
3
Problem Formulation and Protocol Development
As discussed in Chapter 1 of this report, the US Environmental Protection Agency (EPA) is incorporating principles of systematic review as it revises the Integrated Risk Information System (IRIS). Critical elements of a systematic review include formulating the specific question that will be addressed and developing the protocol that specifies the methods that will be used to address it. The National Research Council (NRC) report that reviewed the IRIS formaldehyde assessment (NRC 2011) did not provide any specific recommendations regarding those elements, but the present committee found that some discussion of them is warranted given EPA’s shift toward adopting systematic-review principles. Therefore, this chapter discusses problem formulation and protocol development as parts of the IRIS process and systematic review as shown in Figure 3-1. The committee distinguishes between the scoping exercise described in Chapter 2 and problem formulation described here. The scoping exercise involves soliciting input from EPA program and regional offices to determine the bounds of the assessment—such as exposure pathways and specific exposed groups to consider—that will help EPA with its decision-making, whereas problem formulation is intended to frame the specific scientific questions for the systematic reviews in the IRIS-assessment process. That distinction is consistent with the NRC report Science and Decisions: Advancing Risk Assessment (NRC 2009).
The risk-assessment paradigm that dates to the 1983 report Risk Assessment in the Federal Government: Managing the Process (NRC 1983) and has long been used by EPA has four components: hazard identification, dose-response assessment, exposure assessment, and risk characterization. Two components are encompassed by the IRIS assessment process: identifying potential hazards related to a chemical by using the available literature as a source of information (hazard identification) and characterizing the dose-response relationship (dose-response assessment) (see Figure 3-2). Thus, problem formulation in the IRIS process is restricted to scientific questions that pertain only to those two elements of the risk-assessment paradigm. Although the committee’s review of the problem-formulation step focuses mainly on searching available literature, seeking stakeholder input and advice is an integral part of the process and should not be minimized.
Evidence Used for IRIS Assessments
As indicated in Figure 3-2, evidence typically used by EPA for IRIS assessments comes from human studies, animal studies, and mechanistic studies. Those study types are briefly discussed below to set the stage for further discussion in the present report.
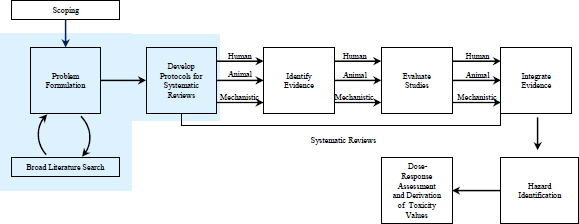
FIGURE 3-1 The IRIS process; problem formulation and protocol development are highlighted. The committee views public input and peer review as integral parts of the IRIS process, although they are not specifically noted in the figure.
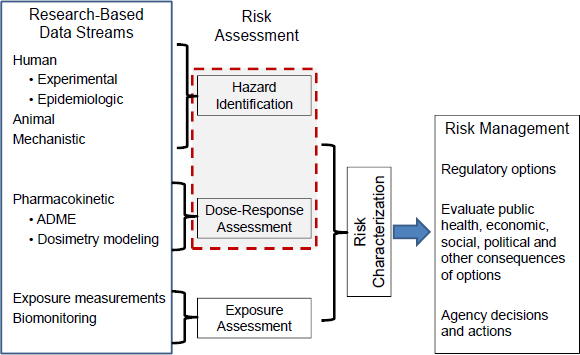
FIGURE 3-2 The risk-assessment, risk-management paradigm. The box outlined in red shows the information included in IRIS assessments. Source: Adapted from NRC 1983.
Human Studies
Human studies of health can be divided into ones that control exposure (experimental studies) and ones that do not (observational studies). Experimental human studies with potentially toxic chemicals are performed infrequently, so most human studies of adverse outcomes are observational epidemiologic studies in which exposure is not controlled, but rather the consequences of inadvertent human exposures.
Broadly speaking, most observational epidemiologic study designs can be categorized as cross-sectional, cohort, or case-control. In cross-sectional studies, measurements of a variety of factors are recorded at a particular time; cross-sectional studies that are considered in an IRIS assessment typically involve assessment of one or more health outcomes in relation to current or past exposure. In cohort studies, persons exposed or nonexposed to a given factor are observed over a period for the onset of health effects related to the exposure. A case-control study compares persons who have a given disease (cases) with those who do not have the disease (controls) with regard to their history of exposure. Each design has appropriate analytic strategies, and each has its own strengths and weaknesses. There are variations of each approach and other designs as
well. Epidemiologic study designs are well described in standard epidemiologic references (see, for example, Rothman et al. 2012).
The general difficulty of observational epidemiologic studies, regardless of design, is that exposure is not randomized but rather is determined by where people live or work, what they eat, what social group they belong to, or a host of other factors that can affect disease risk. As a result, associations between exposure and disease risk can occur even if the exposure does not cause the disease. And it is possible that no association is measured when the exposure does cause disease because confounding factors act to reduce or even cancel the effect of the exposure that is being investigated or the study is too small (underpowered) to see the effect against the background rate of disease.
Despite the inherent weaknesses, epidemiologic studies present a number of advantages for chemical risk assessment. For example, the exposure-effect relationships studied are in the target species, humans; the exposure-effect relationships can be studied in heterogeneous human populations, and it is possible to study the interactions between a chemical exposure and other factors, such as genes and lifestyle; and they provide data on relevant exposure conditions and routes of exposure (Nachman et al. 2011).
For some agents, data might be available from controlled experimental exposures of small groups of people. Of necessity, such studies are limited to brief exposures that are expected to cause no lasting harm and to acute responses. For example, volunteers have been exposed to formaldehyde and other gases. Beyond assessing short-term responses, such studies might be used to improve understanding of dosimetry and to assess biomarkers. They can be a useful bridge to the findings of animal studies.
Animal Studies
Using laboratory animals, primarily rats and mice, to assess chemical hazard remains an essential component of toxicologic and chemical risk assessments (Beyer et al. 2011). Animal studies often provide critical qualitative and quantitative information on the types of adverse effects to expect in humans and some general indication of the amount, frequency, and timing of exposure or dose that could be associated with a particular adverse outcome. Animal testing can be divided into two broad approaches: identification of toxic hazards (discussed here) and mechanistic studies, including pharmacokinetic studies (discussed in the next section).
In vivo animal tests are used to determine the nature of adverse responses (toxicity) that a chemical can produce and then to characterize the dose-response relationship between a chemical and particular types of adverse responses; each type of response will have its own dose-response relationship. Many animal-testing protocols have been developed for specific hazard end points, such as acute organ toxicity, reproductive and developmental (including teratogenic) effects, carcinogenesis, neurologic and behavioral effects, immune-system effects, and eye and skin irritation (see, for example, Eaton and Gilbert 2013). Such tests have rigorous design elements and, if used for regulatory purposes, must follow good laboratory practice guidelines. Many national regulatory agencies—such as EPA, the US Food and Drug Administration (FDA), and the US Department of Agriculture—and international regulatory agencies—such as the European Commission and the Japanese Ministry of Agriculture, Forestry, and Fisheries—have developed such toxicity-testing protocols and have expended substantial efforts to harmonize guidelines (Ertz and Preu 2008). Regardless of the particular end point or type of study being conducted, the ultimate purpose is usually the same: to identify toxic hazards and to characterize the shape of the dose-response curve for a given end point. The doses identified in animal studies with specific response levels can then be modeled to predict minimal response levels, often referred to as benchmark doses (BMDs) at a specified level of response, such as 5% (BMD5) and 10% (BMD10) (Filipsson et al. 2003; Davis et al. 2011). The values derived from the animal studies can then be used to derive toxicity values (reference concentrations, reference doses, and unit risk values) when suitable data from human studies are unavailable for this purpose.
Animal experiments have at least one important advantage over human studies: exposure can be experimentally controlled. Several other sources of variation besides exposure can also be controlled, and the ability to control exposure and other factors eliminates most of the risk of confounding. However, the use of experimental animal data for predicting human health risk is subject to multiple sources of uncertainty, especially uncertainty regarding the relevance of animal-model findings to humans. Species differences in response to toxic chemicals can be highly variable, and reliance on animal studies alone for predicting human health risks can lead to false positives and false negatives. For example, benzene and arsenic were identified as human carcinogens on the basis of epidemiologic data at a time when animal data failed to identify the carcinogenic risks; later refinement of animal models and a better mechanistic understanding of how these chemicals cause cancer have made it possible to explain the reasons for the disparate results of early studies.
Mechanistic Studies
For purposes of this report, mechanistic data come from a wide variety of studies that are not intended to identify an adverse outcome. The committee notes that it is using the term mechanism of action (or simply mechanism) in this report rather than mode of action simply for ease of reading; it recognizes that these terms can have different meanings. This third source of experimental data includes in vitro and in vivo laboratory studies directed at the cellular, biochemical, and molecular mechanisms that explain how a chemical produces particular adverse effects. These studies increasingly take advantage of new “-omics” tools, such as proteomics and metabolomics, to identify early biomarkers of effect. In vitro studies that use cells and tissues derived from humans and animals can provide information on the relative sensitivity of human and animal cells and can identify critical differences in how a chemical is metabolized and eliminated from the body.
Another broad class of mechanistic data is related to the toxicokinetics of a chemical. Physiologically based pharmacokinetic (PBPK) models are increasingly used by EPA and other agencies to support risk assessments (Lipscomb et al. 2012). PBPK models integrate mechanistic absorption, metabolism, distribution, and excretion (ADME) data and can be used to predict the time course of a parent chemical, metabolites, or biomarkers in the exposed organism under various exposure conditions. Thus, they can provide critical insights into potential differences in the dose-response relationship between species or between different groups within a species (for example, sex, race, or ethnicity differences related to genomic variation). PBPK models can also be used to support quantitative extrapolation of in vitro to in vivo data (Yoon et al. 2012) and are used by EPA and others to support extrapolations between species, exposure routes (for example, inhalation to oral), and exposure durations (Barton et al. 2007; Kenyon 2012). The recent methanol IRIS assessment (EPA 2013a) is an excellent example of how PBPK modeling and related mechanistic data can be used to understand species differences in response to different exposures.
Use of mechanistic information in the IRIS process has been focused on supporting the biologic plausibility of in vivo observations in animal or human studies. In some cases, in vitro results can substantially influence hazard identification and dose-response assessment. For example, EPA’s Guidelines for Carcinogen Risk Assessment (EPA 2005) require that a chemical that is associated with an excess incidence of cancers in animal bioassays or human epidemiologic observations be treated as a genotoxic carcinogen if there are largely positive in vitro mutagenesis or genotoxicity studies. That classification will result in low-dose linear extrapolations in dose-response modeling. In contrast, under those same EPA guidelines, a chemical for which in vitro mutagenesis or genotoxicity assays are largely negative might be classified as a nongenotoxic carcinogen; such carcinogens are often modeled by using nonlinear approaches at low doses. Mechanistic data, including data from in vitro studies, can also be used in interpreting discrepancies between results of human and in vivo animal studies. For example, chronic animal
bioassays of the widely used dietary sweetener saccharin found an increased incidence of bladder carcinomas in rats, although extensive use in human populations failed to identify any risks. Mechanistic studies demonstrated that the bladder carcinogen was secondary to a phenomenon peculiar to male rats, and the FDA later removed saccharin from the list of potential food carcinogens. Likewise, EPA has developed guidance documents to evaluate chemicals that induce accumulation of the low-molecular-weight protein alpha2u-globulin (α-2U) in the male rat kidney (EPA 1991). Renal accumulation of α-2U in male rats initiates a chain of events that lead to renal tubule tumor formation. Unlike male rats, female rats and other laboratory mammals do not accumulate α-2U in the kidney and do not develop renal tubule tumors. Humans appear to respond more like female rats than like male rats; thus, the male rat in this case is not a good model for evaluating human risk (Rodgers and Baetcke 1993; McClellan 1996). Conversely, studies might also show that human responses that lead to increased susceptibility to a risk are different from animal responses. For example, the human teratogen thalidomide failed to induce phocomelia and other birth defects in laboratory rats and mice at equivalent doses (Collins 2006).
For a chemical hazard evaluation, there might be hundreds of in vitro and other mechanistic studies of a given chemical and only one or a few in vivo animal or human epidemiologic studies. Although EPA would be unlikely to initiate an IRIS review of a chemical on which the only available data are from in vitro or mechanistic studies, a well-designed systematic review of all the mechanistic information available is an important element of the IRIS process for chemicals on which in vivo animal or human epidemiologic data are available. Kushman et al. (2013) describe a process for conducting systematic reviews of mechanistic data in human-health assessments. Using diethylhexylphthalate as an example, they provide a process that includes all the basic elements of systematic review (defined literature search, inclusion and exclusion criteria, and evidence tables) for evaluation of mechanistic data.
Development of Systematic-Review Questions
A major challenge in the problem-formulation step is determining what adverse outcomes are of potential concern.1 To identify the potentially relevant outcomes associated with exposure to a given chemical, the IRIS chemical-assessment team needs to conduct an initial broad search of the literature and toxicology databases by using the procedures described in the draft handbook for IRIS assessments (EPA2013b, Appendix F). The initial search provides the foundation for constructing well-defined questions and for constructing the protocol for each targeted systematic review for a particular outcome. The thorough and systematic literature search that is conducted for each systematic review (as described in Chapter 4) should not be confused with the broad literature search conducted for problem formulation. The recently revised IRIS process that is described in the preamble of each assessment (see, for example, EPA 2013c) should differentiate better the sequence of steps taken to survey the literature, develop the focused questions for each identified putative outcome, and identify and assess the evidence that addresses the questions.
The committee suggests the following process for conducting problem formulation under the assumptions that the outcomes are broadly defined and that all putative toxicological outcomes are considered.
_____________________________
1The committee is using the term outcome to refer to a disease phenotype—for example, various cancer types, asthma, or diabetes—or specific tissue or organ system damage or dysfunction, such as liver damage, kidney damage, perturbed neurologic function, or altered reproductive function. The adverse outcome might be identified by functional end points (for example, altered liver function or metabolic changes), anatomic end points (for example, histopathologic changes or fetal resorptions), or behavioral end points.
Step 1: With the support of an information specialist trained in conducting systematic reviews, a broad literature search should be designed and performed to identify possible outcomes associated with the chemical under investigation. The term information specialist (or informationist) was developed and is commonly used in the context of clinical medicine (Davidoff and Florance 2000; Whitmore et al. 2008; Grefsheim et al. 2010). In the context of the IRIS process, an information specialist would be a person trained in toxicology and risk assessment, able to interact with the chemical-assessment team, and having expertise in information science and systematic-review methods.
Step 2: A table (see Table 3-1) might be constructed to guide the formulation of specific questions that would then be the subjects of specific systematic reviews. Each study that is identified in the initial search would be included in one or more of the cells in the table. As noted, for simplicity, the committee is not distinguishing between the terms mechanism of action and mode of action and is using mechanism of action (or simply mechanism) throughout the report.
Step 3: The completed table would document which toxicological outcomes have been examined scientifically and warrant formulation of a specific research question and a systematic review of the available evidence. For example, if the search identifies articles that examined the mutagenicity of chemical X in animals, the articles would be listed in the row labeled “Genotoxicity or mutagenesis” under the column labeled “Animal (in vivo) studies.” The articles would lead to a research question (problem formulation): “Is there scientific evidence that chemical X is mutagenic in animals or humans?” The research question would then be addressed by a systematic review, which would require a separate formal search for evidence (see Chapter 4).
TABLE 3-1 Outcomes for Consideration in Problem Formulation
| Outcome | Human (in vivo) Studiesa | Animal (in vivo) Studies | In vitro, Mechanistic Studies |
| Genotoxicity or mutagenesis | |||
| Oncogenesis | |||
| Reproductive | |||
| Developmental, teratogenesis | |||
| Pharmacokinetics | |||
| Neurologic and sensory systems | |||
| Hepatic | |||
| Renal | |||
| Gastrointestinal | |||
| Endocrine | |||
| Metabolic disease | |||
| Respiratory | |||
| Cardiovascular | |||
| Hematopoietic | |||
| Immunologic | |||
| Musculoskeletal | |||
| Dermal | |||
| Other |
aHuman in vivo studies might embody an array of experimental designs, including controlled human exposure studies in chambers, case reports, and epidemiologic studies, including ecologic, cohort, cross-sectional, and case-control studies.
As noted earlier, the approach recommended by the committee appears to be similar to the revised process used in the draft IRIS assessment for benzo[a]pyrene (EPA 2013c). In that document, the literature search (summarized in Figure LS-1) identified 700 potentially relevant publications among 21,000 hits in the search and categorized them by target organ and outcome. The summary of the search did not include the number of articles identified for the study categories listed in Table 3-1 above (human, animal, and mechanistic), and this omission is acceptable in this early step. However, the chemical-assessment team would be expected to expand on that classification for the systematic reviews.
The completed table (Table 3-1) constitutes the basic starting point for careful definition of the hazard-specific questions that can be subjected to systematic review. Each hazard-specific question should specify (1) the specific chemical, process, or mixture being evaluated (and possibly the sources and pathways of exposure), (2) the general types of studies of interest (for example, in vitro, animal in vivo, human clinical, and epidemiologic studies), and (3) the outcomes of interest and the organ system potentially affected.
Questions should be formulated for systematic review if an outcome is deemed to be of possible importance regardless of the amount of evidence that is thought to exist. That approach allows the hazard-identification process to be structured to minimize the chances of incorrectly assuming that a risk does not exist (a false negative). Even if the evidence on a hazard-specific question appears minimal initially, a systematic review can be undertaken if the question is deemed worthy of investigation. Decisions as to which specific outcomes should be further evaluated by specific systematic reviews require careful consideration of numerous factors, including whether the potential outcome is likely to occur at doses encountered by the general population and what the significance of the outcome will be if the potential association suggested in the screening review is real. Expert judgment will play an important role in this step of the IRIS process. The decision process for determining which outcomes should be subjected to a systematic review should be carefully described, and the description should be subject to peer review by experts along with the rest of the IRIS document. The committee recognizes that some chemicals will have numerous “positive” end points and a large database of studies and that multiple systematic reviews could be resource-intensive and challenging to complete in a timely matter. In such circumstances, EPA might need to establish an additional prescreening process to ensure that efforts are focused on the most relevant public-health end points. Additional guidelines should be established that will ensure consistency in the approach for all chemicals.
The committee notes that the preamble appears to merge scoping and problem formulation by stating that “the IRIS Program discusses the scope with other EPA programs and regions to ensure that the assessment will meet their needs. Then a public meeting on problem formulation invites discussion of the key issues and the studies and analytic approaches that might contribute to their resolution” (EPA 2013c, p xiv). It does not explicitly describe how the question is formulated, and it is unclear whether the process described is used for specific systematic-review questions for each relevant outcome. A properly formulated question is important in setting eligibility criteria for the review and designing the literature search strategy. Once the questions for the systematic reviews are specified, the protocol for each review can be developed.
When the systematic-review questions have been specified, a protocol for each review should be developed. A protocol makes the methods and the process of the review transparent, can provide the opportunity for peer review of the methods, and stands as a record of the review. The protocol also minimizes bias in evidence identification by ensuring that inclusion of studies in the review does not depend on the findings of the studies. Any changes made after the protocol is in place should be documented and justified in the final report. Box 3-1 lists the common
BOX 3-1 Systematic-Review Protocol Elements
A. Systematic review question (for example, is benzo[a]pyrene exposure of adult animals associated with neurotoxic effects?)
B. Methods
1. Inclusion and exclusion criteria for studies:
a. Types of studies or participants (for example, experimental animal, observational human, or in vitro mechanistic).
b. Types of exposures (for example, oral or inhalation).
c. Types of outcome (for example, neurotoxic or developmental).
2. Search methods for identification of studies.
3. Assessment of risk of bias and other methodologic characteristics of included studies.
4. Data-collection methods.
5. Analysis.
elements of a systematic-review protocol. The plan for completing each step should be described in the protocol. Further discussion of protocol elements is provided where appropriate in the chapters that follow.
Finding: The materials provided to the committee by EPA describe the need for carefully constructed literature searches but do not provide sufficient distinction between an initial survey of the literature to identify putative adverse outcomes of interest and the comprehensive literature search that is conducted as part of a systematic review of an identified putative outcome.
Recommendation: EPA should establish a transparent process for initially identifying all putative adverse outcomes through a broad search of the literature. The agency should then develop a process that uses guided expert judgment to identify the specific adverse outcomes to be investigated, each of which would then be subjected to systematic review of human, animal, and in vitro or mechanistic data.
Recommendation: For all literature searches, EPA should consult with an information specialist who is trained in conducting systematic reviews.
Finding: A protocol is an essential element of a systematic review. It makes the methods and the process of the review transparent, can provide the opportunity for peer review of the methods, and stands as a record of the review.
Recommendation: EPA should include protocols for all systematic reviews conducted for a specific IRIS assessment as appendixes to the assessment.
Barton, H.A., W.A. Chiu, R. Woodrow Setzer, M.E. Andersen, A.J. Bailer, F.Y. Bois, R.S. Dewoskin, S. Hays, G. Johanson, N. Jones, G. Loizou, R.C. Macphail, C.J. Portier, M. Spendiff, and Y.M. Tan. 2007. Characterizing uncertainty and variability in physiologically based pharmacokinetic models: State of the science and needs for research and implementation. Toxicol. Sci. 99(2):395-402.
Beyer, L.A., B.D. Beck, and T.A. Lewandowski. 2011. Historical perspective on the use of animal bioassays to predict carcinogenicity: Evolution in design and recognition of utility. Crit. Rev. Toxicol. 41(4):321-338.
Collins, T.F. 2006. History and evolution of reproductive and developmental toxicology guidelines. Curr. Pharm. Des. 12(12):1449-1465.
Davidoff, F., and V. Florance. 2000. The informationist: A new health profession? Ann. Intern. Med. 132(12):996-998.
Davis, J.A., J.S. Gift, and Q.J. Zhao. 2011. Introduction to benchmark dose methods and U.S. EPA’s benchmark dose software (BMDS) version 2.1.1. Toxicol. Appl. Pharmacol. 254(2):181-191.
Eaton, D.L., and S.G. Gilbert. 2013. Principles of toxicology. Pp. 13-48 in Casarett and Doull’s Toxicology: The Basic Science of Poisons, 8th Ed., C.D. Klaassen, ed. New York: McGraw-Hill.
EPA (U.S. Environmental Protection Agency). 1991. Alpha2u-Globulin: Association with Chemically Induced Renal Toxicity and Neoplasia in the Male Rat. EPA/625/3-91/019F. Risk Assessment Forum, U.S. Environmental Protection Agency, Washington, DC [online]. Available: http://www.epa.gov/raf/publications/alpha2u-globulin.htm [accessed December 17, 2013].
EPA (U.S. Environmental Protection Agency). 2005. Guidelines for Carcinogen Risk Assessment. EPA/630/P-03/001F. Risk Assessment Forum, U.S. Environmental Protection Agency, Washington, DC. March 2005 [online]. Available: http://www.epa.gov/raf/publications/pdfs/CANCER_GUIDELINES_FINAL_3-25-05.PDF [accessed October 3, 2013].
EPA (U.S. Environmental Protection Agency). 2013a. Toxicological Review of Methanol (Noncancer) (CAS No. 67-56-1) in Support of Summary Information on the Integrated Risk Information System (IRIS). EPA/635/R-11/001Fa. U.S. Environmental Protection Agency, Washington, DC. September 2013 [online]. Available: http://www.epa.gov/iris/toxreviews/0305tr.pdf [accessed November 13, 2013].
EPA (U.S. Environmental Protection Agency). 2013b. Part I: Status of Implementation of Recommendations. Materials Submitted to the National Research Council, by Integrated Risk Information System Program, U.S. Environmental Protection Agency, January 30, 2013 [online]. Available: http://www.epa.gov/IRIS/iris-nrc.htm [accessed October 22, 2013].
EPA (U.S. Environmental Protection Agency). 2013c. Toxicological Review of Benzo[a]pyrene (CAS No. 50-32-8) in Support of Summary Information on the Integrated Risk Information System (IRIS), Public Comment Draft. EPA/635/R13/138a. National Center for Environmental Assessment, Office of Research and Development, U.S. Environmental Protection Agency, Washington, DC. August 2013 [online]. Available: http://cfpub.epa.gov/ncea/iris_drafts/recordisplay.cfm?deid=66193 [accessed October 22, 2013].
Ertz, K., and M. Preu. 2008. International GLP: A critical reflection on the harmonized global GLP standard from a test facility viewpoint. Ann. Ist Super Sanita. 44(4):390-394.
Grefsheim, S.F., S.C. Whitmore, B.A. Rapp, J.A. Rankin, R.R. Robison, and C.C. Canto. 2010. The informationist: Building evidence for an emerging health profession. J. Med. Libr. Assoc. 98(2):147-156.
Filipsson, A.F., S. Sand, J. Nilsson, and K. Victorin. 2003. The benchmark dose method--review of available models, and recommendations for application in health risk assessment. Crit. Rev. Toxicol. 33(5):505-542.
Kenyon, E.M. 2012. Interspecies extrapolation. Methods Mol. Biol. 929:501-520.
Kushman, M.E., A.D. Kraft, K.Z. Guyton, W.A. Chiu, S.L. Makris, and I. Rusyn. 2013. A systematic approach for identifying and presenting mechanistic evidence in human health assessment. Regul. Toxicol. Pharmacol. 67(2):266-277.
Lipscomb, J.C., S. Haddad, T. Poet, and K. Krishnan. 2012. Physiologically-based pharmacokinetic (PBPK) models in toxicity testing and risk assessment. Adv. Exp. Med. Biol. 745:76-95.
McClellan, R.O. 1996. Reducing uncertainty in risk assessment by using specific knowledge to replace default options. Drug Metab. Rev. 28(1-2):149-179.
Nachman, K.E., M.A. Fox, M.C. Sheehan, T.A. Burke, J.V. Rodricks, and T.J. Woodruff. 2011. Leveraging epidemiology to improve risk assessment. Open Epidemiol. J. 4:3-29.
NRC (National Research Council). 1983. Risk Assessment in the Federal Government: Managing the Process. Washington, DC: National Academy Press.
NRC (National Research Council). 2009. Science and Decisions: Advancing Risk Assessment. Washington, DC: National Academies Press.
NRC (National Research Council). 2011. Review of the Environmental Protection Agency’s Draft IRIS Assessment of Formaldehyde. Washington, DC: National Academies Press.
Rodgers, I.S., and K.P. Baetcke. 1993. Interpretation of male rat renal tubule tumors. Environ. Health Perspect. 101(suppl. 6):45-52.
Rothman, K.J., T.L. Lash, and S. Greenland. 2012. Modern Epidemiology, 3rd Ed. Philadelphia, PA: Lippincott Williams & Wilkins.
Whitmore, S.C, S.F. Grefsheim, and J.A. Rankin. 2008. Informationist programme in support of biomedical research: A programme description and preliminary findings of an evaluation. Health Info Libr. J. 25(2):135-141.
Yoon, M., J.L. Campbell, M.E. Andersen, and H.J. Clewell. 2012. Quantitative in vitro to in vivo extrapolation of cell-based toxicity assay results. Crit. Rev. Toxicol. 42(8):633-652.










