
Important Points Highlighted by Individual Speakers
• The draft guidance on companion diagnostics from FDA recommends developing both the test and the drug together, which creates a new opportunity for developers to work and learn together to more clearly define a process and to coordinate a timeline for development.
• The performance of a companion diagnostic is closely tied to the performance of the associated drug, and this relationship is essential for determining the safety and effectiveness of the products for patient use.
• The major challenge for co-development is more commercial than regulatory because there are inherent differences in developing tests and drugs, including mismatched markets and resources.
• Combining the cost of a test and a drug may provide a solution for aligning market differences and accelerating regulatory approval and reimbursement decisions for two different products.
Speakers discussed regulations from the perspective of FDA and of an entrepreneur with a focus on personalized medicine, in the context of the current co-development process for tests and drugs. Elizabeth
Mansfield, director of the personalized medicine staff in the Office of In Vitro Diagnostics and Radiological Health at FDA, described the historical development of FDA’s policies for companion diagnostics and the main features of the companion diagnostic draft guidance. Felix Frueh, entrepreneur-in-residence at Third Rock Ventures, commented on some of the issues those policies raise. Together, they presented an overview of co-development of tests and therapeutics and outlined the challenges of and potential solutions to these issues.
OVERVIEW OF CO-DEVELOPMENT AND COMPANION DIAGNOSTIC POLICY
The concept of companion diagnostics is not new, Mansfield said; testing for estrogen and progesterone receptor expression has been done since the 1990s to determine if a patient would benefit from hormone therapy in treating breast cancer. In 1998 FDA approved the use of the drug Herceptin in patients with breast cancer who tested positive for human epidermal growth factor receptor 2 (HER2), one of the earliest examples of a co-development companion diagnostic model before there was a formal process in place. When Herceptin was approved for use in patients with metastatic breast cancer, FDA also approved a test to examine HER2 levels. One reason for having a test was to decrease risks, Mansfield said, and Herceptin’s cardiotoxic side effects are now well known. In the case of the drugs Selzentry and Tykerb, which were approved in 2007 and whose use depends on test results, a companion diagnostic policy was not yet in place when they were approved and FDA did not apply the policy retroactively.
Recognizing that tests can be drivers of therapy, FDA began to develop guidance to reflect drug development strategies that account for genetic information. It held public discussions about pharmacogenomics, requested voluntary genomic data submissions, and addressed other issues concerning the use of genomic data to guide drug development, Mansfield said. FDA realized that a policy was needed to protect patients while also allowing companies to plan for the development of tests that would support therapeutic approval, she said. FDA also realized that companies want predictability in the regulatory process.
Companion diagnostics are tests, Mansfield emphasized, but the test performance is closely tied to the performance of the associated drug. Thus, knowledge about the test is essential to understand the safety and effectiveness of the drug. Tests for the same analyte can differ, sometimes significantly. The technology, cut-off levels, and performance can all vary, and different tests are likely to identify different populations. “You want to know all of these parameters before you decide on which test is going to be used,” Mansfield said, “and if you want to use multiple tests [you will need
to know] how these different performance parameters compare and affect the outcome [because] test performance actually makes the drug performance.” All of this information is needed to determine which patients will benefit from the drug and how to adequately label a drug.
In July 2011 FDA released a draft guidance document for industry and FDA staff on IVD companion devices and held a 90-day comment period, Mansfield said. At the time of the workshop, FDA expected to release the final version of the guidance soon. Mansfield reviewed a few key pieces of the policy. First, the policy defines an IVD companion device as “an in vitro diagnostic device that provides information that is essential for the safe and effective use of a corresponding therapeutic product.” Such a diagnostic could identify a population for efficacy, for safety, or for other purposes. The document also differentiates companion diagnostics from diagnostics used for other purposes. Thousands of diagnostic tests have been cleared or approved, but only perhaps 15 companion diagnostics had been approved (see Table 2-1) at the time of the workshop, Mansfield said.
TABLE 2-1 FDA-Approved Companion Diagnostic Devices
|
|
||
| Drug (trade name) |
Device (trade name) |
Intended Use |
|
|
||
| Erbitux | therascreen KRAS RGQ PCR Kit | Real-time qualitative PCR assay used for the detection of seven somatic mutations in the KRAS in colorectal cancer tissue. |
| Erbitux, Vectibix | DAKO EGFR PharmDx Kit | Qualitative immunohistochemical assay to identify EGFR expression in normal and neoplastic tissues and as an aid in colorectal cancer tissue. |
| Exjade | Ferriscan | Measures liver iron concentration to aid in the identification and monitoring of non-transfusion dependent thalassemia patients. |
| Gilotrif | therascreen EGFR RGQ PCR Kit | Real-time PCR test for exon 19 deletions and exon 21 (L858R) substitution mutations of EGFR in NSCLC tumor tissue. |
| Gleevec/Glivec | DAKO C-KIT PharmDx |
Immunohistochemical assay for the identification of c-kit protein/CD117 antigen expression in normal and neoplastic tissues and as an aid in diagnosing gastrointestinal stromal tumors. |
| Herceptin | INFORM HER-2/ neu |
FISH DNA probe assay for HER2/neu gene amplification in human breast tissue as an aid to stratify breast cancer patients. Also indicated for use in breast cancer in patients. |
|
|
||
| Drug (trade name) |
Device (trade name) |
Intended Use |
|
|
||
| Herceptin | PathVysion HER-2 DNA Probe Kit |
Detects amplification of the HER2/neu gene via FISH in breast cancer tissue specimens. |
| Herceptin | PATHWAY ANTI-HER-2/NEU (4B5) Rabbit Monoclonal Primary Antibody | IHC test for c-erbB-2 antigen in normal and neoplastic tissue. Indicated as an aid in the assessment of breast cancer patients. |
| Herceptin | InSite HER2/neu kit | IHC assay for the over-expression of HER2/neu (i.e., c-erbB-2) in normal and neoplastic tissue sections. Indicated as an aid in the assessment of breast cancer patients. |
| Herceptin | SPOT-Light HER2 CISH Kit | CISH for HER2 gene amplification in breast carcinoma tissue. |
| Herceptin | Bond Oracle HER2 IHC System | Immunohistochemical assay to determine HER2 oncoprotein status in formalin-fixed, paraffin-embedded breast cancer tissue. |
| Herceptin | HER2 CISH PharmDx Kit | In situ hybridization assay for the HER2 gene and centromeric region of chromosome 17 for breast cancer tissue specimens. |
| Herceptin | INFORM HER2 DUAL ISH DNA Probe Cocktail | In situ hybridization assay for HER2 gene status by enumeration of the ratio of the HER2 gene to Chromosome 17 in breast cancer tissue specimens. |
| Herceptin, Perjeta | HERCEPTEST | Immunocytochemical assay to determine HER2 protein overexpression in breast and gastric cancer. |
| Herceptin, Perjeta | HER2 FISH PharmDx Kit | FISH assay for HER2 gene amplification in breast cancer tissue specimens and specimens from patients with metastatic gastric or gastroesophageal junction adenocarcinoma. |
| Mekinist; Tafinlar | THxID™ BRAF Kit | Qualitative detection of the BRAF V600E and V600K mutations in melanoma tissue. |
| Tarceva | cobas EGFR Mutation Test | Real-time PCR test for exon 19 deletions and exon 21 (L858R) substitution mutations of EGFR in NSCLC tumor tissue. |
| Xalkori | Vysis ALK Break Apart FISH Probe Kit | FISH for ALK gene rearrangments in NSCLC tissue specimens. |
| Zelboraf | cobas 4800 BRAF V600 Mutation Test | Real-time PCR test for BRAF V600E mutation in melanoma tissue. |
|
|
||
NOTE: CISH, chromogenic in situ hybridization; EGFR, epidermal growth factor receptor; FISH, fluorescence in situ hybridization; IHC, immunohistochemistry; NSCLC, non-small-cell lung cancer; PCR, polymerase chain reaction.
SOURCE: Modified from FDA Companion Diagnostic Devices: In Vitro and Imaging Tools, http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm301431.htm (accessed October 16, 2013).
Second, the policy calls for contemporaneous approval of the therapeutic and the companion diagnostic, Mansfield said. “If we approve a test without drug approval, then the test has no legitimate intended use. If a drug is approved without the test, then it is likely that the test wasn’t needed.” The exception to this policy, she said, is that if a therapy is meeting an unmet need, the drug can be approved first with the test approval following soon after so as to not hold up the approval of therapies for diseases that do not have alternative treatment options. FDA has no preference for the manufacturer of a particular test; sponsors determine which test will be submitted for approval. This has led to a learning experience for both test developers and drug developers, Mansfield said, because “these two sectors have not been very familiar with each other.” But the situation is changing, in part because some pharmaceutical companies are creating small diagnostic enterprises to support their personalized medicine efforts.
Mansfield further clarified the labeling policy for therapeutic products. The label refers to “a type of approved or cleared IVD companion diagnostic device, not a specific one by name,” she said. While the test can be named elsewhere in the label, it will not be named in the indications, warnings, or precautions sections. This is to account for the fact that better tests may be approved at a later date, a possibility that FDA wanted to account for, Mansfield said. As the policy states, “This will facilitate the development and use of more than one approved or cleared IVD companion diagnostic device of the type described in the labeling for the therapeutic product.” Also, putting the test name in the label would essentially make the drug–test pair a combination product, which falls under a different category of regulation at FDA. A rare exception to this would be when only one test can be used with the drug.
FDA is also considering what the process will be for follow-on tests and how to account for new information for already-approved therapies. It does not foresee being able to apply a single approach to all follow-on tests, Mansfield said. The biggest concern for such tests is that the population tested in seeking approval for the test would be biased to make the test perform better than it would in practice. Generally, the process for follow-on tests involves an analytical comparison, but samples with clinical outcomes typically are not available. Once a drug is approved, it is unethical or unworkable to run a trial to generate clinical trial specimens with clinical trial outcomes. “Those specimens are essentially gone, and you have to start with a different type of specimen,” she said. As a result, many of the considerations in approving such a test involve the specific test and specific drug. Mansfield added that regardless of whether the test is a follow-on or the initial development, premarket approval is still needed because of the high risk of harm. In contrast, the labeling of the companion diagnostic will list the name of the drug, Mansfield said. The users of the drug need
to know which test to use, and the performance characteristics of the test in the label are generally derived from the therapeutic trial of the drug.
The use of a test in a therapeutic trial is often investigational. In such cases, the risk of use must be determined, and significant risk requires a submission to FDA to ensure safety, regardless of the manufacturer of the test or whether the test is in use. The development of a test is often exempt from investigational regulations, but when the test is used in therapeutic trials, it may not be, Mansfield said. This risk assessment applies to LDTs and tests made by IVD manufacturers, she added.
Mansfield also addressed why certain drugs did not have a companion diagnostic approved together under the policy. She explained that, for example, Kalydeco, a drug that works well in patients with cystic fibrosis who have a particular mutation, was approved by FDA without a companion diagnostic. In the case of cystic fibrosis, 95 percent of patients have a genetic mutation panel performed at the time they are diagnosed, so they already know their mutation status and therefore do not need to be retested to determine whether they should be placed on Kalydeco (ACOG, 2011). Even for Kalydeco’s clinical trial, the patient’s medical record was used to determine their mutation status.
FDA also has been working on guidance for the co-development process, Mansfield said, but the guidance has been difficult to write because of the programs that the agency has reviewed so far, no two co-development programs are the same. Both industry and FDA are gaining experience as the policy is defined, Mansfield said. Most of the general principles in the guidance have been drafted, and Mansfield said she hoped that it would be out to the public within the year. The guidance will likely discuss both therapeutic and diagnostic programs with an emphasis on the diagnostic process.
Other non-IVD diagnostics are also being considered as companion diagnostics. An example is the recent approval of Exjade, a drug used to treat non-transfusion-dependent thalassemias and its companion radiological test to measure liver iron concentration.
As FDA and industry have increased their knowledge and experience with the co-development of diagnostics and therapeutics, the approval process has become smoother, Mansfield concluded, but questions remain, and new ones arise every day.
In the context of co-development, effectiveness means that a drug or test is adequate to accomplish a specific purpose, produces the intended or desired result, and is actually in operation as opposed to just having the potential for use, Frueh said. For a product to be approved by FDA, effectiveness needs to be demonstrated, so it is primarily a regulatory concern.
By contrast, efficacy refers to the power or capacity to achieve the desired effect under ideal conditions, and it is more of a concern for payers. For example, Frueh said, in a clinical trial inclusion and exclusion criteria create a more idealized situation than would be encountered outside of this environment. Co-development is the “development of a test with a drug to make the use of the drug more effective or safer,” he said. “It really is a method to make medicine more precise.” Co-development is not an approach to make a clinical trial less costly, nor is it a way to accelerate the time it takes to bring a product to market, Frueh said. Co-development is also not a biomarker discovery tool, he said, because “by definition you have to know your marker and you have to know what you’re using it for; only that allows you to create a strategy to align the development of the marker with the drug.”
Hurdles to Co-Development
The co-development of a diagnostic and a therapeutic is not a regulatory issue, Frueh said. “I do not believe these regulatory hurdles exist.” In fact, he said, FDA is helping with accelerated approvals, as in the cases of Xalkori, Kalydeco, and Incivek. The most significant obstacles that need to be dealt with involve business development and reimbursement issues. “Drug development and test development are inherently difficult to coordinate,” Frueh said. “For the most part, the timelines of the two businesses really do not align. The resources are completely mismatched, and the market protection between the drug and the diagnostic is entirely different.” The underlying differences result from the differences in markets between drugs and tests. Drugs typically involve high-risk investments and high rewards, he said, while tests make up a significantly smaller fraction of the market and come with moderate to high risk and low rewards. As a result, Frueh said, there is less interest in investing in medical devices and IVDs because they are not as lucrative as making an investment in pharmaceuticals.
In order to explain the return on investment for a test, Frueh provided a detailed scenario. A $500 test intended to be used by a population of 1 million represents a potential market of $100 million if the test is able to reach 20 percent of the population. If the profit margin is 50 percent, then an initial investment of $100 million would be recovered in 2 years. If such a test took 4 years to develop, it would start to make a profit after 6 years. A modest return on investment would be three times the original amount invested, Frueh said, or $300 million in the case of this hypothetical test. Thus, this test would take 12 years to produce the desired return on investment, which represents a 9.5 percent annual rate of growth of capital. “This doesn’t necessarily sound unreasonable,” Frueh said, “but it’s not really
something that gets venture capitalists overly excited.” Because these tests produce a relatively low return on investment—at least from the point of view of venture capitalists—the diagnostic market is relatively unattractive to investors.
Some new drugs are currently priced at record levels; for example, the price of Xalkori is approximately $115,000 per patient per year, and other new drugs are also extremely expensive, Frueh said. Yet the cost of the EML4-ALK companion diagnostic test that is required for the use of Xalkori is reimbursed at $128.48,1 and diagnostic companies are under pressure to reduce prices further. As a result, the disparity between drug and diagnostic prices is increasing, and the goals of drug development tend to dictate the goals of the test development in a co-development effort. Not every diagnostic will progress all the way to the market, nor will the price for the test necessarily be the same 12 years after development begins. “The reality is that the rewards and stakes for drug development are significantly higher than they are for test development,” Frueh said. “This is not a relationship between two equal partners.” As knowledge grows, new markers might be developed, requiring the development of a new test. But the developer does not necessarily have market exclusivity or intellectual property protection; another group could develop a different diagnostic for the same marker and acquire part or all of the market.
Potential Solutions for Co-Development
The critical consideration for a test or drug is the resulting health outcomes, Frueh said. If a test adds to the effectiveness of a therapy, the test has inherent value. Furthermore, the difference in the effectiveness of a drug with the test versus without the test is a measure of the value of the test. Co-development also is aligned with payers’ current demand for more evidence of positive outcomes. Furthermore, payers view such tests as a way to control costs. To capture the value of the test in the payment, Frueh said, “if you get a product that doesn’t work, you also don’t really want to pay for it.… Yet we do it every day in the health care system. I think we need to get out of this loop and really look at what works.”
If payments were for outcomes rather than the products, then the value, taking into account both the test and the drug, could be reflected in the payment. In that case, Frueh said, it would make sense to pay for the test and drug together, which then raises a reimbursement issue. Combining the cost of a test and a drug (see Box 1-2, individual participant submission)
____________________
1CGS Administrators, Molecular Pathology Reimbursement for Dates of Service 01/01/13–09/30/13, http://www.cgsmedicare.com/ohb/coverage/mopath/mopath_reimbursement.html (accessed October 16, 2013).
would make it easier to justify the initial expense to develop the tests. This approach would provide the outcome evidence that payers need, and it also would accelerate regulatory approval and reimbursement.
Such a change would not be easy to make, Frueh acknowledged. But combining the costs for a test and a drug would contribute to an ongoing shift in thinking about regulatory and payer issues from a static paradigm to a more dynamic state, he said (see Figure 2-1). Incorporating reimbursement considerations into strategies for trial designs would improve both efficacy and effectiveness, thus serving the interests of both regulators and payers. The drug industry and diagnostic industry would be more equal, so that they could continue to interact together with FDA during the approval process. This team approach to co-development would help align the two industries, Frueh said.
Two models could incentivize co-development of drugs and diagnostics, Frueh said. The first would be for those involved in early test development so that their investment would be fully compensated by pharmaceutical companies. In this scenario, the drug company would assume the financial responsibility for the test development. Second, a revenue-sharing model could be considered in which a percentage of the drug revenue is generated from sales based on decisions made using the test, Frueh said. This model would reflect a partnership between the two development sectors.
During the discussion, Mansfield, Frueh, and individual workshop participants considered the potential roles of next-generation sequencing in disease diagnosis and in affecting outcomes. Genomic information could have both prognostic value and, in some cases, predictive value for a therapeutic. The use of next-generation sequencing as a diagnostic will require work and discussion with potential sponsors, Mansfield said. Some devices are complicated in that they have more than one indication, such as prostate-specific antigen (PSA) tests for monitoring cancer, which are classified as a Class II device, and PSA tests for diagnosis, which are classified as a Class III device. In the case of molecular diagnostics, genomic information will point toward therapies for which no indication in a drug label exists, which is a much bigger regulatory challenge, Frueh said. “The sheer magnitude of the information that we’ll find on the genetic and molecular level is going to far surpass our capacity to run clinical trials,” he said. In fact, perhaps clinical trials will not be needed, especially because clinical trials cannot be run for every marker and every condition. But the same mutation is not always the driving factor for different cancers, said Walter Koch, vice president of global research at Roche Molecular Systems, and a workshop speaker. In contrast to Frueh’s view, Koch said that only through clinical
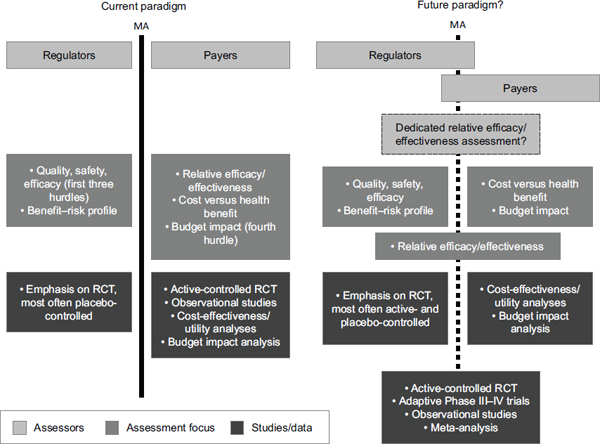
FIGURE 2-1 Integrating regulatory agencies and payers for product development.
NOTE: MA, marketing authorization; RCT, randomized controlled trial.
SOURCE: Reprinted by permission from Macmillan Publishers Ltd.: Nature Reviews Drug Discovery (Eichler et al., 2010), copyright 2010.
trials can a drug target be validated by showing that the drug produces better outcomes. Next-generation sequencing will find many variants, but they will not always be targets for a particular disease, he said.
Speaking for herself and not on behalf of FDA, Mansfield stated that medicine is heading toward next-generation sequencing, which is “the ideal place to be,” she said. “When you get diagnosed, you get a test, and we have all the information on the table from a single test.” In that case, the test may be quite general as opposed to being used as a companion diagnostic to provide usage information about a single targeted therapy. In the future, next-generation sequencing could report only mutations that have known drug safety or efficacy correlations, Mansfield said, with additional data being retained for investigational use. As new information becomes available, new drugs could be approved, which would greatly increase the efficiency of the approval process. But, she said, “even in co-development … not everybody who has [a particular] marker actually benefits from the drug. So we’re still not there yet, even with next-generation sequencing. To the degree that subclassification actually improves that, that’s great. But I think we can’t just assume that because you have a mutation, you’re going to get benefit from the drug because we know the opposite.”
This page intentionally left blank.












