current Practices in Data Disclosure
During the course of a clinical trial, different types of data are collected, transformed into analyzable datasets to address specific research questions, and used to generate various publications and reports for different audiences (Drazen, 2002) (see Figure 1).
Publication in peer-reviewed scientific journals is currently the primary method for sharing clinical trial data with the scientific and medical communities, as well as the public (often through media coverage of published findings). These publications, however, contain only a small subset of the data collected, produced, and analyzed in the course of a trial (Doshi et al., 2013; Zarin, 2013). Scientific journal articles generally contain a brief summary of the trial background, research question(s), methodology, results, figures and tables, and discussion.
Clinical trial sponsors seeking regulatory approval from authorities such as the FDA and the EMA must submit detailed CSRs (discussed below) and IPD as required, which forms the basis of the marketing application for a product. In trials that are not conducted for regulatory approval of a product, detailed CSRs may or may not be prepared (Doshi et al., 2012; Teden, 2013).
Beyond the selected clinical trial data that are disclosed in journal publications, IPD and more detailed clinical datasets have not been routinely shared with the broader scientific community or the public. Some sponsors in both industry and academia have shared IPD and summary data reports upon request and on a case-by-case basis (Rathi et al., 2012). There are a number of initiatives at the NIH to share clinical trial data from the time of publication (Immune Tolerance Network, 2013; NHLBI, 2007). Recently, proposals have been put forth for more proactive sharing of both CSRs and IPD, and several plans have been announced or implemented (EMA, 2013; Krumholz and Ross, 2011; Kuntz, 2013; Loder, 2013; Nisen and Rockhold, 2013; PhRMA and EFPIA, 2013; YODA, 2013).
Many discussions of clinical trial data sharing thus far have not been specific regarding which of many possible clinical trial data elements or datasets might be shared. This framework articulates more specific defi-
nitions and descriptions of data that might be shared to help facilitate more focused discussions among the public and various stakeholders.
The committee has identified various types of clinical trial data (containing differing levels of detail) that might be included in a data sharing activity (see Figure 1).
Raw data Sometimes called source data, raw data are observations about individual participants. These data might be collected specifically for the study protocol, or as part of routine care and used by the investigators. At the source, data might be in the form of measurements of participant characteristics such as weight, blood pressure, or heart rate, and can be associated with the baseline (or initial) visit or subsequent followup visits.
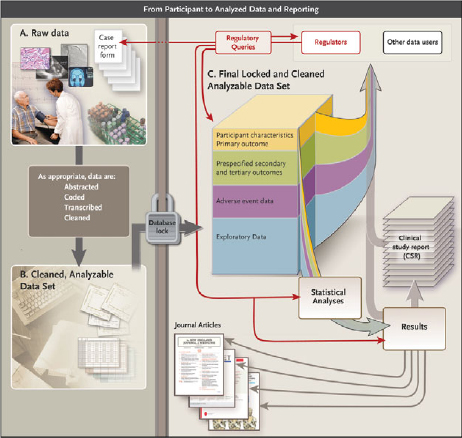
FIGURE 1 Data flow from participant to analyzed data and reporting.
NOTE: See Appendix C for more detail about each element of the figure.
Raw data might also include a baseline description of the participant’s medical history, physical exam information, clinical laboratory results (e.g., serum lipid values, hemoglobin levels), whole exome or genome sequences, or imaging (e.g., X-ray, magnetic resonance imaging). Depending on the trial, demographics, clinical data, and other appropriate raw source information are entered into case report forms. Some data must be abstracted and/or interpreted for the purposes of the protocol, for example, reading the X-ray for tumor size or evaluating the electrocardiogram for evidence of a heart attack. Data might also include assessments by clinical study staff or adjudication committees to determine whether specific clinical end points or adverse events in the participant’s profile (e.g., heart attack, death) meet protocol-specified criteria. In addition to “traditional” clinical trial data, other types of health data are increasingly being collected, including self-reported measures (e.g., quality of life), quantified sensor data (e.g., readings from remote monitoring devices, including smartphone apps and geolocation data), consumer genomics data (e.g., from companies like 23andMe), and community-level self-reported data (e.g., from sites like PatientsLikeMe).
Data entry into the database After collection (and assessment, abstraction, or adjudication as appropriate), source data typically must be entered into an organized data management system (i.e., database) for further evaluation and processing. Data typically undergo a process of cleaning, quality assurance, and quality control to detect inconsistent, incomplete, or inaccurate entries, and to confirm that the data were collected and evaluated according to the protocol and that they match the source data. This process continues throughout the course of the trial as data are collected.
Analyzable dataset Typically, after data are entered in computerized form, new variables are mathematically generated to serve as the basis for later analyses. These variables are sometimes called “derived” variables. For example, patient age might not be entered directly, but calculated by subtracting the birthdate from the date of a given clinic visit. “Treatment response” could be entered as a mathematical comparison of lesion sizes recorded from two images. After the trial is declared complete, the edited and cleaned data are moved into an analyzable data file and locked (i.e., no further changes may be made). If the study is blinded (or masked), then after the database is locked, the treatment code file is merged with the analyzable data file, and the data are unblinded to the investigators. Some or all of this now-unblinded analyzable dataset will then be used for data analyses.
A statistical analysis plan (SAP) is finalized before the trial is completed and unblinded; the SAP drives the initial analyses of the analyzable dataset. The trial protocol should contain both a basic SAP and a more detailed SAP that includes the analyses and any interim analyses to be conducted, as well as the statistical methods that will be used, as determined by the protocol. The full SAP includes, for example, plans for analysis of baseline descriptive data, adherence to the intervention, primary and secondary outcomes, definition of adverse events and serious adverse events, as well as the comparison of these measures across interventions for pre-specified subgroups. The analysis may be very extensive and can result in several dozen tables and figures.
For many clinical trials, the SAP-defined analysis might not use all of the data available in the analyzable dataset. Moreover, peer-reviewed journal publications of clinical trials generally draw on only part of the analyzable dataset. Supplemental data are often collected to permit exploration of ancillary questions not directly related to the primary purpose of the protocol, and researchers might conduct exploratory and post hoc analysis not defined in the SAP to answer additional questions.
Box 3 lists some of the types of reports that are commonly generated from the analyzable dataset. In addition to preparing peer-reviewed journal publications describing the primary and major secondary outcomes specified in the protocol, trial sponsors prepare various additional reports, including results summaries for registries, lay summaries, and clinical study reports.
Publications Several scientific journal publications are commonly derived from the analyses driven by the SAP and from post hoc analyses. Typically, a primary publication will address the primary and possibly the leading
BOX 3
Types of Summary Reports
• Lay-language summary
• Registry results summary (e.g., for ClinicalTrials.gov)
• Publications (including peer-reviewed scientific journals)
• Clinical Study Report (CSR)
![]() Full CSR, with or without appendixes
Full CSR, with or without appendixes
![]() CSR synopsis (executive summary)
CSR synopsis (executive summary)
![]() Redacted CSR
Redacted CSR
![]() Abbreviated CSR
Abbreviated CSR
secondary outcome measures specified in the protocol. The primary publication would also include the baseline measures to demonstrate participant comparability between intervention arms and comparisons of any adverse events of major interest or frequency. Subsequent journal publications might address in more detail a specific aspect of the primary analysis that was not included in the primary publication or analyze outcomes in particular prespecified subgroups of participants. Ideally, each journal publication should have a specific dataset corresponding exclusively to the data used to generate the tables and figures in the publication (which would be a subset of the full analyzable dataset). Each specific dataset is typically stored in separate sets of data files to document the data used for each journal publication.
Registry results summary and lay-language summary Many clinical trials are subject to a requirement that their results be reported to one or more registries, in formats specified by the particular registry (for example, results of trials of FDA-regulated products must be reported to ClinicalTrials.gov in the United States). These summaries are publicly available on the registry website and are generally limited to major outcomes and adverse events.
A lay-language summary is a brief, nontechnical overview written for the general public and trial participants. Lay summaries of the clinical trial protocol are often required by institutional review boards (IRBs) to assist nonscientist members of the IRB in the protocol review and approval process; however, the preparation of lay-language summaries of clinical trial results is uncommon. A recent study suggests that trial participants would value such summaries and that provision of lay-language results summaries to participants is feasible (Getz et al., 2012).
Clinical Study Report (CSR) When a clinical trial is submitted to regulatory agencies as part of an application for marketing approval of an intervention or approval of a new indication, trial sponsors usually submit a detailed CSR. Specifications for CSRs were defined by the ICH and adopted by the FDA, the EMA, and the Japanese Ministry of Health, Labor, and Welfare in an effort to simplify the application process for new interventions globally (ICH, 1995). According to the FDA guidance, the CSR is an
integrated full report of an individual study of any therapeutic, prophylactic or diagnostic agent … conducted in patients. The clinical and statistical description, presen-
tation, and analyses are integrated into a single report incorporating tables and figures into the main text of the report or at the end of the text, with appendices containing such information as the protocol, sample case report forms, investigator-related information, information related to the test drugs/investigational products including active control/comparators, technical statistical documentation, related publications, patient data listings, and technical statistical details such as derivations, computations, analyses, and computer output. (FDA, 1996, p. 1)
Although a CSR contains mainly summary data and summary tables and graphs, it also usually contains considerable additional information (often thousands of pages), including, as described in the definition above, numerous large appendixes. Supplemental information can include detailed narratives describing individual participants. In some instances, the CSR and/or its appendixes might include identifiable participant or commercially confidential information or other protected health information or intellectual property. A CSR synopsis (i.e., executive summary) is sometimes drafted to accompany a full CSR. Some of the supporting clinical trials included in a regulatory submission do not directly contribute to the evaluation of effectiveness of the intervention; for these studies, sponsors may be permitted to submit an abbreviated CSR (FDA, 1999). Some CSRs may also be redacted before they are shared to remove any commercially confidential information and personally identifiable information.
Metadata and Additional Documentation
In order for researchers to make use of clinical trial data that is shared with them (e.g., to perform confirmatory analyses or carry out exploratory analyses), they need further information or metadata (i.e., “data about the data”) in addition to the data elements or datasets described above. Box 4 summarizes some of the metadata that might be needed to facilitate full use of shared data. Supporting documentation critical to interpretation of shared clinical trial data include the full protocol, manual of operations, consent form, case report forms, and the SAP.
The trial protocol describes the trial rationale; the eligibility and exclusion criteria for participants; the primary and secondary hypotheses
and the corresponding primary and secondary outcome measures; the methods used to gather and adjudicate adverse events; other measures intended to evaluate the intervention; and a full description of the intervention and how it is administered. While a trial protocol provides the overall experimental design, a detailed manual of operations describes how the trial was conducted. A copy of the template for the informed consent form describes what participants agreed to, what hypotheses were included, and the additional purposes for which their data might be used. Case report forms capture precisely what measures were made, and at what time points during the trial, as defined in the protocol. The SAP sets out how each data element was analyzed, what specific statistical method was used for each analysis, and how adjustments were made for testing multiple variables. If some analysis methods required critical assumptions, data users would need to understand how those assumptions were verified. For key analyses, full use of the shared data would be aided by providing the computer software and version used, as well as the statistical programming code for the statistical software used for each analysis.
BOX 4
Metadata and Additional Documentation to Support the Use of Shared Clinical Trial Data
• Clinical trial registration number and dataset (available through ClinicalTrials.gov and other registries)
• Full protocol (e.g., all outcomes, study structure), including first version, last version, and all amendments
• Manual of operations (e.g., assay method) and standard operating procedures, including names of parties involved, specifically:
![]() names of persons on the clinical trial team, trial sponsor team, data management team, and data analysis team; and
names of persons on the clinical trial team, trial sponsor team, data management team, and data analysis team; and
![]() names of members of the steering committee, clinical events committee (CEC, which adjudicates end points), data and safety monitoring board (DSMB) or data monitoring committee (DMC), as well as committee charters
names of members of the steering committee, clinical events committee (CEC, which adjudicates end points), data and safety monitoring board (DSMB) or data monitoring committee (DMC), as well as committee charters
• Details of study execution (e.g., participant flow, deviations from protocol)
• Case report forms, informed consent forms, biospecimen information
• SAP, all amendments, and all documentation for additional work processes (including codes, software, and audit of the statistical workflow)
• Publications and report documents (see Box 3)
Summaries and reports (e.g., publications from the trial, lay-language summary of trial results, trial registry results summary, CSR synopsis) would also help shared-data recipients understand and make the most efficient use of shared data.
There are variations in how these data elements are defined and in the terminology used to describe them. In its future deliberations, the committee will seek clarity and consistency in use of the terms. The final report will include discussion of how the information in the analyzable dataset, CSR, and IPD differ and which types of analyses, either confirmatory or exploratory, require which level of data sharing.
Who Are the Providers of Shared Data?
Data are generated at almost every step in the clinical research process, from the initial collection of baseline participant data to the analysis of the analyzable dataset. Different individuals or organizations hold or control the data at different times during the course of the trial (e.g., laboratory technicians, investigators, database administrators, statisticians, DSMBs, sponsors, and regulatory agencies). A data holder may or may not have legal authority to share the data with others. In contrast, an individual or organization with the authority to share the data might not have physical possession of the data at a particular time. Thus, responsibility for providing data for sharing might need to be coordinated among several entities. Potential entities that are likely to be data providers include (but are not limited to) the following:
• Individual participants in a clinical trial (the initial “providers” of data to researchers). Some participants might hold data to the extent that they self-generate and transmit the data (from self-quantifying devices), retain copies of the data, or receive information from investigators. Participants could, in turn, share their information with organizations that aggregate data from many participants (e.g., disease advocacy groups, research platforms such as PatientsLikeMe, Reg4ALL, or Sage Bionetwork’s Bridge).
• Clinical trial funders (e.g., government, industry, foundations, or advocacy organizations).
• Contract research organizations that collect source data from participants on behalf of sponsors.
• Principal investigators or their institutions.
• Site principal investigators of a multisite trial or their institutions.
• The data or biostatistics coordinating center or the institution hosting the center.
• Regulatory agencies to which data are submitted.
• Systematic reviewers and guideline developers.
Potential users of shared data will need to know whom to contact to obtain the data they seek, who owns the data, who controls the data, and where they can get answers to questions that will inevitably arise about the dataset. As such, data providers might have an ongoing resource role, beyond simply sending data and metadata to recipients. The final report will include analysis of the advantages and disadvantages of various actors having responsibility for providing data to be shared.
Who Are the Recipients of Shared Data?
Many individuals and entities could be recipients of shared data from clinical trials. These include (but are not limited to) the following:
• Individuals participating in the trial, as a part of the agreement for participation, for a variety of reasons described above (e.g., trust, transparency, respect, engagement).
• Researchers seeking to reanalyze a study or explore new scientific questions.
• The institutions supporting the researchers.
• Funding agencies (e.g., government, private sector).
• IRBs or scientific peer review committees reviewing a new study of the same or a similar intervention in order to have a more comprehensive safety profile of the intervention.
• The DSMB or DMC for another clinical trial, whose decision to recommend continuing or stopping that trial can be informed by the results of a completed trial that has not yet been or will never be published.
• Educators requesting to use a dataset for teaching purposes (e.g., in a biostatistics class).
• A disease advocacy group seeking to advance research.
• Prospective plaintiffs or attorneys seeking information that could be used in current or future litigation.
• Competitors of the industry sponsor of the intervention studied in the trial.
• Members of the media.
• Interested members of the public.
Potential data recipients are interested in different types of data for potentially very different purposes.
When Might Clinical Trial Data Be Shared?
Data might be shared at various points in the timeline of a clinical trial, for instance,
• After publication of the primary results of the clinical trial in a peer-reviewed journal.
• After discontinuation of development of an intervention by a sponsor.
• After completion or early termination of a clinical trial.
• After regulatory approval of a new intervention or a new indication for the intervention.
• Following the occurrence of serious adverse events.
• Earlier, at the discretion of the data provider or generator.
The final report will include consideration of the advantages and disadvantages associated with disclosure of clinical trial data at various time points. The timing of data sharing could have very important implications that need serious consideration. For example, timing of release of the data will have consequences for the interests of the clinical trial team (e.g., the impact of data sharing on the timing of further publications from the data). Timing is also of great interest to the trial sponsor (e.g., relative to the timing of securing intellectual property rights or regulatory approval).
A variety of models for clinical trial data sharing have been proposed, planned, or implemented (select examples are summarized in Box 5). The types of data that are shared differ across the models. Proposed models of data sharing have generally imposed some sort of restriction on the sharing of data that could directly or potentially identify trial partici-
pants, as well as data that reveal CCI or trade secrets or might result in inaccurate analysis. Access to clinical trial data in current models ranges from essentially full access to de-identified data to fully restricted or no access.
In an open or public access model, data are made available, at a defined time, to any party who seeks them, for any purpose. For example, the EMA has announced that it will release, to any data requester that is a known entity to the agency, both summary and participant-level data (excluding, for example, personally identifiable data and information the EMA deems to be CCI) immediately after a regulatory decision about a new drug (Eichler et al., 2013; EMA 2013; Immport, 2013; Immune Tolerance Network, 2013; NHLBI, 2007).
In some models of data sharing, access is restricted to specific classes of user or for specific purposes. Requestors might need to demonstrate that they meet specified eligibility criteria. Some models require only the name and contact information of the requestor, while others require information about the proposed use of the requested data or how the data will be analyzed. Some models might also impose conditions relating to whether the data generators would receive credit in publications.
In some cases, the actual data are not provided to the requestor. Instead, data holders might run specific data analyses for approved requestors and deliver to the requestors only the results of the requested analyses. In another model, recipients receive credentials to access and run queries on the data, but are not able to download or obtain copies of the data.
Data sharing can also take place indirectly, through a “trusted intermediary” or “honest broker,” who either negotiates the conditions for data sharing (with the data provider retaining control over the data and its release) or takes full control of the data and brokers both the conditions for data release and the delivery to recipients (Mello et al., 2013). Trusted intermediaries might also accept and facilitate data analysis queries from secondary investigators, as mentioned above. The use of a trusted intermediary raises a number of issues, including selection, administration, funding, and compensation.
A controlled-access data sharing model could implicitly or explicitly address issues such as specification of data that will be shared; any categories of data that are to be specifically restricted; and how to address risks of breaches of confidentiality, attempts to re-identify trial participants, data retention periods, scientifically inappropriate analyses, and the need for timely responses to data requests.
To obtain shared data under a controlled-access model, a recipient typically must execute a data use agreement (DUA). Conditions in the DUA might include
• prohibitions on re-identification or contact of individual trial participants;
• requirements to acknowledge the providers of the data in any publications resulting from the shared data;
• requirements to send copies of submitted manuscripts and publications to the trial investigators or study sponsor;
• restrictions on further sharing of the data with additional parties or using the data for purposes other than originally proposed;
• assignment of intellectual property rights for discoveries from the shared data;
• requirements to publish or post findings from the data; and
• requirements to notify industry sponsors of the trial of any findings that raise safety concerns.
There might also be limits on the length of time that a recipient may use or access shared data (ADNI, 2013; Harvard University MRCT, 2013; Nisen and Rockhold, 2013; PhRMA and EFPIA, 2013; YODA, 2013).
Selected Set of Clinical Trial Data Sharing Activities
The possible models and approaches to clinical trial data sharing, and the purposes motivating that sharing, are extensive. The rationale, benefits, risks, and burdens associated with one particular data sharing activity could differ from those of another data sharing activity, depending on the data elements and parties involved.
To stimulate public comments and to provide heuristic organizational structure to its work, the committee has described a selected set of data sharing activities as examples of the types of arrangements or approaches under which clinical trial data might be shared (see Box 5). To derive this selected set, the committee reviewed a range of existing and pro-
posed data sharing activities and distilled them into four conceptual categories that represent broad “families” of activities that have key features in common. No single proposed or enacted data sharing activity has all of the characteristics in the familial description, and different data sharing activities in the same category can have important differences. The categories therefore are derived from, but do not describe any specific, data sharing activities currently underway or proposed. To the extent there are redundancies in characteristics within the models, the committee will address them or take them into account in its forthcoming analysis.
Each activity is described according to the type(s) of data that could be shared; provider(s) of data; recipients of data; timing of data sharing; conditions or qualifications for access; and conditions of data use. The descriptive characteristics are an illustrative but not exhaustive list. Detailed descriptions and, particularly, conclusions and recommendations regarding those descriptive characteristics or strategies and approaches for sharing will be included in the final report.
BOX 5
Set of Clinical Trial Data Sharing Activities
1. Open Access
A data sharing program or system in which data are made broadly available to the public through an open-access website. Data might be aggregated from multiple sources (i.e., more than one institution, company, or researcher), or the website might provide open access to data from one trial or one institutional or individual researcher. Aggregated data might require trial- or institution-specific queries. An open-access data sharing activity could have some or many of the following descriptive characteristics:
a. Type of data: summary data or anonymized IPD
b. Providers of data: as determined by the organizational structure of the data sharing activity. Providers could include sponsors of applications for regulatory approval of products, researchers funded by a government source, or other data generators or aggregators (such as regulatory agencies) required or agreeing to share data
c. Recipients of data: all members of the public via a website
d. Timing of sharing: upon publication or after regulatory decision or abandonment of drug development
e. Conditions or qualifications for access: none; no or minimal identification/log-in requirements; no governing body to make access determinations
f. Conditions of data use: no conditions of use or requirement for signed data use agreement; expectation to abide by applicable
law (e.g., intellectual property protections, privacy protections)
a. Example activities:a EMA Category 2, National Institute of Allergy and Infectious Diseases’ Trial Share, Immunology Database and Analysis Portal (ImmPort); National Heart, Lung, and Blood Institute’s BioLINCC
2. Controlled Access to Individual Company, Institution, or Researcher Data
A data sharing program or system in which data are made available to requestors on a controlled-access basis, pursuant to defined restrictions or conditions. Data are from one institution or researcher. Data might require or permit trial- or product/intervention-specific queries. A controlled access–individual entity data sharing activity could have some or many of the following descriptive characteristics:
a. Type of data: summary data, anonymized, or limited dataset
b. Providers of data: an individual company, institution, researcher, or other entity generating or assembling data (e.g., a patient advocacy organization)
c. Recipients of data: members of the public via a website
d. Timing of sharing: upon publication or after regulatory decision or abandonment of drug development
e. Conditions or qualifications for access: access controlled by the institution/data generator or by a specified governing body such as a learned intermediary. Controls could include requirements to register, have specified expertise (e.g., statistics), successfully complete a test to demonstrate expertise, disclose funding sources, provide a purpose for the data request that meets specified criteria (e.g., related to public health or patient care), or provide an analysis plan (which might or might not need to meet specified criteria)
f. Conditions of data use: restrictions or conditions on data use might include a requirement to sign a data use agreement, requirement to provide and adhere to a publication plan (which might need to meet specified criteria), prohibition on use of data for commercial purposes, prohibition on attempts to re-identify data, requirement to inform data generator about any safety concerns identified, agreement that the data generator retains exclusive rights to inventions or other intellectual property generated by the data recipient, or requirement to acknowledge the source of the shared data and the investigators in the original clinical trial
g. Example activities: PhRMA/EFPIA commitments, Harvard Multi-Regional Clinical Trials Center proposal, Yale Open Data Access
3. Controlled Access to Pooled or Multiple Data Sources
A data sharing program or system in which data are made available to requestors on a controlled-access basis pursuant to defined restrictions or conditions. Data are aggregated from multiple sources (i.e., more than one institution, company, or researcher). Aggregated data might require or permit trial- or institution-specific queries. A controlled access–multiple entity data sharing activity could have some or many of the following descriptive characteristics:
a. Type of data: summary data or anonymized IPD
b. Providers of data: as determined by the organizational structure of the data sharing activity. Providers could include sponsors of applications for regulatory approval of products, researchers funded by a government source, or other data generators as required or agreed on
c. Recipients of data: members of the public via a website
d. Timing of sharing: upon publication or after regulatory decision or abandonment of drug development
e. Conditions or qualifications for access: access controlled by the institution/data generator or by a specified governing body, such as a learned intermediary. Controls could include requirements to register, have specified expertise (e.g., statistics), successfully complete a test to demonstrate expertise, disclose funding sources, provide a purpose for the data request that meets specified criteria (e.g., related to public health or patient care), or provide an analysis plan (which might or might not need to meet specified criteria)
f. Conditions of data use: restrictions or conditions on data use might include a requirement to sign a data use agreement, requirement to provide and adhere to a publication plan (which might need to meet specified criteria), prohibition on use of data for commercial purposes, prohibition on attempts to re-identify data, requirement to inform that data generator about any safety concerns identified, agreement that the data generator retains exclusive rights to inventions or other intellectual property generated by the data recipient, requirement to acknowledge the source of the shared data and the investigators in the original clinical trial
g. Example activities: GSK, EMA Category 3 (clinical trial data with Protection of Personal Data concerns), Alzheimer’s Disease Neuroimaging Initiative
4. Closed Partnership/Consortium
A data sharing program in which data are shared between or among two or more parties. Arrangements could be between or among public, nonprofit, or for-profit entities. Data are not intended to be shared with individuals or others who are not party to the partnership or consortium arrangement. A closed partnership/consortium data sharing activity could have some or many of the following descriptive characteristics:
a. Type of data: summary data, IPD (anonymized or not)
b. Providers of data: as determined by the parties to the arrangement or initiators of the data sharing activity. Providers could include companies or other product development organizations, individual researchers or institutions, or other entities generating data (e.g., a patient advocacy organization) as agreed upon
c. Recipients of data: members of the partnership or consortium
d. Timing of sharing: as agreed upon by the terms of the partnership or consortium arrangement
e. Conditions or qualifications for access: access controls determined by partnership/consortium arrangement
f. Conditions of data use: restrictions or conditions on data use might include a requirement to sign a partnership/consortium agreement, an agreement governing rights to inventions or other intellectual property generated by the data sharing activity, or an agreement dictating whether and how future publications will be undertaken
g. Example activities: PatientsLikeMe
____________________
aActivities listed here are considered examples of the described data sharing activity because they have some or many of the listed descriptive characteristics. No one activity would necessarily have all of the listed characteristics (and some of those characteristics might be mutually exclusive). The activities cited as examples could vary substantially from each other depending on their descriptive characteristics.
This page intentionally left blank.




































