
Microbial Science: Ecology, Diversity, and Characterizing the Microbial World
One might think that the age of great discoveries in biology is past—that humans have been everywhere and seen everything on Earth. But in fact, biology still boasts some formidable unknowns, and many are now being explored in the once invisible world of microbes. In the last 20 years, technological advances have made it possible to explore a microbial world that has proven vastly more extensive, important, and diverse than previously imagined. Analyses of microbial communities in the soil, in the ocean, and even in the human body have shown that previous methods detected only a tiny percentage of the different microbes in these environments. It seems that each technological advance and every new environment sampled reveal even greater diversity in the microbial world. Is there a limit? How can the nature and extent of microbial diversity be satisfactorily characterized?
Source: AAM, 2011a.
Although the world of living things is dominated by microbes, very little is known about the vast majority of them. According to the American Academy of Microbiology, “There are ten-million-fold more bacterial and archaeal cells on our small planet than there are stars in the visible universe, and they may contain as much carbon as all plant and animal life put together” (AAM, 2011b). Much of what is known is based on the very few microorganisms that are culturable in laboratories. Prior to the advent of nucleic acid sequencing, only the phenotypes of culturable microorganisms could be readily studied using tools that had been available for the previous 150 years—growth on selective and differential media,
Gram stains, serotyping with reference antisera, and clinical microbiology techniques—and these are inadequate for the purpose of identifying and attributing the source of the material used in a biological attack because they cannot produce sufficiently fine-grained detail on the nature of the organism (Clements, remarks at Zagreb workshop, 2013).
As noted in Chapter 1, much of the work of microbial forensics today is based on studying biodiversity, phylogenetics,1 phylogeography,2 and genomics. The ability to sequence the genomes of microbes has provided an enormous amount of new knowledge about some selected bacteria and viruses. However, even the most basic information remains unknown for most microorganisms and “knowledge of the evolution and ecology of microbial communities lags far behind cellular microbiology” (NRC, 2007:33). Until recently there have been few systematic efforts to collect and describe the microbes living in soil, seawater, freshwater lakes and streams, plants, and even in the guts and on the bodies of humans and other animals. The new techniques of “metagenomics” circumvent the unculturability of the majority of microbes by enabling the study of microbial genes and sequences from entire communities directly from environmental samples. The use of metagenomic techniques, however, is still an area of active development (Darling presentation, 2013). Meanwhile, knowledge of the natural microbial communities residing throughout most of the world is scarce and vastly incomplete. Although the biology of the small fraction of bacteria and viruses that are pathogenic to human beings, livestock and companion animals, and crop or forestry plants is somewhat better known and understood, there is still much to be learned about their evolution, how many strains of pathogen species exist in nature, what their distribution is throughout the world, and how this distribution affects, and is affected by, ecological conditions.
BIOSURVEILLANCE
Biosurveillance is “the process of active data-gathering with appropriate analysis and interpretation of biosphere data that might relate to disease activity and threats to human or animal health—whether infectious, toxic, metabolic, or otherwise, and regardless of intentional or natural origin—in order to achieve early warning of health threats, early detection of health events, and overall situational awareness of disease activity”
_______________
1 The study of evolutionary relatedness among various groups of organisms through molecular sequencing data and morphological data matrices.
2 A field of study concerned with the principles and processes governing the geographic distributions of genealogical lineages, especially those within and among closely related species.
(Homeland Security Presidential Directive, HSPD-21, 2007).3 In addition to the scientific benefits that will derive from an improved understanding of the microbial world, the application of that knowledge through improved international biosurveillance capabilities for earlier detection and reporting of new and reemerging infectious diseases underscores the fundamental connections between microbial forensics and public health. As the experience with Severe Acute Respiratory Syndrome (SARS) in 2002 and 2003 demonstrated, the global reach of trade and travel, especially by air, means that diseases with pandemic potential can now reach around the world within hours or days rather than the weeks or months of prior years.4 Subsequent outbreaks related to various strains of avian influenza (H1N1, H5N1, and H7N9) as well as the Middle East Respiratory Syndrome (MERS) that emerged in 2012 underscored the importance of improving national and international disease surveillance. Many of these diseases originated with animals, emphasizing the need for greater understanding of zoonoses as threats to health:
The WHO is engaging in an ever-increasing number of cross-sectoral activities to address health threats at the human-animal-ecosystem interface. These threats include existing and emerging zoonoses as well as antimicrobial resistance, food-borne zoonoses, and other threats to food safety.5
A major breakthrough came in 2005 when the World Health Assembly of the World Health Organization (WHO) adopted revisions to the International Health Regulations (IHR), changing the basis for reporting disease outbreaks from a list of specific diseases to a set of characteristics that constitute a “Public Health Emergency of International Concern.” The IHRs reflect more than a decade of international effort to achieve global consensus on the need to improve disease surveillance capacities and to accept international requirements to do so in a timely manner. Much remains to be done, however; as of the initial July 2012 deadline for reporting progress, fewer than 20 percent of WHO member countries reported that they had been able to meet the IHRs, and a majority requested a 2-year extension.6 The international assistance being provided to enable countries to achieve the core surveillance and response compe-
_______________
3http://www.fas.org/irp/offdocs/nspd/hspd-21.htm.
4 Between November 2002 and July 2003, the World Health Organization recorded 8,096 probable cases of SARS in 29 countries, with 774 deaths (http://www.who.int/csr/sars/country/table2004_04_21/en/; accessed April 6, 2014). One estimate put the economic impact of the outbreak at $30 billion (http://www.globalhealth.gov/global-health-topics/global-health-security/index.html; accessed April 6, 2014).
5http://www.who.int/zoonoses/en/; accessed April 6, 2014.
6 See http://www.who.int/ihr/about/en/ for more information about the IHR.
tencies needed to meet their IHR requirements also serves the development of capacities for microbial forensics.
In addition, national and nongovernmental efforts have already demonstrated the value of increased reporting capabilities. For example, the Global Public Health Intelligence Network (GPHIN), which was originally developed by Public Health Canada in collaboration with WHO, continuously searches media sources for information about infectious disease outbreaks. Pro-Med mail, which in addition to monitoring the media and other sources, relies on reports from its network of 60,000 subscribers in 185 countries, is now a project of the International Society of Infectious Diseases that was begun in the mid-1990s by two U.S. nongovernmental organizations.7 According to WHO, “more than 60% of the initial outbreak reports come from unofficial informal sources, including sources other than the electronic media, which require verification.”8
WHO’s own primary surveillance mechanism is the Global Alert and Response Network (GOARN), “a technical collaboration of existing institutions and networks who pool human and technical resources for the rapid identification, confirmation and response to outbreaks of international importance.”9 Also relevant to microbial forensics is the Emerging and Dangerous Pathogens Laboratory Networks, another WHO effort to support the development of increased readiness and capacity in high-security public health and veterinary laboratories for detection and response related to both human and animal disease. This network is supported by a number of international working groups, some of which focus on developing common procedures and protocols that could be relevant for microbial forensics as well.10
As mentioned above and described in the cases in this chapter and elsewhere in the report, animal, plant, and foodborne diseases can also pose threats to public health and challenges for microbial forensics. This has led to increasing cooperation among WHO, the World Organisation for Animal Health (OIE), and the U.N. Food and Agriculture Organization (FAO). For example, in 2006 the three organizations launched the Global Early Warning System for Major Animal Diseases, Including Zoonoses (GLEWS), which seeks to gain the added value of
_______________
7 More information about GPHIN can be found at http://www.who.int/csr/alertresponse/epidemicintelligence/en/ and http://www.hc-sc.gc.ca/ahc-asc/pubs/_intactiv/gphin-rmisp/index-eng.php. Information about Pro-Med mail can be found at http://www.promedmail.org/aboutus/.
8http://www.who.int/csr/alertresponse/epidemicintelligence/en/; accessed April 4, 2014.
9http://www.who.int/csr/outbreaknetwork/en/; accessed April 4, 2014.
10 Further information can be found at http://www.who.int/csr/bioriskreduction/laboratorynetwork/en/; accessed April 4, 2014.
combining and coordinating the alert and disease intelligence mechanisms of OIE, FAO and WHO for the international community and stakeholders to assist in prediction, prevention and control of animal disease threats, including zoonoses, through sharing of information, epidemiological analysis and joint risk assessment.11
In the United States, the White House issued the National Strategy for Public Health and Medical Preparedness in response to Homeland Security Presidential Directive-21 (HSPD-21) in 2007. HSPD-21 called for the United States to
establish a biosurveillance capability, with connections to international disease surveillance systems, that can provide “early warning” of a bioattack or naturally occurring outbreak, and can provide ongoing “near real-time” information about an outbreak as it unfolds. This system should be designed to provide a national “common operating picture” and better situational awareness for state, local, and federal officials, as well as for public and private sector healthcare providers.
In 2008, the Centers for Disease Control and Prevention (CDC) was tasked to lead the establishment of the National Biosurveillance Advisory Subcommittee (NBAS). NBAS provides advice to the Advisory Committee to the Director as well as “leadership and guidance” for implementing the National Biosurveillance Strategy. It issued its first report in 2009 (National Biosurveillance Advisory Subcommittee, 2009), calling for strategic goals and investments in biosurveillance activities and technologies, hiring and retaining trained personnel, and electronic health records and databases. In a second report in 2011, NBAS made further recommendations on governance, information exchange, workforce, and research and development (National Biosurveillance Advisory Subcommittee, 2011). In 2012, the White House issued its National Strategy for Biosurveillance.
More recently, the Global Health Security Agenda, announced in February 2014, is a new partnership created by the United States among WHO, OIE, and FAO along with more than 25 countries to
“…accelerate progress toward a world safe and secure from infectious disease threats and to promote global health security as an international security priority, to
- Prevent and reduce the likelihood of outbreaks—natural, accidental, or intentional;
- Detect threats early to save lives;
_______________
11http://www.glews.net/; accessed April 6, 2014.
- Respond rapidly and effectively using multisectorial, international coordination and communication.”12
Again, success in achieving a number of the Agenda’s goals would also mean improved international capabilities for microbial forensics.
The U.S. microbial forensics community has already intensified its efforts to partner with the public health sector, particularly to improve and increase the use of biosurveillance information. Such information would help microbial forensics investigators to better understand the context of an active outbreak and better interpret microbial forensics findings.
The rest of this chapter explores several actual microbial forensics cases to illustrate the techniques that have been developed over the last 10-12 years, some of the relationships between public health and microbial forensics, and the unmet needs that remain in the area of understanding microbial ecology, diversity, and characterization of strains and other genetic variants. Other aspects of public health and microbial forensics, including unmet needs related to technologies and processes, are taken up in subsequent chapters.
THE AMERITHRAX CASE
Although several attempts to use pathogens as weapons occurred prior to 2001, the most comprehensive major case involving the use of a pathogen as a biological weapon occurred with the anthrax letters (or “Amerithrax”) case. In fall 2001, less than a month after the September 11, 2001 attacks on the World Trade Center and the U.S. Pentagon, letters containing spores of the anthrax disease-causing bacterium (Bacillus anthracis, or B. anthracis) were sent through the U.S. mail. Between October 4 and November 20, 2001, 22 people were sickened by anthrax, with 5 tragic fatalities (Jernigan et al., 2002). In addition, there was extensive environmental contamination caused by the anthrax mailings in U.S. Postal Service buildings, Senate office buildings, and in many other places that processed or received the contaminated letters. The FBI led the effort to characterize the material contained in the letters and identify the individual or individuals responsible for the mailings. This investigation involved extensive scientific study spanning almost 9 years (NRC, 2011). Over the 9 years, the FBI devoted 600,000 investigator hours to the case, which involved 17 Special Agents as well as 10 U.S. Postal Inspectors. During the investigation, 10,000 witnesses were interviewed, 80 searches conducted, 4 million megabytes of computer memory were analyzed,
_______________
12 Additional information about the Agenda may be found at http://www.globalhealth.gov/global-health-topics/global-health-security/ghsagenda.html; accessed April 6, 2014.
and 5,750 grand jury subpoenas were served. Twenty-nine government, university, and commercial labs assisted with the scientific analyses that were a central aspect of the investigation (U.S. Department of Justice, 2010). Note that, at the time of the anthrax letters mailings, the tools and technologies that were readily available were not adequate and the science of microbial forensics was in its infancy and limited to a few pioneering laboratories (Clements remarks, 2013). The Amerithrax investigation accelerated the development of microbial forensics, resulting in remarkable development and applications of new techniques and approaches for using laboratory tools to pinpoint the genetic identity of a microbial agent (Tucker and Koblentz, 2009). Microbial forensics became an essential part of the scientific investigation, which was combined with physicochemical analyses and other evidence to narrow the search for the source of the B. anthracis used in the attacks.
An important point to be made here is that B. anthracis is extremely stable genetically. One reason for this stability is that the organism’s life cycle includes long periods of dormancy in the form of spores, rendering the genome highly homogeneous (Pilo and Frey, 2011). Paradoxically, reconstruction of the evolutionary history of B. anthracis has been challenging because of the same stability that allowed investigators to track its use as a biological weapon (Van Ert et al., 2007). Very early in the investigation the anthrax spores in the letters and the environmental and clinical isolates were identified as the “Ames strain” by Dr. Paul Keim and members of his team at Northern Arizona University (NAU). At the Zagreb workshop, Keim summarized what enabled them to accomplish this identification in the era before whole-genome sequencing (WGS) became commonplace. Research in the mid-1990s to differentiate B. anthracis strains had been limited to working with small sections of genomic DNA. The genomes of very few bacteria had, at that point, been fully sequenced. Keim and his colleagues had focused on hypervariable regions in the genetic material of B. anthracis called variable number tandem repeat (VNTR) regions. His team was able to identify eight loci in B. anthracis that had multiple alleles (i.e., different versions of the same genetic marker). They developed a typing system called multiple-locus VNTR Analysis, or MLVA. In fact, B. anthracis was one of the first bacteria upon which this subtyping was performed. In 2001, NAU had a database of about 400 different B. anthracis strains and could differentiate these isolates into approximately 90 different genotypes. Both Keim’s lab at NAU and the CDC lab independently produced the same result: the MLVA8 genotype was found to be consistent only with the Ames strain genotype that Keim and colleagues had identified earlier (Keim et al., 2000).
The Ames strain was a known laboratory strain used by the U.S. military and other vaccine development teams. The strain was origi-
nally isolated from a dead cow in Texas in 1981 and was shipped by Texas A&M University to the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID) at Fort Detrick in Frederick, Maryland (NRC, 2011). Over time, it was shared with other labs in the United States and in Canada, Sweden, and the United Kingdom. Thus, although the identification of the Ames strain narrowed the possibilities, making the source likely to be a laboratory that had access to the strain, MLVA was insufficient to identify its source unequivocally. The evidence, therefore, needed to be more painstakingly examined for additional unique and distinguishing genetic and other features that could be compared to samples obtained from laboratories holding the Ames strain to narrow the search for the source and perpetrator(s). Scientists from the Department of Defense had identified several colony morphology variants in the samples from the letters (Figure 2-1). With funding from the National Institutes of Health, the National Science Foundation, and other government agencies, FBI scientists worked with The Institute for Genomic Research (TIGR) in Rockville, Maryland, to identify the genetic basis of the altered appearance of the bacterial colonies (Read et al., 2002). Some of these were determined to be insertions or deletions, while others were associated with single nucleotide polymorphisms (SNPs) (NRC, 2011). FBI investigators then contracted with four laboratories to develop highly specific molecular genetic assays to try to detect some of these variants in the anthrax powder evidence. These assays were used in the examination of the samples the FBI had collected to form its repository of known Ames strain samples.
Unfortunately, the rigor used in collecting and processing samples did not match the rigor of the powerful and validated assays. Validating an evidence-handling stream is very difficult. Achieving high confidence
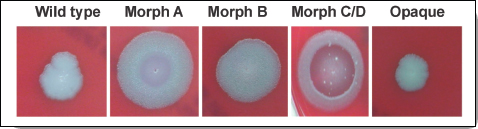
FIGURE 2-1 Variants of representative colonies of each of the morphotypes of B. anthracis Ames.
SOURCE: Rasko et al. (2011).
would entail processing a large number of blank samples,13 which is very expensive. In the anthrax letters case, this could not be performed in real time and was not done. A “false positive” result that was uncovered during early evidence handling led to the unnecessary and expensive draining of a Maryland pond. This evidence proved not to be consistent with the Ames strain (NRC, 2011). Trace evidence errors can lead to costly mistakes. Nevertheless, the analysis of the repository samples using the then newly created assays eventually led the FBI to focus attention on a particular flask at USAMRIID containing a spore preparation known as RMR-1029.
In addition, physical science analytical approaches, such as scanning transmission electron microscopy, energy-dispersive X-ray analysis, carbon dating by accelerator mass spectrometry, and inductively coupled plasma-optical emission and mass spectrometry, were used to try to determine the chemical and elemental profiles of the spore powders (NRC, 2011). These kinds of testing were designed to answer such questions as how the material was grown and processed, when the anthrax preparation might have been made, whether there were contaminants or trace elements that would provide a clue to the production location or materials used, and whether there was evidence of an effort to deliberately include additives to improve dispersal of the anthrax spores.
Early in the investigation, elemental analysis of the letter spores detected silicon, leading some to conclude that this material had been deliberately added to aid aerosol dispersal of the anthrax spores. The conclusion that silicon was present was correct, but the interpretation that it had been added to weaponize the spores was not supported. A sensitive and sophisticated analysis performed at the Sandia National Laboratories showed that the silicon signal was inside, not outside the spore coat, which meant the spores had not been coated to decrease clumping (Michael and Kotula, 2009). However, because the erroneous conclusion was leaked to the media, it was never possible to completely mitigate this false conclusion. In fact, controversy concerning the source of the silicon content in the attack material remains active in the literature (Bhattacharjee, 2010; Hugh-Jones et al., 2011).
Two other analytical paths proved to be ineffective. One was stable isotope analysis. Because rainfall can have different oxygen isotope ratios, this analysis was performed to try to identify the region of origin of the
_______________
13 “Blanks are analytical quality control samples analyzed in the same manner as site samples. They are used in the measurement of contamination that has been introduced into a sample either (1) in the field while the samples were being collected or transported to the laboratory or (2) in the laboratory during sample preparation or analysis” (U.S. Environmental Protection Agency, 2004; http://www.epa.gov/oswer/riskassessment/ragsa/pdf/ch5.pdf).
water used to produce the spores. However, rainfall composition was not sufficiently predictive; in addition, autoclaving versus filtering water for sterilization can change isotope ratios. The second path was predicated on the fact that early nuclear weapons testing had caused a change in atmospheric carbon-14 ratios. This phenomenon suggested a potential way to date the production of the spores and showed that the spores in the letter sent to Senator Leahy were produced between 1998 and 2001. However, this date range did not prove to be useful information.
Keim emphasized, during his presentation at the Zagreb workshop, that one of the most powerful tools in forensics is one that has the ability to differentiate between inclusion and exclusion. The term “inclusion” means that an association or match might exist; that is, it is a failure to exclude some putative relationship. There might be an exact match—as might occur in some cases in human DNA testing—or the sample might be identified as a co-member of the same phylogenetic group. (The term “member” is preferred to “match” because in phylogenetic analysis a “member” may not be identical but would still be part of the same strain.) Conclusions as to whether two things are the same—inclusion—tend to be weak for use for attribution in court, because the clonal nature of microorganisms and unknown history or other factors can be used to discount or reduce the significance of such evidence. In contrast, exclusion can serve as very powerful evidence (Budowle et al., 2008). When it is concluded that something is not the same—not the Ames strain—based on the evolutionary models and assays, these conclusions are very strong. Exclusionary conclusions do not get newspaper headlines, but they are very important as they reduce the potential list of candidates and allow investigators to further focus efforts. The hypothesis testing used in molecular forensics and epidemiology can be summarized as follows:
- “Inclusion” (failure to exclude). Might be an exact match or at least co-members of the same phylogenetic group that are descended from a most recent common ancestor. Conclusions may be weakly supported.
- “Exclusion” (failure to include). Different from the comparator and very unlikely to be the same or from the same source. Conclusions may be very strong.
Three examples of the power of exclusion are based on the analyses of the B. anthracis strains obtained from (1) the biological-weapons lab accident in Sverdlovsk in the former Soviet Union in 1979 (Meselson et al., 1994), (2) the Aum Shinrikyo terrorist attack in Tokyo in 1993 (Danzig and Hosford, 2012; Keim et al., 2001), and (3) a biological-weapons production facility in Al Hakam, Iraq (analyzed soon after the 2001 U.S. anthrax
letters attack). None of these were consistent with the Ames strain in the anthrax letters. Links to the Japanese terrorist group and the Russian and Iraqi biological weapons programs were therefore ruled out.
Keim named the following as the key challenges associated with the anthrax letters investigation:
- Initial genetic conclusions about what the anthrax letters strain was and where it originated were based upon limited databases, which investigators recognized. Despite this, the information was shared with policy makers, who proceeded as though these initial conclusions were strong conclusions.
- Throughout the investigation, genomic technology was undergoing rapid developments from relying on an 8 locus MLVA system to 15- and 60-locus MLVA systems, to SNP analysis, and finally to WGS—which made trying to validate a system in real time very difficult.
- Trace evidence was analyzed using partially validated systems, leading to costly investigative errors.
- Scientific and peer-reviewed publication was limited during the entire process. To this day, critical data generated by the investigation remain unpublished.
- A “kitchen sink” approach to forensics was used. There was strong pressure to “try my method,” including pressure exerted from very high political levels.
- The public media added to confusion and disruption. There was too much coverage, for example, of the incorrect interpretation of the presence of silicon, yet too little when the false interpretation was corrected.
- It was a politically charged environment. Certain individuals sought the limelight, and their decision making affected the process of the science.
- Subject-area experts were also criminal suspects. Those performing analyses were simultaneously being investigated.
Obviously some of these circumstances are unavoidable in today’s environment. However, Keim provided two strong suggestions for improving the forensic approach to microbial attribution. The first is that investigators should define a specific hypothesis or hypotheses. Hypothesis building before obtaining results can reduce bias by averting any tendency to try to fit the results to a desired interpretation. If investigators focus their questions around hypotheses to be tested, it is possible to develop yes/no or inclusion/exclusion criteria in an investigation. The second recommendation is that investigators define a relevant reference
population for genetic analyses. Early in the anthrax investigation, there was inadequate information about both North American and worldwide B. anthracis strains.
THE IMPORTANCE OF PHYLOGENETIC INFORMATION
The Amerithrax case abundantly illustrated the importance of having adequate phylogenetic information about B. anthracis Ames, the agent involved in the case. Fortunately, Paul Keim and others had developed a rather extensive, although still incomplete, picture of the evolution of B. anthracis and the distribution of major strains throughout the world (Figure 2-2). This knowledge of strains of B. anthracis helped to pinpoint quickly the identity of the attack agent as the Ames strain because its genetic characteristics could be compared with other strains in the reference database. It should be noted, however, that the Ames strain is unusual in having a short history and a limited distribution. If the attack strain had been something other than Ames, its identity might not have been as simple to pinpoint.
In a more recent case dealing with anthrax, Dr. Richard Vipond, Operations Manager for the Rare and Imported Pathogens Laboratory at Porton Down in the United Kingdom, reviewed the forensic work employed during a 2009-2010 anthrax outbreak involving contaminated heroin (Price et al., 2012; Vipond presentation, 2013). In the United Kingdom and Scotland, most rare diseases are imported. Porton Down, part of the U.K. public health system, has substantial expertise in this area and is a WHO collaborative center for arbovirus, viral hemorrhagic fevers, and special pathogens, including B. anthracis. Historically, anthrax disease in the United Kingdom has been linked to importation and to contamination by animal products of “brownfield” (contaminated land previously used for industrial purposes) sites. Outbreaks in cattle occur in contaminated burial pit sites and imported materials; people renovating buildings may be infected via handling materials such as contaminated horsehair plaster.
Because of their experience with the drumthrax cases, the Porton Down investigators had the tools Keim’s team had developed for the Amerithrax case in place and thus were able to use 13 canonical SNPs (Van Ert et al., 2007) and 8 MLVA loci (Keim et al., 2000) to establish quickly that the heroin-associated strains were all the same, including the one in Germany. The strains matched the Trans-Eurasian group of strains, which is widely distributed, most likely through trade routes (Figure 2-2). The strain had not previously been seen in the United Kingdom, nor is it shown there on the map in Figure 2-2. With real-time PCR methods, resolution down to the single copy is possible, and so detection is not a major issue. There were 53 confirmed cases, and Vipond suspects there
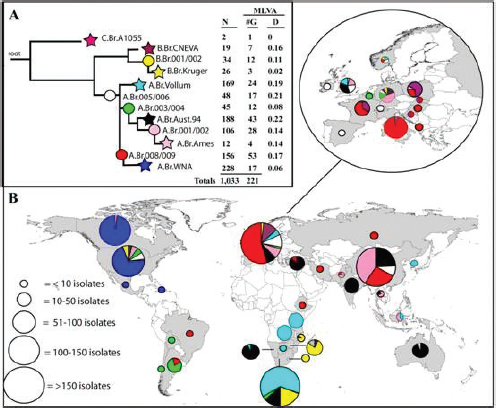
FIGURE 2-2 Worldwide distribution of B. anthracis clonal lineages: phylogenetic and geographic relationships among 1,033 B. anthracis isolates. (Panel A) Population structure based upon analysis of data from 12 canonical SNPs. The number of isolates (N) and associated MLVA genotypes (G) within each sublineage are indicated as well as the average Hamming distance (D) as estimated from VNTR data. The major lineages (A, B, C) are labeled, as are the derived sublineages (1-12), which are also color-coded. (Panel B) Frequency and geographic distribution of the B. anthracis sublineages. The colors represented in the pie charts correspond to the sublineage color designation in panel A.
SOURCE: Van Ert et al., 2007.
may have been 117 cases in total. Grunow et al. (2013) also reported that in 2012, additional cases occurred in Germany, France, Denmark, and the United Kingdom.
Vipond does not believe that the heroin-related anthrax outbreak represented a biocrime or bioterrorist attack. One argument against the outbreak being due to bioterrorism is that drug users are not an ideal target population for capturing the public’s interest. In addition, when the police stepped up their operations in Glasgow in hopes of confirm-
ing the source, they may have contributed to the spread of the disease by driving distributors to sell the heroin farther afield, resulting in cases emerging elsewhere in Scotland and down into England. The German case is thought to have emerged as a result of the normal trafficking route.
Vipond also pointed out that assays are becoming progressively more sensitive and are picking up natural background. A key need is to know what is there already, so one can spot the difference between a deliberate release and a new natural outbreak. One can live with natural background levels of B. anthracis in the soil and not become infected. So as detection gets better, one will be able to identify what has been there since time immemorial. The key is being able to distinguish between an anomaly and a deliberate release so no one hits the panic button every time something unusual is seen.
Dr. Ruifu Yang of the Beijing Institute of Microbiology and Epidemiology in China is a recognized expert in, among other things, the phylogeny and evolution of Yersinia pestis, the organism that causes plague. At the Zagreb workshop, he reviewed his work with genome sequences of Y. pestis, samples of which have been collected from around the world, to show how phylogenetics can aid microbial forensics. He also discussed the effect of environment on a pathogen’s ecology and evolution. A pathogen’s ecology can strongly determine its genomic diversity. For some microorganisms, ecological diversity is high because the organisms may inhabit ecological niches in soil, the ocean, and even in the human body, in which there are 10 times the number of microbial cells as human cells (Koeppel et al., 2008).
Yang classified bacterial pathogens into three groups based on the rate of a pathogen’s genomic recombination, the acquisition of alleles. A genomically monomorphic pathogen, such as Y. pestis or B. anthracis, rarely exhibits recombination, and thus may be relatively easy to source (see Figure 2-3). A genomically intermediate diversified pathogen (GIDP), such as Escherichia coli or Klebsiella pneumoniae, undergoes frequent recombination, but this does not necessarily affect the identification of phylogenetic relationships across samples. But a genomically highly diversified pathogen (GHDP), such as Helicobacter pylori or Vibrio parahaemolyticus, has a high frequency of recombination, and the phylogenetic relationship, especially in the deep branches, is distorted, which makes tracing a source more difficult, although it is still possible to distinguish among populations (e.g., Asian vs. U.S. strains).
An understanding of a pathogen’s ecology and genomic diversity will aid in developing microbial forensics strategies to trace a pathogen’s source. For example, it would be appropriate to use SNPs to trace all three of the above categories of pathogens; to use VNTRs to trace GIDP and GHDP pathogens; and to use loss or gain of genomic fragments, as well
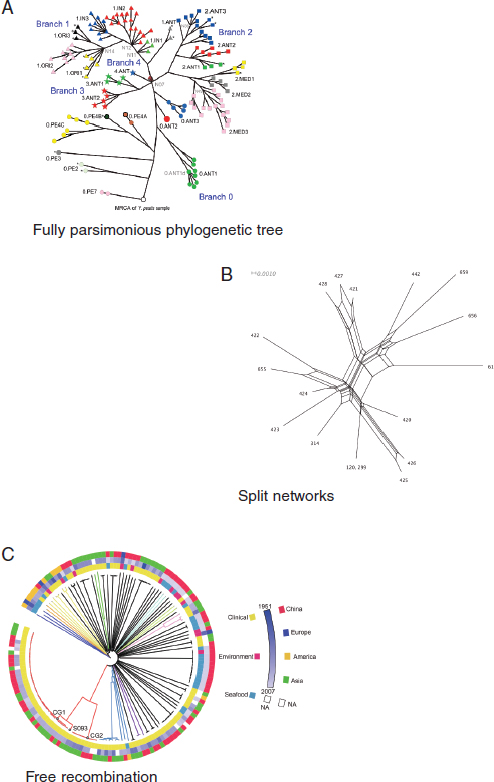
FIGURE 2-3 Three classifications of bacterial pathogens. (A) Y. pestis: Rare recombination, with parsimonious phylogenetic tree. (B) Intermediate level of recombination. (C) A neighbor-joining tree of 157 Vibrio parahaemolyticus genomes. V. parahaemolyticus is a highly diversified pathogen in which high-frequency homologous recombination leads to a radiated phylogenetic relationship.
SOURCES: A, Cui et al. (2013); B and C, Yang presentation, 2013.
as combined markers, to trace GIDP and GHDP pathogens. Databases can be built to accommodate these different target markers.
Y. pestis is widely distributed in China. As of 2010, there were 296 counties with 1.437 million km2 of natural plague foci. In different landscapes, climates, and ecologies, the hosts (e.g., various species of marmot, squirrel, rat) and bacterial strains vary. Dr. Yang and his colleagues are trying to understand the complex interactions between the pathogens, hosts, insect vectors, and environment. They have performed WGS on single strains and employed comparative genomics to understand pathogen diversity, using SNP analysis to understand the transmission of Y. pestis through history and the phylogenetic differences in strains. Collaboration with other investigators has enabled them to track the historical transmissions of Y. pestis plagues, which have repeatedly emerged from China, to trade routes such as the Silk Road (see Figure 2-4; Morelli et al., 2010). These analyses enabled them to track the evolution of different strains. The Black Plague strain of Y. pestis was different from strains before and
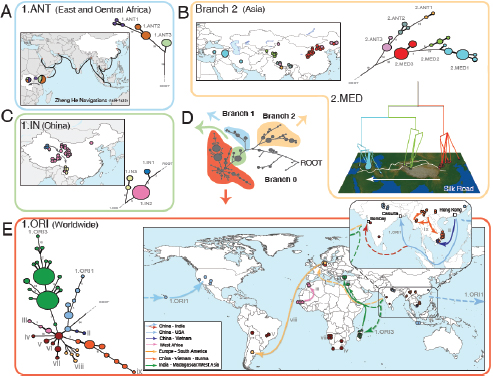
FIGURE 2-4 Distribution of Y. pestis.
SOURCE: Morelli et al. (2010).
after it, and the ecological context in which the strains developed helped shape these differences.
Unfortunately, having information at the level of detail that is available for anthrax and plague is unusual for most microorganisms (AAM, 2009a). As noted in the opening of this chapter, the microbial world has proven vastly more extensive and diverse than previously imagined. Metagenomic techniques now offer the possibility of determining the presence of new microorganismal genes directly from environmental samples or from the bodies of humans, other animals, and plants. To take advantage of this technique, the U.S. National Institutes of Health established a major project on the Human Microbiome and there are comparable European programs in existence. These efforts have produced vast quantities of new information on the bacteria, viruses, and microbial eukaryotes living in or on the human body (e.g., see Human Microbiome Jumpstart Reference Strains Consortium, 2010). One researcher, Rob Knight, has even investigated the possibility that human skin bacteria, which appear to be unique to individuals, can be used as a forensic identification tool. Knight and his team found that residual skin bacteria left on objects could be matched to the skin bacteria of the individual who touched the object. Furthermore, these bacteria can be recovered from objects, such as a computer mouse, for up to 2 weeks (Fierer et al., 2010). The human microbiome is, however, influenced by a wide diversity of factors—for example, health, environment, diet, hygiene—which present many challenges.
Other groups are seeking to characterize new microorganisms, such as viruses associated with particular animals. Anthony et al. (2013) recently reported on a rigorous and systematic 5-year effort to discover the viruses associated with a single bat, the Indian Flying Fox, which carries Nipah and Hendra viruses, among others. Fifty-five putative viruses (50 of which were new to science) from nine virus families were identified by repeated sampling from this one species. Extrapolating from the numbers derived from the Indian bat to the rest of the roughly 5,500 known mammalian species, the group estimated that there are a minimum of 320,000 undiscovered mammalian viruses waiting to be described, assuming that all mammalian species harbor a similar number of viruses. The importance of this work was characterized as enabling the establishment of preliminary bounds on the potential size of the pool of zoonotic viruses in mammalian wildlife. Other “virus hunters” are working to develop an early warning system to monitor the transmission of infectious diseases from animals to humans. Nathan Wolfe’s work in Africa and Southeast Asia, for example, is designed to develop forecasting capabilities that would identify and mitigate potential new zoonotic disease outbreaks before they become serious pandemics (Wolfe et al., 2007).
Such efforts are, however, barely scratching the surface of the microbial world. Even phylogenetic studies of organisms that are relatively well understood, such as B. anthracis, are still incomplete, and there is as yet no worldwide repository of information on pathogen species and strains or any coordinated effort to develop information exchange mechanisms. John Clements pointed out that if public health agencies are not routinely collecting surveillance data (e.g., on influenza cases), data will not exist for law enforcement to use for microbial forensics purposes. Such data could be useful to both sectors.
Dr. Aaron Darling further reviewed microbial ecology and diversity in the context of forensics, focusing on metagenomics, which he defines as the study of DNA from uncultured organisms. He believes metagenomics has enormous potential to help in microbial forensics but that challenges remain for optimization of the technology. A major advantage of using metagenomics is that one can avoid culture bias (some organisms reproduce more quickly than others). There are also, however, disadvantages to metagenomics. One is DNA extraction bias. An organism cannot be sequenced if its DNA cannot be extracted. For example, if the DNA is trapped in spores with hard, uncrackable walls, extraction may not be successful. Therefore, the nucleic acids of an organism that are easily extractable in a sample may suggest that that organism is in greater abundance whereas another organism—perhaps of forensic interest—seems to be of low abundance. A second limitation is that a small number of species greatly dominate most ecosystems. It is difficult to study rare taxa, which comprise the majority of species, and the organism of interest may be naturally present at very low abundance. This situation would require a great amount of sequencing for characterization, higher throughput, or better targeting of informative genetic markers.
Darling stated that most of what is known about the ecology and evolution of pathogens comes from the study of isolates. Like Dr. Yang, he and his colleagues have categorized organisms by their recombination propensity, dividing them into organisms that tend to be monomorphic, those that are intermediately clonal, and those that recombine heavily. Even highly recombinant organisms, however, can reflect obvious phylogenetic structure.
Virologists have led the way in culture-independent forensic analyses, and there have been a number of biocrime cases where phylogenetic evidence was used to support prosecution of individuals for deliberately infecting others. In one such case, an HIV-infected individual was accused of intentionally infecting victims. Analyses of samples of DNA sequences of human immunodeficiency virus 1 (HIV-1) taken from the victims and the alleged suspect were performed in order to reconstruct the phylogenies of the HIV retrovirus polymerase (pol) and envelope (env)
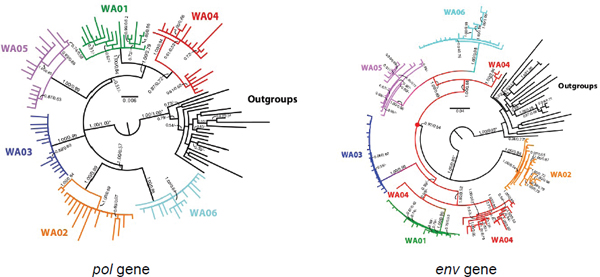
FIGURE 2-5 Red lineages represent HIV sequences from the suspected perpetrator. These lineages are ancestral in the env genes but not the pol genes in victims’ samples.
SOURCE: Scaduto et al. (2010).
genes (Scaduto et al., 2010).14 However, Darling noted with concern that the analysis of the pol gene shows a cluster of the victims’ pol sequences that does not include any of the suspect’s sequences, whereas analysis of the env gene shows the suspect’s sequences are ancestral to most of the victims’ sequences (see Figure 2-5). The env gene was used as evidence to support association, but seemingly discordant results from two genes with different evolutionary rates should be understood and appreciated.
Darling noted the previously mentioned methods focused on SNPs as the basis for inferring phylogenies and performing forensic analyses. He pointed out that WGS, however, has revolutionized the understanding of bacterial evolution. In one study, after sequencing three E. coli genomes, researchers compiled a list of all genes in the genomes; only about 40 percent of genes were common to all three strains (Perna et al., 2001; Welch et al., 2002). Although it has been known for some time that bacteria exhibit heterogeneity among strains (e.g., Bergthorsson and Ochman, 1995), it had long been assumed that members of the same species generally have highly homogeneous genomes and are largely identical because they are
_______________
14 The paper by Scaduto et al. presents the molecular evidence used in two U.S. criminal cases: State of Washington v Anthony Eugene Whitfield, case number 04-1-0617-5 (Superior Court of the State of Washington, Thurston County, 2004) and State of Texas v Philippe Padieu, case numbers 219-82276-07, 219-82277-07, 219-82278-07, 219-82279-07, 219-82280-07, and 219-82705-07 (219th Judicial District Court, Collin County, TX, 2009). The discrepancy noted by Darling comes from the Texas case.
the same species. This belief proved untrue, sparking a controversy over the nature of the definition of a species in bacteria. But it also suggested that one might be able to use gene content, instead of just nucleotide sequences, for forensic characterization because the gene content appears to be evolving almost as fast, if not faster, than the nucleotides.
Darling related information about a joint pilot project by the University of California, Davis and the JGI, that sought to characterize the genetic diversity of microbial organisms worldwide.15 As noted earlier, if one wishes to build a database of background information about different microbial ecosystems around the globe, an understanding of what exists is needed. The researchers gathered cultures of approximately 100 organisms they deemed the most diverse from culture collections around the world and sequenced their genomes (Wu et al., 2009). They then measured the rate at which they discovered new genes, which was very high. Moreover, novelty in gene content correlates highly with novelty in the 16S rRNA16 sequence (Wu et al., 2009). Even within a single species, with each additional genome sequenced, discovery of new genes occurs.
As the discovery of additional 16S rRNA diversity continues, it becomes ever clearer that an immense, and probably unachievable, amount of sequencing would be required to characterize all the microbial DNA and RNA on the planet. The amount of 16S rRNA diversity that has been described in cultured samples is dwarfed by the amount of diversity that has been described in uncultured or cloned cultures. Uncultured bacterial diversity greatly exceeds known isolate diversity. How do we gain access to all this uncultured diversity if one cannot grow it? There is a need to be able to build up databases on background microbial communities and use them in forensics.
Although metagenomics has been posed as a solution for this task, Darling indicated that metagenomics in its current form is far from adequate. There are two problematic technical issues. First, samples are complex mixtures. When extracted, the fragmented DNA from the chromosomes from the various microbes in a sample are comingled and they cannot readily be pieced back together. Second, as part of the sample preparation for sequencing, the already fragmented DNA is broken down further to much smaller sizes so the sequencing chemistries can read them. In breaking up the longer molecules, information is lost. While assembly of short read lengths has been possible with homogeneous samples, the assemblies are problematic with complex samples and some genes or anonymous sequences may not be detected. Furthermore, it is
_______________
15 For more information see http://www.jgi.doe.gov/programs/GEBA/index.html. Accessed November 13, 2013.
16 The 16S rRNA gene is a section of prokaryotic DNA found in all bacteria and archaea.
difficult to place separate genes, if detected, on the same microbial chromosome even if they originally were syntenic. It becomes unclear which chromosomes are which, which chromosomes were in which cells, and which genes were in which chromosomes. This is a tremendously difficult dataset to analyze, and currently most analyses do not do a good job of reconstructing the cellular origins of the genomic material. To solve this problem, Darling suggests (1) physically “dissecting” microbial communities to preserve information, (2) developing better inference methods with the help of bioinformaticians, and (3) developing alternative sequencing technologies.
Darling has been investigating amplification and sequencing of DNA from single cells obtained directly from environmental samples (i.e., single-cell genomics). He and his colleagues sequenced 201 uncultivated archaeal17 and bacterial cells from nine different habitats. These organisms belonged to 29 major, but mostly unfamiliar, phylogenetic branches (Rinke et al., 2013). The additional genomic information enabled the researchers to resolve many intra- and interphylum-level relationships and to propose two new superphyla. However, the microbial diversity they were able to capture by this method was only a tiny fraction of organisms in existing genome databases. Therefore this method currently does not scale up for application to microbial forensics.
Darling believes that the most important metagenomic analyses in microbial forensics are phylogenetic analyses. He explained that while the classic, isolate-based genome alignment provides one full-length DNA sequence per isolate, metagenomic alignments provide many DNA fragments, with no information on which fragments originated from the same cell. Current methods for phylogenetic inference assume that complete linkage information is available. In particular, they assume that a full-length DNA sequence for the gene under study is available from each organism being analyzed. However, metagenomic data do not satisfy these requirements because the data frequently contain small nonoverlapping fragments of each organism’s gene sequence. Two inferential approaches that can be used to create phylogenetic trees from metagenomic data include phylogenetic placement, in which the read fragments are “mapped” into a reference phylogeny tree; and co-estimating the read clusters. This process is predicated on the assumption that there is a limited amount of diversity in a metagenomic sample: even if one has 20 million reads, they must combine into a finite number of species. Two ways to implement this approach include Bayesian evolutionary analysis
_______________
17 Archaea constitute a domain of single-celled microorganisms. Both the bacteria and archaea domains comprise prokaryotic organisms, but the two domains have different evolutionary histories.
by sampling trees (BEAST), which is a software architecture for analyzing molecular sequences related by an evolutionary tree, and PhyloSift.
Phylogenetics, which provides information on evolutionary relationships based on genetic data, can be used to determine what may be present in a sample. The greater the amount of genetic data, the deeper the resolution of the information on the microbe(s) present. With limited information, such as a single marker, phylum level may be the best that can be determined, which is likely to be of little use for attribution and determining the presence of, for example, a Select Agent. With a whole genome or a number of genes, deeper classification may be feasible. Bayesian hypothesis testing can be applied to any gene or genetic marker phylogeny, including toxin genes of interest. Metagenomic-derived phylogenetic data also can be used to answer the question, “What is the closest match between a sample of interest and a database of related samples?” This can be accomplished using phylogenetic placement data and the “phylogenetic Kantorvich–Rubinstein distance” (Evans and Matsen, 2012).
However, these methods can be applied only to a single gene at a time or to a set of phylogenetically coherent genes. The capability to analyze all genes simultaneously is needed. Given current technology, the information that is contained in whole genomes is not going to be entirely available and some information will not be captured. The analytical challenge of accomplishing this corresponds to reconstructing the histories of coevolution of all genes in a phylogenetic pattern. Evolution is complex and reticulate. Within a species tree, there are phylogenies of individual genes, which include gene duplication, gene conversion, lateral gene transfer, and gene death. The target areas that define a species or strain and distinguish it from near neighbors must be well defined. One or more sites may be required depending on phylogenetic resolution. Efforts are under way using informatics and phylogenetic reconstruction to meet the challenges (Bérard et al., 2012; Boussau et al., 2013). But scaling these methods presents technical, cost, and training challenges and is likely to remain difficult.
THE ISSUE OF DATA SHARING
Even if there is a major effort to characterize microbial diversity comprehensively through nucleic acid sequencing, unless the data that are collected are shared in such a way that scientists around the world have access to them, they will not provide optimal benefit for either public health or microbial forensics. During the discussion following the workshop session on microbial ecology and diversity, various workshop participants pointed out a variety of problems involved with data sharing.
Paul Keim pointed out that language (e.g., English vs. Chinese) and export-control laws can pose problems for sharing data, particularly for organisms that are considered potential bioweapons. He noted that export control laws can sometimes be circumvented by publication of the data, but Dr. Yang stated that the decision to share is not always left up to the scientists, who are not always permitted to publish everything. Yang said that the worldwide scientific community could press governments to address the need for data sharing, given the bioweapons context, citing the needs of public health and microbial forensics. Yang’s database of Y. pestis, for example, could aid a traceback in the event of a Y. pestis outbreak anywhere in the world. A major theme among the workshop participants was that solutions to data sharing are severely needed. Ideally, countries would have their own microorganism collections and genomic databases and share them. But standards for the technology used would be needed so that all collections would have the same level of testing, accuracy, and quality. In addition, large developed countries have an advantage over small developing countries when it comes to data-generating capacity. It was suggested that an international framework is needed both to encourage and to reward data sharing.
























