
1
Introduction
BACKGROUND
Characterization, or ''typing," of blood, semen, and other body fluids has been used for forensic purposes for more than 50 years.1 It began with blood groups, such as those of the ABO system, and later was extended to serum proteins and red-cell enzymes and in some forensic applications, particularly paternity testing, to human leukocyte antigens (HLA), which are associated with tissue types. The genetically determined person-to-person variation revealed by such typing was used mainly to include or exclude suspects, that is, to determine whether a person showed a combination of genetically determined characteristics consistent with having been the source of an evidence sample in a criminal case or having been the father of a child in a paternity case. Except when HLA testing was used, the chance that a randomly chosen person would be excluded by the tests was about 98%; that left a 2% chance that the test would "include" an innocent person.
In the last decade, methods have become available for deoxyribonucleic acid (DNA) typing, that is, for showing distinguishing differences in the genetic material itself. Advances in DNA technology in the 1970s paved the way for the detection of variation (polymorphism) in specific DNA sequences and shifted the study of human variation from the protein products of DNA to DNA itself. By analyzing a sufficient number of regions of DNA that show much person-to-person variability, one can reduce the probability of a chance match (inclusion) of two persons to an extremely low
level. Indeed, the probability can, in principle, be made so low that DNA typing becomes not simply a method for exclusion or inclusion, but a means of absolute identification.
The potential applicability of DNA typing to forensic samples was demonstrated during the mid-1980s by laboratories in the United Kingdom, United States, and Canada. Their work established that DNA was present in forensic samples in sufficient quantity for testing (see Table 1.1) and that it survived in a state that allowed it to be typed. In the publications in 1985 by Jeffreys and colleagues,2,3 the term ''DNA fingerprint" carried the connotation of absolute identification. The mass-media coverage that accompanied the publications fixed in the general public's minds the idea that DNA typing could be used for absolute identification. Thus, the traditional forensic paradigm of genetic testing as a tool for exclusion was in a linguistic stroke changed to a paradigm of identification. (See Box 1 for a contrasting of dermatoglyphic fingerprints with "DNA fingerprints.")
Forensic DNA typing, first used in casework in 1985 in the United Kingdom, was initiated in the United States in late 1986 by commercial laboratories and in 1988 by the Federal Bureau of Investigation (FBI) and is now being used by dozens of state and local crime laboratories. Because of its great potential benefits for criminal and civil justice, but also because of the possibilities for its misuse or abuse, forensic DNA typing has been subjected to special scrutiny. Important questions have been asked about reliability, validity, and confidentiality:4-6
TABLE 1.1 DNA Content of Biological Samples
|
Type of Sample |
Amount of DNAa |
|
Blood |
20,000-40,000 ng/ml |
|
stain 1 cm2 in area |
ca. 200 ng |
|
stain 1 mm2 in area |
ca. 2 ng |
|
Semen |
150,000-300,000 ng/ml |
|
postcoital vaginal swab |
0-3,000 ng |
|
Hair: |
|
|
plucked |
1-750 ng/hair |
|
shed |
1-12 ng/hair |
|
Saliva |
1,000-10,000 ng/ml |
|
Urine |
1-20 ng/ml |
|
a The amount of DNA is given in nanograms (ng); 1 ng = one-billionth of a gram (10-9 g). |
|
|
FINGERPRINTS IN PERSONAL IDENTIFICATION: DIFFERENCES BETWEEN DNA TYPING AND DERMATOGLYPHICS Because of the central role of dermatoglyphic fingerprints in human identification, arising out of the personal uniqueness of the patterns, it is useful to compare and contrast traditional fingerprints with "DNA fingerprints". Fingerprints were described as an individualizing characteristic as early as 1892. The use of fingerprints in forensic science (and in relation to chromosonal abnormalities, such as Down syndrome, and other clinical disorders) was developed empirically without reference to the specific genetic basis of patterns. Ridge count is a polygenetic or multifactoral trait. The close correlation for ridge counts with that expected for an almost exclusively genetic trait, when "identical" and fratenal twins and other relatives are compared, supports polygenetic inheritance. In the forensic application, minutiae in the fingerprint patterns, not ridge counts, are used for personal identification. The minutiae result from random nongenetic events during embryonic development of the fingerpads. As a consequence, the patterns even of "identical" twins are distinguishable. Indeed, it appears that the fingerprint pattern of each human being is unique. The distinction between two types of fingerprints is illustrated by prints from "identical" twins shown here. The dermatoglyphic fingerprints shown in figure B-a (Twin A) and figure B-B (Twin B) are distinguished by the patterns of loops and whorls.
However, typing of DNA from the blood of these twins in three laboratories showed a match for all tests. One laboratory, testing for variation in four chromosones, estimated the population frequency of |
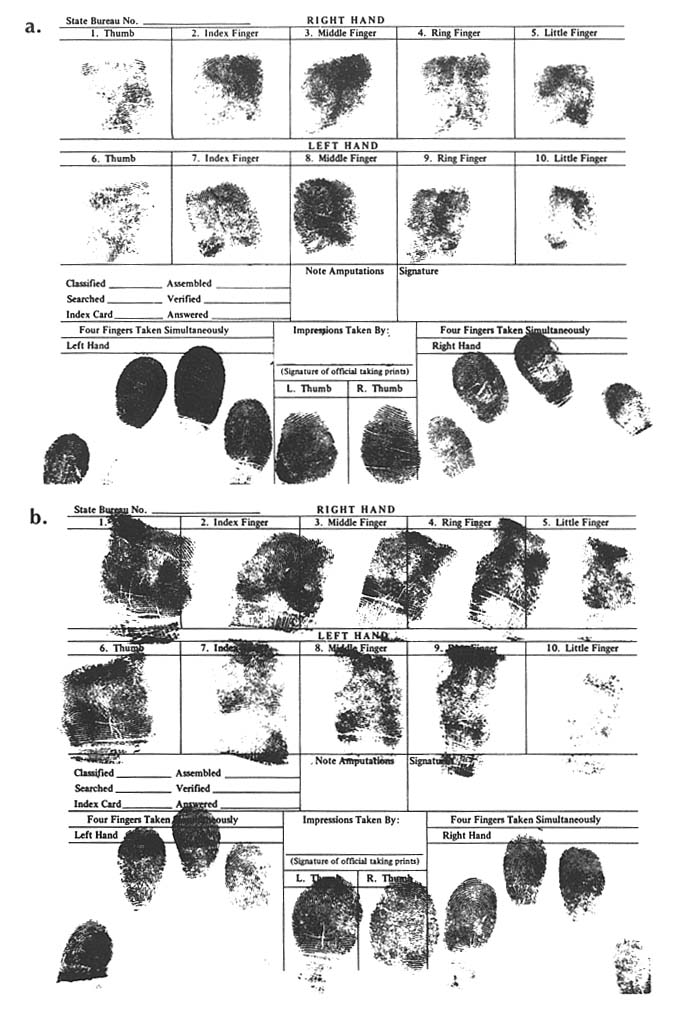
the particular DNA patterns to be 1 in about 700,000. A second laboratory used four other probes and estimated the chance of a random match as 1 in 1.8 million. For illustrative purposes, the patterns obtained with a "cocktail" of four signal-locus probes are shown in figure B-c. Twin A gave sample B, twin B gave sample E, and samples A, C, and D were from unrelated males of the same ethnic group. All five samples were submitted and tested in a blind manner. The other lanes show controls. (Courtesy of Robin W. Cotton and Matthew John McCoy, Cellmark Diagnostics.)
The uniqueness of the "DNA fingerprint" is based on genetic variation, whereas that of the dermatoglyphic fingerprint is based largely on nongenetic variation.
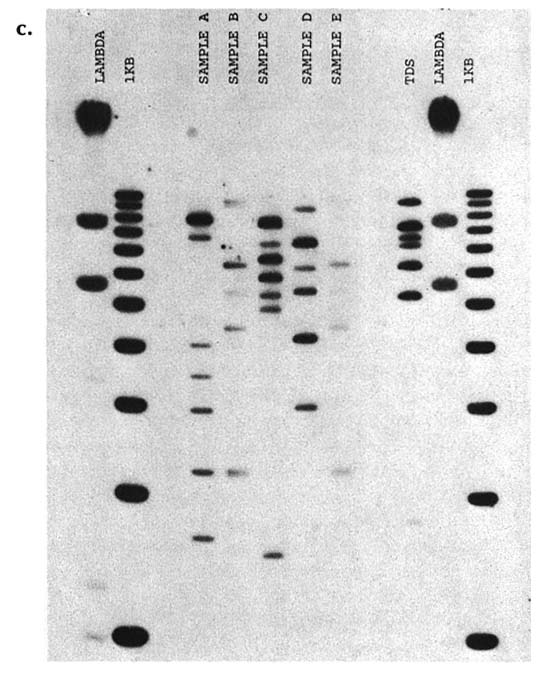
FIGURE B-c DNA types of identical twins are indistinguishable. Samples in lines A-E are from different white males; samples B and E are from identical twins.
-
Criticisms were raised concerning the reliability of the technical methods, including the criteria for identifying DNA patterns and declaring matches, as well as quality control.
-
Questions were raised about the validity of estimates of probability of random inclusion that were being presented in courts. Were the individual components of a specific DNA pattern statistically independent, so that it was proper to multiply their frequencies together in calculating the chance of a match? What population databanks were appropriate?
-
Because DNA can be used to derive medical and other personal information, questions of confidentiality and privacy have assumed greater importance in DNA typing than in the use of non-DNA tests.
By the summer of 1989, because of questions concerning DNA typing raised in connection with some well-publicized criminal cases, the scientific and legal communities had called for an examination of the issues by the National Research Council of the National Academy of Sciences.5,7,8 As a response, the Committee on DNA Technology in Forensic Science was formed, and its first meeting was held in January 1990. The committee was to address the general applicability and appropriateness of the use of DNA technology in forensic science, the need for standards in data collection and analysis, the need for advances in technology, management of DNA typing data, and legal, societal, and ethical issues surrounding DNA typing.
GENETIC BASIS OF DNA TYPING
Genetics is the science of biological variation. The fundamental basis of genetics and the essence of Mendel's discovery in 1865 is that inheritance is particulate and that the inherited factors (genes) that determine visible traits exist in pairs of alleles (i.e., alternative forms of a gene at a given site)—one on a chromosome inherited from the father and one on a chromosome from the mother. Chromosomes that contain genes are threadlike or rodlike structures in the cell nucleus. An organism's particular combination of alleles is referred to as the organism's genotype; the collection of traits resulting therefrom is referred to as the organism's phenotype. Most markers (i.e., identifiable physical locations on a chromosome) used in forensic DNA typing are not parts of expressed genes (i.e., genes that code for products like proteins); they are in noncoding portions of DNA. Hence, they are not associated with a phenotype.
A trait that differs among individuals is referred to as a polymorphism.9 In DNA typing, that term is used interchangeably with "variation." The variations in blood groups, serum protein types, and HLA tissue types used for forensic testing in the pre-DNA era were polymorphisms in the protein product; these proteins contain variations that reflect variations in DNA. But DNA technology makes it possible to study the variations directly.
STRUCTURE AND FUNCTION OF DNA
A human has 22 pairs of nonsex chromosomes (autosomes) and two sex chromosomes—two X chromosomes in a female or an X chromosome and a Y chromosome in a male. Each autosome or X or Y chromosome is composed of a long DNA molecule constructed as a double helix (Figure 1-1). Each component strand of the double helix is a chain of nucleotides of four types designated by the names of the bases adenine (A), cytosine (C), guanine (G), and thymine (T). The nucleotides bond, A to T and C to G, between the two strands of the helix like the rungs of a ladder or, better, the steps in a spiral staircase. A pair of complementary nucleotides (or bases)—A-T, G-C, T-A, or C-G—is called a basepair (bp). DNA replication, which takes place in association with cell division, involves the separation of the two strands of the double helix and the synthesis of a new strand of nucleotides complementary to each strand.
Genes are segments of the DNA molecule. They constitute the blueprint for the structure of proteins of various types that are responsible for the makeup and function of cells and the body as a whole. A human has 50,000-100,000 genes, each occurring in every nucleated body cell. Chromosome 1, the largest, might, for example, have about 5,000 genes spaced at intervals along the DNA molecule that it consists of.
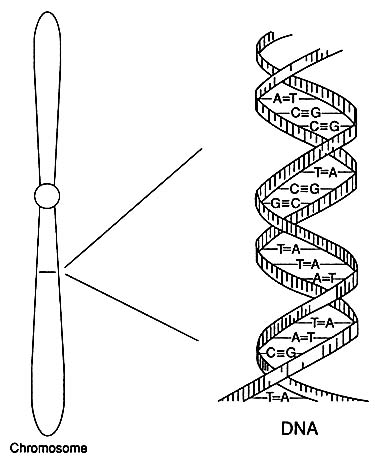
FIGURE 1-1 Diagram of the double-helical structure of DNA in a chromosome. The line shown in the chromosome is expanded to show the DNA structure.
Individual Variation in DNA
DNA technology has revealed variations in the genome, the total genetic makeup of the members of a species: single-nucleotide differences, deletions, and insertions. In noncoding regions of DNA, which are less constrained by forces of selection, it is estimated that at least one nucleotide per 300-1,000, on the average, varies between two people.10 The nucleotide difference might change the recognition site for a particular site-specific endonuclease (restriction enzyme) so as to keep the DNA from being cut at that site by that enzyme. For example, in Figure 1-2 note that a single nucleotide change from C to T has eliminated a restriction enzyme cutting site. In addition, some regions of DNA contain repetitive units, multiple identical strings of nucleotides arranged in tandem. In VNTRs (variable
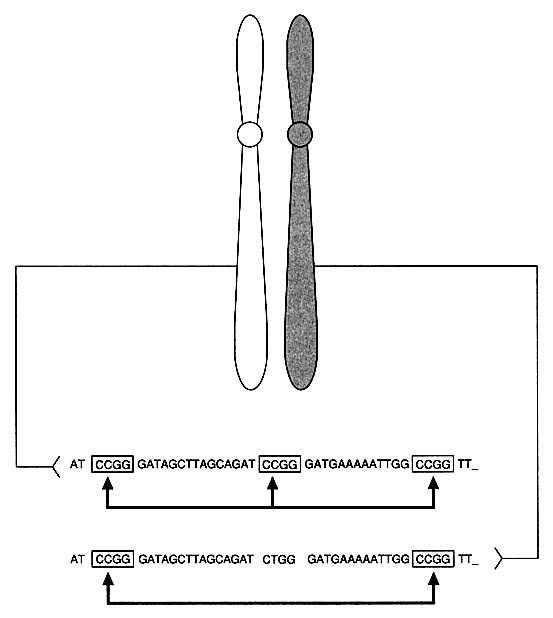
FIGURE 1-2 Basis of restriction fragment length polymorphism (RFLP). Diagram indicates heterozygosity at a "restriction site": chromosome on left has sequence CCGG that is recognized and cut by specific restriction enzyme, whereas chromosome on right has one-nucleotide difference that results in sequence CTGG, which is not recognized and cut by enzyme.
number tandem repeats), the number of repetitions of a sequence can vary from person to person. VNTRs are a leading form of variation used currently in forensic DNA typing. The repeating unit can be as small as a dinucleotide—e.g., the (TG)n polymorphism—or as large as 30, or even more, nucleotides. Tandem repeats are not limited to noncoding segments of DNA, although they are found less frequently in coding segments.
The two main types of variation—single-nucleotide differences and VNTRs—are both potentially recognizable by change in the lengths of fragments that result when DNA is cut with a restriction enzyme. Variation in the lengths of fragments can result from a change in the cluster of four, five, or six nucleotides that is the specific cutting site of the particular restriction enzyme (Figure 1-2). Or the variation can result, not from a change in the cutting site of the enzyme, but from the existence of different numbers of tandem repeats between two cutting sites. Figure 1-3 diagrams the major characteristics of the two forms of variation and their use in DNA typing.
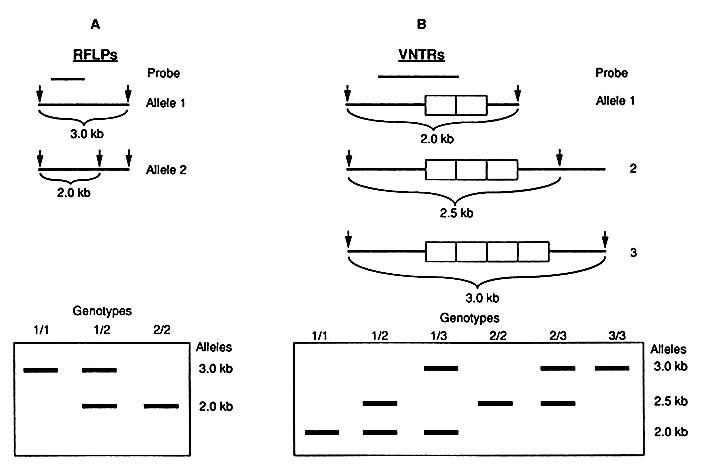
FIGURE 1-3 Two types of RFLPs. Structure of alleles in chromosome is diagrammed at top: arrows indicate sites of cutting by enzyme; lengths of fragments demonstrated by probe (short line above) are given. Electrophoretic patterns are diagrammed below. A. Diallelic RFLP system resulting from single nucleotide change as diagrammed in Figure 1-2. Electrophoretic patterns are those of three genotypes: homozygotes for either allele 1 or allele 2 and 1/2 heterozygote. B. Multiallelic VNTR system. With three alleles as diagrammed, there are six possible genotypes as demonstrated by electrophoresis.
As shown in Figures 1-2 and 1-3A, variation at the cutting site of a restriction enzyme can result in two alternative forms (alleles): the enzyme cuts or it does not. That is called a diallelic system; three genotypes are possible. If a person received the same allele at a particular site (locus) from both parents, the genotype (or person) is said to be homozygous for that allele; if different alleles were inherited from the two parents, the person is heterozygous at that locus. (Some use the term locus, rather than site. Others reserve locus for use in relation to expressed genes.)
As also shown in Figure 1-3B, when the variation is in the number of tandem repeats, there can be many alleles, of which a given person can have only two. That is called a multiallelic, or hypervariable, system. The number of genotypes possible is the sum of the positive integers from 1 to the number of alleles; e.g., in a three-allele system, as shown in Fig. 1-3B, there are expected to be 6 genotypes (1 + 2 + 3). The number of genotypes is also given by the formula n(n + 1)/2, where n is the number of alleles.
Inheritance of variation in the noncoding segments of DNA follows the same rules that Mendel inferred for expressed genes. A given individual inherits one of the father's two alleles and one of the mother's two alleles. When two variable sites, each on a different chromosome, are examined, the inheritance at one site is independent of that at the other; i.e., which paternal allele is inherited at site 1 bears no relation to which paternal allele is inherited at site 2. When the two sites are on the same chromosome, they might also be transmitted independently, if they are sufficiently far apart. When they are very close on the same chromosome, the phenomenon of linkage disequilibrium can result—a deviation from independent inheritance in which particular alleles at the two sites tend to be transmitted together.
TECHNOLOGICAL BASIS OF DNA TYPING
Forensic DNA typing usually consists of comparing "evidence DNA" i.e., DNA extracted from material—most often semen—left at a crime scene with "suspect DNA" (i.e., DNA extracted from the blood of a suspect). The tools of DNA typing include restriction enzymes, electrophoresis, probes, and the polymerase chain reaction.11,12,13
Restriction Fragment Length Polymorphisms
In the RFLP approach shown in Figure 1-4, DNA is subjected to controlled fragmentation with restriction enzymes that cut double-stranded DNA at sequence-specific positions. The long DNA molecules are thereby reduced to a reproducible set of short pieces called restriction fragments (RFs), which are usually several hundred to several thousand basepairs long. Many hundreds of thousands of fragments are produced by digestion of human
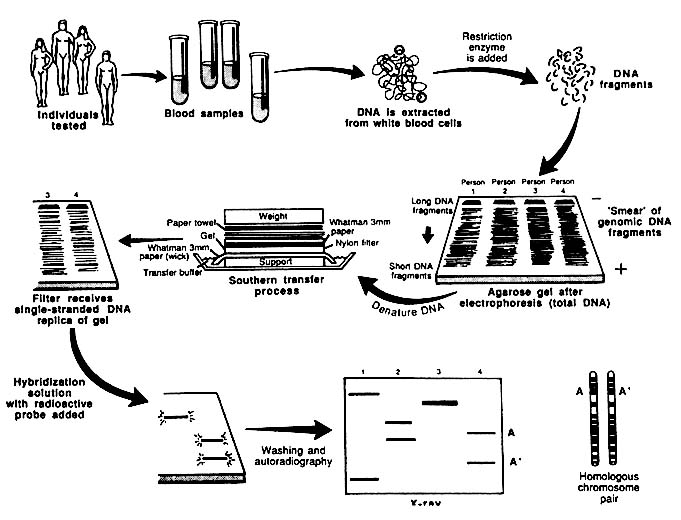
FIGURE 1-4 Schematic representation of Southern blotting of single-locus, multi-allelic VNTR. In example shown here, DNA from four persons is tested. All have different patterns. Three are heterozygous and one homozygous, for a total of seven different alleles. From L. T. Kirby, "DNA Fingerprinting: An Introduction," Stockton Press. New York, 1990. Copyright © 1990 by Stockton Press. Reprinted with permission of W.H. Freeman and Company.
DNA with a single restriction enzyme; each fragment has a distinct sequence and length. For analysis of RFs to demonstrate RFLPs, the fragments are separated electrophoretically on the basis of size. Electrophoresis, typically performed on agarose or acrylamide gels, results in large fragments at one end and small fragments at the other; the small fragments migrate farthest in the electric field. The fragments are then denatured (i.e., rendered single-stranded), neutralized, and transferred from the gel to a nylon membrane, to which they are fixed; this facilitates detection of specific RFLPs and VNTRs.
RFLPs that are defined by specific sequences are detected by hybridization with a probe, a short segment of single-stranded DNA tagged with a group such as radioactive phosphorus, that is used to detect a particular complementary DNA sequence. The nylon membrane is placed in a bath that contains the probe, and the probe hybridizes to the target denatured RF. Nonspecifically bound probe is washed off. Hybridization probes have
conventionally been labeled with radioactive isotopes, but attention is increasingly being given to nonisotopic labeling. When isotopically labeled probes are used, the pattern of probe binding is visualized with autoradiography (see examples in Box 1-A and Figure 1-5).
The complete process—DNA digestion, electrophoresis, membrane transfer, and hybridization—was developed by Edwin Southern in 1975;14 in its present modified form, it is still usually referred to as Southern blotting. These procedures are routinely used in molecular biology, biochemistry, genetics, and clinical DNA diagnosis; there is no difference in their forensic application. Differences among individuals are expressed as differences in the lengths of RFs.15 RFLPs can result from several kinds of differences at the level of the genome:
-
Mutations that alter the base sequence at a restriction-enzyme recognition, or cleavage, site can result in a loss of the cutting site or the generation of a cutting site that was not present before. Insertion or deletion of nucleotides between two cleavage sites also changes RF lengths. Variation of these sorts is generally associated with a small number of alleles. For example, the loss or gain of a particular cleavage site might be responsible for only two alleles.
-
Some regions of DNA contain multiple segments of short-sequence repeats. Consequently, there is a class of RFs that differ in the number of repeated segments present. Some VNTR polymorphisms have a small number of alleles, and the patterns of RFs that represent each of the alleles at a given locus can be readily distinguished. But highly polymorphic VNTR loci have 50-100 alleles or even more. In that situation, the distribution of RF size is essentially continuous; alleles with RFs close in size might not be resolvable with electrophoresis, and the limit of resolution must be defined operationally. Because of the extensive variability, the VNTR class of RFLPs has proved the most informative in distinguishing among persons.
RFLP analysis with single-locus probes is usually designed to result in a simple pattern of one or two RFLP bands, depending on whether the
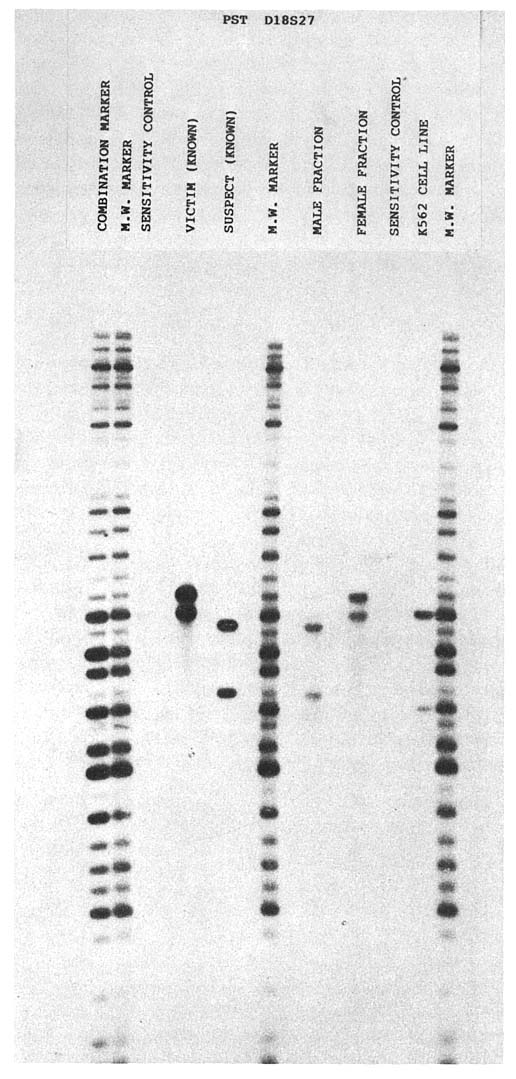
FIGURE 1-5 One of several autoradiographs generated during course of investigation of actual rape-murder case. Law enforcement officials submitted blood samples from victim and suspect and vaginal swabs from victim. DNA was isolated from stains found on vaginal swabs, digested with restriction enzyme PstI, and hybridized to probes at genetic loci D2S44, D17S79, D14S13, and D18S27 and monomorphic locus DXZ-1. Known samples are represented in lanes labeled ''victim (known)" and "suspect (known)." Lanes labeled "male fraction" and "female fraction" represent DNA resulting from differential lyses of vaginal swabs. "Sensitivity control" lane and "K562 cell line" lane contain 50 ng and 1 g, respectively, of DNA from immortalized cell line. "Combination market" lane contains "cocktail" of molecular-weight marker and adenovirus (to monitor migration during electrophoresis).
person is homozygous or heterozygous, respectively. The range of variation shown in the patterns from different persons depends on how many different alleles exist at the particular target locus, e.g., how many different tandem repeats are in the population as a whole (see Figure 1-3B).
An alternative to the use of a single-locus probe is the use of a multilocus probe that hybridizes to many different VNTR sites in the genome. The resulting patterns in a single person contain many bands of varied intensity; the patterns have been compared with barcodes. The approach was developed by Jeffreys and colleagues.2,3,16,17 Because of the complexity of the patterns, interpretation can be difficult. Consequently, the use of multiple single-locus probes is favored.
Polymerase Chain Reaction for Amplifying DNA
Use of the polymerase chain reaction (PCR) allows a million or more copies of a short region of DNA to be made. It is a method of DNA amplification. For DNA typing, one amplifies a genetically informative sequence, usually 100-2,000 bp long, and detects the genotype in the amplified product. Because many copies are made, genetic typing can rely on nonisotopic methods. With PCR amplification, very small samples of tissue or body fluids—theoretically even a single nucleated cell—can be used to study DNA.18,19
The PCR process (Figure 1-6) is simple; indeed, it is analogous to the process by which cells replicate their DNA.20,21,22,23 Two short oligonucleotides are hybridized to the opposite strands of a target DNA segment in positions flanking the sequence region to be amplified; the two oligonucleotides are oriented so that their 3' ends point toward each other. (The ends of a DNA segment are referred to as 5' and 3'; synthesis of new chains proceeds from the 3' end.) The two oligonucleotides serve as primers for an enzyme-mediated replication of the target sequence. The PCR amplification process itself consists of a three-step cycle:
-
The double-stranded template DNA is dissociated into single strands by incubation at high temperature, typically 94°C.
-
The temperature is lowered to allow the oligonucleotide primers to bind to their complementary sequences in the DNA that is to be amplified.
-
A DNA polymerase extends the primers from each of the two primer-binding sites across the region between them, with the target sequence as template.
Because the extension products of one primer bind the other primer in successive cycles, there is in principle a doubling of the target sequence in each cycle. However, the efficiency of amplification is not 100%, and the yield from a 30-cycle amplification is generally about 106-107 copies of the
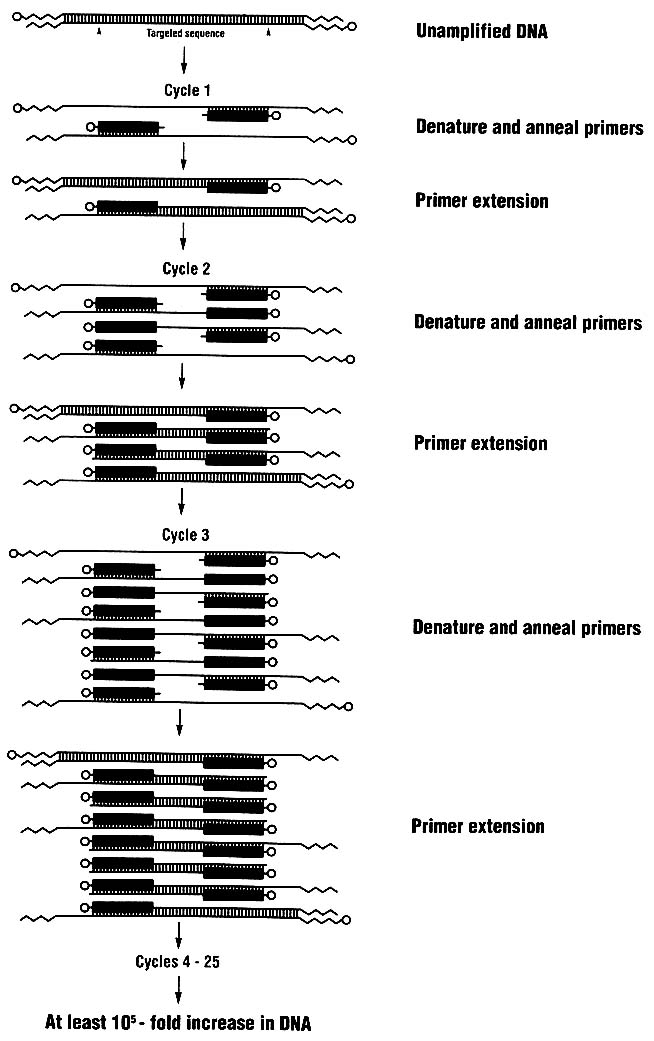
target sequence. The primers become physically incorporated into the amplification products.
Both the amplification and the genetic typing can be completed in a day. The efficiency can be improved by amplifying several different products in the same reaction mix; this is termed multiplex amplification.
Several methods have been coupled with PCR for the detection of genetic variation in the amplified DNA; some are listed in Table 1.2. Detection of variation in DNA with PCR-amplified material is fundamentally no different from detection with unamplified samples. The difference is technical: the availability of the larger quantity of pure material produced with PCR affords more options for means of detection.
The use of allele-specific oligonucleotide (ASO) probes is the most generalized approach to the detection of alleles that differ in sequence.33 The sequence-specific probe is usually a short oligonucleotide, 15-30 nucleotides long, with a sequence exactly matching the sequence of the target allele. The ASO probe is mixed with dissociated strands of PCR reaction product under such conditions that the ASO and the PCR product strands hybridize if there is perfect sequence complementarity, but do not if there are mismatches in sequence.
The usual format for the use of ASO probes is to spot dissociated PCR product strands onto a nitrocellulose or nylon membrane and probe the membrane with labeled ASO. That is analogous to Southern blotting; because the samples are spotted as a "dot" on the membrane, the method is referred to as "dot blotting" (Figure 1-7). An alternative method uses an array of ASO probes immobilized on a test strip.34 The test strip is immersed in a solution of labeled PCR product; the PCR product hybridizes only to its complementary probe. This procedure has been called ''reverse dot blotting" or "blot dotting." A commercial kit based on the reverse dot blot principle has been released (Cetus Corporation).
TABLE 1.2 Some PCR-Based Systems for the Detection of Genetic Variation
|
Sequence-based detection systems |
|
|
Allele-specific oligonucleotide (ASO)24 |
|
|
Oligonucleotide-ligation assay (OLA)28 |
|
|
Restriction-site-specific cleavage (Amp-FLPs)29 |
|
|
Denaturing-gradient gel electrophoresis30 |
|
|
Chemical cleavage of mismatched heteroduplexes31 |
|
|
Length-variation systems |
|
|
Simple insertions and deletions |
|
|
VNTR polymorphisms32 |
|
|
Analysis of nucleotide sequences |
|
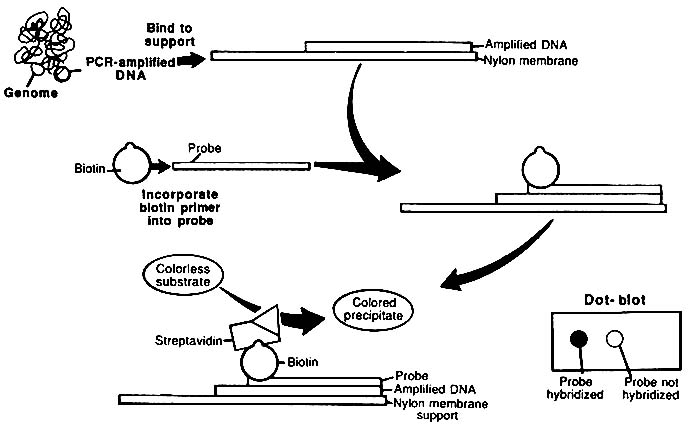
FIGURE 1-7 Diagrammatic representation of dot-blot procedure. PCR-amplified target DNA is immobilized on nylon membrane, and biotinylated probe hybridizes to the target if there is no nucleotide mismatch. Avidin-horseradish peroxidase (HPR) conjugate that binds to biotinylated probe is added. HPR converts colorless substrate to colored product. From L. T. Kirby, "DNA Fingerprinting: An Introduction," Stockton Press, New York, 1990. Copyright © by Stockton Press. Reprinted with permission of W.H. Freeman and Company.
Amplification of DNA regions that contain inserts or deletions yields products of different size, which are readily detected with electrophoresis. It has proved possible to amplify some of the discrete alleles that make up VNTR polymorphisms;19 these typing systems combine convenience with the potential for good discrimination. A number of PCR-VNTR typing systems are in development, and it is anticipated that they will come into greater use over the next few years.35 Indeed, the whole technology of DNA typing can be expected to evolve continually in the next decade. A recent description of PCR-based "digital" DNA typing by Jeffreys is but one example of what might come.36 In his method, PCR is used to amplify tandem repeats in a portion of DNA. These repeats are then analyzed for the presence of nucleotide polymorphism and assigned numerically (digits 1,2.3, etc.). Thus, by analyzing the repeats (50 or more repeats can easily be analyzed), one is able to assign a specific digital code to each sample (e.g., 1122113111221112…) and unambiguously distinguish one sample from another. The method is simple, avoids match criteria, and requires no side-by-side comparison of DNA samples. The Jeffreys "digital" DNA typing method is still in the research stage, but, if perfected and adapted for foren
sic samples, it might be possible to achieve absolute identification of persons by analysis of a few such repeats.
PCR provides excellent starting material for direct DNA sequencing, and sequence analysis might ultimately be the approach used for personal identification. But it will require improvements in automated sequencing technology and the generation of larger databases on sequence variability.
POPULATION GENETICS RELEVANT TO THE INTERPRETATION OF DNA TYPING
The finding that two samples of human tissue differ in their DNA patterns leads to the conclusion that the two came from different persons. However, if the two samples are indistinguishable with regard to the detected DNA patterns, two possibilities exist: the two samples came from the same person (or from identical twins), or the two samples came from different persons whose DNA patterns in the target regions investigated are the same. Those two possibilities cannot be distinguished. To provide the trier of fact—a judge or jury, for example—with information for weighing the two possibilities, it has been traditional to us statistics from population genetics to estimate the fraction of people in the population who have the particular combination of DNA patterns. What is the chance of picking at random a person who has the same genetic patterns as found in the evidence sample? Obviously, the lower the probability, the stronger the inference that the evidence sample is associated with a particular person who has those patterns.
Thus, the central question in relation to forensic DNA typing is, What is the probability that a person picked at random would match the evidence sample in DNA patterns? Or, What proportion of persons in the same population as a suspect have the same combination of DNA patterns as the evidence sample? In answering the questions, the population frequencies of the patterns at the several (often four) loci tested are multiplied together, on the assumption that they are independent. Indeed, the alleles (two in a diallelic system) at each locus are assumed to vary independently. Estimation of the frequencies of specific alleles needed to answer the questions is based on population genetics.37 Experience with blood groups, enzyme markers, and HLA types—including the genetic markers used for forensic personal "identification" in the pre-DNA era—indicates that the frequency of specific allele can vary widely among populations. That is demonstrated in Figure 1-8 for the B blood-group allele; maps of the A and O blood-group alleles show similar variability. It is to be expected that the frequencies of the various alleles for the DNA polymorphisms also show differences among populations. Thus, it might be critically important to pick the right population with which to compare a given suspect's DNA.
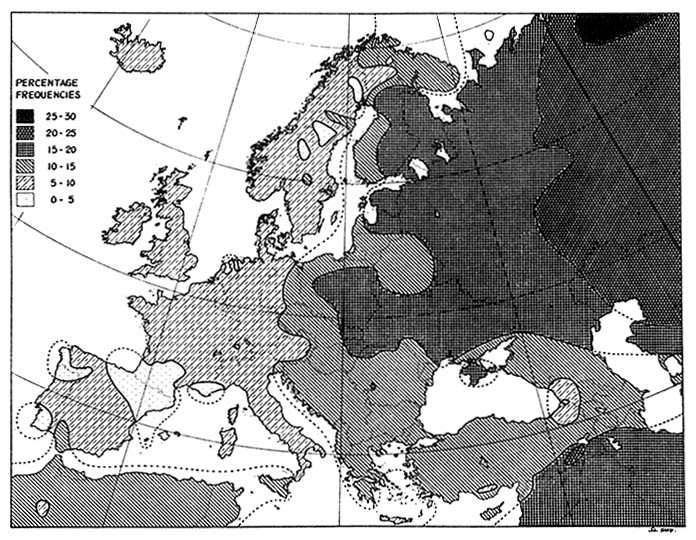
FIGURE 1-8 Distribution of gene for blood group B in Europe. Similar wide variation is observed with genes for blood groups A and O. From Mourant et al.,38 p. 266.
Estimating the Frequency of Alleles in Populations
As diagrammed in Figure 1-9, a diallelic system such as represented by many RFLPs (Figure 1-3) gives rise to three genotypes that in the state of Hardy-Weinberg equilibrium (see Chapter 3) are expected to have the following frequencies: p2 (homozygotes for allele A1), 2pq (heterozygotes), and q2 (homozygotes for allele A2), where p is the frequency of allele A1 and q is the frequency of allele A2. The frequency of each of the two alleles can be derived by counting: e.g., each A1 homozygote has 2 A1 alleles, and each A1/A2 heterozygote has 1 A1 allele. Or the proportion of the A1 allele can be taken as the square root of the frequency of A1 homozygotes. The two methods should give closely similar results, if the population is in Hardy-Weinberg equilibrium.
A multiallelic system like that represented by VNTRs (Figure 1-10) gives rise to many genotypes, as diagrammed in Figure 1-11; with five alleles, there are 15 genotypes [n(n + 1)/2]. Again, the frequency of each allele can be determined by counting or, more easily, by taking the square
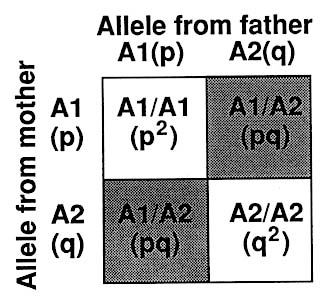
p,q = proportion of A1 and A2 alleles, respectively, in the population -- p + q = 1
p2, 2pq, q2 =proportion of 3 genotypes -- p2 + 2pq + q2 = 1
where p2 = proportion of A1 homozygotes
2pq = proportion of heterozygotes
q2 = proportion of A2 homozygotes
FIGURE 1-9 Punnett square (named for British geneticist) indicating frequency of genotypes in diallelic RFLP system.
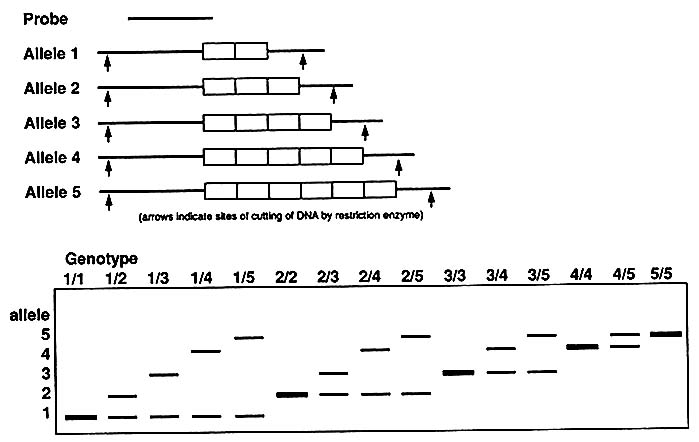
FIGURE 1-10 Schematic representation of alleles and genotypes in five-allele VNTR system.

FIGURE 1-11 A. Punnett square showing genotypes in single locus, five-allele VNTR system, as schematized in Figure 1-10. B. A worked example: frequency of each of 15 genotypes with allele frequencies indicated.
root of the frequency of the homozygotes for the particular allele or half the summed frequencies of the heterozygotes, which, in the case of the five-allele system, will be of four types for each allele.
Population Substructure
It is intuitively obvious that relatives have genes in common. Thus, the chance that DNA typing will yield a match with a suspect when the evidence sample in fact came from a brother or other relative is considerable, especially if only a few loci are tested.
The U.S. population is a conglomerate of many different population groups, which might be viewed as extended families derived from all parts of the world. The major ethnic groupings—white, black, Hispanic, Asian, etc.—are each composites of many different subpopulations, which might have quite different frequencies of the alleles used in forensic DNA typing. Allele frequencies estimated from sampling of an overall ethnic group represent weighted averages. Some of the component subpopulations might have allele frequencies quite different from the mean values of the whole population. Further discussion of this problem and recommendations for handling it are given in Chapter 3.
CHARACTERISTICS OF AN OPTIMAL FORENSIC DNA TYPING SYSTEM
The methods of DNA typing continue to evolve as new ways to detect individual variation are developed. Sequencing of DNA might ultimately be the optimal method of personal identification, but that is still far from practical. It is important that the flexibility to adopt new methods be retained as standardization of DNA technology is developed (see Chapter 4) and databanks are created (see Chapter 5).
Any method of forensic DNA typing, like methods for medical DNA and other testing, should be rapid, accurate, and inexpensive. In addition, to achieve maximal discrimination among individuals, forensic DNA typing requires the use of markers with a high level of variability or polymorphism. Ideally, the high degree of variability would be found in all the world's populations. The markers and the probes used to detect them should have a unique sequence, so that each probe hybridizes with only one part of the genome. Single-locus probes should be used. The loci of the markers should be independent, e.g., on separate chromosomes. The markers should, furthermore, come from noncoding and therefore presumably nonfunctional parts of the genome, to avoid claims, spurious or otherwise, of association of particular markers with particular behavioral traits or diseases.
The automation of DNA typing might help to reduce its time and expense. An advantage of speed and low cost is that one can test more parts
of the genome. Even if a locus is only modestly polymorphic, its use in DNA typing could have other advantages, such as complete unambiguity of scoring; used in combination, such loci could demonstrate that the chance of a random match is extremely low.
It must be emphasized that new methods and technology for demonstrating individuality in each person's DNA continue to be developed. The methods outlined in this chapter are likely to be superseded in efficiency, automatability, economy, and other features by new methods. Care must be taken to ensure that DNA typing techniques used for forensic purposes do not become "locked in" prematurely. Otherwise, society and the criminal justice system will not be able to derive maximal benefit from advances in the science and technology.
REFERENCES
1. Gaensslen RE. Sourcebook in forensic serology, immunology, and biochemistry. Washington, D.C.: U.S. Government Printing Office, 1983.
2. Jeffreys AJ, Wilson V, Thein SL. Individual-specific "fingerprints" of human DNA. Nature. 316:75-79, 1985.
3. Gill P, Jeffreys AJ, Werrett DJ, Forensic application of DNA "fingerprints." Nature. 318:577-579, 1985.
4. Evett IW, Werrett DJ, Gill P, Buckleton JS. DNA fingerprinting on trial. Nature. 340:435, 1989.
5. Lander ES. DNA fingerprinting on trial. Nature. 339:501-505, 1989.
6. U.S. Congress, Office of Technology Assessment. Genetic witness: forensic uses of DNA tests. OTA-BA-438. Washington, D.C.: U.S. Government Printing Office, 1990.
7. Thompson L. A smudge on DNA fingerprinting? The Washington Post, Monday, June 26, 1989.
8. Barinaga M. DNA fingerprinting: pitfalls come to light, Nature. 339:89, 1989.
9. Roychoudhury AK, Nei M. Human polymorphism genes: world distribution, New York: Oxford University Press, 1988.
10. Cooper DN, Smith BA, Cooke HJ, Niemann S, Schmidtke J. An estimate of unique DNA sequence heterozygosity in the human genome. Hum Genet. 69:201-205, 1985.
11. Kirby LT. DNA fingerprinting: an introduction, New York: Stockton Press, 1990.
12. Farley MA, Harrington JJ, eds.: Forensic DNA technology. Chelsea, Michigan: Lewis Publishers, 1991.
13. U.S. Department of Justice, Federal Bureau of Investigation. Proceedings of the international symposium on the forensic aspects of DNA analysis. Washington, D.C.: U.S. Government Printing Office, 1991.
14. Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol. 98:503-527, 1975.
15. Botstein D, White RL, Skolnick M, Davis RW. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet. 32:314-331, 1980.
16. Helminen P, Ehnholm C, Lokki ML, Jeffreys A, Peltonen L. Application of DNA "fingerprints" to paternity determinations. Lancet. 1:574-576, 1988.
17. Jeffreys AJ, Wilson V, Thein SL, Weatherall DJ, Ponder BAJ. DNA "fingerprints" and segregation analysis of multiple markers in human pedigrees. Am J Hum Genet. 39:11-24, 1986.
18. Higuchi R, yon Beroldingen CH, Sensabaugh GF, Erlich HA. DNA typing from single hairs. Nature. 332:543-546, 1988.
19. Jeffreys AJ, Wilson V, Neumann R, Keyte J. Amplification of human minisatellites by the polymerase chain reaction: towards DNA fingerprinting of single cells. Nucleic Acids Res. 16:10953-10971, 1988.
20. Erlich IIA, ed. PCR technology: principles and applications for DNA amplification. New York: Stockton Press, 1989.
21. Rose EA. Applications of the polymerase chain reaction to genomc analysis. FASEB J. 5:46-54, 1991.
22. Amheim N, Levenson CH. Polymerase chain reaction. Chem Eng News. 68:36-47, October 1, 1990.
23. Mullis KB. The unusual origin of the polymerase chain reaction. Sci Am.262:56-65, 1990.
24. Conner B J, Reyes AA, Morin C, Itakura K, Teplitz RL, Wallace RB. Detection of' sickle-cell beta-S-globin allele by hybridization with synthetic oligonucleotides. Proc Natl Acad Sci LISA. 80:278-282, 1983.
25. Chehab FF, Kan YW. Detection of specific DNA-sequences by fluorescence amplifica-tion--a color complementation assay. Proc Natl Acad Sci USA. 86:9178-9182, 1989.
26. Newton CR, Graham A, Heptinstall LC, Porvell SI, Summers JC, Markham AF. Analysis of any point mutation in DNA—the amplification refractory mutation system (ARMS). Nucleic Acids Res. 17:2503-2516, 1989.
27. Wu DY, Ugozzoli L, Pal BK, Wallace RB. Allele-specific enzymatic amplification of beta-globin genomic DNA for diagnosis of sickle-cell anemia. Proc Natl Acad Sci LISA. 86:2757-2760, 1989.
28. Nickerson DA, Kaiser R, Lappin S, Steward J, Hood L, Landegren U. Automated DNA diagnostics using an ELISA-based oligonucleotide ligation assay. Proc Natl Acad Sci LISA. 87:8923-8927, 1990.
29. Kasai K, Nakamura Y, White R. Amplification of a variable number of tandem repeats (VNTR) locus (pMCT118) by the polymerase chain reaction (PCR) and its application to forensic science. J For Sci. 35:1196-1200, 1990.
30. Myers RM, Maniatis T, Lerman I,S. Detection and localization of single base changes by denaturing gradient gel electrophoresis. Methods Enzymol. 155:501-527, 1987.
31. Cotton RGH, Rodriques NR, Campbell RD. Reactivity of cytosine and thymine in single-base-pair mismatches with hydroxylamine and osmium-tetroxide and its application to the study of mutations. Proc Natl Acad Sci USA. 85:4397-4401, 1988.
32. Nakamura Y, Leppert M, O'Connell P, Wolff R, Holm T, Culver M, Martin C, Fujimoto E, Hoff M, Kumlin E, White R. Variable number of tandem repeat (VNTR) markers for human gene mapping. Science. 235:1616-1622, 1987.
33. Saiki R, Bugawan TL, Horn GT, Mullis KB, Erlich HA. Analysis of enzymatically amplified [3-globin anti HLA-DQ DNA with allele-specific oligonucleotide probes. Nature. 324:163-166, 1986.
34. Saiki R, Walsh PS, Levenson CH, Erlich HA. Genetic analysis of amplified DNA with immobilized sequence-specific oligonucleotide probes. Proc Natl Acad Sci USA. 86:6230-6234, 1989.
35. liagelberg E, Gray IC, Jeffreys AJ. Identification of the skeletal remains of a murder by DNA analysis. Nature. 352:427-428, 1991.
36. Jeffreys A, MacLeod A, Tamaki K, Nell D, Monckton D. Minisatellite repeat coding as a digital approach to DNA typing. Nature. 354:204-209, 1991.
37. Hartl DL, Clark AC. Principles of population genetics. 2nd ed. Sunderland, Massachusetts: Sinauer Associates, 1989.
38. Mourant AE, Kopec AC, Domaniewska-Sobczak K. The distribution of the human blood groups and other polymorphisms. 2nd ed. Oxford: Oxford University Press, 1976.
























