
3
Research Opportunities in Catalytic Science
INTRODUCTION
For viable commercial application, catalysts of any type—heterogeneous, homogeneous, or enzymatic—must exhibit a number of properties, the principal ones being high activity, selectivity, durability, and, in most cases, regenerability. The activity of a catalyst influences the size of the reactor necessary to achieve a given level of conversion of reactants, as well as the amount of catalyst required. The higher the catalyst activity, the smaller are the reactor size and the inventory of catalyst and, hence, the lower are the capital and operating costs. High catalyst activity can also permit less severe operating conditions (e.g., temperature and pressure), and this too can result in savings in capital and operating costs. The amount of reactant required to produce a unit of product, the properties of the product, and the amount of energy required to separate the desired product from reactants and by-products are all governed by catalyst selectivity. As a consequence, catalyst selectivity strongly influences the economics of a process. Catalyst productivity and the time on-stream are dictated by catalyst durability. All catalysts undergo a progressive loss in activity and/or selectivity with time due to chemical poisoning, denaturing, thermal deactivation or decomposition, and physical fouling. When the loss in performance becomes too severe, the catalyst must be either regenerated or replaced. In view of this, high durability and ease of regeneration become important properties.
All of the performance characteristics described above are intimately connected to catalyst structure and composition. It is, therefore, highly desirable to develop a detailed understanding of the relationships among a catalyst's structure, its physical and chemical properties, and its activity,
selectivity, and stability. To achieve this goal it is often necessary to characterize catalyst composition and structure at the atomic level. Ultimately, it is most desirable to identify and characterize the catalytic site at the atomic level. Because many catalysts are known to undergo physical and chemical changes under reaction conditions, catalyst characterization should preferably be carried out in situ, or at least under conditions relevant to actual catalytic processes. Knowledge of the interactions between a catalyst and reactants, intermediates, and products is needed to understand the influence of the catalyst on the structure and bonding of species involved in catalyzed reactions. The dynamics of chemical transformations occurring under the influence of the catalyst is yet another area in which information is needed.
The development of new or improved catalysts is complex, involving extensive testing and evaluation. Because the range of variables is often very large, and the relationships between changes in these variables and catalyst performance are often not clearly identified, catalyst development can be a tedious and expensive task. Knowledge derived from scientific studies provides a basis for conceiving new catalysts and catalytic reactions, and for interpreting the results of experimental observation. Moreover, many of the analytical techniques developed in the pursuit of catalytic science can be used effectively to elucidate cause-effect relationships and, thereby, to accelerate the process of catalyst development.
Opportunities for advancing the frontiers of catalytic science exist in four areas: the synthesis of new classes of catalytic materials, catalyst characterization, the mechanism and dynamics of catalytic reactions, and the theory of catalysis. Each of these areas is highlighted below, with indications given for future research directions.
SYNTHESIS OF CATALYTIC MATERIALS
There are three reasons for pursuing research on the synthesis of catalytic materials. The first is to find new or improved catalysts for a desired reaction (e.g., efficient production of high-quality fuels, the decomposition of nitric oxide, the direct conversion of methane to methanol, the synthesis of homochiral or enantiomerically pure drugs). In this instance, either new classes of materials or modifications of existing materials are sought to achieve the desired increase in activity or selectivity. The second reason for studying catalyst synthesis is to establish the relationships between preparative procedure and final catalyst structure and properties. The objective in this case is to understand how the choice of starting materials and synthesis conditions influences catalyst composition and structure. Success in this endeavor can lead to the identification of principles and strategies for preparing catalysts with specified properties. The third reason
is to reduce manufacturing costs by reducing raw material or processing costs.
The importance of catalyst synthesis is well illustrated by the recent development of high-activity catalysts for the reduction of nitrogen oxide (NOx) emissions from power plants. The material of choice for this application is titania-supported vanadia. In currently practiced technology, vanadia is dispersed into the pores of a titania monolith. A reaction engineering analysis of the performance of such catalysts has revealed that the catalyst operates in the diffusion-influenced regime and that the pore structure of the support can be optimized for maximum performance. Further research has shown that the desired pore structure cannot be achieved by using bulk titania because of physical strength constraints, but can be achieved by using silica. To obtain the chemical properties of titania required for high intrinsic activity, titania is dispersed into the pores of a silica monolith, and vanadia is then deposited on the titania particles. Based on laboratory-scale tests, the resulting material exhibits a catalytic activity 50% higher than that previously available and promises improved poison resistance due to its bimodal pore structure. This illustration shows the manner in which knowledge of material properties can be combined with an analysis of reaction dynamics and mass transfer to design a catalyst with optimal performance characteristics for a targeted application.
High selectivity in combination with high activity can often be achieved with homogeneous catalysts. These properties are influenced by the nature of the transition metal situated at the catalytically active center of the complex. Variations in the composition of the ligands and the solvent in which the complex is dissolved can influence the catalytic properties of the complex. Strategic manipulation of these variables can be used to obtain useful catalysts. A recent illustration of this point is the synthesis of naproxen, an anti-inflammatory drug. As currently manufactured, this drug is expensive because the synthesis procedure results in a mixture of the two optical isomers that must be separated, because S-naproxen is the desired product but R-naproxen is a liver toxin. To reduce the costs of production, one wants to increase catalyst selectivity for the S-isomer. Recent research has shown that naproxen can be produced with high selectivity by asymmetric hydrogenation of α-(6-methoxy-2-naphthyl) acrylic acid using a soluble ruthenium complex containing a chiral phosphine ligand. This advance holds promise not only for reducing the cost of production, but also for eliminating potentially harmful by-products.
Molecular sieves, of which zeolites are a special class, offer extensive opportunities for the design of new catalysts. These materials are characterized by a crystalline framework containing cavities and channels of molecular dimensions (0.3-1.0 nm). Catalytic activity is typically due to acidic
sites in the framework. Multifunctional catalysis can be achieved by dispersing small metal particles within the cavities of the molecular sieve.
What makes molecular sieves such interesting materials is their wide range of compositions and topologies. Prior to 1982, most molecular sieves were based on aluminosilicates. These materials are commonly known as zeolites. However, recent developments at Universal Oil Products (UOP) Inc. have demonstrated the feasibility of producing molecular sieves based on aluminophosphates. By substitution of various metallic elements for either aluminum or phosphorus, a broad range of materials can be produced that vary in their catalytic properties. Although about 60 different molecular sieve topologies are known today, tens of thousands of structures are theoretically possible, but knowledge of how to synthesize specific structures is not yet available. An issue of particular interest is to find ways of producing molecular sieves with openings in excess of 0.7 nm. Such materials would be able to accept the large reactant molecules found in heavy petroleum and in other heavy fossil liquids. Progress toward this goal has recently been achieved by the discovery of a large-pore molecular sieve (VPI-5) that contains 18-membered oxygen rings with an opening of 1.2 nm. The challenge now is to find ways to make VPI-5 catalytically active and stable under reaction conditions.
Molecular sieve crystals provide not only uniform intracrystalline pores, but also uniform and well-defined tetrahedral coordination for framework metal ions. The discovery of a great variety of structures and chemical compositions in the aluminophosphate molecular sieve family of crystals opens opportunities to prepare molecular sieves with transition metal cations in the crystal framework. This unique, predictable positioning of transition metal ions in microporous crystal lattices offers opportunities in oxidation, redox, or other transition-metal-related catalysis.
Clays, phosphonates, and other lamellar structures represent yet another class of materials that are of growing interest to the field of catalysis. By introducing either organic or inorganic ''pillars" between the lamellae, it is possible to create galleries of molecular dimensions. Through the control of pillar height, uniform interlamellar spacings of 0.5-4.0 nm can be achieved. Such pillared layered structures can be used as catalysts if the pillars contain acidic groups or as supports for transition metals or their complexes. In the latter case, the support can serve to orient the reactant so that unique reactant and product selectivities can be achieved. A significant challenge for the future is to find ways to improve the thermal and hydrothermal stability of such pillared lamellar structures.
Enzymes are highly efficient catalysts that have great potential industrially, especially for the synthesis of chiral compounds. Until the advent of recombinant DNA technology, this potential could not be realized. Previously, only those few enzymes that were obtainable in large amounts from
|
THE POLYETHYLENE STORY: CUT-RESISTANT SURGICAL GLOVES AND LAUNDRY BAGS What do cut-resistant surgical gloves and laundry bags have in common? They are both made from polyethylene. Polyethylene was discovered in the late 1930s by a group of chemists who were studying the properties of ethylene, a gas, at high pressure and temperature. They discovered that under certain conditions, the gas turned into a flexible white solid that proved to be an excellent insulator. (It has since found favor as a food wrapper.) World War II created an urgent demand for large quantities of such an insulator to coat the wires of radar sets and electronic weaponry. Without that demand, in fact, polyethylene might have been abandoned as too dangerous to manufacture: the early production plants often blew up. As it turned out, World War II also resulted in a safer way to make polyethylene. Germany, cut off from the world's oil fields by the Allied blockade, mounted a crash research program to develop synthetic fuel and lubricating oils. Karl Ziegler, one of the chief chemists in that effort, continued this research after the war, using ethylene as his raw material. One day in 1953, he was surprised to find the reaction vessel full of polyethylene, although he had not been using the high temperature and pressure then thought necessary to make it. The secret proved to be a metal catalyst. Unlike the high-temperature variety, this new polyethylene was rigid, not flexible, and it differed in other ways as well. (It is now used to make milk bottles.) Further research showed that the two types of polyethylene grow in very different ways. Ziegler's catalyst selectively stitches ethylene molecules together end-to-end to make long, straight chains of polyethylene. These linear molecules stack readily, like cordwood, to give the rigid, crystalline, high-density material that is used for milk bottles. The high-temperature process, however, proceeds through "radical" intermediates—unstable species that react indiscriminately with anything in sight, including their own polymer chains. When this happens, the once-linear chain develops branches and, just as a pile of brushwood does not stack neatly, so neither do the branched polymers, which results in a soft, amorphous, low-density material. Modern catalysts can be used to polymerize ethylene into an ultra-high-molecular-weight linear polymer. This material can be spun into extremely strong fibers that can be used in cut-resistant surgical gloves. Today's catalysts have been much improved since Ziegler's discovery, so that very selective processes now produce very specific products in very high yields. Further improvements will result not only in milder processing conditions but also in superior material properties for a broader range of uses. |
natural sources could be considered for commercialization. Even some natural enzymes had properties not directly suitable for a particular industrial application. Now it is possible to produce any enzyme in industrially useful form by cloning the gene and expressing it in a suitable production organism. The properties of the enzyme, such as stability, selectivity, and catalytic efficiency, are controlled by its three-dimensional structure. This structure is in turn determined by the sequence of amino acids in the enzyme, which can now be altered by changing the gene that codes for the enzyme. The researcher can fit the desired application by changing the amino acid sequence in the enzyme. This has been done for several enzymes in commercial use and will lead to the increased use of enzymes as catalysts for industrial processes.
Another area of rapidly emerging opportunity is the formation of catalytically active antibodies targeted for specific reactions. Various approaches are available. These include the raising of antibodies to structures resembling those of the transition states of the reactions for which a catalyst is desired. Recent studies have demonstrated rate accelerations for a number of antibody-catalyzed reactions of up to 106 times the uncatalyzed rate. A development in the design of catalytic antibodies involves the introduction of catalytic activity into antibodies via either molecular biological or chemical means. These strategies allow for the a priori evolution of catalytic activity in an antibody combining site and also allow incremental increases in the rates of catalytic antibodies generated by other means. Examples of such approaches include introduction of catalytic groups via hapten-antibody complementarity, chemical modification, generation of cofactor binding sites, and site-directed mutagenesis. These systems offer catalysts for reactions beyond those available from enzymes.
Within the United States, the design and synthesis of new catalytic materials have been pursued largely in industry but have received much less attention in government and university laboratories. This is not the case in Japan and the European Community, where considerable research on catalyst synthesis is evident, and in fact targeted, in many academic research institutions. The absence of a strong effort toward the synthesis and evaluation of all types of novel materials is a weakness in the catalysis program in the United States. Not only does this sharply limit the techniques that can be brought to bear on problems in this area, but it also curbs the opportunities for discovery.
As illustrated by the examples presented above, many exciting research opportunities are available for the design and synthesis of all types of new catalytic materials, and strong support should be given to this important area.
CATALYST CHARACTERIZATION
Knowledge of catalyst composition and structure is critical to a fundamental understanding of the chemistry actually occurring in catalysis. From such information it is possible to determine which portions of a catalyst are active and how changes in catalyst synthesis and structure affect the properties of such sites. Catalyst characterization is also vital to understanding the changes that occur in the structure and composition of a catalyst following pretreatment, during induction periods, in use under reaction conditions, and during regeneration.
Over the past two decades, advances in analytical instrumentation have led to significant improvements in sensitivity and resolution. This has enabled researchers to obtain detailed structural and compositional analyses, often with atomic spatial resolution. Techniques currently available for catalyst characterization are listed in Table 3.1.
Table 3.1 Experimental Techniques for Characterizing Catalysts and Adsorbed Species
|
Technique |
Acronym |
Type of Information |
|
Low-energy electron diffraction |
LEED |
Two-dimensional structure and registry with metal surface |
|
Auger electron spectroscopy |
AES |
Elemental analysis |
|
X-ray photoelectron spectroscopy |
XPS |
Elemental analysis and valence state |
|
Ion scattering spectroscopy |
ISS |
Elemental analysis |
|
Ultraviolet photoelectron spectroscopy |
UPS |
Electronic structure |
|
Electron energy loss spectroscopy |
EELS |
Molecular structure |
|
Infrared spectroscopy |
IRS |
Molecular structure |
|
Laser Raman spectroscopy |
LRS |
Molecular structure |
|
X-ray diffraction |
XRD |
Bulk crystal structure |
|
Extended x-ray absorption fine structure |
EXAFS |
Bond distance and coordination number |
|
Transmission electron microscopy |
TEM |
Crystal size, shape, morphology, and structure |
|
Scanning transmission electron microscopy |
STEM |
Microstructure and composition |
|
Scanning tunneling microscopy |
STM |
Microstructure |
|
Ultraviolet spectroscopy |
— |
Electronic state |
|
Mössbauer spectroscopy |
— |
Ionic state |
|
Nuclear magnetic resonance spectroscopy |
NMR |
Molecular structure and motion |
|
STUDIES OF AMMONIA SYNTHESIS OVER IRON SINGLE CRYSTALS Since 1913, ammonia has been produced in tonnage quantities by direct combination of nitrogen and hydrogen at high temperature and pressure. Most of it is used to make nitrogen-rich fertilizers, and the balance to manufacture explosives, of use in peace as in war. Ammonia synthesis occurs at the surface of iron catalysts. Because ammonia is a low-priced commodity chemical, the catalyst must be cheap and durable, and its activity as high as possible, so that temperature and pressure can be kept low to minimize the size and cost of huge industrial reactors. Factors responsible for the activity and durability of commercial iron catalysts have been elucidated in the 1980s by surface science studies conducted with model catalysts consisting of single crystals of iron approximately 1 cm in size. The rate of ammonia synthesis at high temperature and pressure was measured over five of these large crystals of pure iron, cut in five different ways so as to expose five different facets. Two facets were found to be almost equally active, but more active in producing ammonia than the other three. It is logical to expect that the best commercial iron catalysts should expose one or both of the best facets. Is it so? The answer seems to be yes, for a very simple reason. When the activity of a commercial iron catalyst is compared to that of single crystals under identical conditions, it is found that both values per iron atom exposed are about the same and equal to those found on the best two facets. Thus, it appears that the commercial catalyst has been optimized. How did this happen? The answer to this second question is more subtle and related to the other desirable feature of a catalyst, namely, durability. Commercial iron catalysts consist of very small crystals of iron, a few millionths of a centimeter in size. As a result, the catalyst exhibits a very large surface area per unit volume, a most desirable characteristic to minimize reactor size. However, tiny crystals of pure iron fuse under the harsh temperatures of ammonia synthesis, a phenomenon that leads to catalyst sintering and the death of the catalyst. Indeed, a commercial iron catalyst is not made of pure iron metal, but of iron to which certain oxides have been added. Because the added oxides are beneficial, they are called promoters. One of these promoters is aluminum oxide, alumina. Alumina by itself or on an iron surface is totally inactive in ammonia synthesis. Its role as a promoter has been ascribed in the past to its ability to prevent sintering of iron particles (i.e., to ensure catalyst durability). Recent work with large single crystals does not refute this interpretation, but adds a significant dimension to the role of alumina as a promoter. Thus, surface science studies have revealed that the addition |
|
of alumina to the most inactive facet of a large iron single crystal, followed by heating under appropriate conditions, causes its recrystallization to form ultimately the two most active facets during ammonia synthesis. The conclusion is that alumina brings about the restructuring of any iron crystal face to produce and maintain those facets that are most active for ammonia synthesis. It has been said that surface science work with large metallic single crystals provides the standards by which work with commercial metallic catalysts can be assessed and understood. This vignette provides a striking illustration of that statement. |
Experience has demonstrated that no single technique can provide a complete picture but that, instead, a combination of techniques is necessary. The characterization of zeolites is a case in point. The lattice structure of a zeolite can be determined from x-ray diffraction by using either single-crystal or powder-diffraction techniques. The correctness of the lattice structure may be confirmed by atomic resolution electron microscopy, which also provides evidence of lattice defects or of the presence of minority impurity phases. The distribution of the elements constituting the lattice framework can be established by 29Si magic angle spinning (MAS) nuclear magnetic resonance (NMR) spectroscopy, and the nature and strength of Brønsted acid sites can be determined by using 1H NMR spectroscopy or a variety of sorption techniques. The size, crystallographic perfection, and defects of cavities inside the zeolite can be probed by 129Xe NMR spectroscopy. If metal particles are occluded within the zeolite cavities, the average size of these particles can be established by extended x-ray absorption fine structure (EXAFS). By combining information from the above techniques, it is possible to develop a detailed atomic view of a zeolite catalyst that can serve as the starting point for developing theoretical models of catalytic sites (see below) and identifying strategies to improve catalyst performance.
Acid catalysis applied both in homogeneous solution and over solid catalysts is the largest branch of industrial catalysis. In homogeneous solution the understanding of reaction mechanisms is aided greatly by the broad knowledge of the chemistry of homogeneous acid solutions. Understanding the chemistry over solid acids is hampered by a lack of absolute acidity measurements or of sound comparisons between liquid and solid acids. The recent application of 1H NMR in combination with other methods promises to provide an absolute scale and measurement of the acid strength of solid acid catalysts. If successfully developed, such techniques could be applied to the development of a variety of practical solid acid catalysts.
Heterogeneous noble metal catalysts are generally prepared by dispersing the metal in the form of small crystallites onto the surface of a metal
oxide such as alumina or high-surface-area porous carbon. The size and morphology of metal particles can be established from transmission electron microscopy. Electron microscopy can also provide information on the nature of the interaction between the metal particles and the support. EXAFS can be used to determine the average metal-metal bond length and coordination number. For bimetallic catalysts (e.g., platinum-iridium, platinum-rhenium), the degree of interaction and the distribution of the two components can also be established.
A large body of evidence acquired in recent years has shown that the properties of metal catalysts can be modeled by using single-crystal metal surfaces. This method has proved invaluable for the advancement of catalyst science because of the large array of techniques developed by surface scientists for the characterization of single-crystal surfaces. The atomic composition of such surfaces can be analyzed by using Auger electron spectroscopy (AES), x-ray photoelectron spectroscopy (XPS), and ion scattering spectroscopy (ISS). The structure of the exposed surfaces and the presence of terraces and defects can be established by low-energy electron diffraction (LEED). An exciting recent development, scanning tunneling microscopy (STM), has made it possible to obtain atomic-resolution views of surfaces and to identify the influence of thermal and chemical pretreatment on surface structure.
Insights into structure-function relationships can be obtained through the use of model catalyst systems. Examples of such systems include metals deposited on oxides, oxides on oxides, and oxides on metals. After initial preparation by means of vapor deposition, the sample can be characterized by AES and XPS. The catalytic properties of the catalyst can be determined by transferring the sample to a high-pressure chamber. Characterization of the sample after reaction reveals changes that have occurred in the catalyst composition and structure. Model catalyst systems are also well suited for examination by means of scanning transmission spectroscopy (STEM) and controlled atmosphere electron microscopy (CAEM). These techniques can provide detailed evidence for surface reconstruction, sintering, alloying, and phase separation.
Significant advances have also been made in the characterization of enzymes, catalytic antibodies, and homogeneous catalysts. Where such catalysts can be obtained in the form of single crystals, the analysis of x-ray diffraction patterns can provide a complete atomic structure. However, although such information is quite useful, it does not indicate structural changes that may occur when the catalyst is saturated and surrounded by solvent. Recent advances in multidimensional (i.e., two-, three-, and four-dimensional) NMR spectroscopy are making it possible to obtain this information in solution, and enzyme structures containing as many as 153 amino acids have been analyzed completely.
The detailed mechanism of some homogeneous transition metal catalysts has been elucidated by work in the closely allied field of organometallic chemistry. In many cases, intermediates can be trapped and models for catalytically active species can be prepared and studied for kinetic competence in the catalytic process. These complexes can be characterized completely by small-molecule crystallography and examined in solution by high-field NMR techniques.
Because the advancement of catalysis along the experimental front depends heavily on the availability of advanced instrumental characterization techniques, it is important that research on the development of new techniques be well supported. The United States has traditionally been a leader in the field of instrumental analysis, and many of the techniques in use today originated in academic laboratories. This position is currently in jeopardy, because of strong competition from the European Community and Japan. Particular emphasis should be placed on the development of techniques that can be used to characterize catalysts in situ. Examples of such techniques include STM, atomic force microscopy (AFM), NMR spectroscopy, and EXAFS.
MECHANISM AND DYNAMICS OF CATALYTIC REACTIONS
Knowledge of catalytic reaction mechanisms—in particular the structure, dynamics, and energetics of reaction intermediates formed along the catalytic reaction path—can provide insights for developing new catalysts and improving existing ones. In situ observation, in turn, is the most effective technique for elucidating reaction mechanisms, the dynamics of molecular interactions with catalysts, and the structure of stable intermediates. Such investigations also provide information on the thermodynamics of catalyst-substrate interactions and the associated activation barriers. Homogeneous catalysts can also be studied in solution by NMR techniques; these studies have resulted in new homogeneous catalytic reactions, and they provide models for related heterogeneous processes. Several examples will serve to illustrate the importance of a multitechnique approach.
L-Dopa, used in the treatment of Parkinson's disease, is prepared by asymmetric hydrogenation. The catalysts used in this process are homogeneous rhodium complexes. When synthesized with chiral phosphine ligands, these complexes enable the production of the desired enantiomer with greater than 95% selectivity. Detailed mechanistic studies involving NMR spectroscopy have permitted the identification of catalytic intermediates and have led to the design of new asymmetric hydrogenation catalysts and to the selection of optimal reaction conditions. As the emphasis on producing optically pure drugs increases, determination of the details of homogeneous asymmetric catalysts will become increasingly important.
|
CONTROLLING BUGS WITH BUGS Are there ways to control gypsy moths, cabbage loopers, and other agricultural annoyances without using chemical pesticides? The answer is yes. There are now available a number of biological products registered with the Environmental Protection Agency (EPA) that can be used to control a fairly wide array of insects, weeds, and other plant pests (Figure 3.1). Most of these products are based on microorganisms (''bugs") that are the pest's natural enemies—disease-causing "pathogens" that kill the pest outright or foul-smelling, worse-tasting "antagonists" that drive the pest away. These biopesticides have several advantages over chemical pesticides. Biopesticides act through mechanisms specific to the target organism and nonexistent in other organisms, thus sabotaging the target's metabolism without poisoning every other creature as well. Biopesticides are fully biodegradable, leaving no residues behind, and biopesticides can be applied with the same equipment used to apply conventional pesticides, at the same intervals, and with essentially equivalent results. If these marvelous products are truly available, why isn't there a greater awareness of them? There are several reasons. Although some of these products have been around for 20 years or more, they did not work as well in the past as they do now, thanks to continuing research. Nor have they ever been price competitive with conventional pesticides. Where they have been used extensively, for example, in eastern U.S. oak forests to control gypsy moths, they have been well received. They have, however, generally been seen as specialty products for "niche" markets, rather than as alternatives to chemical pesticides in the broadest sense. This view may be changing. Opposition to chemical pesticides continues to increase with increasing awareness of their environmental consequences. The cost of developing new chemical pesticides and of registering them with the EPA—a precondition to their sale in the United States that requires extensive environmental and toxicological studies—has led to a decline in the rate of new product development. Furthermore, many insects and plant diseases have shown an amazing ability to develop resistance to an ever-broadening array of pesticides, limiting the useful life of even newly developed products. Biopesticides, on the other hand, have received increasing attention over the past five years. Several new biotechnology companies are focusing specifically on biopesticide development, and even old-line agricultural chemical companies have begun to shift their pesticide lines toward environmentally compatible products. Biotechnology—biocatalysis—is indispensable to biopesticide development, as the story of the development of insecticides from the soil |
|
bacterium Bacillus thuringiensis shows. Over the past 10 years, scientists have found that this bug's insecticidal properties are encoded by a diverse array of genes that direct the synthesis of insecticidal proteins. These proteins act as selective poisons in the stomachs of insect larvae. Different genes produce poisons specific to various plant-attacking beetle larvae and caterpillars, as well as to the larvae of such carriers of human disease as mosquitoes and blackflies. By isolating and analyzing the genes involved, scientists can now "engineer" new gene combinations that improve on their naturally occurring counterparts, creating more potent weapons against a broader range of pests. Such engineering is an exquisite example of biocatalytic synthesis, using the bacterium's metabolic machinery to create proteins that would be prohibitively expensive to manufacture by industrial chemical processes. The new genes can be transferred to other bacterial hosts. Plants can also be "vaccinated" by endowing other organisms that grow in or on them with the gene. This scheme, which would provide a steady supply of the biopesticide to the plant, may prove more efficient than conventional spraying. Those who protest the continued use of pesticides should be gladdened to know that there is hope for alternative products. Many are available now. Others will become available in the near future, if industry continues to support their development. For these new products to make their contribution, however, regulatory agencies and the public must be willing to agree that genetically engineered products are safe for use in the environment. The benefit to be derived from these products must be shown clearly to outweigh any perceived risk. This is the challenge to be faced if we are to continue to maintain and increase our agricultural productivity. |
To produce high-octane components for gasoline, normal paraffins must be converted into branched paraffins, olefins, and aromatic compounds. Such transformations can be carried out over a platinum catalyst under a hydrogen atmosphere in a process called reforming. Extensive efforts have been undertaken to understand the fundamental chemistry involved in the reforming process. Vibrational spectroscopies, such as electron energy loss spectroscopy (EELS) and infrared spectroscopy, and 13C NMR spectroscopy have revealed that paraffins are only weakly adsorbed but undergo a loss of hydrogen on heating. The exact amount of hydrogen lost can be determined from temperature-programmed desorption spectroscopy, whereas the structure of the surface species formed as the temperature increases can be determined from both infrared and NMR spectroscopies. These techniques are particularly valuable because they can be used at el-

evated temperature and pressure. Additional information on the structure of adsorbed hydrocarbons has been obtained from LEED studies. More recently, molecular beam investigations have proved useful in determining the dynamics of hydrocarbon adsorption and decomposition on metal surfaces. Because the thermal energy of the hydrocarbon can be controlled in such experiments, it is possible to determine the kinetics of hydrocarbon reactions in great detail. Studies carried out with well-defined single-crystal surfaces of platinum have demonstrated the sensitivity of reaction dynamics to the crystallographic planes exposed and to the presence of defects such as steps and kinks.
Ethylene oxide and propylene oxide are critical intermediates in chemical manufacture. Ethylene can be catalytically oxidized by molecular oxygen over a silver catalyst to give an excellent yield of ethylene oxide. The synthesis of propylene oxide requires the catalytic transfer of oxygen from an alkyl peroxide to propylene. Transition metal catalysts, usually based on molybdenum, are essential for the reaction. These two reactions have a major impact on the commodity chemical industry. Recently, the synthesis of epoxides has been modified to yield enantiomerically pure products, an advance that has had a major impact on the manufacture and synthesis of bioactive molecules. Barry Sharpless' group, at MIT, has shown that titanium complexed to tartrate esters (obtained from nature as one optical isomer) will transfer oxygen from tertiary-butyl hydroperoxide to allyl alcohols to give enantiomerically pure epoxides. Such studies have provided the paradigm for the role of structure, mechanism, and dynamics in the discovery of selective catalysts for the production of enantiomerically pure products.
The conversion of methanol to gasoline over ZSM-5, a medium-pore zeolite, has also been investigated extensively as part of the effort to develop alternatives to petroleum as a source of transportation fuels. Both 1H and 13C NMR have been useful techniques for characterizing the intermediates formed between olefins and protic sites inside the zeolite and for characterizing the structure of coke, which builds up slowly with time and contributes to catalyst deactivation. This type of information has proved useful in understanding how zeolite acidity and pore size influence the rate at which deactivation occurs.
The selective catalytic reduction of nitric oxide by ammonia over titania-supported vanadia catalysts provides an effective means for reducing NOx emissions from stationary sources. Isotopic tracers have revealed that this reaction is initiated by the formation of an adduct between nitric oxide and ammonia, whereas Raman and NMR studies of adsorbed ammonia have shown how ammonia complexes with vanadium ions present on the catalyst surface. Such investigations suggest that oxygen atoms bound to vanadium ions play an important role in activating the adsorbed ammonia for reaction. The role of additive oxides that are known to enhance catalyst activity and
selectivity can be investigated by using spectroscopic techniques. Such promoters interact with the reactants and suppress the oxidation of sulfur dioxide to sulfur trioxide, an undesired side reaction.
In summary, investigations of reaction mechanisms and kinetics and, especially, in situ observations of catalytic reaction intermediates are essential for advancing the science of catalysis, inasmuch as the results of such studies provide an overall view of catalysis and help elucidate the relationships between catalyst structure and function. The current interest in developing catalysts for the production of environmentally benign gasoline, the abatement of air pollution from mobile and stationary sources, the synthesis of enantiomerically pure drugs, and the synthesis of novel polymers all benefit from studies of the relevant reaction mechanisms and kinetics. The successful advancement of knowledge in this area requires the further development of techniques for characterizing adsorbed species, particularly their structure and bonding; for identifying the connectivity between species in terms of a reaction network; and for characterizing the dynamics of elementary chemical transformations occurring under the influence of the catalyst.
THEORY OF CATALYSIS
The science of catalysis has traditionally advanced as a consequence of new experimental techniques, but theory has begun to play an increasingly important role. This change is the result of recent advances that have occurred in theoretical and computational chemistry, reaction engineering, and the availability of powerful supercomputers for extensive calculations, which enable graphical display of results. Information gained from theoretical studies is becoming helpful in guiding the design of novel catalysts, interpreting experimental measurements, and understanding the way in which catalyst composition and structure affect its activity and selectivity.
Considerable progress has been made in modeling and calculating the relative energies of intermediates in homogeneous transition metal catalysis. These systems are small and generally involve one metal and one constant ligand set. However, even for relatively simple systems, major approximations are required for ab initio calculations. The results obtained from these systems in which detailed molecular structures can be determined and systematic structural changes can be made will serve as guides for the more complex heterogeneous systems.
Theoretical calculations of catalyst structure can provide a useful basis for assessing the stability of particular structures as a function of composition and surrounding environment. For example, in the area of zeolite catalysis, it is possible to predict the stability of channel openings as a function of the ratio of silicon (Si) to aluminum (Al) and the preferred location of aluminum in the framework. Similarly, theoretical models of
![]()
suggest a direct relationship between Si-O-Al bond angles and zeolite acid strength, consistent with experimental observation. In the case of bimetallic catalysts, the spatial distribution of each component in small particles can be computed for a given particle size and composition. Computational chemistry has also proved useful in examining the influence of site-specific modifications and solvent effects on the structure of active sites in enzymes.
The interpretation of spectroscopic analyses of catalysts can be facilitated with the aid of appropriate computations. Calculations of the occupation of valence electrons in d-orbitals have proved useful in the proper interpretation of x-ray absorption near-edge structure (XANES) measurements on small metal particles. For zeolitic materials, theoretical predictions of infrared can be made. Comparison of calculated and experimentally observed spectra can be used as a sensitive test of atom-atom potentials. With accurate knowledge of such potentials, it is possible to make predictions about phase transformations and other structural changes occurring at elevated temperature.
There are an increasing number of examples in the literature indicating the feasibility of computing the potential energy curves for elementary steps in catalyzed reactions of small molecules. Figure 3.2 illustrates a potential energy diagram obtained from an ab initio quantum chemical study of ethylene hydrogenation by using a model Wilkinson catalyst, RhCl(PH3)3. Such calculations are useful, as a first step, for identifying the most energetically demanding (i.e., rate-limiting) steps in a reaction sequence; however, future efforts along these lines must be carried out by using realistic phosphine ligands so that meaningful comparisons can be made with experimental observations. Prediction of the influence of solvents on the energetics and dynamics of homogeneous catalytic reactions also remains an important challenge. Some initial attempts to address such problems have been carried out in the area of enzyme catalysis, where the properties of the solvent are known to influence the rate of substrate-enzyme association.
Progress has also been made in evaluating the energetics of atoms and small molecules interacting with metal surfaces. Atomic heats of adsorption can now be calculated by using ab initio techniques, which are in very good agreement with experiment. Studies of molecular adsorption have identified the preferred sites for adsorption and the structure and bonding of the adsorbed molecule. To date, however, ab initio techniques have proved too expensive to permit full exploration of the multidimensional potential surface governing reactions. As a consequence, less accurate, semiempirical methods have been explored. Such techniques have provided information regarding the dissociation path of diatomic molecules on transition metal surfaces and the activation barriers for various elementary reactions. Cal-
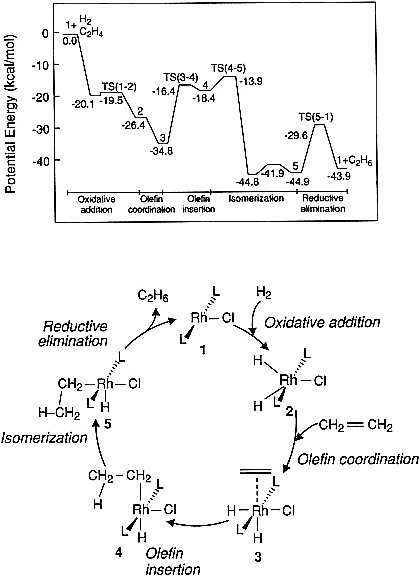
Figure 3.2 Potential energy diagram obtained from an ab initio quantum chemical study of ethylene hydrogenation by using a model Wilkinson catalyst. (Reprinted, by permission, from N. Koga, C. Daniel, J. Han, X. Y. Fu, and K. Morokuma, 1987, pp. 3455-3456 in Journal of the American Chemical Society, Vol. 109. Copyright © 1987 by the American Chemical Society.)
culations of the effect of poisons (e.g., sulfur) and promoters (e.g., K+) have yielded information on the manner in which the components affect the adsorption of molecules. Computed potential energy surfaces can be used to calculate the rate coefficients for elementary processes such as adsorption, diffusion, desorption, and reaction. By use of dynamic simulation techniques and dynamically corrected transition state theory, estimates of rate parameters have been obtained that are in reasonable agreement with experimental observation.
The application of theoretical methods to zeolites has proved fruitful. Advances in theory have contributed to understanding the thermodynamics of adsorption in zeolites and the dynamics of diffusion. Monte Carlo calculations can now provide accurate predictions of the isotherms and heats of adsorption of hydrocarbons in a number of zeolites. Molecular dynamics calculations can be used to describe the motion of molecules through the void space of zeolites. Such calculations have been used to determine the molecular diffusion coefficients, various properties of motion, and the spatial distribution of sorbates within the zeolite void space. Promising progress has also been made recently in the use of ab initio quantum chemical techniques to characterize the interaction of reactant molecules with acid sites within a zeolite. Such calculations can provide a description of the reactant-zeolite potential surface, from which it will then be possible to define the reaction intermediates and to determine the rate coefficients for chemical transformations.
Yet another area in which progress is being made is the modeling of catalyst particle and reactor performance. Such models combine information about intrinsic catalyst activity and selectivity, intraparticle mass and heat transfer, and heat, mass, and fluid transport within the reactor to predict product conversion and yield, and catalyst performance with time on-stream. Models of this type can be used to determine the extent to which transport phenomena affect catalyst or reactor performance and the mode by which catalyst deactivation occurs (e.g., sintering, poisoning, fouling).
It has also become possible to use catalyst and reactor models to determine the optimal design of catalyst particles and structures. In such cases, calculations are made to maximize or minimize a desired objective function for a given set of design and operating parameters. Typical catalyst design parameters include descriptors of the structure and composition of the catalyst surface, the pore structure, the activity distribution in the catalyst particle, and the size and shape of the catalyst particle. Typical operating parameters are reactor type (fixed bed, ebullated bed, fluid bed, and so on), inlet conditions (temperature, pressure, composition, flow rate), and parameters related to heat or solid flow management. The objective function (or functions) to be maximized (or minimized) provides a quantitative measure by which the performance of the catalyst, catalytic reactor, or catalytic process is to be judged
|
SOLID-STATE HIGH-TEMPERATURE FUEL CELLS An anonymous wit once paraphrased the three laws of thermodynamics thus: First, you can never win, you can only break even. Second, you can never break even. Third, you can't get out of the game. The second law limits the efficiency of a fossil-fuel-fired power plant—only a fraction (typically less than 40%) of the chemical energy released by burning the fuel is converted to electricity. The balance is dissipated in various unavoidable ways—as friction between moving parts, as waste heat up smokestacks and cooling towers, and so forth. Fuel cells, however, translate chemical energy directly into electrical energy without any mechanical or thermal intermediaries. Fuel cells can have efficiencies as high as 90%, depending on their applications. With such high efficiencies, power plants based on fuel cell technology would consume much less fossil fuel and emit proportionally fewer pollutants than would conventional power plants. All fuels burn by reacting with oxygen to release energy, and the key to a fuel cell's efficiency lies in using catalysts to control that reaction. There are a number of different types of fuel cells. One promising variety is the high-temperature, solid-state fuel cell (Figure 3.3), which is essentially a barrier made of ceramic, typically zirconium oxide doped with traces of yttrium oxide, whose structure conducts oxygen ions (negatively charged oxygen atoms). On one side of the barrier—the fuel cell's negative terminal—the fuel, typically a mixture of carbon monoxide and hydrogen gas created by gasifying coal or steam-refining natural gas, reacts catalytically with oxygen ions to liberate water, carbon dioxide, and electrons. The electrons go out over the wire as an electric current. On the other side of the barrier—the positive terminal—the electrons returning through the wire are catalytically added to molecules of oxygen from the atmosphere to create more oxygen ions that diffuse through the barrier and perpetuate the cycle. The yttria-doped zirconia fuel cell typically operates at around 1000° C. So what are we waiting for? Why aren't power plants based on fuel cells? Unfortunately, several technological hurdles must be overcome before this catalytic technology can be a commercial success. A worldwide effort has been under way for more than two decades to clear these hurdles, which include devising ways to keep the catalyst from breaking down at such high temperatures, avoiding cracks and leaks in the ceramic structure, and designing a ceramic that conducts enough oxygen ions through a sufficiently small volume to make the size of the equipment economical. Some current efforts are focused on so-called cross-flow monolithic designs in which a solid ceramic block is laced with sets of fuel channels perpendicular to, and alternating with, sets of air channels. This design provides a high interior surface area upon which reactions can occur and thus occupies a very compact volume per unit of energy generated. |
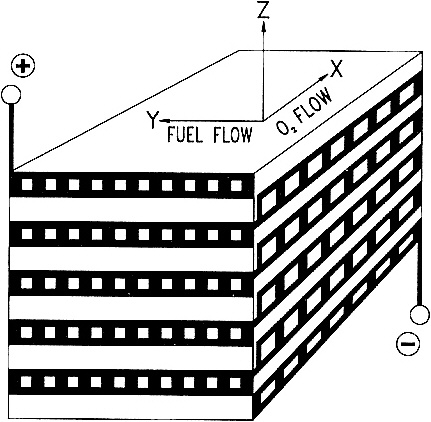
Figure 3.3 Schematic of a cross-flow, solid-state, high-temperature fuel cell. (Figure courtesy of W. R. Grace & Company.)
(e.g., maximum feed conversion, maximum time on-stream, minimum coke loading). As advances are made in the theoretical prediction of transport and reaction in catalysts, one can envision the development of a hierarchy of algorithms for describing catalyst performance, starting at the molecular level and progressing upward to the level of a complete reactor.
In summary, the application of theoretical methods to describe and predict the properties of catalysts is a rapidly developing area of research that can contribute much to the scientific understanding of catalysts and the chemical reactions occurring under their influence. Critical areas for future research include the computation of accurate catalyst-substrate potential surfaces by using ab initio quantum chemical techniques, the evaluation of rate and diffusion coefficients via molecular dynamics and transition state theory, the application of Monte Carlo techniques for describing the dynamics of
complex reaction systems, and the investigation of stochastic methods for simulating catalyst particle and catalytic reactor performance. The successful advancement of such efforts requires adequate availability of supercomputer time and access to appropriate types of workstations. With the increasing complexity of the systems studied, it is also necessary to have capabilities for graphical representation of the results of computations.
Conclusions
Catalysis is a complex, interdisciplinary science. Therefore, progress toward a substantially improved vision of the chemistry and its practical application depends on parallel advances in several fields, most likely including the synthesis of new catalytic materials and recognition of the reaction path of catalytic reactions. For this reason, future research strategies should be focused on developing methods with the ability to observe the catalytic reaction steps in situ or at least the catalytic site at atomic resolution. There is also a need to link heterogeneous catalytic phenomena to the broader knowledge base in solutions and in well-defined metal complexes.
Substantial progress and scientific breakthroughs have been made in recent years in several fields including atomic resolution of metal surfaces, in situ observation of an olefin complexed to zeolite acid sites by NMR spectroscopy, and in situ characterization of several reaction intermediates by a variety of spectroscopic techniques. Theoretical modeling is ready for substantial growth as a result of progress in computer technology and theory itself. For these reasons, it is desirable to focus on areas in which the extensive scientific and technological resources of academe and industry may lead to the fastest practical results. In order of priority, these areas are
-
in situ studies of catalytic reactions;
-
characterization of catalytic sites (of actual catalysts) at atomic resolution (metals, oxides);
-
synthesis of new materials that might serve as catalysts or catalyst supports; and
-
theoretical modeling linked to experimental verification.
The development of new characterization tools, particularly in spectroscopy, has been mainly the province of academic research, and thus is expected to continue because industry is finding it increasingly difficult to justify the costs associated with technique development. To maintain the present worldwide leadership of the United States in both the science of catalysis and catalyst technology, it is essential to provide additional support for academic research focused on items 1-4 above. Furthermore, additional steps must be taken to facilitate interaction and, in fact, cooperation between industry, dealing with proprietary catalysts, and academe, developing advanced characterization tools and theory for catalysis.






















