
Page 109
5
Atmospheric Chemistry of Ozone and Its Precursors
Introduction
Large quantities of chemical compounds are emitted into the atmosphere as a result of anthropogenic and biogenic activities. These emissions lead to a complex spectrum of chemical and physical processes that result in such diverse effects as photochemical air pollution (including the formation of ozone in urban, suburban, and rural air masses), acid deposition, long-range transport of chemicals, stratospheric ozone depletion, and accumulation of greenhouse gases. Over the past 15 to 20 years, many laboratory and ambient atmospheric studies have investigated the physical and chemical processes of the atmosphere. Because these processes are complex, computer models are often used to elucidate and predict the effects of anthropogenic and biogenic emissions—and of changes in these emissions—on the chemistry of the atmosphere.
In this chapter, the gas-phase chemistry of the relatively unpolluted, methane-dominated troposphere is summarized, and the additional complexities of the chemistry of polluted atmospheres are discussed. The tropospheric chemistry of organic compounds of anthropogenic and biogenic origin, respectively, is discussed in detail, and the calculated tropospheric lifetimes of these compounds are presented. The formulation and testing of chemical mechanisms for use in urban and regional airshed computer models are discussed briefly, and the reactivities of organic compounds with respect to ozone formation, as calculated using these models, are discussed.
Page 110
General Schemes of Tropospheric Chemistry
Ozone is present in the natural, unpolluted troposphere, and its tropospheric column density is approximately 10% of the total atmospheric (troposphere + stratosphere) ozone column density. (Logan, 1985; Brühl and Crutzen, 1989; Fishman et al., 1990). The ozone present in the stratosphere absorbs short-wavelength radiation (![]() nm (nanometers or 10-9 meters)) from the sun and allows only those wavelengths
nm (nanometers or 10-9 meters)) from the sun and allows only those wavelengths ![]() nm to penetrate into the troposphere (Peterson, 1976; Demerjian et al., 1980). The sources of ozone in the natural troposphere are downward transport from the stratosphere and in situ photochemical production. Losses result from photochemical processes and from deposition and destruction at the earth's surface. The rates of downward transport, production, and losses are estimated to be of the same order of magnitude (Logan, 1985). The ozone present in the troposphere is important in the atmospheric chemistry because the OH radical is generated from the photolysis of ozone at wavelengths <319 nm (Levy, 1971; DeMore et al., 1990). The formation of OH radicals leads to cycles of reactions that result in the photochemical degradation of organic compounds of anthropogenic and biogenic origin, the enhanced formation of ozone, and the atmospheric formation of acidic compounds (see, for example, Heicklen et al., 1969; Stedman et al., 1970; Finlayson-Pitts and Pitts, 1986; WMO, 1986). The generation of the OH radical from ozone is shown in the following reactions:
nm to penetrate into the troposphere (Peterson, 1976; Demerjian et al., 1980). The sources of ozone in the natural troposphere are downward transport from the stratosphere and in situ photochemical production. Losses result from photochemical processes and from deposition and destruction at the earth's surface. The rates of downward transport, production, and losses are estimated to be of the same order of magnitude (Logan, 1985). The ozone present in the troposphere is important in the atmospheric chemistry because the OH radical is generated from the photolysis of ozone at wavelengths <319 nm (Levy, 1971; DeMore et al., 1990). The formation of OH radicals leads to cycles of reactions that result in the photochemical degradation of organic compounds of anthropogenic and biogenic origin, the enhanced formation of ozone, and the atmospheric formation of acidic compounds (see, for example, Heicklen et al., 1969; Stedman et al., 1970; Finlayson-Pitts and Pitts, 1986; WMO, 1986). The generation of the OH radical from ozone is shown in the following reactions:
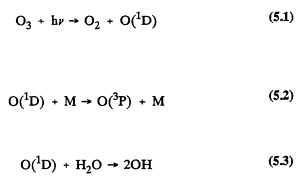
Energy from solar radiation is represented by hv, the product of Planck's constant, h, and the frequency, v, of the electromagnetic wave of solar radiation. O(1D) is an excited oxygen atom, and M is an inert compound, such as N2 or O2. O(3P) is a ground state oxygen atom. The chemistry of the clean, unpolluted troposphere is dominated by the chemistry of methane (CH4) and
Page 111
its degradation products, formaldehyde (HCHO) and carbon monoxide (CO) (see, for example, Levy, 1972; Crutzen, 1973; Fishman and Crutzen, 1977; Logan et al., 1981).
Tropospheric Methane Oxidation Cycle
A sequence of reactions (Ravishankara, 1988; Atkinson et al., 1989a; Atkinson, 1990b) starts with the reaction of the OH radical with methane
![]()
The tropospheric lifetime of methane, ![]() , is controlled by reaction with the OH radical,
, is controlled by reaction with the OH radical,
![]()
where ![]() is the rate constant for the reaction of the OH radical with methane and [OH] is the OH radical concentration. It should be noted that
is the rate constant for the reaction of the OH radical with methane and [OH] is the OH radical concentration. It should be noted that ![]() depends on temperature, and hence on altitude, and that the OH radical concentration is temporally and spatially dependent. The lifetime of methane in the troposphere is long enough that a diurnally and annually averaged concentration of global tropospheric OH radical can be used to calculate the lifetime of methane (and of other similarly long-lived trace species). Based on methylchloroform (CH3CCl3) emissions and atmospheric budgets and an equation analogous to Equation 5.5, Prinn et al. (1987) derived a tropospheric lifetime for methylchloroform of 6.3 years and a globally averaged tropospheric OH radical concentration of 7.7 × 105 molecule/cm3. From this OH radical concentration, the methane lifetime is calculated to be approximately 12 years (Vaghjiani and Ravishankara, 1991). For organic compounds that react more rapidly with the OH radical and have much shorter lifetimes (
depends on temperature, and hence on altitude, and that the OH radical concentration is temporally and spatially dependent. The lifetime of methane in the troposphere is long enough that a diurnally and annually averaged concentration of global tropospheric OH radical can be used to calculate the lifetime of methane (and of other similarly long-lived trace species). Based on methylchloroform (CH3CCl3) emissions and atmospheric budgets and an equation analogous to Equation 5.5, Prinn et al. (1987) derived a tropospheric lifetime for methylchloroform of 6.3 years and a globally averaged tropospheric OH radical concentration of 7.7 × 105 molecule/cm3. From this OH radical concentration, the methane lifetime is calculated to be approximately 12 years (Vaghjiani and Ravishankara, 1991). For organic compounds that react more rapidly with the OH radical and have much shorter lifetimes (![]() year), the temporal and spatial variations of the OH radical concentrations need to be considered in the calculation of tropospheric lifetimes.
year), the temporal and spatial variations of the OH radical concentrations need to be considered in the calculation of tropospheric lifetimes.
Page 112
Under tropospheric conditions, the methyl radical rapidly, and solely, adds oxygen to form the methyl peroxy radical (CH3O2):
![]()
which can then react with nitric oxide (NO), nitrogen dioxide (NO2), hydroperoxyl radical (HO2), and organic peroxy radicals (RO2)
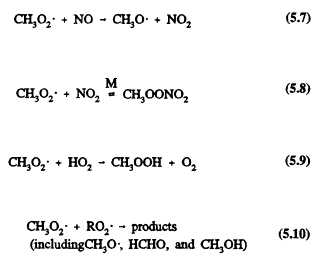
Methyl peroxynitrate, CH3OONO2, thermally dissociates back to the reactants with a lifetime of methyl peroxynitrate with respect to thermal decomposition of ˜1 sec at room temperature and atmospheric pressure, which increases to ˜2 days for the temperature and pressure conditions in the upper troposphere (Atkinson et al., 1989a; Atkinson, 1990b). Because the reactions of the CH3O2 radical with NO and NO2 have comparable rate constants for the temperatures and pressures encountered in the troposphere (Atkinson, 1990a), methyl peroxynitrate can act as a temporary reservoir of NO2 and CH3O2 radicals in the upper troposphere.
The reaction of the methylperoxy radical with NO will dominate over reaction with the HO2 radical for tropospheric NO mixing ratios equal to or great-
Page 113
er than approximately 10-30 parts per trillion (ppt) (Logan et al., 1981). However, in the clean, unpolluted lower troposphere, NO mixing ratios are generally <30 ppt (see, for example, Kley et al., 1981; Logan, 1983; Ridley et al., 1987, 1989; Drummond et al., 1988; and Chapter 8), and under these conditions the HO2 radical reaction to form methyl hydroperoxide, CH3OOH, is important.
The subsequent reactions of CH3OOH under tropospheric conditions are photolysis and reaction with the OH radical (Ravishankara, 1988; Atkinson, 1989, 1990a, b)
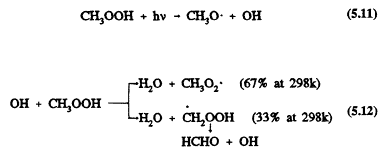
These two processes are comparable in importance, and they reform the CH3O and CH3O2 radicals. Wet deposition of methyl hydroperoxide and its incorporation into cloud, fog, and rain water also could be important (Hell-pointner and Gäb, 1989). For a discussion of cloud chemistry, see, for example, Chameides (1984), Jacob (1986), Jacob et al. (1989), and Pandis and Seinfeld (1989).
The sole loss process for the methoxy radical in the clean troposphere is through reaction with oxygen to generate formaldehyde (Atkinson et al., 1989a; DeMore et al., 1990)
![]()
The HO2 radical can lead to the regeneration of the chain-carrying OH radical by reaction with NO or react with peroxy (RO2) radicals (including HO2) or ozone
Page 114
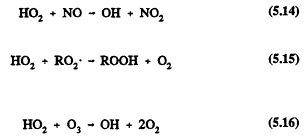
The self-reaction of HO2 radicals forms hydrogen peroxide, which, like methyl hydroperoxide, can undergo wet deposition and incorporation into cloud, fog, and rain water.
![]()
When enough NO is present that the reactions of CH3O2 and HO2 radicals with NO dominate over the reactions of these peroxy radicals with HO2 (or other peroxy radicals) or of HO2 radicals with ozone, then the overall methane photooxidation reaction is given by
![]()
with methane being degraded to formaldehyde, two molecules of NO being converted to NO2, and the OH radical being regenerated. (An equal sign is used instead of an arrow to indicate that the net overall process shown as Reaction 5.18 represents many individual reactions.) When the reaction of the CH3O2 radical with HO2 dominates, then the oxidation of methane becomes a net sink for OH and HO2 radicals, with approximately
![]()
The NOx concentrations in the atmospheric boundary layer over continental areas in the northern hemisphere are generally high enough that the reactions
Page 115
of RO2 and HO2 peroxy radicals with NO dominate over the reactions of the RO2 and HO2 radicals with HO2 and the reaction of the HO2 radical with ozone. The result is net ozone formation. Only in remote locations such as the mid-Pacific Ocean and portions of the southern hemisphere are the NOx concentrations low enough that the reactions of HO2 radicals with ozone (Reaction 5.16) and other peroxy (RO2) radicals dominate, leading to net ozone removal.
Formaldehyde also undergoes reaction in the troposphere by photolysis and reaction with the OH radical (Atkinson et al., 1959a; Atkinson, 1990a)
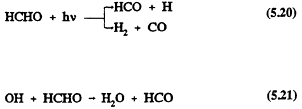
followed by
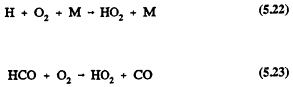
and
![]()
For HCHO, photolysis dominates over reaction with the OH radical (Atkinson, 1988), and the calculated lower tropospheric lifetime of HCHO due to photolysis and, to a lesser extent, reaction with the OH radical is ˜4 hours at the sun's zenith angle of 0º (Rogers, 1990). The tropospheric removal of CO is by reaction with the OH radical, with a calculated lower tropospheric lifetime of ˜2 months. The tropospheric lifetimes of HCHO and CO are thus both much shorter than that of methane.
Page 116
Photochemical Formation of Ozone
In the troposphere, ozone formation occurs to any significant extent only from the photolysis of NO2 at-wavelengths <424 rim, when sufficient solar energy is absorbed by NO2 to cause it to photodissociate
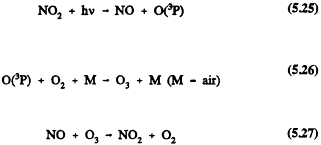
In the absence of other processes that convert NO to NO2, and assuming steady-state conditions, then
![]()
and the ozone concentration is linked to the NO2/NO concentration ratio during daylight hours. (Here j1 is the diurnally, seasonally, and latitudinally dependent rate of photolysis of NO2, and k2 is the rate constant for Reaction 5.27.) For an NO2/NO concentration ratio of one, a reasonable mid-day value in the dean lower troposphere, and a temperature of 298 K, the resulting ozone concentration is ˜ 5 × 1011 molecule/cm3 (20 parts per billion (ppb) mixing ratio).
As discussed above for the methane oxidation cycle, the presence of volatile organic compounds (VOCs) causes enhanced NO-to-NO2 conversion and hence the production of concentrations of ozone that exceed those encountered in the dean background troposphere (see, for example, Parrish et al., 1986). This is discussed further below. For example, for the OH radical-initiated reaction of methane in the presence of NO given above, the overall reaction is
Page 117
![]()
This leads to a net reaction of
![]()
Other Reactions in the Tropospheric Nitrogen Cycle
In addition to Reactions 5.25 and 5.27 and the reaction of the HO2 radical with NO to regenerate the OH radical,
![]()
other tropospherically important reactions involve oxides of nitrogen (Finlayson-Pitts and Pitts, 1986; WMO, 1986; Atkinson et al., 1989a; DeMore et al., 1990). The recombination reactions

to form nitrous acid (HONO) and pernitric acid (HOONO2) are of little importance because of the rapid photodissociation of HONO
Page 118
![]()
and the thermal decomposition of HOONO2 back to reactants. However, the combination reaction of the OH radical with NO2
![]()
is the major gas-phase route to the formation of nitric acid (HNO3), and it is the major homogeneous gas-phase sink for NOx (oxides of nitrogen) in the troposphere. This reaction also serves as a sink for OH and HO2 radicals (odd hydrogen) for NOx mixing ratios ![]() ppb, and under these conditions the removal of OH radicals by Reaction 5.35 balances the formation of HOx (oxides of hydrogen) radicals from the photolysis of ozone and HCHO.
ppb, and under these conditions the removal of OH radicals by Reaction 5.35 balances the formation of HOx (oxides of hydrogen) radicals from the photolysis of ozone and HCHO.
The major reactions involved in the oxidation of methane in the presence of NOx are diagrammed in Figure 5-1, which emphasizes the chain-cycle nature of this overall reaction process.
Ozone also reacts with NO2 to form the nitrate (NO3) radical,
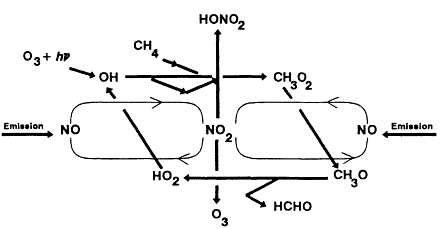
Figure 5-1
Major reactions involved in the oxidation
of methane (CH4) in the presence of NOx.
Page 119
![]()
and the NO3 radical is interconverted with NO2 and dinitrogen pentoxide (N2O5) through the reactions
![]()
Because NO3 radicals rapidly photolyze (with a photolysis lifetime of ˜5 seconds at a solar zenith angle of 0º)
![]()
and react rapidly with NO,
![]()
concentrations of the NO3 radical, and hence of N2O5, remain low during the daytime but can increase during evening and nighttime hours (Platt et al., 1981, 1984; Pitts et al., 1984a).
The homogeneous gas-phase reaction of N2O5 with water vapor to form nitric acid
![]()
is slow enough that only an upper limit can be placed on the rate constant (Atkinson et al., 1989a; Hatakeyama and Leu, 1989), but the wet and dry deposition of N2O5 or of NO3 radicals provides a potentially important nighttime route to the removal of gas-phase NOx and the formation of acid deposi-
Page 120
tion (see, for example, Heikes and Thompson, 1983; Chameides, 1986; Mozurkewich and Calvert, 1988).
![]()
The formation of aqueous-phase nitric acid subsequent to wet deposition of the NO3 radical is expected to proceed via the intermediate formation of the nitrate (NO3-) ion (Chameides, 1986). In addition to this nighttime heterogeneous (involvement of at least two physical phases) removal process for NOx through the intermediary of NO3 radicals and N2O5, heterogeneous chemistry, including cloud chemistry, could be important in the chemical processes that occur in the troposphere (see, for example, Chameides, 1984; Jacob, 1986; Jacob et al., 1989; Pandis et al., 1989; Lelieveld and Crutzen, 1990). For example, Lelieveld and Crutzen (1990) have postulated that in the presence of clouds the formation of ozone in the troposphere is significantly diminished by the scavenging of HO2 radicals and HCHO from the gas phase into cloud water. Clearly, further work is necessary to elucidate the role of heterogeneous reactions and aqueous-phase reactions in the chemistry of the troposphere and in the formation and destruction of ozone.
As noted above, nitrous acid (HONO) photolyzes to generate the OH radical
![]()
and this photolysis reaction is rapid (˜ 10-3 s-1 at a 0º zenith angle of the sun). In urban areas, HONO is formed at night, probably by the heterogeneous hydrolysis of NO2 (Sakamaki et al., 1983; Pitts et al., 1984b; Akimoto et al., 1987; Svensson et al., 1987; Jenkin et al., 1988; Lammel and Perner, 1988).
![]()
Under laboratory conditions, this heterogeneous formation of HONO is first-order in the NO2 concentration. Because comparable amounts of nitric acid
Page 121
are not seen in the gas phase, nitric acid is thought to remain on the reaction vessel surfaces. Direct emission of HONO from combustion sources (Pitts et al., 1984c, 1989) also could contribute to the presence of HONO in a polluted atmosphere. The build-up of HONO at night can lead to substantial predawn concentrations of HONO—up to ˜10 ppb (Harris et al., 1982; Winer et al., 1987; Rodgers and Davis, 1989). The rapid photolysis of HONO in the early morning can then lead to a pulse of OH radicals and to rapid initiation of photochemical activity (Harris et al., 1982; Lurmann et al., 1986a).
Chemistry of the Polluted Troposphere
In the lower troposphere, and especially in polluted urban areas, the chemical reactions of biogenic and anthropogenic VOC and anthropogenic NOx emissions dominate over those of methane and its degradation products (Logan et al., 1981; Brewer et al., 1983; Finlayson-Pitts and Pitts, 1986; Seinfeld, 1989). Although in principle an extension of the chemistry of the dean, methane-dominated troposphere, the chemistry of the polluted troposphere, including urban and rural air masses, is significantly more complicated because of the presence of many VOCs of various classes (alkanes, alkenes, and aromatic hydrocarbons) and the added complexities in the chemistry of these organic species (see, for example, Atkinson, 1990a).
In the troposphere, VOCs undergo photolysis and reaction with OH and NO3 radicals and ozone (and, for some aldehydes, also with HO2 radicals) (Finlayson-Pitts and Pitts, 1986; Atkinson, 1988, 1990a). As with methane (Equation 5.5), the lifetime, ![]() , of a chemical with respect to reaction with a species X is given by
, of a chemical with respect to reaction with a species X is given by
![]()
and depends on the rate constant kx for reaction with X and the ambient tropospheric concentration of X ([X]). The OH and NO3 radical and ozone concentrations vary temporally and spatially, and hence the ''instantaneous' lifetime ![]() and the loss rate,
and the loss rate, ![]() , of a chemical also vary with space and time. The variations in the OH radical, NO3 radical, or ozone concentrations at any given time and place translate directly into variations in the instantaneous loss rate and lifetime of a chemical.
, of a chemical also vary with space and time. The variations in the OH radical, NO3 radical, or ozone concentrations at any given time and place translate directly into variations in the instantaneous loss rate and lifetime of a chemical.
Direct ambient measurements of the OH radical in the lower troposphere (Hübler et al., 1984; Perrier et al., 1987; Platt et al., 1988) give concentrations
Page 122
that range from < 5 × 105 to 9 × 106 molecule/cm3, and these data are reasonably consistent with indirect measurements of OH radical concentrations (Roberts et al., 1984; Ayers and Gillett, 1988; Arey et al., 1989a) and with the diurnally and annually averaged concentration of global tropospheric OH radical (Prinn et al., 1987). Although at a given time and place, it might be possible to specify the concentrations of OH radical and ozone reasonably well, this is not the case for the NO3 radical. Nighttime maximum NO3 radical mixing ratios measured in the lower troposphere over continental areas range from <2 ppt to 430 ppt; the mixing ratio in marine air masses has been measured to be <0.5 ppt (Atkinson et al., 1986, and references therein). Nighttime concentrations of NO3 in the troposphere are uncertain to at least an order of magnitude. Furthermore, as discussed by Winer et al. (1984), reaction with the NO3 radical can be a removal process for the reacting organic compound or NOx, depending on the relative strengths of the emission rates or the formation rates of the VOCs and NO3 radicals.
In the remainder of this chapter, lifetimes are calculated assuming specified ambient concentrations of OH and NO3 radicals and ozone. Table 5-1 gives the calculated tropospheric lifetimes of selected organic compounds from anthropogenic and biogenic sources with respect to the reactions that degrade them.
| TABLE 5-1 | ||||
| Lifetime due to reaction with | ||||
| VOC | OH | NO3 | O3 | hu |
| Methane | ˜12 yearsb | > 120 years | >4,500 years | |
| Ethane | 60 days | > 12 years | >4,500 years | |
| Propane | 13 days | > 2.5 years | >4,500 years | |
| n-Butane | 6.1 days | ¢2.5 years | >4,500 years | |
| n-Octane | 1.8 days | 260 days | >4,500 years | |
| Ethene | 1.8 days | 225 days | 9.7 days | |
(Table continued on next page)
Page 123
(Table continued from previous page)
| Lifetime due to reaction with | ||||
| VOC | OH | NO3 | O3 | hv |
| Propene | 7.0 hours | 4.9 days | 1.5 days | |
| Isoprene | 1.8 hours | 50 rain | 1.2 days | |
| a-Pinene | 3.4 hours | 5 rain | 1.0 days | |
| Acetylene | 19 days |
| 5.8 years | |
| Formaldehyde | 1.6 days | 77 days | >4.5 years | |
| Acetaldehyde | 1.0 days | 17 days | > 4.5 years | 4 hours |
| Acetone | 68 days | c | > 4.5 years | 15 days |
| Methyl ethyl ketone | 13.4 days | c | > 4.5 years | |
| Methylglyoxal | 10.8 hours | c | >4.5 years | 2 hours |
| Methanol | 17 days | >77 days | c | |
| Ethanol | 4.7 days | > 51 days | c | |
| Methyl t-butyl ether | 5.5 days | c | c | |
| Benzene | 12.5 days | > 6 years | > 4.5 years | |
| Toulene | 2.6 days | 1.9 years | > 4.5 years | |
| m-Xylene | 7.8 hours | 200 days | >4.5 years | |
| aOH, 12-hour average concentration of 1.5 × 106 molecule/cm3 (0.06 ppt) (Prinn et al., 1987); NO3 12-hour average concentration of 5 × 108 molecule/cm3 (20 ppt) (Atkinson, 1991); O3 24-hour average concentration of 7 × 1011 molecule/cm3 (28 ppb) (Logan, 1985). Calculated from room temperature rate data, except for methane, of Atlkinson (1988, 1990a, 1991), Plum et al. (1983), and Rogers (1990). | ||||
| bFrom Vaghjiani and Ravishankara (1991). | ||||
| cExpected to be of negligible importance. | ||||
Page 124
All of the tropospheric processes represented in Table 5-1 lead to the formation of organic peroxy radicals (RO2). For example, for the reactions of OH and NO3 radicals with alkanes (Reactions 5.45 and 5.46) where RH represents an alkane, and with alkenes (Reactions 5.47 and 5.48) where >C=C< represents an alkene
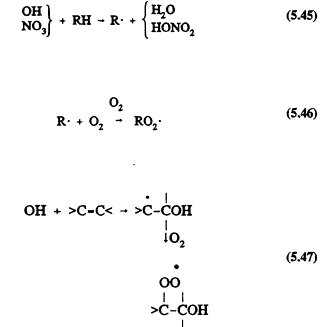
Page 125
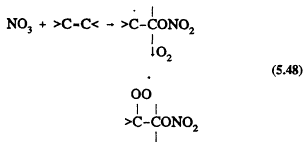
As with the CH3O2 radical formed from methane, these more complex RO2 radicals react with NO, NO2, and HO2 radicals. The difference is that for the RO2 radicals with more than two carbon atoms, the reaction with NO also can lead to the formation of organic nitrates,
![]()
with this organic nitrate formation increasing with increasing pressure, decreasing temperature, and (for the n-alkane series) the carbon number of the alkane (see, for example, Harris and Kerr, 1989; Carter and Atkinson, 1989a). At 298 K and atmospheric pressure the alkyl nitrate yields from the OH radical-initiated reactions of the n-alkanes increase from ˜4% for propane to ˜33% for n-octane (Carter and Atkinson, 1989a).
The alkoxy or substituted alkoxy (RO2) radicals can react with O2 (as for the CH3O radical formed from methane); they can undergo unimolecular decomposition; or, for the alkoxy radicals with four or more carbon atoms, they can isomerize (Atkinson, 1990a). For example, neglecting the combination reactions with NO and NO2, which are generally of negligible importance under tropospheric conditions (Atkinson, 1990a), the following reactions are possible for the 2-pentoxy radical formed from n-pentane,
Page 126
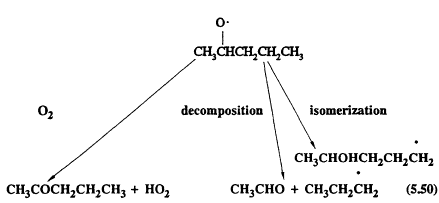
The alkyl radicals formed (C3H7 and CH3CHOHCH2CH2CH2, in this case) then react further.
The reaction mechanisms of the aromatic hydrocarbons are not well understood (Atkinson et al., 1989b; Atkinson, 1990a). In the troposphere, benzene and the alkyl-substituted benzenes react only with the OH radical (Atkinson 1988, 1990a), and the kinetics and initial reaction mechanisms of these OH radical reactions are well understood (Atkinson, 1989). The major pathway of the OH radical reaction involves initial OH radical addition to the aromatic flag to yield a hydroxycyclohexadienyl-type radical (Atkinson, 1989). The subsequent reactions of these hydroxycyclohexadienyl radicals under tropospheric conditions are not well understood (see, for example, Atkinson et al., 1989b). Laboratory studies show that the hydroxycyclohexadienyl radicals react rapidly with NO2 (Zellner et al., 1985; Knispel et al., 1990; Zetzsch et al., 1990) and that the reactions of these radicals with oxygen are slow (Knispel et al., 1990). At present, the relative importance of the reactions of the hydroxycyclohexadienyl radicals with oxygen and NO2 under ambient tropospheric conditions is not totally clear.
The degradation reactions for all classes of VOCs, in addition to the conversion of NO to NO2 and the formation of ozone, lead to the formation of carbonyl compounds (aldehydes, ketones, hydroxycarbonyls, and dicarbonyls), organic acids, organic nitrates (including peroxyacyl nitrates), and the inorganic acids, HONO2 and (in the presence of SO2) H2SO4. In most cases these first-generation products undergo further tropospheric degradation reactions leading to a further spectrum of organic products, NO-to-NO2 conversion, and ozone formation. Because the carbonyl compounds are the major first-generation products, their subsequent reactions are important.
The simplest aldehyde, formaldehyde (HCHO, the tropospheric reactions
Page 127
of which have been presented above regarding the tropospheric methane oxidation cycle), has chemistry that is somewhat different from the higher aldehydes, such as acetaldehyde (Atkinson, 1990b). In the troposphere the photolysis of HCHO is calculated to be more important than reaction with the OH radical, in contrast to the higher aldehydes for which the OH radical reactions are more important than photolysis (Atkinson, 1990a). Furthermore, the HCO radical formed from the photolysis and OH radical reaction of HCHO reacts with oxygen to form the HO2 radical and CO,
![]()
whereas the acyl (RCO) radicals formed from the higher aldehydes
![]()
react with O2 by addition to form the corresponding acylperoxy (RC(O)OO) radicals
![]()
These acylperoxy radicals react with NO, NO2, or HO2 radicals
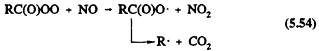
Page 128
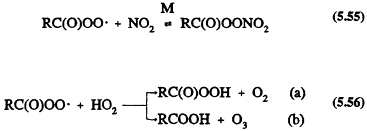
Reaction 5.55 leads to the formation of peroxyacyl nitrates, the simplest member of which is peroxyacetyl nitrate (PAN, CH3C(O)OONO2), which thermally decomposes back to the reactants with a lifetime of ˜30 min at 298 K and atmospheric pressure (Atkinson et al., 1989a). PAN and certain of its homologues, such as peroxypropionyl nitrate (PPN) and peroxybenzoyl nitrate (PBzN), have been observed in ambient air (Roberts, 1990, and references therein).
Until recently, the temperature dependence of the ratio of the rate constants for Reactions 5.54 and 5.55 was not well known (Atkinson et al., 1989a). That uncertainty led to different temperature dependencies assumed for those reactions for R = CH3 in the chemical mechanisms developed for use in airshed computer models (Carter et al., 1986a; Gery et al., 1988a, 1989). The output of such models led to widely differing predictions for ozone (and PAN) formation at temperatures below 298 K (Dodge, 1989). Experimental data of Kirchner et al. (1990) and Tuazon et al. (1991) show that the ratio of rate constants for Reactions 5.54 and 5.55 (R = CH3) is 2.2, independent of temperature over the range ˜280-320 K. For the acetylperoxy radical, the ratio of the rate constant for Reaction 5.56a divided by the sum of rate constants for Reactions 5.56a and 5.56b is 0.67, independent of temperature (Moortgat et al., 1989).
The small (![]() ) alkanes (RH) have fairly simple reaction schemes after their initial reactions with the OH radical (their only significant tropospheric removal process). For example,
) alkanes (RH) have fairly simple reaction schemes after their initial reactions with the OH radical (their only significant tropospheric removal process). For example,
![]()
Page 129
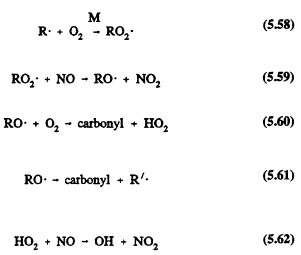
where R', an alkyl radical with fewer carbon atoms than the parent RH alkane, then undergoes an analogous series of reactions that lead to the formation of carbonyl compounds (which react further in the atmosphere by photolysis and reaction with the OH radical), the conversion of NO to NO2, and the regeneration of OH radicals.
It should be noted that, apart from the losses of certain product species onto surfaces through wet and dry deposition (for example, N2O5, HONO2, aldehydes, H2O2, hydroperoxides, and SO2) (Heikes and Thompson, 1983; Leuenberger et al., 1985; Betterton and Hoffmann, 1988; Mozurkewich and Calvert, 1988), heterogeneous reactions of intermediate radical species have generally not been considered important in the chemistry of the troposphere. However, there is a growing appreciation of the importance of heterogeneous scavenging reactions that involve radical species in the global budgets of ozone and of the various NOx species (Chameides, 1986; Lelieveld and Crutzen, 1990).
The general reaction scheme for the degradation of a VOC in the troposphere can be written in a very approximate way as
![]()
where RO2 can also be HO2, followed by
Page 130
![]()
Reaction (5.63) includes all loss processes of the VOC under atmospheric conditions, and a, b, g, and d are coefficients (which can be greater than or less than one, including zero) that generally depend on the relative importance of the various loss processes and on the VOC/NOx concentration ratio. Reaction process 5.63 determines the lifetime of an organic compound in the troposphere (refer to Table 5.1). Subsequent reactions (Reaction 5.64) lead to conversion of NO to NO2, to the generation or regeneration of OH radicals, and to the formation of ozone.
Atmospheric Chemistry of Anthropogenic Vocs
The general features of the atmospheric chemistry of alkanes, alkenes, and aromatic hydrocarbons emitted from anthropogenic sources are understood, although there are still some significant uncertainties (Atkinson, 1990a). The kinetics of the initial reactions of the majority of anthropogenic VOCs with OH and NO3 radicals and ozone, and their photolysis rates, have either been determined experimentally or can be calculated reliably (Atkinson, 1989, 1990a, 1991). Table 5-1 lists the calculated lifetimes of a series of anthropogenic VOCs with respect to reaction in the troposphere with the important reactive species.
In the sections below, the salient features of the atmospheric chemistry of the alkanes, alkenes, aromatic VOCs, and oxygenates are briefly discussed, including the chemistry of the potential alternative fuels. This discussion is largely based on the recent review and evaluation of Atkinson (1990a), which should be consulted for more detail.
Alkanes
In the troposphere, the alkanes react essentially only with the OH radical; the nighttime NO3 radical reaction is of minor significance in terms of the overall removal of the alkanes (Atkinson, 1990a). The OH (and NO3) radical reactions proceed by H-atom abstraction,
Page 131
![]()
followed for the simple alkanes (those with fewer than four carbon atoms) by the sequence of reactions (for example, for a secondary alkyl radical R1R2CH in the presence of NOx)
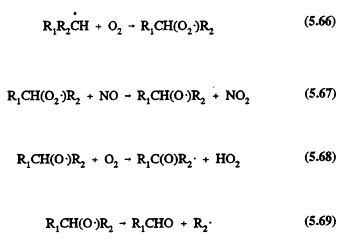
The carbonyl compounds R1CHO and R1C(O)R2 and the fragment alkyl radical R2 undergo further reactions. For the alkanes composed of more than three carbon atoms, alkyl nitrate formation from the reactions of the alkyl peroxy radicals with NO,
![]()
in competition with the formation of NO2 and the corresponding alkoxy radical, becomes increasingly important, and the alkyl nitrate formation yields at 298 K and 760 Torr total pressure increase from ˜4% from propane to ˜33% from n-octane (Carter and Atkinson, 1989a).
The isomerization of alkoxy radicals involving a six-membered transition state (thus requiring a carbon chain of four or more), also is expected to become important for the ![]() alkanes; for example,
alkanes; for example,
Page 132
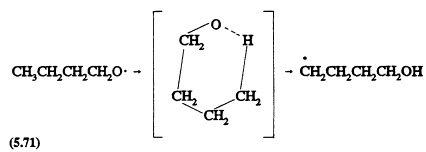
followed by a sequence of reactions (Atkinson, 1990a) that leads to the formation of d-hydroxycarbonyl compounds, for example, HOCH2CH2CH2CHO from the above n-butoxy radical. This isomerization reaction is in competition with unimolecular decomposition of the alkoxy radical or reaction of the alkoxy radical with O2 (Atkinson, 1990a).
The major uncertainties in the atmospheric chemistry of the alkanes concern the alkyl nitrate formation yields from the reactions of the various alkyl peroxy and substituted alkyl peroxy radicals with NO, and the importance of, and reactions subsequent to, alkoxy radical isomerization. A further important area of uncertainty concerns the atmospheric chemistry of the carbonyl compounds formed as first-generation products from the alkanes.
Alkenes
In the troposphere, the chemical removal of the alkenes proceeds by reaction with OH and NO3 radicals and ozone, and all removal pathways must be considered. The rate constants for the initial reactions of these species are reasonably well defined and the initial steps of the reaction mechanisms are known (Atkinson, 1990a). The major uncertainties in the alkene chemistry (apart from the chemistry of isoprene and the monoterpenes discussed below) involve
• The reaction mechanism and the products formed from the long-chain alkenes, such as the 1-alkenes composed of more than four carbon atoms. For example, it is not known whether isomerization of the b-hydroxyalkoxy radicals occurs (Atkinson and Lloyd, 1984).
• The reaction mechanisms of the ozone reactions and the radical formation yields in these reactions. The only alkene for which the reaction mechanism appears to be reasonably well understood is ethene; the experimental data are much less definitive for the higher alkenes (Atkinson, 1990a). Exper-
Page 133
imental data on the reaction of propene with ozone in air lead to a yield of radical species that is significantly higher than the yield estimated from computer modeling of environmental chamber data (Carter et al., 1986a; Carter, 1990a). For the alkenes composed of four or more carbon. atoms, few experimental data on reaction mechanisms or products are available, and further studies are needed.
• The reaction mechanisms and products formed from the reactions of the NO3 radical with the alkenes under tropospheric conditions (Atkinson, 1991), although these reactions generally are important only for the internal alkenes, such as the 2-butenes.
Aromatic Vocs
The greatest uncertainties in the atmospheric chemistry of anthropogenic VOCs concern the aromatic compounds. The aromatic hydrocarbons react only with the OH radical under tropospheric conditions, by two pathways, one involving H-atom abstraction from the substituent groups (or, for benzene, from the aromatic ring C-H bonds)
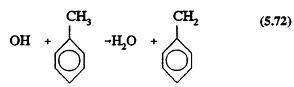
and the other involving initial OH radical addition to the aromatic ring to form a hydroxycyclohexadienyl radical
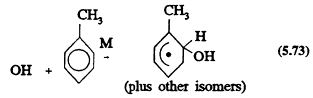
The rate constants for Reactions 5.72 and 5.73 and the ratios of the two rate constants are known (Atkinson, 1989), and the reaction sequence that follows
Page 134
the H-atom abstraction pathway (Reaction 5.72) is reasonably well understood. (It leads to the formation of aromatic aldehydes, benzyl nitrates, and peroxybenzoyl nitrates (Atkinson, 1990a).) It also is known that the OH radical addition pathway (Reaction 5.73) leads to the formation of ring-retaining products, such as phenols and nitroaromatics (the latter in low yield), and to the formation of ring-cleavage products, including ![]() - and g-dicarbonyls. The formation yields of many of these products have been measured (Atkinson, 1990a).
- and g-dicarbonyls. The formation yields of many of these products have been measured (Atkinson, 1990a).
However, the reactions of the hydroxycyclohexadienyl and alkyl-substituted hydroxycyclohexadienyl radicals formed from the initial addition of the OH radical to the ring under tropospheric conditions are not understood. Recent kinetic data (Zellner et al., 1985; Knispel et al., 1990; Zetzsch et al., 1990) show that the hydroxycyclohexadienyl radicals react rapidly with NO2, but that their reactions with NO and O2 are slow. The study of the products formed from the OH radical-initiated reactions of benzene and toluene by Atkinson et al. (1989b) is consistent with these kinetic data, and it leads to the conclusion that in the presence of NO2 concentrations ![]() molecule/cm3 (
molecule/cm3 (![]() ppb) the hydroxycyclohexadienyl radicals react with NO2 and not with O2. It is possible that this is also the situation under conditions that are representative of less-polluted areas. This finding, that the hydroxycyclohexadienyl-type radicals react rapidly with NO2
ppb) the hydroxycyclohexadienyl radicals react with NO2 and not with O2. It is possible that this is also the situation under conditions that are representative of less-polluted areas. This finding, that the hydroxycyclohexadienyl-type radicals react rapidly with NO2
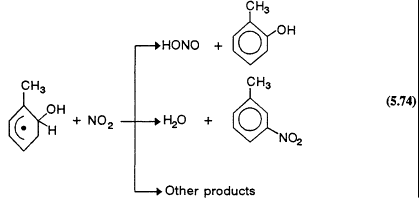
and only very slowly with O2, differs from the reaction sequences in the current chemical mechanisms of Gery et al. (1988a, 1989) and Carter et al. (1986a). Clearly, further experimental and mechanism development work on the tropospheric chemistry of the aromatic hydrocarbons is necessary.
Page 135
Carbonyl Compounds
It is evident from the discussions of the atmospheric chemistry of VOCs of anthropogenic and biogenic origin that carbonyl compounds are formed during the atmospheric degradation of all VOCs. The chemistry of these various carbonyl compounds needs to be known. Unfortunately, there are several areas of uncertainty concerning the atmospheric chemistry of all of the carbonyl compounds other than formaldehyde and acetaldehyde. In particular, there is a need for data concerning the absorption cross-sections and photolysis products and the photodissociation quantum yields (as a function of wavelength) for these carbonyl compounds. These data are necessary to assess the importance of photolysis as a tropospheric degradation route for these carbonyl compounds.
Reactions of Organic Peroxy (RO2) Radicals
Under conditions where the mixing ratio of NO is less than approximately 30 ppt, the reactions of organic peroxy radicals with HO2 radicals and other peroxy radicals dominate over reaction with NO (Logan et al., 1981). To date, however, there are few data concerning the kinetics and products of the reactions of the HO2 radical with organic peroxy radicals or of the various combination reactions of organic peroxy radicals (Atkinson, 1990a).
Oxygenates Proposed as Alternative Fuels
Oxygenated organic compounds are being investigated as alternative fuels, either as single compounds or as blends with present gasolines. Methanol (CH3OH), ethanol (CH3CH2OH) and methyl t-butyl ether [CH3OC(CH3)3] are now used as additives to gasoline, and the alcohols could also be used alone. The chemistry of these compounds is briefly discussed below. Alternative fuels are discussed further in Chapter 12.
Methanol
The only important gas-phase reaction of methanol is with the OH radical, with a rate constant at 298 K of 9.3 × 10-13 cm3/molecule-sec (Atkinson, 1989). This reaction proceeds by H-atom abstraction (the percentages are for room temperature),
Page 136
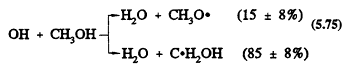
followed by the reactions
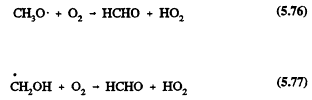
Hence the overall reaction of the OH radical with methanol under atmospheric conditions leads to the formation of HCHO and the HO2 radical.
![]()
Ethanol
As with methanol, the only important reaction for ethanol under tropospheric conditions is with the OH radical. This reaction has a rate constant at 298 K of 3.3 × 10-12 cm3/molecule-s (Atkinson, 1989). The OH radical reaction can proceed by three channels (the percentages are for room temperature):
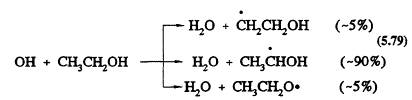
Page 137
Under tropospheric conditions, the major reactions of these initially formed radicals are:
for CH2CH2OH
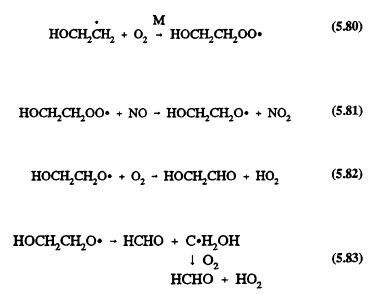
for CH3CHOH
![]()
for CH3CH2O
Page 138
![]()
At room temperature and 760 Tort total pressure of air in the presence of NO, the overall OH radical reaction is
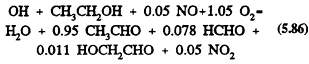
Methyl t-butyl ether
The only significant reaction under tropospheric conditions is with the OH radical, with a rate constant at room temperature of 2.8 × 10-12 cm3/molecule-s (Atkinson, 1989). This reaction proceeds by H-atom abstraction
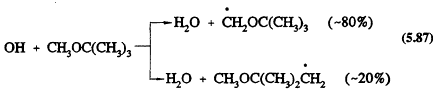
The subsequent reactions will involve addition of O2 to form the peroxy radicals, followed by, in the presence of NO, the conversion of NO to NO2 to yield the alkoxy radicals OCH2OC(CH3)3 and CH3OC(CH3)2CH2O. The OCH2OC(CH3)3 radical then reacts with O2 to generate mainly t-butyl formate [(CH3)3COCHO] (Japar et al., 1990; Tuazon et al., 1991).
![]()
Page 139
Biogenic VOCs
Measurements of the ambient concentrations of isoprene and other VOCs that are known to be emitted by vegetation, as well as estimates of the total inventory of biogenic VOC sources, suggest that these compounds help foster episodes of high concentrations of ozone in areas affected by anthropogenic NOx (Lamb et al., 1987; Trainer et al., 1987; Chameides et al., 1988; Sillman et al., 1990b). This section focuses on the atmospheric chemistry of biogenic VOCs; they are discussed again in Chapters 8 and 9. The atmospheric chemistry of isoprene and most of the monoterpenes observed as vegetative emissions has been investigated over the past 10 years. In general, isoprene and the monoterpenes can be regarded as alkenes or cycloalkenes, and their gas-phase atmospheric reactions are generally analogous to those for the alkenes such as propene and trans-2-butene. Rate constants have been determined at room temperature for the gas-phase reactions of isoprene, a series of monoterpenes, and related compounds with OH and NO3 radicals and ozone; these data are given in Table 5-2.
| TABLE 5-2 | ||||
| Rate constant, cm3/molecule-s, for reaction with | ||||
| VOC | Structure | OHa | NO3b | O3c |
| Isoprene |
| 1.0 × 10-10 | 5.9 × 10-13 | 1.4 × 10-17 |
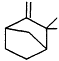
| Camphene |
| 5.3 × 10-11 | 6.5 × 10-13 | 9.0 × 10-19 |
(Table continued on next page)
Page 140
(Table continued from previous page)
| Rate constant, cm3/molecule-s, for reaction with | ||||
| VOC | Structure | OHa | NO3b | O3c |
| 2-Carene |
| 8.0 × 10-11 | 1.9 × 10-11 | 2.4 × 10-16 |
| D3-Carene |
| 8.8 × 10-11 | 1.0 × 10-11 | 3.8 × 10-17 |
| d-Limonene |
| 1.7 ×10-10 | 1.3 × 10-11 | 2.1 × 10-16 |
| Myrcene |
| 2.2 × 10-10 | 1.1 × 10-11 | 4.9 × 10-16 |
| Ocimene |
| 2.5 × 10-10 | 2.2 × 10-11 | 5.6 × 10-16 |
| a-Phellandrene |
| 3.1 × 10-10 | 8.5 × 10-11 | 1.9 × 10-15 |
| a-Pinene |
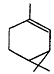
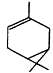
| 5.4 × 10-11 | 5.8 × 10-12 | 8.7 × 10-17 |
| b-Pinene |
| 7.9 × 10-11 | 2.4 × 10-12 | 1.5 × 10-17 |
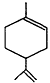
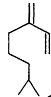
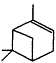
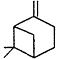
(Table continued on next page)
Page 141
(Table continued from previous page)
| Rate constant, cm3/molecule-s, for reaction with | ||||
| VOC | Structure | OHa | NO3b | O3c |
| Sabinene |
| 1.2 × 10-10 | 1.0 × 10-11 | 8.8 × 10-17 |
| a-Terpinene |
| 3.6 × 10-10 | 1.8 × 10-10 | 8.7 × 10-15 |
| g-Terpinene |
| 1.8 × 10-10 | 2.9 × 10-11 | 1.4 × 10-16 |
| Terpinolene |
| 2.3 × 10-10 | 9.6 × 10-11 | 1.4 × 10-15 |
| 1,8-Cineole |
| 1.1 × 10-11 | 1.7 × 10-16 | <1.5 × 10-19 |
| p-Cymene |
| 1.5 × 10-11 | 9.9 × 10-16 | <5 × 10-20 |
| aFrom Atkinson, 1989; Atkinson et al., 1990a, and Corchnoy and Atkinson, 1990. | ||||
| bFrom Atkinson et al., 1988; Atkinson et al., 1990a; and Corchnoy and Atkinson, 1990. | ||||
| cFrom Atkinson and Carter, 1984 and Atkinson et al., 1990b. | ||||
Page 142
Isoprene and the monoterpenes are highly reactive toward all three of these reactive intermediates. The tropospheric lifetimes due to reaction with OH and NO3 radicals and ozone can be calculated by combining the rate constant data with estimated ambient tropospheric concentrations of OH and NO3 radicals and ozone. The resulting tropospheric lifetimes with respect to these gas-phase reactive loss processes are given in Table 5-3. Obviously, the calculated lifetimes of isoprene and the monoterpenes are short. The OH radical and ozone reactions are of generally comparable importance during the daytime, and the NO3 radical reaction is important at night if NO3 radicals are present at concentrations of > 107 molecule/cm3 (> 0.4 ppt). (Over continental areas, lower tropospheric nighttime NO3-radical mixing ratios range from <2 ppt to 430 ppt [Atkinson et al., 1986]). As noted above, the NO3 radical reactions act as a removal process for either the biogenic VOCs or NOx, depending on the relative magnitudes of the biogenic emission fluxes and the formation rate of the NO3 radical from the reaction of ozone with NO2 (Winer et al., 1984).
| TABLE 5-3 | |||
| Lifetime due to reaction with | |||
| VOC | OHa | O3b | NO3c |
| Isoprene | 1.8 hr | 1.2 days | 1.7 days |
| Camphene | 3.5 hr | 18 days | 1.5 days |
| 2-Carene | 2.3 hr | 1.7 hr | 36 min |
| D3-Carene | 2.1 hr | 10 hr | 1.1 hr |
| d-Limonene | 1.1 hr | 1.9 hr | 53 min |
| Myrecene | 52 rain | 49 rain | 1.1 hr |
| Ocimene | 44 min | 43 min | 31 min |
| a-Phellandrene | 35 min | 13 min | 8 min |
| a-Pinene | 3.4 hr | 4.6 hr | 2.0 hr |
| b-Pinene | 2.3 hr | 1.1 days | 4.9 hr |
| Sabinene | 1.6 hr | 4.5 hr | 1.1 hr |
(Table continued on next page)
Page 143
(Table continued from previous page)
| a-Terpinene | 31 min | 3 min | 4 min |
| g-Terpinene | 1.0 hr | 2.8 hr | 24 min |
| Terpinolene | 49 min | 17 min | 7 min |
| 1,8-Cineole | 1.4 days | > 110 days | 16 yr |
|
| 1.0 days | > 330 days | 2.7 yr |
| aFor a 12-hr daytime average OH radical concentration of 1.5 × 106 molecule/cm3 (0.06 ppt) (Prinn et al., 1987). | |||
| bFor a 24-hr average O3 concentration of 7 × 1011 molecule/cm3 (30 ppb) (Logan, 1985). | |||
| cFor a 12-hr average NO3 radical concentration of 2.4 × 107 molecule/cm3 (1 ppt) (Atkinson et al., 1986) | |||
Few definitive data are available concerning the products formed from the atmospheric reactions of isoprene and the monoterpenes. The most studied of the biogenic compounds have been isoprene and its major degradation products methacrolein and methyl vinyl ketone (Arnts and Gay, 1979; Kamens et al., 1982; Niki et al., 1983; Gu et al., 1985; Tuazon and Atkinson, 1989, 1990a,b; Paulson et al., 1992a,b); these two degradation products have recently been observed and measured in ambient air (Pierotti et al., 1990; Martin et al., 1991). However, the products and reaction mechanisms of the atmospherically important reactions of isoprene and the monoterpenes are not well understood; for the monoterpenes few products have been identified and even fewer have been quantified. Based on the aerosol formation observed in recent product studies from the OH radical-initiated and ozone reactions with a- and b-pinene (Hatakeyama et al., 1989, 1991; Pandis et al., 1991), it is calculated that the atmospheric degradation reactions of the biogenic monoterpene VOCs can account for a significant, and often dominant, fraction of the secondary aerosol observed in urban and rural areas (Pandis et al., 1991). In contrast, the atmospheric photooxidation of isoprene is expected to be a negligible pathway for the formation of secondary aerosol (Pandis et al., 1991). The product data reported in the literature are summarized below.
NO3 Radical Reaction
Barnes et al. (1990) have used Fourier transform infrared (FT-IR) absorp-
Page 144
tion spectroscopy to investigate the gas-phase reactions of isoprene, a- and b-pinerie, D3-carene, and d-limonene in the presence of one atmosphere of air. Formaldehyde (HCHO), CO, and methacrolein were identified from the NO3 radical reaction with isoprene; the HCHO and CO yields were 11% and 4%, respectively (Barnes et al., 1990). The FT-IR spectra indicated the presence of >C=O and -ONO2 groups, and the intensities of these FT-IR bands allowed an estimated formation yield of ˜80% of nitrate-containing products. The NO3 radical reactions with the monoterpenes led to the formation of aerosols, although for a- and b-pinene, spectral features indicated the presence of >C=O and -ONO2 groups. It should be noted that the initial isoprene and monoterpene concentrations in these experiments were ˜5 × 1014 molecule/cm3 (20,000 ppb), to be compared with ambient concentrations of less than 20 ppb (see Petersson, 1988)).
Kotzias et al. (1989) also used FT-IR absorption spectroscopy and mass spectrometry (MS) to study the reaction of the NO3 radical with b-pinene. Their results are similar to those of Barnes et al. (1990) in that both the FT-IR and MS data indicate the presence of organic nitrates.
The initial reaction steps in the NO3 reactions are expected to involve initial NO3 radical addition to a >C=C< bond
![]()
followed by
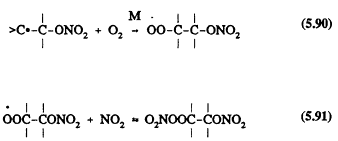
Page 145
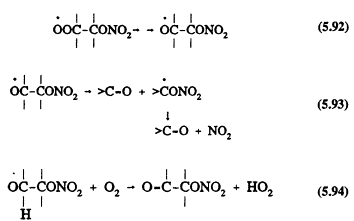
The nitratoperoxynitrate (O2NOROONO2) formed in Reaction 5.91 is thermally unstable, and the organic peroxy radicals undergo radical-radical reactions with other peroxy (RO2) and HO2 radicals (Atkinson, 1991).
Ozone Reactions
There have been few quantitative product studies of the reactions of ozone with isoprene and the monoterpenes. For isoprene, Kamens et al. (1982) and Niki et al. (1983) observed the formation of HCHO, methacrolein, and methyl vinyl ketone. Both groups reported HCHO, methyl vinyl ketone, and methacrolein yields (in molar units) of 85-96%, 13-18%, and 33-42%, respectively. The use of isotope labeling allowed Niki et al. (1983) to conclude that the majority of the HCHO formed arose from secondary reactions. The recent study of Paulson et al. (1992b) has provided evidence that the O3 reaction with isoprene leads to the formation of OH radicals and O(3P) atoms in large amounts, with molar yields of 65% and 45%, respectively. The formation of OH radicals and O(3P) atoms leads to secondary reactions which complicate the analysis of the O3-isoprene reaction. Based on computer modeling of product data, Paulson et al. (1992b) concluded that the products formed from the O3 reaction with isoprene are methacrolein, methyl vinyl ketone, and propene, with yields of 68%, 25%, and 7% respectively.
In general, the initial reaction sequence is expected to be (Atkinson and Lloyd, 1984; Atkinson and Carter, 1984; Atkinson, 1990a),
Page 146
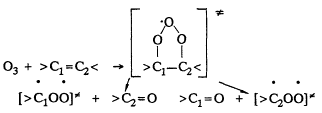
followed by decomposition or stabilization of the initially energy-rich biradicals ![]() , and this accounts for the HCHO; methyl vinyl ketone and methacrolein observed. The reported product distributions account for only ˜60% of the overall products formed.
, and this accounts for the HCHO; methyl vinyl ketone and methacrolein observed. The reported product distributions account for only ˜60% of the overall products formed.
Several studies have investigated the products of the ozone reactions with monoterpenes (see, for example, Wilson et al., 1972; Schwartz, 1974; Schuetzle and Rasmussen, 1978; Hull, 1981; Yokouchi and Ambe, 1985; Hatekayama et al., 1989); the two most recent studies (Yokouchi and Ambe, 1985; Hatakeyama et al., 1989) are the most definitive. Yokouchi and Ambe (1985) used high concentrations, ˜(3-15) × 1015 molecule/cm3 (˜120,000-600,000 ppb), of ozone and the monoterpenes (a- and b-pinene and d-limonene), and observed ready formation of aerosols, as expected from the high concentrations of reactants (Finlayson-Pitts and Pitts, 1986; Izumi et al., 1988). Using gas chromatography (GC) and GC/MS techniques, they identified pinonaldehyde (2' ,2'-dimethyl-3' -acetylcyclobutyl ethanol) and, to a lesser extent, pinonic acid (2',2'-dimethyl-3'-acetylcyclobutyl acetic acid) from a-pinene and 6,6-dimethylbicyclo[3.1.1]heptan-2-one from b-pinene. No products were identified from the d-limonene reaction.
The most recent product study of Hatakeyama et al. (1989) was carried out at much lower reactant concentrations (typically ˜3 × 1013 molecule/cm3 [˜1,000 ppb] ), using FT-IR absorption spectroscopy and GC/MS for analysis. From the a-pinene reaction, CO, CO2, HCHO, pinonaldehyde and nor-pinonaldehyde, were identified, with molar formation yields of 9%, 30%, and 22%, respectively, for CO, CO2, and HCHO; the ''total aldehydes'' yield was ˜51% (mainly pinonaldehyde and nor-pinonaldehyde). The products identified from the b-pinene reaction were CO2, HCHO, and 6,6-dimethylbicyclo-[3.1.1]heptan-2-one with molar yields of 27%, 76%, and 40%, respectively. Aerosol formation accounted for 14-18% of the overall reaction. The products observed in these two recent studies can be explained with the general reaction scheme outlined above, although only a fraction of the overall product distribution has been accounted for.
Page 147
Oh Radical Reactions
Few quantitative studies have dealt with the OH-radical-initiated reactions of isoprene or the monoterpenes. Prior to recent studies on the reactions of isoprene (Tuazon and Atkinson, 1990a; Paulson et al., 1992b), methacrolein (Tuazon and Atkinson, 1990b), methyl vinyl ketone (Tuazon and Atkinson, 1989), and a series of monoterpenes (Arey et al., 1990), the only quantitative product studies were those of Arnts and Gay (1979) for isoprene and monoterpenes and of Gu et al. (1985) for isoprene. Gu et al. (1985) reported the formation of methacrolein, methyl vinyl ketone, and 3-methylfuran from isoprene with formation yields of 23%, 17%, and 6%, respectively. This is in agreement with the yields of 29%, 21%, and 4.4%, respectively, obtained by Tuazon and Atkinson (1990a) and Atkinson et al. (1989c). (These yields require upward revision by ˜10-15% [Atkinson, pers. comm., 1991; Paulson et al., 1992a], because of the neglect of the O(3P) atom reaction in the Tuazon and Atkinson [1990a] study.) Arnts and Gay (1979) used irradiated mixtures of NOx, VOC, and air to generate OH radicals, and secondary reactions involving ozone and possibly NO3 radicals were undoubtedly important. In general, the carbon balances they determined by FT-IR absorption spectroscopy were low; < 17% for the monoterpenes and 44% for isoprene. Only for isoprene did they identify organic products other than HCHO, CH3CHO, CH3C(O)OONO2 [PAN], HCOOH, and CH3COCH3, these being methacrolein and methyl vinyl ketone (Arnts and Gay, 1979).
As shown in Figure 5-2, the major products observed by Tuazon and Atkinson (1989, 1990a,b) and by Atkinson et al. (1989c) from the OH-radical-initiated reactions of isoprene, methyl vinyl ketone, and methacrolein are as follows: from isoprene, methyl vinyl ketone, methacrolein, formaldehyde (HCHO), and 3-methylfuran; from methyl vinyl ketone, glycolaldehyde (HOCH2CHO), methylglyoxal (CH3COCHO), HCHO, and peroxyacetyl nitrate (CH3C(O)OONO2, PAN); and from methacrolein, hydroxyacetone (HOCH2COCH3), CH3COCHO, a peroxyacyl nitrate identified as CH2= C(CH3)C(O)OONO2, CO2, and HCHO. Other unidentified products were observed from isoprene, and these were characterized from their IR spectra as organic nitrates (yield ˜10-15%) and as carbonyls or hydroxycarbonyls (yield ˜25%)
These product yield data show (Tuazon and Atkinson, 1990a; Paulson et al., 1992b; and Figure 5-2) that the OH radical reaction with isoprene in the presence of NOx leads to the formation of methacrolein + HCHO (˜24%), methyl vinyl ketone + HCHO (˜34%), and 3-methylfuran (˜5%); the HCHO yield is equal to the sum of the methacrolein and methyl vinyl ketone yields. For the OH radical-initiated reaction of methyl vinyl ketone, the reaction pathways are essentially totally accounted for; the OH radical addition to
Page 148
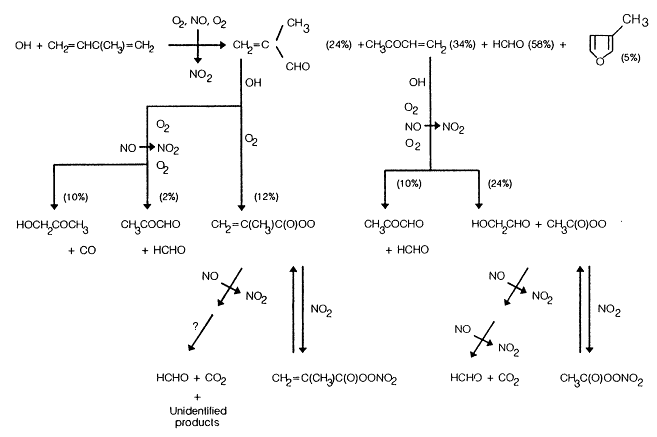
Figure 5-2
Overall reaction scheme
Page 149
the terminal carbon atom of the > C = C < bond (which leads to the formation of HOCH2CHO) accounts for 72 ± 21% of the overall reaction. The product yield data for the OH radical reaction with methacrolein show that OH radical addition to the >C=C< bond accounts for approximately 50% of the overall reaction; the remaining ˜50% proceeds by H-atom abstraction for the -CHO group. In particular, the OH radical addition pathway leads to the formation of hydroxyacetone in high yield, in contrast to expectations provided in the literature (Lloyd et al., 1983) that assume CH3COCHO + HCHO + HO2 would be the major product.
A reaction scheme for the OH radical reaction of isoprene in the presence of NOx, including the subsequent reactions of methyl vinyl ketone and methacrolein, is shown in Figure 5-2.
Arey et al. (1990) have investigated the products of the OH radical-initiated reactions of a series of monoterpenes. 6,6-Dimethylbicyclo[3.1.1]heptan-2-one was observed as a product from the gas-phase reaction of b-pinene with the OH radical in the presence of NOx, with a formation yield of 30.0 ± 4.5%, and 4-acetyl-1-methylcyclohexene was observed as a product from the OH radical reaction of d-limonene with a formation yield of 17.4 ± 2.8%. On the basis of mass spectral data, a number of ketone and keto-aldehyde products were tentatively identified in irradiated CH3ONO-NO-air-monoterpene mixtures of d-limonene, a-pinene, D3-carene, sabinene, and terpinolene. The estimated formation yields of the observed products ranged from a low of ˜ 29% for a-pinene to a high of ˜45% for d-limonene. No significant products were observed from the OH radical-initiated reaction of myrcene. Other unidentified products are obviously formed from these reactions in large overall yield.
The major features of the atmospheric degradations of anthropogenic and biogenic VOCs are shown in Figure 5-3.
Development and Testing of Chemical Mechanisms
Computer models that incorporate emissions of VOCs and NOx, meteorology, and the chemistry of VOC/NOx mixtures simulate the complex physical and chemical processes of the atmosphere and predict the effects of changes of emissions of anthropogenic VOCs, biogenic VOCs, or NOx on photochemical air pollution. An essential component is the chemical mechanism that describes the series of reactions in the troposphere subsequent to emissions of VOCs and NOx.
In general, chemical mechanisms are assembled using the available literature on kinetic, mechanistic, and product data for the atmospherically impor-
Page 150
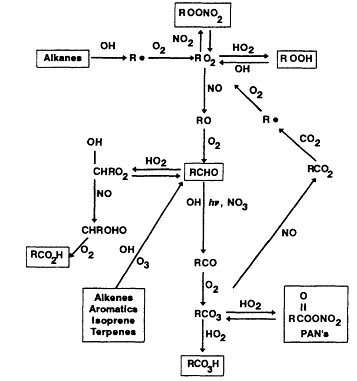
Figure 5-3
Simplified diagram of the chemical processing that occurs among Vocs.
tant inorganic and organic reactions, often using relevant reviews and evaluations (for example, the series of the National Aeronautics and Space Administration [NASA] and International Union of Pure and Applied Chemistry [IUPAC] data evaluation panels, with the most recent evaluations from these groups being those of DeMore et al. [1990] and Atkinson et al. [1989a], respectively). Apart from the initial reactions of VOCs with OH and NO3 radicals and ozone, the vast majority of the tropospheric degradation reactions of VOCs are not well understood with regard to rate constant or products, so there are large areas of uncertainty, and reaction sequences are formulated by analogy.
The initial formulation(s) of the chemical mechanisms are then tested against environmental chamber data concerning the irradiations of single VOCs or of mixtures of VOCs in air in the presence of NOx, and the mecha-
Page 151
nism predictions are compared with the experimental data. However, there are significant problems associated with the direct comparison of chemical mechanism predictions against environmental chamber data, because of the effects of the chamber itself (see, for example, Bufalini et al., [1977]; Carter et al., [1982a]; Joshi et al., [1982]; and Killus and Whitten, [1990]). It is necessary to take into account several chamber effects, such as light intensity and spectral distribution, sorption of chemicals to walls, degassing of chemicals from the chamber walls, and heterogeneous reactions that lead to the formation (upon irradiation) of radical species (Carter et al., 1982a; Killus and Whitten, 1990; Carter and Lurmann, 1991). (The radicals appear to be formed, at least in part, through the heterogeneous generation of nitrous acid.) Some of these effects are not completely understood at a physical or chemical level, and the necessity of including the array of chamber effects introduces additional uncertainties into the direct comparison of mechanism predictions with experimental data.
Through comparison of experimental and predicted data, the initial chemical mechanism is refined and adjusted to provide a good fit between the experimental data and mechanism predictions. In some areas of the chemical mechanism, such as the degradation reaction schemes of the aromatic VOCs, the chemical mechanism has been derived by providing the best fit of an assumed mechanism to the experimental data.
Two chemical mechanisms have been developed recently for use in urban airshed simulation models, and predictions of these two mechanisms (Carter et al., 1986a; Gery et al., 1988a, 1989) are in reasonably good agreement at room temperature (Dodge, 1989). Although there are significant areas of uncertainty in the chemical mechanisms for the tropospheric oxidation of VOCs, this general agreement arises because the chemical mechanisms have been tested against, and revised to be in agreement with, a common data base of environmental chamber experiments conducted at the University of North Carolina and the University of California, Riverside (Carter et al., 1986a; Gery et al., 1988a, 1989; Carter, 1990a).
Hence, although the unknown aspects of the atmospheric chemistry of alkanes, alkenes, and aromatics have in some cases been treated differently, the resulting chemical mechanisms are constrained to be in close agreement with one another and with the environmental chamber data (which are mostly available at room temperature). This agreement of the two most recent chemical mechanisms at room temperature does not guarantee their correctness; indeed, one or both mechanisms could be incorrect in their treatment of various aspects of the chemistry of the alkanes, alkenes, and aromatic hydrocarbons, and this could well be the case for the aromatic hydrocarbon chemistry. However, because the chemical mechanisms are tested against
Page 152
environmental chamber data, it is not clear to what extent even a totally incorrect treatment of the chemistry of a specific class of VOCs will result in incorrect predictions of the airshed model under actual atmospheric conditions.
The chemical mechanisms of Gery et al. (1988a) and Carter et al. (1986a) do, however, lead to significantly different predictions of ozone formation at temperatures below ˜298 K for VOC/NOx ratios <10 (Dodge, 1989); the CB-IV mechanism of Gery et al. (1988a) predicts the formation of less ozone at lower temperatures than does the mechanism of Carter et al. (1986a). This discrepancy arises because of different assumptions for the temperature dependence of the ratio of the rate constants for Reactions 5.96 and 5.97, where the acetyl peroxy radical reacts with NO and NO2, respectively.
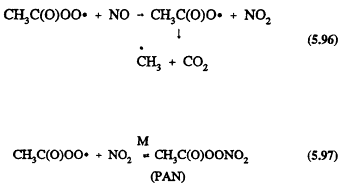
Gery et al. (1988a) assume a temperature dependence in the ratio of the rate constants for Reactions 5.96 and 5.97 of e-5250/T, and Carter et al. (1986a) use a value of the rate constant ratio that is independent of temperature. At room temperature the rate constant ratio for Reactions 5.96 and 5.97 used in both mechanisms is similar. The more sensitive temperature dependence used by Gery et al. (1988a) leads to NOx being sequestered as PAN at lower temperatures and not being available to participate in the formation of ozone.
The data of Kirchner et al. (1990) and Tuazon et al. (1991) show that the rate constant ratio for Reactions 5.96 and 5.97 is equal to 2.2, independent of temperature over the range 283-313 K at 740 Torr total pressure of air. This will require revision of the Gery et al. (1988a) CB-IV mechanism and of the Carter et al. (1986a) mechanism.
Page 153
Ozone Formation Potential of Various Vocs
VOCs emitted from anthropogenic and biogenic sources react in the troposphere in the presence of NOx and sunlight to lead to the photochemical formation of ozone. For two decades it has been known that VOCs vary widely in the speed with which they react in the troposphere and in the extent to which they promote or inhibit ozone formation (see, for example, Altshuller and Bufalini, 1971, and references therein; Dimitriades, 1974; Pitts et al., 1977). Several reactivity scales have been proposed to define the potential of VOCs to form ozone in the atmosphere, and these have included the maximum amount of ozone generated in irradiated mixtures of a single VOC, NOx, and air for several hours (Wilson and Doyle, 1970; Laity et al., 1973; Dimitriades and Joshi, 1977; Joshi et al., 1982); the rate of NO photooxidation (Heuss and Glasson, 1968; Glasson and Tuesday, 1970a,b, 1971), the rate of ozone formation (Heuss and Glasson, 1968; Winer et al., 1979), the rate of VOC consumption (Heuss and Glasson, 1968), all in irradiated mixtures of VOCs and NOx; and the rate constants for the reaction of the VOC with the OH radical (Darnall et al., 1976; Pitts et al., 1977). Although there are differences among these reactivity scales, the reactivities of VOCs as obtained from these scales are in a general order of increasing reactivity: alkanes and monoalkylbenzenes < 1-alkenes and dialkylbenzenes < trialkylbenzenes and internal alkenes (Hems and Glasson, 1968; Darnall et al., 1976).
There are problems associated with basing reactivity scales on smog chamber experiments because of chamber effects (see, for example, Bufalini et al., 1977; Joshi et al., 1982; Carter et al., 1982a), and experimental chamber data are not directly applicable to ambient atmospheric conditions because they generally do not take into account the dilution and continuous input of VOCs and NOx in ambient air (Carter and Atkinson, 1989b). The use of the OH radical reaction rate constant scale does not suffer from these effects (Darnall et al., 1976), but this scale does not take into account the reactions subsequent to the initial OH radical reaction and it ignores other tropospheric loss processes, such as photolysis and reaction with NO3 radicals and ozone. Thus, for example, the formation of photoreactive products, such as formaldehyde, leads to increased overall reactivity with respect to ozone formation, whereas the generation of products such as organic nitrates, which act as sinks for NOx and radical species, leads to a decreased ozone-forming potential (Carter and Atkinson, 1987, 1989b).
A useful definition of reactivity is that of incremental reactivity, defined as the amount of ozone formed per unit mount (as carbon) of VOC added to a VOC mixture representative of conditions in urban and rural areas in a given air mass (Dodge, 1984; Carter and Atkinson, 1987, 1989b; Carter, 1991),
Page 154
![]()
where D[ozone] is the change in the amount of ozone formed as a result of the change in the amount of organic present, D[VOC] (note that Carter and Atkinson [1989b] used the quantity D([ozone] - [NO]) rather than D[ozone] under conditions where the maximum ozone was not attained and NO was not fully consumed). This concept of incremental reactivity corresponds closely to control strategy conditions, in that the effects of reducing the emission of a VOC or group of VOCs, or of replacing a VOC or group of VOCs with other VOCs, on the ozone-forming potential of complex mixture of VOC emissions are simulated.
The theoretical and experimental studies of Bufalini and Dodge (1983), Dodge (1984), and Carter and Atkinson (1987, 1989b) show that, in agreement with previous scales, VOCs exhibit wide variations in reactivity with respect to ozone formation. Furthermore, the absolute and relative calculated incremental reactivities of VOCs depend on VOC/NOx ratios (Bufalini and Dodge, 1983; Dodge, 1984; Carter and Atkinson, 1989b) and on the "scenario" used (i.e., the VOC mix to which incremental changes are made and associated physical factors, such as the amount of dilution [Carter and Atkinson, 1989b]). Table 5-4 shows, as an example, the incremental reactivities calculated when selected VOCs are added to an eight-component urban VOC mixture at various VOC/NOx ratios (Carter and Atkinson, 1989b). These data are in general agreement with the earlier modeling study of Dodge (1984) and show that the incremental reactivities of VOCs depend on the particular VOC and vary with the VOC/NOx ratio. In particular, the incremental reactivities are generally independent of, or increase with, the VOC/NOx ratio up to a VOC-/NOx ratio of ˜6-8. At higher ratios, the absolute magnitude of the incremental reactivities decreases with increasing VOC/NOx ratio, becoming dose to zero (or more negative) at high VOC/NOx ratio, where the formation of ozone is NOx-limited and VOC control becomes irrelevant (Carter and Atkinson, 1989b; Sillman et al., 1990b). The observed negative incremental reactivities are due to the presence of NOx or radical sinks in the chemistry of the VOC, with the NOx sinks being most important at high VOC/NOx ratios and the radical sinks most important at low VOC/NOx ratios (Carter and Atkinson, 1989b).
Page 155
| TABLE 5-4 | ||||
| VOC/NOx, ppbC/ppb | ||||
| Compound | 4 | 8 | 16 | 40 |
| Carbon monoxide | 0.011 | 0.022 | 0.012 | 0.005 |
| Ethane | 0.024 | 0.041 | 0.018 | 0.007 |
| n-Butane | 0.10 | 0.16 | 0.069 | 0.019 |
| n-Octane | 0.068 | 0.12 | 0.027 | -0.031 |
| Ethene | 0.85 | 0.90 | 0.33 | 0.14 |
| Propene | 1.28 | 1.03 | 0.39 | 0.14 |
| trans-2-Butene | 1.42 | 0.97 | 0.31 | 0.054 |
| Benzene | 0.038 | 0.033 | -0.002 | -0.002 |
| Toluene | 0.26 | 0.16 | -0.036 | -0.051 |
| m-Xylene | 0.98 | 0.63 | 0.091 | -0.025 |
| Formaldehyde | 2.42 | 1.20 | 0.32 | 0.051 |
| Acetaldehyde | 134 | 0.83 | 0.29 | 0.098 |
| Benzaldehyde | -0.11 | -0.27 | -0.40 | -0.40 |
| Methanol | 0.12 | 0.17 | 0.066 | 0.029 |
| Ethanol | 0.18 | 0.22 | 0.065 | 0.006 |
| Urban mixa | 0.41 | 0.32 | 0.088 | 0.011 |
| aEight-component VOC mix used to simulate VOC emissions in an urban area in the calculations. Surrogate composition, in units of ppb compound per ppbC surrogate, was ethene, 0.025; propene, 0.0167; n-butane, 0.0375; n-pentane, 0.0400; isooctane, 0.0188; toluene, 0.0179; m-xylene, 0.0156; formaldehyde, 0.0375; and inert constituents, 0.113. | ||||
| Source: Adapted from Carter and Atkinson (1989b). | ||||
Page 156
Theoretical studies of the ozone-forming potential of VOCs have investigated the factors that influence these reactivities. There are several approaches to dealing with this topic conceptually, but the computer modeling studies of Atkinson and Carter (1989b) and of Carter (1991) provide a useful framework for discussing the various factors involved. A generalized scheme for the degradation of VOCs in the troposphere in the presence of NOx is
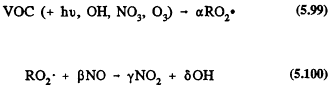
The rate of formation of ozone and oxidation of NO are then determined by the rate of formation of RO2 radicals and the number of molecules of NO converted to NO2 per RO2 radical generated. To a first approximation these processes can be dealt with independently, and the ozone-forming potential of a VOC then depends on
• The rate at which the organic compound reacts in the troposphere. This reaction rate is equal to the inverse of its lifetime (i.e., its decay rate). The quantity of interest is the fraction of the emitted organic that has reacted Coy whatever route, photolysis, reaction with OH radicals, NO3 radicals, ozone, etc.) during the time being considered.
• The reaction mechanism subsequent to the initial reaction(s) of the organic compound. Different aspects of the reaction mechanisms, for example, the VOC/NOx concentration ratio, become important under different conditions.
The following features of a reaction mechanism affect the formation of Ozone:
• The existence of NOx sinks (low values of g in Reaction 5.100) in the reaction mechanism lead to a lowering of reactivity with respect to ozone formation. Examples include the generation of alkyl nitrates from the reaction of alkyl peroxy radicals with NO (which competes with the pathway to form NO2 and the corresponding alkoxy radical),
Page 157
![]()
the formation of peroxyacetyl nitrate (PAN) or its analogues,
![]()
and the formation of other organic nitrogen-containing compounds
![]()
This aspect of the reaction mechanisms becomes important at high VOC/NOx ratios, where the availability of NOx becomes limiting, and the formation of organic nitro compounds competes with the formation of NO2 (with subsequent photolysis to generate ozone).
• The generation or loss of radical species can lead to a net formation or loss of OH radicals (d > 1 or < 1, respectively, in Reaction 5.100), which in turn leads to an enhancement or suppression of OH radicals in the entire air mass and hence to an enhancement or suppression of overall reactivity of all chemicals through the effect on the formation rate of RO2 radicals (Reaction 5.99). The effects of radical formation or loss are most important at low VOC/NOx ratios, where the formation of ozone is determined by the rate at which RO2 radicals are formed.
The alkenes, including isoprene and the monoterpenes of biogenic origin, react with ozone in addition to reacting with OH and NO3 radicals (Atkinson and Carter, 1984; Atkinson, 1991; Atkinson et al., 1990b; Tables 5-1 and 5-2), and this reaction process can act as a sink for ozone, especially under the low NOx conditions encountered in rural and clean atmospheres. However, because the reactions of ozone with alkenes lead to the generation of radicals
Page 158
from biogenic sources will act as ozone sinks unless NOx mixing ratios are < 0.1 ppb.
To a first approximation the overall reactivity of VOC towards ozone formation is
![]()
where
![]()
The mechanistic reactivity then reflects the presence of radical and NOx sources or sinks in the VOC's reaction mechanism subsequent to the initial loss process. An extreme example is benzaldehyde, whose reaction mechanism subsequent to the initial OH radical reaction results in the loss of radical and NOx sinks, with the overall OH radical reaction having d = 0 (no net OH radical formation in Reaction 5.100) and (b-g) = 1 (consumption of one molecule of NOx per molecule of benzaldehyde reacted in Reaction 5.100).
The approach in Chapter 8 to assess the importance of isoprene and other biogenic organic emissions in the formation of ozone in urban-suburban, rural, and remote air masses uses the kinetic reactivities of the VOCs measured in ambient air, and hence is based on the instantaneous rate of formation of RO2 radicals (Reaction 5.99). While the differences in the mechanistic reactivities of the various VOCs are neglected, this simpification is not expected to significantly alter the conclusions drawn in Chapter 8, especially since isoprene has a high positive mechanistic reactivity compared with the reactivities of the alkanes and aromatic VOCs (Table 5-5) that comprise the bulk of anthropogenic emissions observed in ambient air.
Page 159
| TABLE 5-5 | ||||
| Compound | Incremental reactivity, mole O3/mole C | Kinetic reactivity, fraction reacted | Mechanistic reactivity, mole O3/mole C | |
| Carbon monoxide | 0.019 | 0.043 | 0.45 | |
| Methane | 0.0025 | 0.0016 | 1.6 | |
| Ethane | 0.030 | 0.049 | 0.61 | |
| Propane | 0.069 | 0.21 | 0.34 | |
| n-Butane | 0.124 | 0.37 | 0.34 | |
| n-Octane | 0.081 | 0.75 | 0.107 | |
| Ethene | 0.77 | 0.81 | 0.95 | |
| Propene | 0.82 | 0.97 | 0.85 | |
| trans-2-Butene | 0.81 | 0.99 | 0.82 | |
| Benzene | 0.023 | 0.21 | 0.111 | |
| Toluene | 0.106 | 0.64 | 0.17 | |
| m-Xylene | 0.50 | 0.96 | 0.52 | |
| Formaldehyde | 1.26 | 0.97 | 1.30 | |
| Acetaldehyde | 0.70 | 0.92 | 0.77 | |
| Benzaldehyde | -0.29 | 0.95 | -0.31 | |
| Acetone | 0.055 | 0.058 | 0.95 | |
| Methanol | 0.147 | 0.16 | 0.93 | |
| Ethanol | 0.19 | 0.44 | 0.42 | |
| Isoprene | 0.70 | 1.00 | 0.70 | |
| a-Pinene | 0.21 | 0.99 | 0.21 | |
| Urban mixa | 0.28 | |||
(Table continued on next page)
Page 160
(Table continued from previous page)
| aAll-city average urban VOC mix. |
| Source: Adapted from Carter (1991) |
Table 5-5 shows the calculated incremental reactivities and kinetic and mechanistic reactivities for selected VOCs for conditions that simulate those in 12 U.S. cities, with an all-city average VOC mix and the NOx concentrations varied to yield the maximum amount of ozone (Carter, 1991).
Summary
The general features of the atmospheric chemistry of ozone and its precursors are well understood. The chemistry of the polluted troposphere is considerably more complex than that of a less polluted, methane-dominated troposphere because of the presence of many VOCs of various classes. VOCs in the troposphere are photolyzed and react with OH and NO3 radicals and ozone to form organic peroxy radicals (RO2). Subsequent reactions lead to the conversion of NO to NO2, the generation of OH radicals, and the formation of ozone.
The atmospheric chemistry of anthropogenic VOCs, including alkanes, alkenes, and aromatic hydrocarbons, is generally understood. The kinetics of the initial reactions of the majority of anthropogenic VOCs, and the photolysis rates of these VOCs, have been determined experimentally or can be reliably calculated. However, there are many uncertainties concerning the chemistry of aromatic hydrocarbons, carbonyl compounds, and long-chain alkanes and alkenes.
Biogenic VOCs, including isoprene and the monoterpenes, are believed to foster episodes of high ozone concentrations in the presence of anthropogenic NOx. These VOCs are highly reactive toward OH and NO3 radicals and ozone. The mechanisms and products of the important reactions of these compounds are not well understood.
An essential component of air quality models is the chemical mechanism that describes the series of reactions in the troposphere subsequent to emissions of VOCs and NOx. Chemical mechanisms are constructed using kinetic, mechanistic, and product data and refined by comparison with environmental chamber data. Two mechanisms recently developed for use in urban airshed models (those of Gery et al. [1988a, 1989] and Carter et al. [1986a]) have been tested against a common base of environmental chamber data; hence their close agreement does not guarantee their correctness.
VOCs vary widely in the speed with which they react in the troposphere
Page 161
and the extent to which they promote or inhibit ozone formation. Several VOC reactivity scales have been proposed. One useful measure is incremental reactivity, defined as the amount of ozone formed per unit amount of VOC added to a VOC mixture representative of conditions in urban and rural areas. The ozone-forming potential of a VOC depends on the rate at which it reacts and on the reaction mechanism subsequent to the initial reaction. VOC reactivities generally decrease with increasing VOC/NOx ratios; at high ratios, ozone formation is NOx-limited and VOC control is irrelevant.
The following areas of uncertainty need to be addressed before the role of VOCs in the formation of ozone can be assessed in detail:
• The detailed tropospheric degradation reaction mechanisms of the aromatic VOCs are not well understood. In particular, the reactions of the hydroxycyclohexadienyl-type radicals under ambient tropospheric conditions require study.
• The tropospheric reaction mechanisms of biogenic VOCs (for example, isoprene and the monoterpenes) must be investigated, and the gas- and aerosol-phase products determined under realistic atmospheric conditions.
• Reactions under low-NOx conditions, such that reactions of organic peroxy radicals with HO2 and other RO2 radicals and of HO2 with ozone dominate over the reactions of HO2 and RO2 radicals with NO, require study. In addition to the need for further kinetic and mechanism data, environmental chamber studies carried out at low VOC/NOx ratios, close to rural ambient ratios, would be extremely useful.
• The chemistry of long-chain alkanes and long-chain alkenes (for example, the ![]() 1-alkenes) needs to be elucidated.
1-alkenes) needs to be elucidated.
• The formation of carbonyl compounds during the atmospheric degradation reactions of VOCs and the atmospheric chemistry of these carbonyl compounds require further study. In particular, there is a need for absorption cross-sections, photodissociation quantum yields, and photodissociation product data (as a function of wavelength) for the carbonyl compounds.
• The role of heterogeneous reactions in the chemistry of tropospheric ozone needs to be clarified, expanding on issues raised by Lelieveld and Crutzen (1990).
Long-term research (over a period of 5 years or more) is needed to obtain the basic kinetic and product data required for formulation of detailed chemical mechanisms for the atmospheric degradation of anthropogenic and biogenic VOCs. Short- to medium-term programs (2-5 years) are needed to determine the reactivities of VOCs, either singly or as mixtures, with respect to ozone formation. Short-term studies (1-2 years) will be particularly useful
Page 162
for assessing the effects on ozone formation of conversion from gasoline to alternative fuels and for quantifying the effects of biogenic VOCs on urban, suburban, and rural ozone.






















































