
3
Hematologic Cancer
A steadily increasing number of clinically useful genetic alterations have been identified in various forms of cancer. The following portion of this report will discuss many of these alterations. Understanding of these changes is generally more advanced in hematologic cancers relative to solid tumors. Furthermore, solid tumors represent a much broader category of disease in which research has tended to focus on genetic alterations that appear in several different types of tumors. For these reasons, discussion of genetic changes in hematologic cancers is addressed on a disease-by-disease basis, whereas discussion of solid tumors is organized around certain general major issues in the genetics of solid tumors and a number of key alterations found among selected solid neoplasms. Table 2–1 provides an overview of the utility of a number of techniques in genetic testing of both hematologic cancers and solid tumors.
Neoplasms of blood-forming tissues are among the most easy to study, because tumor cells are often readily accessible in peripheral blood and are relatively easy to work with in cell culture systems. For these reasons, the genetic basis for a number of these diseases is now understood in considerable detail (Tables 3–1 and 3–2).
Chronic Myelogenous Leukemia
Chronic myelogenous leukemia (CML) is a clonal myeloproliferative disorder that affects all hematopoietic cell types. Clinically, the disease usually
Table 3-1 Nonrandom Chromosomal Abnormalities in Malignant Myeloid Diseases
|
Diseasea |
Chromosomal Abnormality |
|
CML |
t(9;22)(q34;q11) |
|
CML blast phase |
t(9;22)(q34;q11) with +8, +Ph1, +19, or i(17q) |
|
AML-M2 |
t(8;21)(q22;q22) |
|
APL-M3, M3V |
t(15;17)(q22;q11–12) |
|
AMMoL-M4Eo |
inv(16)(p13q22) or t(16;16)(p13;q22) |
|
AMMol-M4 |
Translocations or deletions of 11q23 |
|
AMoL-M5 |
t(9; 11) (p22;q23), t(11q13 or q23) |
|
AML |
+8 |
|
|
-7 or del(7q) |
|
|
-5 or del(5q) |
|
|
del(20q) |
|
|
t(3;3)(q21;q26) or inv(3)(q21q26) |
|
|
t(6;9)(p23;q34) |
|
Therapy-related AML |
-7 or del(7q) and/or -5 or del(5q) |
|
|
der(I)t(1;7)(p11;p11) |
|
|
t(9;11)(p22;q23) |
|
a CML, chronic myelogenous leukemia; AML, acute myeloblastic leukemia; AML-M2, AML with maturation; AMMoL, acute myelomonocytic leukemia; AMMoL-M4Eo, acute myelomonocytic leukemia with abnormal eosinophils; AMoL, acute monoblastic leukemia; APL-M3, M3V, hypergranular (M3) and microgranular (M3V) acute promyelocytic leukemia. |
|
follows a well-defined course. Most patients present with a high white blood cell count, mild constitutional complaints, and splenomegaly. Many are asymptomatic at this stage of their disease, which is referred to as the chronic phase. Inevitably and tragically, after a period of months to years, most untreated patients with CML develop blast crisis, a phase that resembles acute leukemia. In some patients, blast crisis is presaged by a prodromal phase of disease acceleration. The only curative therapy for CML is bone marrow transplant, which has a good outcome when performed during the stable phase, but a poor outcome in patients in blast crisis.
Table 3-2 Cytogenetic-Immunophenotypic-Genomic Correlations in Malignant Lymphoid Diseases
|
Phenotype |
Rearrangement |
Involved Genes |
|
|
Acute lymphoblastic leukemia |
|||
|
Pre-B |
t(1;19)(q23;p13) |
PBX |
E2A |
|
B(SIg+) |
t(8;14)(q24;q32) |
MYC |
IGH |
|
|
t(2; 8)(p11–12;q24) |
IGK |
MYC |
|
|
t(8;22)(q24;q11) |
MYC |
IGL |
|
|
t(5;14)(q31;q32) |
IL3 |
IGH |
|
|
dic(9;12)(p11;p12) |
|
|
|
B or B-myeloid |
t(9;22)(q34;q11) |
ABL |
BCR |
|
|
t(4;11)(q21;q23) |
|
MLL |
|
Other |
hyperdiploidy |
|
|
|
|
(50–60 chromosomes) |
|
|
|
|
del(9p),t(9p) |
IFNA/B |
|
|
|
del(12p),t(12p) |
|
|
|
|
t(8;17)(q24;q22) |
MYC |
BCL3a |
|
T |
t(11;14)(p15;q11) |
RBTN1 |
TCRD |
|
|
t(11;14)(p13;q11) |
TCL2b |
TCRD |
|
|
t(8;14)(q24;q11) |
MYC |
TCRA |
|
|
inv(14)(q11q32.3) |
IGH |
TCRA |
|
|
inv(14)(q11q32.1)/ |
TCL1 |
TCRA |
|
|
t(14;14)(q11;q32) |
|
|
|
|
t(10;14)(q24;q11) |
TCL3a |
TCRD |
|
|
t(1;14)(p32;q11) |
TCL5 |
TCRD |
|
|
t(7;9)(q35;q32) |
TCRB |
SUP-T3c |
|
|
t(7;9)(q35;q34) |
TCRB |
TAN1 |
|
|
t(7;7)(p15;q11) |
TCRG |
|
|
|
t(7;14)(p15;q11) |
TCRG |
|
|
|
t(7;14)(q35;q11) |
|
|
|
|
t(7;19)(q35;p13) |
TCRB |
LYL1 |
|
Non-Hodgkin's lymphoma |
|
|
|
|
B |
t(8;14)(q24;q32) |
MYC |
IGH |
|
|
t(2;8)(p11–12;q24) |
IGK |
MYC |
|
|
t(8;22)(q24;q11) |
MYC |
IGL |
|
|
t(14;18)(q32;q21) |
IGH |
BCL2 |
|
|
t(11;14)(q13;q32) |
BCL1 |
IGH |
|
T |
see T-cell ALL |
|
|
|
Chronic lymphocytic leukemia |
|
|
|
|
B |
t(11;14)(q13;q32) |
BCL1 |
IGH |
|
|
t(14;19)(q32;q13) |
IGH |
BCL3a |
|
|
t(2;14)(p13;q32) |
IGH |
|
|
|
t(18;22)(q21;q11) |
BCL2 |
IGL |
|
|
t(14q32) |
|
|
|
|
+12 |
|
|
In 1960, Nowell and Hungerford described the presence of an abnormal chromosome in blood cells of patients with CML (21). Named the Philadelphia chromosome (abbreviated Ph1) after the city of its discovery, this chromosome was subsequently shown by Rowley (22) with improved staining techniques to be the product of a translocation event involving the long arm of chromosome 9 (9q34) and the long arm of chromosome 22 (22q11), leading to the formation of a translocation [t(9;22)(q34;q11)] (Figure 1-1). Molecular analysis has localized the breakpoint on chromosome 9 to ABL (23), a gene previously identified in modified form in a mouse leukemia virus, and the breakpoint on chromosome 22 to BCR (24,25), a gene of unknown function (Figure 3-1) (26). The rearrangement results in a fusion gene on the Ph1 chromosome, which contains the 5´ portion of BCR and the 3´ portion of ABL, and encodes a unique, chimeric protein with a molecular weight of 210 kilodaltons (27,28). Experimental studies and the analysis of samples from patients have shown that the BCR sequence increases tyrosine kinase enzyme activity associated with ABL (28). Although the precise mechanism is unknown, it is clear that this unique form of ABL serves as a major factor in the transformation of hematopoietic cells (29). When analyzed at the molecular level, all patients with CML have a BCR-ABL rearrangement; consequently, molecular diagnosis of CML depends on demonstration of the chimeric gene in patients with a clinical picture consistent with CML.
CML presents many options with respect to genetically based approaches for diagnosis. The genetic defect can be detected at the level of the chromosomal translocation, the fusion gene, the chimeric RNA, or the altered structure or activity of the protein. Choices among available forms of tests involve considerations of reliability, cost, and convenience.
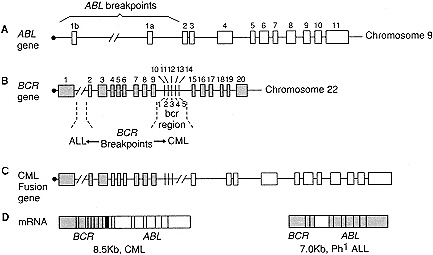
Figure 3-1 A.Normal structure of ABL gene on chromosome 9. Exons are represented by boxes. B. Normal breakpoint cluster region (BCR) gene structure on chromosome 22, showing the location of breakpoints that occur in chronic myelogenous leukemia (CML) within the bcr region and those that occur in Ph+ acute lymphocytic leukemia (ALL) between exons 1 and 2. C. A fusion gene is created as a result of ABL sequences being spliced to BCR sequences in the bcr region (CML). D. mRNA transcripts that are a result of the translocation in CML (left) and ALL (right). (Modified, from Carroll, W.L., and Schwartz, A.L. Molecular oncology. In Fernbach, D.J., and Vietti, T.J., eds., Clinical Pediatric Oncology, 5th ed. St. Louis: C.V. Mosby, in press. Used with permission.)
Cytogenetics will likely remain in the near future the standard method of diagnosis. About 95% of patients with CML have the Ph1, which is usually demonstrable in karyotypes prepared from peripheral blood cells. However, the remaining 5% of patients are Ph1-, having either more complex translocations involving multiple chromosomes that obscure the presence of the 9;22 translocation (30) or cryptic BCR-ABL rearrangements that are detectable only at the submicroscopic level (31). In these patients, other tests must be used.
Regardless of the karyotype, in most instances the breakpoints in BCR are clustered in a fairly small region. By using probes that span this region, most rearrangements involving BCR can be detected by Southern blot analysis. In one recent analysis of 68 patients with CML, use of two probes and several different restriction enzymes led to detection of BCR rearrangement in 61 of 61 Ph1+ cases and 3 of 7 Ph1-cases (32). In another series, 190 of 191 cases of Ph1+ CML (99.5%) and 12 of 27 cases of Ph1-CML (44.4%)
had BCR rearrangements by Southern blotting (33). Clearly, this type of analysis represents a generally suitable alternative to cytogenetics.
Rearrangements involving BCR can also be directly visualized in inter-phase cells by in situ hybridization using probes for ABL and BCR together (34), or by staining concomitantly with two BCR probes that flank the breakpoint region (Figure 2-1). In the former instance, one would look for co-localization of the BCR and ABL probes and, in the latter, the movement apart of the two probes. This method is not widely available at present, but likely will come into widespread use as kits for in situ hybridization become commercially available.
Conceivably, the chimeric p210 protein could also be detected, either by looking for a protein of 210 kilodaltons with tyrosine kinase activity or by using antibodies directed at the unique amino acid sequence generated by the BCR-ABL fusion (35). The assay for aberrant p210 kinase activity is possible but may be too cumbersome for widespread use, however, and it is unclear whether antibody reagents sufficiently specific for BCR-ABL can be produced.
Once CML has been diagnosed, the disease is typically well controlled initially with palliative chemotherapy and/or biologic therapy such as interferon. If the patient is a suitable candidate of an appropriate age, and if an available donor exists, bone marrow transplantation is the treatment of choice. The optimal timing of transplant, a procedure with 20–30% mortality, is, however, currently not settled. Patients may persist in chronic phase for years with a good quality of life, but then suddenly develop blast crisis, a phase of the disease with very poor outcome, even with transplantation. A test that would predict when blast crisis is likely to occur might therefore help to determine the timing of transplantation, while maximizing the interval of good-quality life in the chronic phase prior to transplantation.
Accelerated phase and blast crisis are often associated with the appearance of new karyotypic abnormalities, most commonly the addition of a second Ph1 chromosome, i(17q), or trisomy 8, all of which may be observed alone, in combination with one another, or together with other, less frequently acquired aberrations (36,37). Detection of these chromosomal markers either cytogenetically or with fluorescence in situ hybridization (FISH) is not likely to be sufficiently sensitive to be clinically useful, but supports the idea that disease progression is caused by acquisition of additional mutations. Correlation of i(17q) with disease progression is interesting, since an important tumor suppressor gene, TP53, maps to 17p13 and has been found to be mutated in a wide range of human tumors. This gene is usually found to be normal when CML cells from patients in stable phase are studied. In contrast, 20–25% of cases of blast crisis are associated with mutations in TP53, implying a role for such mutations in a subset of blast crises (38,39).
At present, there is no evidence to suggest that detection of TP53 ab-
normalities will be useful in predicting blast crisis, possibly because the lead time is too short. Several other molecular markers have recently been proposed as potential predictors of disease acceleration. Some studies have found an association between the relative position of breakpoints within the BCR breakpoint cluster region and the time to progression of CML (40,41). Other workers have failed to detect this relationship (42,43). Its role in patient management therefore remains uncertain.
Hypermethylation of cytosine residues within the calcitonin gene has also been reported to correlate with disease status (44,45). In one study following 27 patients with stable phase CML, 6 of the 8 patients who progressed demonstrated hypermethylation prior to transformation, with a median lead time of six months before the appearance of clinical disease acceleration (45). This approach shows promise, but it does not appear to be a 100% sensitive predictor of disease progression and needs further evaluation. Nonetheless, these studies give credence to the idea that molecular tests will permit detection of impending disease progression while the patient is in stable phase.
Following bone marrow transplant, a substantial fraction of patients relapse, often several years after the procedure. It is estimated that at the time of bone marrow transplant, most patients in chronic phase CML have a tumor burden of about 1012 cells. Cytogenetics can be used to detect persistence of the Ph1 at a frequency of 1 in 1000 cells (at best). Therefore, a patient in cytogenetic remission could have a tumor burden of as much as 109 cells. More sensitive tests to detect relapse at an earlier stage can identify patients who need additional therapy.
Polymerase chain reaction (PCR) directed at the DNA of the Ph1 breakpoint is not a diagnostic option because of the variability in the positions of the breaks in the BCR and ABL genes. In contrast, the structure of the BCR-ABL messenger RNA is conserved in almost all cases, so that PCR of cDNA synthesized from the chimeric messenger RNA represents one such highly sensitive test (46). In principle, PCR analysis should be capable of detecting 1 tumor cell in 105 or 106 normal cells and is thus 100-to 1000-fold more sensitive than cytogenetic analysis. Scattered initial reports suggest that PCR analysis may indeed be able to detect patients destined to relapse following transplant, sometimes years before clinical disease becomes evident (47–49), but it is too early to determine the value of PCR in this setting (50). PCR has also played a role in evaluating patient response to new forms of therapy, such as alpha interferon (51,52). Finally, PCR amplification of RNA has recently been utilized to define BCR-ABL breakpoints in leukemic cells of CML patients (53). Synthesis of antisense oligodeoxynucleotides complementary to the breakpoints led to suppression of leukemia cell colonies in culture. The ramifications of this work are impressive; the potential exists for in vitro or in vivo modulation of BCR-ABL expression in cells of CML patients through the use of antisense technology.
Chronic Lymphocytic Leukemia
Chronic lymphocytic leukemia (CLL) is a neoplasm characterized by proliferation of mature-appearing lymphocytes in bone marrow, lymph nodes, spleen, and liver. It is typically an indolent disease of older individuals. Treatment is palliative, being aimed at alleviation of symptoms. However, CLL occasionally occurs in younger individuals and in some cases pursues a more aggressive course. Also, in 5–10% of cases it terminates in conversion to an aggressive large cell lymphoma, an event known as Richter's syndrome (54–56), or less frequently into prolymphocytic leukemia.
Early identification of individuals with aggressive disease, particularly younger individuals or individuals undergoing Richter's transformation, would identify a high-risk population requiring more aggressive therapy. Recent evidence increasingly suggests that trisomy 12 is an independent predictor of poor outcome (57,58). Because of the low proliferation rate characteristic of this tumor, it is frequently difficult to perform adequate karyotypic analysis. Approximately 35% of cases have only normal karyotypes and 20% are inadequate (58). Anastasi et al. (59) reexamined forty cases of CLL, previously studied by standard cytogenetics (60), with FISH, using a probe for the centromere of chromosome 12 (Plate 2). The new analysis revealed that twelve patients had trisomy 12, only seven of whom had been identified by using conventional cytogenetics. Of the five new cases detected with FISH, two had previously unidentified chromosomal abnormalities, one was thought to be normal, and two were inadequate. Similar results on 15 patients were reported recently (61). These studies suggest that in situ hybridization will become the method of choice for detection of trisomy 12 and other cytogenetic abnormalities in CLL.
Acute Lymphocytic Leukemia
Acute lymphocytic leukemia (ALL) is an aggressive neoplasm composed of immature lymphoid cells that overwhelm the bone marrow, peripheral blood, lymphoid tissues, and sometimes the viscera. It occurs throughout life, but is most common in childhood. Particularly in children, treatment of this disease represents one of the triumphs of oncology. Although once always fatal, up to 60% of the children affected are now long-term survivors. Prognosis is dependent upon clinical characteristics, such as patient age, peripheral white blood cell count, the immunophenotype of the tumor cells, and the presence of certain tumor-specific genetic lesions. Each of these prognostic factors tends to correlate with the others to varying degrees.
Several nonrandom chromosomal abnormalities occur in ALL. The Ph1 chromosome is the most frequent rearrangement, comprising at least 15–20% of adult ALL and 3–6% of childhood ALL (62) (Figure 3-1). While
the breakpoints appear identical to those in CML at the cytogenetic level, fine mapping has shown that in about 50% of cases, breakpoints within BCR lie within a different region, resulting in production of a 185-kilodalton chimeric BCR-ABL protein, slightly smaller than that seen in CML (63). This rearrangement is seen very rarely in CML (64) and is associated with an early B cell (pre-B cell) phenotype in most cases. A Southern blot method for detection of this second type of BCR-ABL rearrangement has not yet been fully developed. Pulsed-field gel electrophoresis (PFGE) has the capacity to detect all BCR-ABL rearrangements (65) but, for reasons already discussed, is unlikely to be of general clinical utility. Other alternatives to cytogenetics that are currently available but not yet established include FISH as well as PCR directed at chimeric mRNA (34,46).
Five to ten percent of patients with ALL have a t(4;11)(q21;q23) rearrangement. Of note, among children, almost all patients with this abnormality are less than one year of age and have very high blast counts, both indicators of a poor prognosis. The tumor cells are usually CD19 positive and CD10 (CALLA) negative, consistent with a very early B cell phenotype, but in culture the tumor cells can be induced to differentiate into a monocyte-like cell (66). Several laboratories are close to identification of the breakpoints, but the involved genes remain unknown at present. Detection relies on cytogenetics, with FISH soon to be introduced.
About 25% of patients with pre-B cell ALL have the chromosomal abnormality t(1;19)(q21;p13). Like the Ph1 chromosome, this rearrangement results in a fusion protein composed of portions of the E2A gene on chromosome 19 and the PBX gene on chromosome 1 (67,68). Both of these genes appear to be able to function as transcription factors, and it is presumed that the aberrant fusion protein leads to oncogenesis by altering gene expression in the pre-B cell. Identification of breakpoint sequences will permit detection by PCR, Southern blotting (69), and FISH (70).
B cell ALL is classically associated with the presence of lymphoblasts that have moderately abundant, vacuolated, royal-blue cytoplasm when stained with Wright's stain, a morphology called L3 ALL in the FAB classification. This type of ALL closely resembles Burkitt's lymphoma in terms of its appearance and immunophenotype and, like Burkitt's lymphoma, is typically associated with translocations involving the oncogene MYC located on chromosome 8 (71) and the expression of surface immunoglobulin. Only about 40% of cases with known translocations involving MYC are classified as L3 ALL using strict FAB criteria (greater than 50% vacuolated lymphoblasts), however, with most other cases being classified as L2 ALL. Conversely, cells resembling L3 blasts can be seen in cases of ALL lacking MYC translocation and, more rarely, in non-lymphoid neoplasms (72,73). Therefore, morphology alone is not a completely reliable predictor of the presence of MYC translocations in ALL.
MYC appears to function through binding to DNA as a transcription factor and is assumed to lead to transformation by altering the expression of other genes. In about 75% of cases, MYC is joined to DNA within the immunoglobulin heavy chain locus on chromosome 14, and in the remaining cases, to DNA within the kappa immunoglobulin light chain locus on chromosome 2 or the lambda immunoglobulin light chain locus on chromosome 22 (74). The breakpoints in MYC are variable, lying either 5' of the gene or within a 5' noncoding portion of the gene when juxtaposed to heavy chain locus sequences, and 3' of the gene when the kappa or lambda genes are involved. Regardless of the location of the breakpoint, the net effect appears to be deregulation of normal control of MYC expression. The breakpoints within the immunoglobulin locus are also heterogeneous, occurring at a variety of widespread positions within the gene. Because of the variation in location of the breaks within MYC and the immunoglobulin genes, detection by Southern blotting or PCR, while possible, is somewhat complex and has not been widely used. FISH or PFGE may represent the best alternative approach.
Although fewer leukemias of T cell origin have been studied, a distinct pattern of nonrandom karyotypic abnormalities is emerging. Analogous to the rearrangements involving the immunoglobulin loci in B cell ALL, rearrangements in T cell ALL frequently involve the T cell receptor loci, most commonly the α-δ T cell receptor locus located at band 14q11 and the β T cell receptor locus at 7q35. Approximately 25% of patients with T cell ALL have rearrangements with a gene called TCL5 (alternative names for the gene are tal-1 and SCL) on chromosome 1 (75). The structure of this gene is consistent with function as a transcription factor. In some cases, TCL5 rearranges with the δ T cell receptor locus on chromosome 14. The breakpoints are readily detected by Southern blot analysis and may be homogeneous enough for consistent detection by PCR as well. FISH should also work well. In a significant subset of cases, however, TCL5 rearrangements are associated with a normal karyotype. In these cases, TCL5 has fused with a closely linked gene on chromosome I called SIL (75,76). The resultant interstitial deletion is too small to be seen cytogenetically, but can be demonstrated consistently by using the Southern blot technique. Because of the very tight clustering of breakpoints in both genes, this rearrangement should also be readily detectable by PCR.
In addition to identifying genes of probable importance in normal and abnormal lymphocytic differentiation, detection of chromosomal abnormalities in ALL is of prognostic importance. It was rigorously demonstrated for the first time at the Third International Workshop on Chromosomes and Leukemia (77) that the karyotype, especially in children, is an important independent prognostic factor in ALL. The survival data were updated at the Sixth Workshop (78); among children, the highest percentage of pa-
tients with a 15-year disease-free survival were those whose tumors contained more than 50 chromosomes (71%), contained 47–50 chromosomes (61%), or showed a normal karyotype (58%). Tumors associated with less than a 15% chance of cure included those with t(4;11), abnormalities of 8q24 or 14q+, or a Ph1 chromosome.
In contrast to childhood ALL, treatment of adults with ALL is generally less successful, perhaps in part because of the relatively high incidence of the Ph1 translocation in this patient population. Patients with translocations involving 8q24 rarely go into remission with standard ALL chemotherapy, but have about a 40% long-term remission rate with Burkitt's lymphoma-type therapy. Most other adult patients, including those with the Ph1, frequently achieve remission, but have poor long-term survival with standard therapy and may be candidates for bone marrow transplant in first remission. Only patients with translocations involving 14q11, a finding that usually correlates with a T cell immunophenotype, have long-term remission rates of greater than 50% and appear to do well with more standard forms of ALL therapy (79).
As therapy improves, the importance of various cytogenetic abnormalities in ALL (as well as other forms of cancer) may change or diminish, and in fact there is evidence that this is already occurring. For example, a recent series of papers from a single center suggests that with more intensive chemotherapy, all patients with childhood ALL fall into a single good prognosis group, regardless of ploidy or specific karyotypic abnormalities, except for those patients with the Ph1 chromosome, who have very poor outcomes and clearly require other forms of therapy (80,81).
Ultimately, it is possible that FISH, performed with mixtures of multiple centfomeric probes, may serve as a useful alternative approach to evaluating chromosomal abnormalities in these patients, particularly because ALL cells may be difficult to karyotype. Even in experienced cytogenetic laboratories, 10–40% of cases of ALL yield normal karyotypes or metaphase cells that are of inadequate quality for evaluation.
As in CML, detection of minimal residual disease by PCR directed at tumor-specific nucleotide sequences has great promise for guiding therapy in ALL (82–84). It is likely to be of use in two settings. While frequently effective, therapy in ALL is currently given for two or more years following diagnosis and is associated with significant side effects. It is unclear at present how much therapy is sufficient in individual patients. It is possible that sensitive techniques of tumor detection would permit more accurate assessment of residual disease following treatment or of the kinetics of response during therapy, thereby allowing some patients to receive shorter courses and perhaps identifying patients for whom early bone marrow transplant is indicated. In those patients who do receive transplants, detection of residual disease post-transplant may identify a patient population that re-
quires further treatment. In some cases, detection will assay sequences resulting from chromosomal translocations; in other cases, the target will be unique sequences generated by antigen receptor gene rearrangements, as described in the subsequent discussion of lymphomas.
Acute Myelogenous Leukemia
Like ALL, acute myelogenous leukemia (AML) is associated with a heterogeneous collection of specific chromosomal abnormalities, several of which have now been shown to be of clinical importance. Unlike ALL, many of these chromosomal abnormalities tend to correlate well with tumor phenotype. Subclassification of AML based on standard morphologic criteria can, however, be difficult, and confirmation of AML subtype by genetic testing is likely to aid assignment of prognosis and therapies in many cases.
A t(8;21)(q22;q22) was first identified in AML in 1973 (85). This is the most frequent abnormality in AML in children and occurs in approximately 7–10% of patients with AML in the United States. However, its frequency varies, and it is reported to be particularly common in Japan (37.5%) and in South Africa (62.5%) (86). In more than 90% of patients, it is associated with an M2 phenotype (acute myeloblastic leukemia with maturation). The translocation breakpoint on chromosome 21 has recently been cloned (87), which should permit detection by non-cytogenetic means.
Acute promyelocytic leukemia (APL, M3 AML) is associated with a unique translocation [t(15;17)(q21;q11)] (88). Leukemic cells are frequently difficult to grow in culture, but in experienced laboratories, all patients with APL are seen to possess this translocation. The translocation causes the fusion of a previously uncharacterized gene called MYL on chromosome 15 and the gene for the alpha retinoic acid receptor (RARA) on chromosome 17 (89-91); it is believed to be responsible for a unique feature of APL. This is the only form of leukemia that responds in virtually 100% of cases to therapy with all trans-retinoic acid (RA) (92,93). Therapy with RA is probably not curative, and most patients will relapse eventually. Nonetheless, RA holds great promise as an induction therapy, particularly in patients with life-threatening disseminated intravascular coagulation, an important complication of APL.
While the light microscopic appearance of APL is highly characteristic, it is not an entirely reliable predictor of the presence of t(15;17). In one recent series, one case of morphologically typical APL was found to have a normal karyotype and a normal RARA transcript. Of note, this patient was refractory to RA therapy (93). Moreover, the microgranular variant of APL, which is also associated with t(15;17), may be difficult to diagnose by light microscopy (94).
Since the presence of t(15;17) or, more precisely, a RARA-MYL fusion
gene, appears to correlate with response to RA, its detection is clearly valuable. As noted, cytogenetic detection can be difficult. Biondi et al. (95) have recently demonstrated detection of the t(15;17) by Southern blotting using a combination of probes directed against RARA and MYL in 26 out of 26 cases of APL, and this may become the technique of choice for detecting this rearrangement. In situ hybridization may also prove useful, as well as PCR directed against the chimeric transcript.
Acute monoblastic leukemia (M5) is closely associated with abnormalities involving the long arm of chromosome 11, particularly in children (96). The other chromosome involved in the translocation is variable, with chromosome 9 being most common, and chromosomes 10, 17, and 19 less common. The breakpoint in 11q23 is cytogenetically identical to that seen in ALL associated with t(4;11), suggesting the presence of an important gene that may affect differentiation along myeloid and lymphoid lines. Detection is primarily cytogenetic, but FISH and other approaches will soon be available.
Several other translocations are less commonly associated with other specific morphologies in AML. Inversion of chromosome 16, inv(16)(p13q22), or a translocation involving the two chromosome 16 homologues, t(16;16)(p13;q22), is consistently seen in acute myelomonocytic leukemia with abnormal eosinophils (M4Eo) (97). The tumor is characterized by differentiation along myelocytic and monocytic lines and by the presence of unique eosinophils with large and irregular basophilic granules. Most patients also have an increase in the absolute number of eosinophils in the marrow. The genes involved have not yet been identified.
A translocation involving chromosomes 6 and 9, t(6;9)(p23;q34), is associated with an increase in normal-appearing basophils in the bone marrow (62). This translocation breakpoint has very recently been cloned, which will allow non-cytogenetic diagnosis (98).
Uncommonly, patients with AML present with normal or elevated platelet counts. A high percentage of these patients have an inversion, inv(3)(q21q26.2), or translocation, t(3;3)(q21;q26.2), involving chromosome 3 (99). The genes involved are unknown.
Other abnormalities not associated with a specific tumor phenotype include trisomy 8, monosomy 7, monosomy 5, and deletions of 5q. These abnormalities are more common in patients who have worked in occupations that expose them to potentially mutagenic agents and are also frequently observed in patients who develop AML after cytotoxic chemotherapy. In general, specific translocations tend to be seen much less frequently in this group of patients than in those patients who develop disease in the absence of predisposing factors. Exceptions to this generalization are translocations involving chromosome 11 band q23, which appear to be common in patients who have received high doses of epipodophyllotoxins, especially the drugs teniposide (VM26) and etoposide (VP16) (100).
At present, the most important practical benefit of cytogenetic analysis in AML is in helping to determine prognosis. Classification of karyotypic abnormalities into a favorable group, including t(8;21), inv(16) or t(16;16), and t(15;17), or an unfavorable group, including inv(3) or t(3;3), —7 or del(7q), and —5 or del(5q), has been reported by several investigators (62, 101). More than 100 recurrent chromosomal abnormalities have now been reported in AML, and many other abnormalities will likely also prove to have prognostic importance as additional data accrue. More accurate determination of the prognosis for remission and long-term disease-free survival will, in turn, probably have a major impact on therapeutic recommendations. In the case of APL, patient response to RA therapy appears to be correlated with the presence of the t(15;17). Finally, in principle, the unique DNA sequences generated by each of the translocations specific for AML are potential targets for approaches aimed at detection of minimal residual disease.
Myelodysplastic Syndromes
Myelodysplastic syndromes are a diverse collection of clonal disorders that usually occur in older populations and are characterized by cytopenias; peculiar morphologic abnormalities in myeloid, erythroid, and megakaryocytic precursors; and increased risk of AML. They may appear sporadically or following exposure to environmental mutagens or chemotherapeutic agents, and are associated with the same set of cytogenetic defects seen in individuals with post-therapy AML, most commonly monosomy 5, del(5q), monosomy 7, del(7q), and trisomy 8, as well as del(20q). In many cases, the chromosomal abnormalities are complex.
Cytogenetic studies are of some use in two circumstances. Occasional patients with myelodysplasia have relatively little morphologic dysplasia. Demonstration of a clonal cytogenetic abnormality can help to establish the diagnosis in such patients. Also, in general, the more complex the karyotype, the greater is the risk of transformation to acute leukemia (102). Acquisition of additional abnormalities over time is also an ominous sign.
Several proto-oncogenes have been implicated in the pathogenesis of myelodysplasia. Up to 30-40% of cases are associated with mutations in NRAS (103-106), a gene encoding a guanosine triphosphate (GTP)-binding protein, which likely plays a role in modulating cell response to external stimuli produced through the binding of growth factors or hormones to specific membrane-bound receptors. Interestingly, up to 13% of asymptomatic individuals who have received chemotherapy for non-Hodgkin's lymphoma are found to have detectable RAS mutations when their peripheral blood leukocytes are studied by using sensitive PCR-based assays (107), suggesting that RAS mutations may represent an early event in the patho-
genesis of therapy-related myelodysplasia. It remains to be seen if these types of studies can predict which mutagen-exposed individuals will eventually develop myelodysplasia.
Mutations in the gene FMS, which encodes the receptor for a hematopoietic growth factor, M-CSF, have also been reported in a subset of patients with myelodysplasia, particularly those with peripheral monocytosis (108). These types of studies have yet to have a significant impact on management of patients, but it is hoped that defining the genetic basis of myelodysplasia will lead to new approaches to treatment.
Lymphoma
Non-Hodgkin's lymphomas, almost all of which are composed of malignant lymphocytes, are a diverse group of disorders that often present diagnostic difficulties to the pathologist. Because of this, molecular approaches for differentiation of lymphoma from benign disorders have been widely applied. These approaches are based on unique aspects of lymphocyte biology.
A fundamental feature of the normal immune system is the ability of lymphocytes to recognize and respond to a broad array of antigens. This is accomplished through antigen receptor proteins expressed on the cell surface—immunoglobulins on the surface of B cells, and T cell receptors on the surface of T cells. Although only a few genes exist for these proteins, literally millions of different receptor molecules are produced. This remarkable feat is made possible through gene rearrangement. All antigen receptor genes are composed of three sets of unique and discontinuous gene segments known as V, J, and C segments, and in some genes an additional set, D. During lymphocytic differentiation, single members from each of the V, D, and J sets are joined together by DNA recombination to produce a potentially functional gene (Figure 3-3). DNA located between recombining segments is deleted and lost. In addition, at V-D, D-J, and V-J junctions, the removal of small amounts of DNA and the addition of short random sequences occur. Both recombinational and junctional diversity contribute to the overall diversity of antigen receptors.
In any particular lymphocyte, the configuration of DNA in a rearranged antigen receptor locus appears, for the most part, to be fixed throughout the life of the lymphocyte and its progeny; this is also true of neoplasms derived from transformed lymphocytes. The result is a situation akin to that previously discussed for chromosomal translocations, namely, the creation of unique DNA sequences that can be used as a marker for a clone of cells. Since these sequences are created by DNA changes that occur at submicroscopic levels, techniques other than cytogenetic analysis must be used for detection.
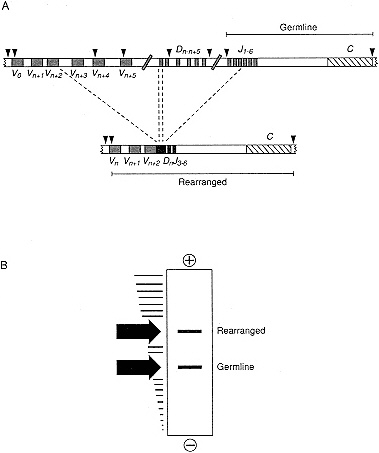
Figure 3-3 A. Diagram of hypothetical rearrangement in the immunoglobulin heavy chain gene. The relative positions of germline V,D,J, and C region segments are shown in the top line; the configuration of V,D,J, and C segments resulting from a rearrangement is shown in the bottom line. Arrowheads indicate the locations of restriction enzyme cleavage sites. Brackets indicate the length of the restriction fragments spanning the J segments before and after gene rearrangement. B. Bands produced in a Southern blot hybridization analysis of tissue containing a clonal rearrangement of an antigen receptor gene, such as that illustrated in A. The rectangle represents an autoradiogram of an agarose gel in which restriction fragments produced from tissue DNA have been electrophoresed from top (anode) to bottom (cathode). To the left of the autoradiogram are shown the positions in the gel from which the autoradiogram was generated of variously sized antigen receptor DNA fragments after electrophoresis. The hybridization probe used to produce the autoradiogram could be derived from any DNA between J6 and the next restriction to the right.
In practice, differentiation of benign and malignant lymphocytic proliferations frequently rests upon analysis of clonality. For many B cell proliferations, this is performed at the level of the antigen receptor protein rather than the gene. Most reactive and neoplastic B lymphocytes express surface or cytoplasmic immunoglobulin, a molecule composed of heavy chains and either kappa or lambda light chains. Reactive proliferations contain a roughly 2:1 ratio of cells expressing kappa or lambda light chain, respectively. In contrast, clonal proliferations, being derived from a single lymphocytic precursor, express only a single light chain type. Antibodies that recognize only lambda or kappa light chains can therefore be used to differentiate polyclonal and monoclonal (neoplastic) proliferations (109). Staining for light chain can be performed in tissue sections or on cells in suspension and represents a valuable method for analyzing the clonality of B cell proliferations. These kinds of studies are now routinely performed in most pathology departments. The only limitation is that they are best performed on unfixed tissue.
This approach, however, is not applicable to all lymphomas. In some cases, B cell lymphomas fail to express immunoglobulin or involve only part of the tissue and are obscured by reactive lymphocytes. While immunophenotyping may be quite helpful in the assessment of T cell neoplasms (15), specific antibody reagents are not available for directly evaluating the clonality of T cell proliferations. In these cases, techniques aimed at studying antigen receptor genes must be used in order to prove clonality (16).
The method most commonly employed for assessing the issue of clonality in such cases is Southern blot analysis using probes specific for antigen receptor genes (110). Polyclonal proliferations contain many different antigen receptor gene rearrangements, each with its own configuration of rearranged segments. The positions of restriction sites in and around these segments vary with the configuration of segments within a rearranged gene, and DNA fragments for any particular antigen receptor gene will be of many different sizes. Too few rearranged fragments comigrate in the gel under these circumstances to produce a signal on a blot. Proliferations containing clonal lymphocytes bearing uniform rearrangements, in contrast, typically yield a dominant fragment of a different size from that of germline, unrearranged DNA and result in an additional distinct band or set of bands. The method is sensitive enough to detect malignant cells present at a frequency of 1–5%. As with all forms of Southern blot analysis, unfixed tissue or tissue fixed in ethanol is required, but this technology is now widely regarded as an important adjunct to the morphologic and immunohistochemical diagnosis of lymphoma.
It is important to recognize that not all clonal lymphocytic proliferations behave in a frankly malignant fashion. For example, B cell proliferations involving the salivary gland of patients with Sjögren's syndrome (111)
and cutaneous T cell infiltrates in the disorder lymphomatoid papulosis (112) are typically clonal by Southern blotting. While a fraction of these patients subsequently develop aggressive lymphomas, most do not and require no treatment for their lymphoproliferative disorder. Therefore, the significance of clonality can currently be interpreted only in light of the morphologic, immunophenotypic, and clinical data available in a given case.
Several specific chromosomal translocations have been described in malignant lymphoma. Most, but not all, involve breakpoints that occur within an antigen receptor gene, leading to the suggestion that these rearrangements result from mistakes made during the process of antigen receptor gene rearrangement.
Small non-cleaved cell lymphoma, Burkitt's-type, like B cell ALL, is associated with rearrangement of the MYC gene and one of the three immunoglobulin loci (113). The resultant t(8;14), t(2;8), or t(8;22) translocations are identical to those seen in B cell (L3) ALL at the cytogenetic level. In Burkitt's lymphoma in Africa and in lymphomas associated with AIDS, the MYC gene is usually translocated to a JH segment (74,114,115), whereas rearrangements involving an immunoglobulin switch region are typically seen in sporadic cases of non-African Burkitt's lymphoma (74,115). As in B cell ALL, due to the variability in the position of the breakpoints in MYC and in the immunoglobulin genes, detection of the rearrangement by Southern blotting and PCR is problematic, and cytogenetic analysis is currently the only reliable way of detecting these translocations. In addition to translocation, structural mutations have been found within regulatory sequences of the MYC gene. In a recent series of Burkitt's lymphomas, 20 of 20 tumors were found to contain mutations in specific 5' regulatory sequences. It is believed that these contribute to deregulation of MYC expression (116,117). These altered sequences also represent potential targets for molecular approaches to diagnosis and monitoring of disease status.
Follicular lymphoma, the most common lymphoma in the United States, is an indolent neoplasm in which a large percentage of cases contain a translocation, t(14;18)(q32;q21) (118), involving the immunoglobulin heavy chain locus on chromosome 14 and a gene on chromosome 18 called BCL2 (119–121). BCL2 appears to be a unique kind of proto-oncogene. It is a mitochondrial protein that may play a role in abrogating the programmed cell death of B lymphocytes within follicular centers (122), and also of T cells in the thymus and epithelial cells throughout the body (123). Cells with a t(14;18) express structurally normal BCL2 protein at high levels, and thereby are thought to gain a survival advantage.
Rearrangements involving BCL2 occur within the 3', noncoding region of the gene within two clusters, a major breakpoint region (MBR) in 65–70% of cases (120,121), and a minor cluster region (MCR) in 20–25% of cases (124). Uncommonly, rearrangements may involve the 5' untranslated
region of the gene (125). Detection is possible by cytogenetics, FISH, or PCR (126,127). It is also possible to look for increased expression of BCL2 protein by immunohistochernistry. Normally, BCL2 is highly expressed by mantle zone cells, but weakly or not at all by follicular center cells (128). The malignant follicles of follicular lymphoma, however, show a high level of BCL2 expression in most cases, and stains for BCL2 may thus have some role in distinguishing follicular lymphoma from reactive follicular hyperplasias (128–130). Moderate to high level expression of BCL2 is also seen in a variety of other lymphoproliferative disorders, including some that have not been associated with t(14;18) (130). This implies that other mechanisms besides t(14;18) may lead to altered BCL2 expression, and suggests that deregulation of BCL2 may prove to be of general importance in hematopoietic malignancies, and possibly in solid tumors as well. Because of this nonspecificity, immunohistochemistry will probably have a limited role in evaluating the presence of this type of translocation.
Detection of t(14;18) by PCR may have a role in monitoring response to therapy (131). Examples include detection of minimal residual disease after bone marrow transplant and evaluation of ex vivo purging of marrow prior to autologous transplant. However, applications of PCR for these purposes may have to take into account recent reports that small numbers of t(14;18) translocations have been detected in benign hyperplastic lymphoid tissues (132). In addition, validation of data gathered by using a PCR approach will be slow, because follicular lymphoma is a very indolent disease and long-term follow-up will be needed.
Lymphocytic lymphoma of intermediate differentiation, sometimes referred to as centrocytic lymphoma or mantle zone lymphoma, is a relatively rare form of non-Hodgkin's B lineage lymphoma (5—10%) that is frequently associated with moderate levels of circulating neoplastic lymphocytes and probably most often follows a chronic course (133). Cytogenetic analyses have shown some cases of this lymphoma to contain a specific chromosomal translocation, t(11;14)(q13;q32) (134). Like many other translocations in B cell tumors, the breakpoint in chromosome 14 occurs within the immunoglobulin heavy chain gene. By analogy to other translocations, a gene termed BCL1 has been hypothesized to lie near the chromosome 11 breakpoint, but this gene has not been well characterized (135). Using DNA probes derived from the region of the chromosome 11 breakpoint, a recent study has described an incidence of at least 50% rearrangement of such DNA in this lymphoma (136).
Many other translocations have been described in a few cases of lymphoma. As a rule of thumb, those cases with B cell phenotype tend to have a rearrangement that involves one of the immunoglobulin genes, and those with T cell phenotype have rearrangements involving T cell receptor genes. These translocations will be important in understanding the various mecha-
nisms of lymphomagenesis, but have limited clinical importance at present because they are individually infrequent.
To summarize, while the majority of specific translocations within lymphomas are of limited diagnostic use at this time, the t(14;18) (119–121), and possibly the mutations involving MYC, have the potential to serve as very useful markers for response to therapy and detection of minimal residual disease. Some data also suggest that the presence of additional karyotypic abnormalities, particularly structural changes involving chromosome 17, identify patients with follicular lymphoma who have a poor prognosis (118,137,138). More data are needed to corroborate this finding.
While unique sequences generated by translocation events can be evaluated in only a subset of patients with lymphoma, unique junctional sequences created by antigen gene rearrangements serve as potential tumor-specific markers in the vast majority of lymphomas and also in most cases of ALL and CLL. These techniques, which include PCR with junction-specific primers (83) and PCR coupled to sensitive ribonuclease (RNase) protection assays (84), may play an important role in monitoring the response of lymphoproliferative disorders to therapy in the future. At the present time, application of this approach is restricted primarily to research laboratories.




















