
4
Solid Tumors
Advances in understanding the genetic changes associated with solid tumors have come more slowly than with hematopoietic tumors, in large part because of difficulties in inducing the neoplastic cells of many tumors to divide in vitro. Moreover, when karyotypes are obtained, they are often found to be of inferior quality. Of 14,000 neoplasms now entered in the cytogenetic data base, only 21% are solid tumors, although these are the most common tumors of man (139). Solid tumor cytogenetics is still in its infancy, but several areas of current and future clinical utility are becoming apparent.
In the past few years, techniques for the culture of solid tumors have improved, and cytogenetic data on these cancers are accruing rapidly (140). The results are comparable to those obtained with the hematopoietic tumors. Most solid tumors have acquired, nonrandom chromosomal abnormalities (Table 4-1), which in some cases are sufficiently specific to be diagnostic. This is especially true for the sarcomas. Of note, simple cytogenetic changes are often found in benign tumors; aberrations are therefore indicative of neoplasia, not malignancy. This generalization may not be entirely accurate, since some studies with fluorescence in situ hybridization (FISH) suggest that normal uncultured stromal cells may contain simple abnormalities such as trisomy 7, monosomy X, or monosomy Y (141,142), and some very aggressive tumors have minimal karyotypic changes.
Among the malignant solid tumors, karyotypic complexity often ex-
Table 4-1 Nonrandom Chromosomal Abnormalities in Solid Tumors
|
Disease |
Chromosomal Abnormalities |
|
A. INVOLVING EMBRYONIC CELLS |
|
|
Neuroblastoma |
del(1)(p36) |
|
Ewing's sarcoma/ peripheral neuroepithelioma |
t(11;22)(q24;q12) |
|
Wilms' tumor |
del(11)(p13)a +1q |
|
Retinoblastoma |
del(13)(q14)a +1q |
|
B. ADULT CANCERS |
|
|
Breast |
del(1p) |
|
Colon |
del(17p),-18 |
|
Small cell lung carcinoma |
del(3)(p13-23) |
|
Non-small cell lung cancer |
del(9p) |
|
Renal carcinoma |
del(3)(p11-22) |
|
Bladder cancer |
i(5p),-9, del(19q) |
|
Prostate |
del(10)(q26) |
|
Liposarcoma |
t(12;16)(q13;p11) |
|
Extraskeletal myxoid chondrosarcoma |
t(9;22)(q31;q12) |
|
Synovial sarcoma |
t(X; 18)(p11.2;q11.2) |
|
Rhabdomyosarcoma (alveolar) |
t(2;13)(q35–q37;q14) |
|
Malignant melanoma |
del(1)(p11–22), del(6)(q11q27), i(6p) |
|
Testicular tumors |
i(12p), +12 |
|
Glioma |
-10, del(9p) |
|
C. BENIGN TUMORS |
|
|
Pleomorphic adenomas of the salivary glands |
t(3;8)(p21;q12) |
|
Meningioma and acoustic neuroma |
-22 or del(22q) |
|
Lipoma |
t(3;12)(q27–28;q13–14) ring chromosome |
|
Ovarian tumors |
+12 |
|
Leiomyoma |
t(12;14)(q14–15;q23–24) |
|
a Observed as a constitutional abnormality as well as in some tumors. |
|
ceeds that seen in hematopoietic tumors and in the most extreme cases virtually precludes accurate karyotypic analysis. In these tumors, genomic instability may be so extreme that almost every metaphase cell is different. However, even karyotypic complexity per se can be useful since only certain tumor types characteristically achieve this degree of chromosomal disarray.
Chromosomal Aberrations of Diagnostic Importance
Some types of solid tumors may be difficult to diagnose by morphology because of an ambiguous appearance or failure to show discernible features of specific differentiation. Examples of tumors in which cytogenetic analysis is particularly useful in resolving this type of diagnostic problem include small round cell tumors of childhood and spindle cell tumors.
Round cell tumors of childhood include lymphoma, neuroblastoma, Ewing's sarcoma/peripheral neuroepithelioma, embryonal rhabdomyosarcoma, and alveolar rhabdomyosarcoma. Lymphoma can usually be excluded from the differential diagnosis by using clinical data and immunohistochemistry. Although peripheral neuroectodermal tumors are similar to neuroblastomas, a number of biological and molecular features suggest that they are also related to Ewing's sarcoma. Both have the same translocation [t(11;22)(q24;q11–12)] and express MYC and neither expresses MYCN. Recent treatment of peripheral neuroectodermal tumors with therapy similar to that used in Ewing's sarcoma appears to be effective (143). Embryonal and alveolar rhabdomyosarcomas are tumors showing differentiation along skeletal muscle lines. While immunohistochemistry or electron microscopy of the round cell tumors is useful, in a significant proportion of cases the final diagnosis is uncertain even after morphologic approaches have been exhausted. Specific diagnosis is not trivial, since prognosis and therapy differ for each one of these tumors.
In this group of tumors, cytogenetics can provide definitive diagnosis, because each of the tumors is associated with distinguishing karyotypic abnormalities. Neuroblastoma often shows deletions in chromosome 1 centered about band p36 (144,145), double minute chromosomes (146,147), and homogeneously staining regions (147,148) due to amplification of the MYCN gene (149). The t(11;22)(q24;q12) is pathognomonic for both Ewing's sarcoma and peripheral neuroepithelioma (150). About 20% of cases of Ewing's sarcoma also show a translocation involving chromosomes 1 and 16 (151). Embryonal rhabdomyosarcoma is frequently associated with trisomy 2 (152) and the presence of double minute chromosomes (153), whereas t(2;13)(q35–q37;q14) (152,154) or, less frequently, t(1;13)(p36;q14) (155,156) translocations are specifically found in alveolar rhabdomyosarcoma. As yet, none of the genes involved in these translocations or in del(1p) associated with neuroblastoma has been identified. The primary mode of detection is karyotyping, with the exception of MYCN amplification or 1p loss of heterozygosity (LOH) in neuroblastoma, which can be detected by Southern blotting (157).
Spindle cell tumors include synovial sarcoma, myxoid liposarcoma, extraskeletal myxoid chondrosarcoma, peripheral neural sheath tumors, and malignant fibrous histiocytoma. Like round cell tumors of childhood, dif-
ferent types of spindle cell tumors cannot always be distinguished from one another morphologically, particularly when poorly differentiated. Synovial sarcoma, myxoid liposarcoma, and extraskeletal myxoid chondrosarcoma are all associated with specific translocations—t(X;18)(p11.2;q11.2) in synovial sarcoma (158); t(12;16)(q13;p11) in myxoid liposarcoma (159); and t(9;22)(q31;q12) in extraskeletal myxoid chondrosarcoma (160). In contrast, malignant fibrous histiocytoma (161) and malignant peripheral nerve sheath tumors (162,163) usually have very complex cytogenetic aberrations. Malignant fibrohistiocytoma also frequently contains a 19q+ marker chromosome (164).
An indication of the value of cytogenetic analysis of sarcomas was provided by a recent study of malignant soft tissue tumors (165). Fifty-five of sixty-one tumors were found to contain clonal chromosomal aberrations; the six negative cases appeared to be due to outgrowth of normal stroma in culture and therefore did not reflect the true tumor karyotype. In forty cases, the aberrations suggested or confirmed a diagnosis and in fifteen cases the findings established a diagnosis. Eight of fourteen round cell tumors in children contained diagnostic abnormalities. These findings strongly support a role for cytogenetic analysis in diagnosis of these types of tumors. As translocation breakpoints are identified, other means of detection will also become available.
It may be very difficult to differentiate mesothelioma from reactive mesothelial cells, particularly when diagnostic material is limited to a small biopsy or to cytologic inspection of pleural fluid. Mesothelioma characteristically is associated with deletions of 1p, 3p, and chromosome 22, and typically demonstrates a relatively simple karyotype (165,166). Detection of such abnormalities provides an objective means to differentiate mesothelioma from benign mesothelial cells. In principle, this same approach can also be applied to other types of cytologic specimens. It is also import to distinguish mesothelioma from adenocarcinoma of the lung involving the pleura. Like mesothelioma, adenocarcinoma may also have deletions involving 3p (167,168), but usually in the context of complex, sometimes bizarre karyotypes (169,170). In most cases, ultrastructural studies, stains for mucins, and immunoperoxidase stains for keratins, carcinoembryonic antigen (CEA), and Leu M1 lead to a definitive diagnosis, but in some cases these studies are ambiguous. In this subset of cases, cytogenetics may help to confirm a diagnosis of mesothelioma.
Chromosomal Aberrations of Prognostic Importance
Neuroblastoma
As previously noted, neuroblastoma, a common childhood tumor of the peripheral nervous system, is associated with deletions of 1p and amplifica-
tion of MYCN (Plate 3). Neuroblastomas can be classified by categorizing the tumors by stage of disease. One method of staging the tumors is the Evans staging system, which was the basis for staging the tumors in Figures 4-1 and 4-2, utilizing the following criteria: Stage I, tumor localized to the structure or organ of origin; Stage II, tumor that extends in continuity beyond the structure or organ of origin but does not cross the midline (may include lymph node involvement); Stage III, as in II but does cross the midline; Stage IV, primary tumor metastasized to distant sites, including bone, bone marrow, organs, soft tissues, and lymph nodes; and Stage IV-S, small primary tumor (such as Stage I or II) with dissemination, but limited to liver, skin, or bone marrow. Plate 3 illustrates the amount of MYCN
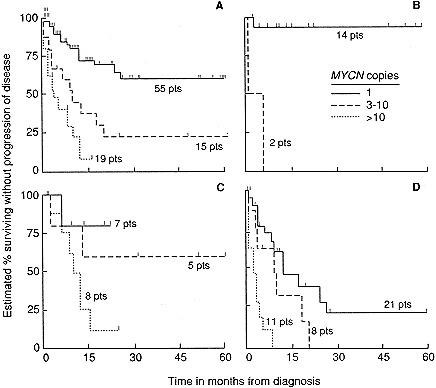
Figure 4-2 Relation between number of MYCN gene copies and progression-free survival. A. Survival in all patients (P < 0.0001). B. Patients with Stage II tumors (P = 0.03; exact life-table test). C. Patients with Stage III tumors (P = 0.1). D. Patients with Stage IV tumors (P = 0.0002). [Seeger, S.C., et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. Reprinted, by permission of the New England Journal of Medicine 1985; 313(18):1111–1116.]
amplification found in tumors in these stages. A significant correlation has been found between amplification of the gene and poor outcome for the patient (Figure 4-2) (171). In patients whose tumors had 1, 3 to 10, or more than 10 copies of MYCN, progression-free survival after 18 months was estimated to be 70, 30, and 5%, respectively.
Based on flow cytometric determination of DNA content, cytogenetic studies, and Southern blot analysis of MYCN amplification, three subsets of patients with distinct prognoses have emerged (172). Patients in the first group have a hyperdiploid or triploid modal chromosome number and lack del(1p) and MYCN amplification. These patients are usually less than one year of age and have a good prognosis. Patients in the second group have near-diploid or near-tetrapiold modal karyotype and also lack del(1p) and MYCN amplification. These patients are older, have more advanced disease at diagnosis, and tend to pursue a slowly progressive course with an eventually fatal outcome. The last group of patients has near-diploid or tetraploid karyotype and del(lp), MYCN amplification, or both. Patients in this group are older and present with advanced stage disease, which is rapidly fatal. These patient groups appear to be stable, with no conversion or progression between groups over the course of the disease.
Multivariate analysis has shown that diploidy and MYCN amplification, which is usually associated with deletion of 1p, are the most reliable prognostic indicators (Figure 4-2), predicting aggressive behavior and poor response to chemotherapy in neuroblastoma. In fact, after these two factors have been taken into account, age and stage appear to be of little additional prognostic significance. It remains to be seen whether early bone marrow transplantation will improve the prognosis of these patients.
Breast Cancer
Breast cancer affects one in eleven American women during their lifetime. An issue of major controversy in treatment of this disease concerns women without evidence of tumor spread to axillary lymph nodes, so-called node-negative cancer. A significant proportion of these women, about 30%, are destined to relapse within five years. On this basis, it has been suggested that all patients with node-negative disease should receive chemotherapy. This situation is far from ideal, since 70% of women are apparently cured by primary excision and do not need this extra therapy, which is associated with significant side effects.
To improve this situation, many investigators have attempted to find markers in node-negative tumors that predict relapse. Some of the newer markers being studied include molecular alterations in proto-oncogenes, growth factors, growth factor receptors, tumor suppressor genes, and genes believed to be associated with the metastatic phenotype. The genes that have
received greatest attention thus far include two proto-oncogene/growth factor receptors, termed HER-2/neu and the epidermal growth factor receptor (EGFR) also known as ERBB (173–176); the cathepsin D gene product (177); and the NM23 gene (178–180). Overexpression of the EGFR gene product has been shown to correlate with a poor outcome in both node-negative and node-positive breast cancer (175,176). One model proposed for this correlation with outcome is that overexpression of the growth factor receptor plays a role in the more aggressive phenotype by increasing growth potential of the tumor cells. The cathepsin D gene is believed to encode a proteolytic enzyme that may be involved in promoting metastasis by breakdown of extracellular barriers. Studies have shown that overexpression of this gene correlates with a worsened prognosis in node-negative breast cancer (177). The NM23 gene encodes a nucleoside diphosphate (NDP)-kinase and was initially discovered by its differential expression in murine melanoma cell lines with high and low metastatic potential. In these studies, NM23 expression was highest in those cell lines with low metastatic potential (178). Subsequent studies evaluating this gene in human breast cancer have demonstrated that its expression may show a similar correlation with prognosis; that is, tumors expressing little or no NM23 were more likely to relapse (179,180). This association held for both node-positive and node-negative disease.
Perhaps the best studied of these molecular markers in human breast cancer is the ERBB2 gene (also known as HER-2/neu). This gene encodes a membrane protein with a structural homology to the epidermal growth factor receptor. This gene is amplified in 25–30% of human breast cancers (173,174). The increase in copy number correlates with elevated levels of ERBB2 mRNA and immunoreactive protein (Figure 4-3). Conversely, 80–90% of tumors with immunohistochemical evidence of ERBB2 overexpression show evidence of gene amplification.
A number of studies have now shown that ERBB2 amplification has prognostic importance in breast cancer. In some of these studies, ERBB2 amplification was the second best predictor, after number of involved lymph nodes, of disease-free survival and overall survival in women with metastases to axillary lymph nodes (173,174). One of these studies systematically compared the results obtained with Southern blotting, Northern blotting, Western blotting, and immunohistochemistry (the various methods used for assessing alterations of the gene in human tissue) in a blinded fashion (173) (Plate 4). The analysis demonstrated very good correlation between overexpression of protein (as detected by immunohistochomistry) and degree of gene amplification (as detected by Southern blotting). The study did not find an association with relapse in node-negative patients; however, this result was based on a small patient sample size. Follow-up studies from this group and others now demonstrate increased risk for relapse and decreased survival if amplification or overexpression is seen in node-negative patients
(181–186). Thus, detection of ERBB2 amplification or overexpression may play an important role in management of patients with breast cancer regardless of nodal involvement.
Because overexpression of ERBB2 appears to correlate best with tumor phenotype and prognosis, immunohistochemistry will likely prove to be the method of choice for analysis, in large part because the method is already in place in most pathology laboratories. Weaknesses of this technique include the current need for frozen tissue for optimal immunostaining and the subjective nature of interpretation of such stains. The former problem may be overcome by the development of new, specific antibodies that work on paraffin-embedded tissue; the latter problem, by application of digital image analysis. Before the test can be widely used, it will also be necessary to evaluate the importance of ERBB2 among the many other biologic markers that have been suggested to predict prognosis in breast cancer. These include tumor grade (187), proliferative rate and ploidy (188,189), and quantitation of tumor-associated angiogenesis (190), as well as the other molecular markers mentioned above (175–180).
ERBB2 amplification and overexpression have also been detected in about 30% of cases of ovarian cancer in which, as in breast cancer, overexpression correlates with poor prognosis (173). Immunohistochemical detection of overexpression of ERBB2 may, therefore, help guide therapy of this cancer in the future.
Lung Cancer
Initial studies have focused on loss of heterozygosity at several positions in the genome. LOH has been reported in the chromosome arms 3p, 13q, and 17p and in chromosome 11 in both small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) (191–196). LOH in chromosome arms 3p, 13q, and 17p occurs in virtually all cases of SCLC and in 15–80% of NSCLC according to various reports.
The areas of LOH on 13q and 17p correspond respectively to the localizations of the RBI and TP53 tumor suppressor genes. Almost all cases of SCLC and many cases of NSCLC show inactivating point mutations in the RB1 gene (197). Most cases of SCLC and about half of the cases of NSCLC have inactivating point mutations of the TP53 gene (198–200).
In addition, mutations of proto-oncogenes occur in approximately 10–15% of all lung cancers. Most frequently mutated is the KRAS gene, which is abnormal in 20–30% of adenocarcinomas (201).
The clinical significance of most of these molecular events is, at present, unknown. One study showed increased survival of patients with resectable (Stage I–IIIa) adenocarcinomas whose tumors lack KRAS mutations compared with patients whose tumors contain KRAS mutations; however, the
plateau of the survival curve of the former group had not yet been reached during the 2-year follow-up period. Thus, with longer follow-up, absence of KRAS mutations may only predict time to relapse and not survival (202).
Other Solid Tumors
A few other tumors show possible associations between karyotype and prognosis. Abnormalities of chromosomes 7 and 11 correlate with a particularly aggressive course in metastatic malignant melanoma (203), but overall prognosis is so poor that this information is not clinically useful. Another small study suggested the presence of a 19p+ marker chromosome correlated with increased risk of local recurrence and/or distant metastasis in malignant fibrohistiocytoma (164). Again, in the absence of effective adjuvant therapy following surgical excision, the clinical utility of such information is limited.
Genetic Markers of Predisposition to Cancer
The identification of familial forms of cancer has further strengthened the notion that cancer is an essentially genetic disorder. More than 50 different tumors have shown a propensity to aggregate in certain families. In some families, specific tumor types tend to occur, whereas other ''cancer families'' are susceptible to more diverse kinds of tumors. In a growing number of instances, susceptibility to familial forms of cancer has been linked to inheritance of constitutional chromosomal abnormalities or to mutations in specific genes. Every cell of an affected individual contains the predisposing abnormality, so any normal tissue can be screened to assess risk. Detection of people who have inherited such an abnormality suggests the need for close observation for development of malignancy and for appropriate early intervention. In a more general sense, understanding these genetic changes in molecular terms may lead to new understanding of mechanisms of tumorigenesis and new approaches to therapy.
Retinoblastoma
Perhaps the best characterized of the constitutional abnormalities are deletions of chromosome 13 centered about band q14, which are found in normal tissues of 3–10% of children with retinoblastoma (204,205), a tumor of embryonic neural retina. About 20% of tumors from patients with normal constitutional or non-tumor karyotypes contain similar deletions (206) or loss of the entire chromosome 13. These deletions define a region of the genome that was recognized as likely to contain a gene or genes important in causing retinoblastoma.
Recognizing that familial disease was multifocal and bilateral, and spo-
radic disease unifocal, Knudson in 1971 proposed a "two-hit" model for retinoblastoma (207). According to this model, patients prone to the disease inherit one mutation that predisposes them to retinoblastoma and then acquire a second mutation in any susceptible target cell, thereby leading to actual tumor development (Figure 4-4). In contrast, normal individuals would have to acquire two mutations within a single cell, a low-probability event. A molecular search eventually identified a gene called RB1 (208), which is always mutated on both alleles in tumors and typically mutated in one allele in the normal tissue of patients with familial retinoblastoma—a refinement of Knudson's hypothesis in that the two mutations involve alleles of the same gene. The precise function of the protein product of this gene is still being worked out, but RB1 can bind to at least one and possibly several transcription factors and can be phosphorylated by p34cdc2 kinase, a
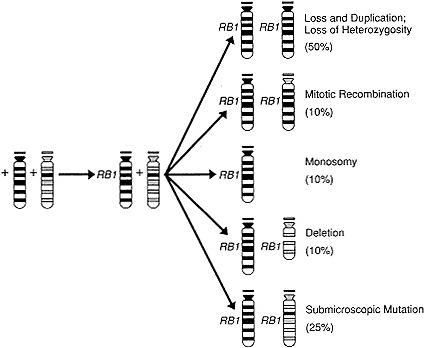
Figure 4-4 Schematic representation of two normal chromosomes 13 with normal alleles at the retinoblastoma (RB1) locus (+). Also shown are the mechanisms whereby a cell with a single mutation at this RB1 locus can become homozygous or hemizygous for the mutated allele. (Gallie, B.L., and Phillips, R.A. Retinoblastoma: A model of oncogenesis. Adapted and published courtesy of Ophthalmology 1984; 91:666–672.)
cell cycle-specific protein kinase (for recent review, see 209). Through these and other yet to be defined interactions, RB1 appears to have an important role in regulating cell proliferation and thus falls into the category of tumor suppressor genes. It is expressed in all cell types. It is therefore surprising that inherited abnormalities in RB1 are associated predominantly with retinoblastoma and, as it happens, with osteosarcoma later in life (210).
Identification of restriction fragment length polymorphisms (RFLPs) linked to RB1 and now within the RB1 gene itself has made prenatal diagnosis of susceptibility to familial retinoblastoma a possibility (211) (Figure 4-5). Isolated cases of retinoblastoma, however, may be sporadic or may be secondary to inheritance of a new germline mutation. In the latter case, the patient is at risk for recurrent tumors and can pass this risk along to offspring. The only way to differentiate between these two possibilities is to scan the RB1 genes of normal tissue for evidence of mutation, either by direct sequencing or by some indirect technique [RNase mismatch, denaturing gradient gel electrophoresis (212), etc.]. This effort is made much easier if the mutation(s) in the tumor have been identified; the normal tissue can then be scanned for the specific mutation(s) observed in the tumor, as opposed to a search for an unknown (and perhaps nonexistent) mutation. Hence, preservation of the original tumor for this type of molecular genetic analysis may be very important in determining the occurrence of a new germline mutation in a patient with no family history of retinoblastoma and unilateral disease. At present, these types of tests are available only at a few reference laboratories, a situation unlikely to change in the near future. However, preservation of tumor tissue could occur at any location.
Acquired mutations in the RB1 gene are now being detected in a variety of sporadic tumors, including some lymphoid neoplasms (213); carcinoma of the lung (197), breast (214), and prostate (214); and certain sarcomas (215,216). In addition, a number of viruses linked to human cancers, most notably human papilloma virus, which has been implicated in cervical carcinoma, have been shown to produce proteins that bind to the RB1 protein and inhibit its function (217). These data indicate that RB1 is likely to play an important role in many human cancers. It remains to be explained why inherited RB1 mutations should be associated principally with retinoblastoma and osteosarcoma, when acquired abnormalities of RB1 appear to be potentially oncogenic in many other tissue types.
Mapping of Other Cancer Predisposition Loci
Elucidation of the genetic basis of familial retinoblastoma demonstrated the utility of RFLP analysis and, in some cases, cytogenetic analysis, in mapping the location of genes that cause familial tumor syndromes. By
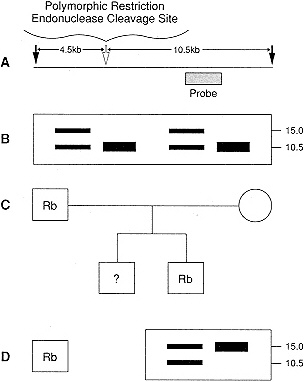
Figure 4-5 Use of restriction fragment length polymorphisms (RFLPs) to track an abnormal chromosome. A. Some restriction enzyme cutting sites are variably inherited by the population (white arrow). In this example, restriction enzyme digestion will lead to a 10.5-kb fragment if the site is present, or a 15-kb fragment if it is absent when the products are detected by a common probe (striped bar). A pedigree with inherited retinoblastoma (Rb) shows the restriction fragments from each family member (B) above the pedigree (C). The father, who had Rb as an infant, is heterozygous for this RFLP, since digestion yields two bands on Southern blot analysis. One son also developed retinoblastoma and his restriction pattern is identical to the father's. Since the mother is homozygous for the presence of the restriction site (she has two 10.5-kb bands), the RB1 gene must have been inherited on the paternal allele that lacks the restriction site (15-kb band). Examination of fetal DNA shows that an unborn son did not inherit the RB1 allele, since a 15-kb band is not present. Therefore, he is not at risk to develop Rb. D. Loss of heterozygosity in the tumor is observed in the son with retinoblastoma. Southern blot analysis shows the presence of both the 10.5-kb fragment and the 15-kb fragment in the blood sample on the left. In the tumor sample on the right only the 15-kb fragment is found. (Modified, from Carroll, W.L., and Schwartz, A.L. Molecular oncology. In Fernbach, D.J., and Vietti, T.J., eds., Clinical Pediatric Oncology, 5th ed. St. Louis: C.V. Mosby, in press. Used with permission.)
using similar approaches, a number of other loci associated with predisposition to a variety of tumors have been determined (Table 4-2). Like retinoblastoma, these familial tumor syndromes have an autosomal dominant pattern of inheritance and are associated with LOH, suggesting the involvement of tumor suppressor genes. In some cases, the affected genes have been identified, including those responsible for neurofibromatosis type 1 (218,219) and Li-Fraumeni syndrome (220). Identification of TP53 mutations in Li-Fraumeni syndrome is particularly interesting, because this gene also appears to be frequently mutated in some of the more common sporadic human tumors.
Li-Fraumeni Syndrome
The Li-Fraumeni syndrome is a rare disorder associated with increased risk of a wide variety of tumors, including childhood sarcomas; carcinomas involving breast, lung, prostate, adrenal gland, and cervix; brain tumors; and acute leukemia (221). The syndrome may account for 1–2% of all childhood tumors and 5–10% of childhood sarcomas (222). Recently, patients with this disorder have been shown to have germline mutations in the TP53 gene (220). Tumor-specific mutations in this gene have also been demonstrated in a wide variety of sporadic tumors; indeed, mutations in this gene may represent the most common genetic change in human cancers (for recent review, see 223). The function of the gene is under intensive investigation, but is not currently well understood. Like RB1, it appears to function as a tumor suppressor gene by regulating the progression of cells through the cell cycle. It is also inactivated by proteins encoded by several tumorigenic viruses, including human papilloma virus (224).
Table 4-2 Summary of Mapping of Cancer Predisposition Loci
|
Chromosomal Region |
Predisposition Syndrome |
Cancers/Tumors |
|
3p |
Von Hippel-Lindau |
Renal cell carcinoma |
|
5q |
Familial adenomatous polyposis |
Colon carcinoma |
|
10q |
MEN-2A syndrome |
Thyroid (MCT), pheochromocytoma |
|
11p13 |
WAGR syndrome |
Wilms' tumor |
|
11p15 |
Beckwith-Wiedemann |
Wilms', hepatoblastoma, others |
|
11q |
MEN-1 syndrome |
Parathyroid, pancreas, pituitary |
|
13q |
Familial retinoblastoma |
Retinoblastoma, osteosarcoma |
|
17p |
Li-Fraumeni syndrome |
Breast, osteosarcoma, soft tissue sarcoma, brain cancer, leukemia |
|
17q |
Neurofibromatosis 1 |
Neurofibroma, pheochromocytoma |
|
22q |
Neurofibromatosis 2 |
Acoustic neuroma, meningioma |
In patients with the Li-Fraumeni syndrome and in sporadic tumors, most mutations in TP53 are due to single base pair substitutions and result in production of an altered protein. The mutations in the gene cluster in three "hot spots." For unknown reasons, the frequency of alterations within hot spots varies widely from tumor to tumor. In contrast, all Li-Fraumeni families thus far analyzed have mutations that fall within a single small region centered about codon 248. These observations suggest that different mutant forms of TP53 may have distinct functional consequences.
As of yet, the prognostic and diagnostic impact of TP53 mutations are limited, except in allowing identification of patients with the syndrome. However, the wide variety of human cancers associated with sporadic and germline TP53 mutations makes this genetic lesion a conceivable target for new therapeutic approaches.
Loss of Heterozygosity in Sporadic Solid Tumors
Comparative RFLP analysis performed on DNA isolated from tumors and normal tissues of individual patients has also been used to look for evidence of LOH in sporadic forms of cancer. Numerous sporadic tumors have now been shown to be associated with LOH within specific chromosomes or portions of chromosomes (225,226). Sometimes the region of LOH has matched the location of known tumor suppressor genes such as TP53; in other cases, the involved loci remain unknown.
In many instances, LOH can be demonstrated at multiple genomic sites within a single tumor sample and is frequently associated with concomitant activation of oncogenes through a variety of mechanisms, a point summarized in Table 4-3. These data suggest that oncogenesis is the product of a series of sequential genetic alterations that affect the function of multiple genes.
Table 4-3 Patterns of Oncogene Activation and Suppressor Gene Inactivation in Selected Human Cancers
|
|
Oncogene Activation |
||
|
Tumor Type |
Point Mutation/Amplification |
Deletion/LOH |
|
|
Small cell lung cancer |
|
MYC, MYCN, MYCL |
3p, 13q (RB1), 17p (TP53) |
|
Colorectal cancer |
KRAS |
|
5q (APC), 17p (TP53), 18q (DCC) |
|
Breast cancer |
HRAS |
ERBB2, MYC, INT2/HST |
1p, 1q, 3p, 11p, 13q (RB1), 17p (TP53) |
|
Neuroblastoma |
|
MYCN |
1p36, 14q |
|
Glioblastoma multiforme |
|
ERBB1, MYCN, GLI |
9p, 10q, 17p (TP53) |
Genetic Changes and Tumor Progression
While individual mutations are frequently characteristic and not uncommonly diagnostic of particular malignancies, it has been appreciated for some time that development of cancer is a multistep process that may proceed through phases of benign neoplasia. According to the genetic theory of oncogenesis, each step of the process corresponds to acquisition of a mutation that results in some additional selective advantage, either by enhancing proliferation or by delaying cell death. This results in waves of clonal expansion and replacement as tumor progression occurs, with cancer resulting only when mutational events have occurred in a set of critical genes. On the basis of mutation rates and the association of cancer with increasing age, it was predicted more than 30 years ago that roughly four to six distinct mutations would be needed to produce a malignancy.
Colorectal Neoplasms
The work of Vogelstein and colleagues has provided considerable molecular evidence to support this view of oncogenesis (227). Their work has focused on three regions of the genome, 5q, 17p, and 18q, that are frequently subject to chromosomal deletion and/or LOH in colonic carcinomas and on the proto-oncogene KRAS. Like other members of the RAS family of proto-oncogenes, KRAS appears to bind guanosine triphosphate (GTP) and likely plays a role in modulating cell response to external stimuli produced through the binding of growth factors or hormones to specific membrane-bound receptors. The approach of these investigators has been to correlate changes in these four loci with stages of tumor progression, resulting in the model shown in Figure 4-6. Briefly, small colonic adenomas are most
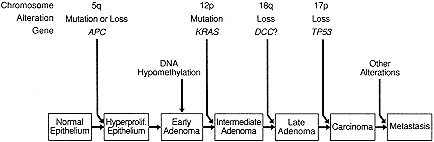
Figure 4-6 A genetic model for colorectal tumorigenesis. See text for explanation. (Modified, from Fearson, E.R., and Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990; 61:759–767. Copyright, Cell Press. Used with permission.)
commonly associated with changes in 5q. Many of these early lesions also demonstrate hypomethylation of cytosine residues within their DNA. Whether this is a purely epigenetic change or a reflection of mutations that directly affect methylation is unclear. In addition to changes in 5q, about 50% of larger adenomas of intermediate stage have point mutations in KRAS. Late adenomas, in turn, have additional alterations in 17p and 18q in about 25 and 50% of cases, respectively. These changes are even more frequent in adenocarcinomas, in which changes in 17p and 18q are each detected in more than 70% of cases. In general, carcinomas usually contain three or four alterations, and early adenomas only one or two changes, with intermediate and late adenomas having a mean number of alterations falling between these two extremes. The overall findings are consistent with a genetic basis for multistep oncogenesis.
Recognition of recurrent 5q, 17p, and 18q alterations led to a search for tumor suppressor genes at each of these sites. The affected gene on 17p appears to be TP53 (228). A gene called DCC (for deleted in colon carcinoma) has been mapped to the site of the 18q deletions (229). This gene encodes a protein expressed in normal colonic mucosa whose structure is consistent with its function as a cell adhesion molecule. Loss of this protein might alter interactions of tumor cells with each other or with surrounding extracellular matrix, and could play a role in tumor invasion or metastasis. Candidate tumor suppressor genes on 5q named MCC and APC have also been identified (230,231). These genes encode coiled-coil proteins with potential homologies to proteins known to interact with GTP-binding proteins. Familial adenomatous polyposis (FAP), an inherited syndrome associated with diffuse neoplasia of the colon and rectum, also maps to 5q, and has been shown in some cases to result from mutation of the APC gene (232).
Several additional aspects of this work deserve comment. Occasional tumors were found to contain areas of adenocarcinoma adjacent to areas of benign adenoma. In each case, the cancer had all of the genetic alterations found in the adenoma plus one additional genetic change. While the changes in these specimens generally occurred in the order shown in the model, there were exceptions, such as the deletion of 17p (presumably by loss or mutation of TP53) in adenomatous areas with del(17p) and del(5q) in the adjacent carcinoma. Thus, the order in which the mutations occur may not be as critical as the total accumulation of changes (227). Furthermore, as noted above, not all carcinomas contained identifiable changes in all four loci. Changes in genes other than those noted above may also collaborate to produce colonic carcinoma. For example, regions from chromosomes 1q, 4p, 6p, 6q, 8p, 9q, and 22q were lost in 22–50% of cases studied, and genes within these regions may represent potential sites for as yet uncharacterized tumor suppressor genes that could play a pathogenic role in a subset of
colorectal carcinomas (233). Finally, it should be remembered that colon cancer, like other malignancies, is itself also susceptible to progression, presumably through further mutation and clonal evolution, to produce increasingly malignant phenotypes. These critical steps, which are responsible for such clinically important properties as invasion and metastasis, remain largely unknown at the molecular level. However, the total number of genetic alterations accumulated in colorectal carcinomas (estimated by allelic losses) is highly correlated with the propensity of the tumors to metastasize and cause death, thereby providing important prognostic information (234).
Practical benefits of results already achieved are on the horizon. For example, recognition of critical early events in colonic neoplasia could lead in the near future to new screening tests designed to look for specific genetic lesions within cells desquamated into the colonic lumen. A recent report describing the detection of mutations within desquamated urothelial cells in urine has demonstrated the feasibility of genetic analysis of body fluid specimens (235), and this type of approach could conceivably be applied to any kind of cytologic specimen. Some (236,237), but not all (238), initial studies also suggest that the presence of aneuploidy in colorectal tumors can be used to predict prognosis more precisely. Finally, discovery of new suppressor genes has produced additional targets for possible therapeutic intervention.
Glial Tumors
Like colorectal tumors, glial tumors also demonstrate histologic and clinical evidence of tumor progression. The histologic grade of these tumors correlates with clinical prognosis, and the progression of tow-grade tumors to higher-grade tumors over time, as well as the coexistence of high-and low-grade histologies within a single biopsy, are frequent occurrences.
Cavenee and coworkers have correlated genotypic changes with phenotypic evidence of tumor progression (239,240). By using probes that recognize RFLPs, LOH of chromosome 17 was found in low-and high-grade gliomas at approximately equal frequencies. In contrast, amplification of the EGFR gene was found more frequently in high-grade than in low-grade lesions, and LOH on chromosome 10 was detected exclusively in high-grade tumors. These data are consistent with a pathway for progression of glial tumors dependent on the accumulation of multiple genomic aberrations, a situation similar to that proposed for colorectal neoplasms. LOH of portions of chromosomes 9, 10, and 22 has also been observed (239,241); the position of these alterations in the above schema is unclear.
Many other carcinomas and sarcomas, and some lymphoid tumors, also have natural histories consistent with a multistep model of oncogenesis.
Approaches similar to those taken by Vogelstein and Cavenee may unravel the critical genetic events that underlie progression in this diverse group of neoplasms.
Cancer-Associated Genetic Changes and Environmental Mutagens
The genetic mechanisms of environmental factors in human tumor development have not yet been investigated extensively. However, in one recent report, replacement of a guanine (G) residue with a thymidine (T) residue at codon 249 of the TP53 gene was detected in a series of hepatocellular carcinomas (242,243), a tumor that is quite rare in the United States, but very common in Africa and parts of Asia. Aflatoxin, a compound produced by fungi found in poorly stored foodstuffs, which has been linked to hepatocellular carcinoma, has been shown to induce G to T mutations, suggesting, therefore, that aflatoxin is specifically responsible for the mutation in TP53. It has not previously been possible to prove that any particular tumor was the direct result of exposure to an environmental carcinogen. The linkage of aflatoxin to a specific type of TP53 mutation raises the possibility that some mutagens may produce genetic changes that are so specific as to constitute a "smoking gun," thereby raising the possibility of a new field in "oncologic forensics."
Viral Markers in Cancer Diagnosis
Four different viruses have been associated with human cancers: Epstein-Barr virus (EBV), human T cell lymphoma/leukemia virus I (HTLV I), human papilloma virus (HPV), and hepatitis B virus (HBV). These viruses are unrelated, differ greatly in complexity, probably participate in tumorigenesis by disparate molecular mechanisms, and are linked to quite different types of tumors.
Of the four oncogenic viruses, EBV has been found in the greatest variety of tumors. The connection of this herpes virus with any cancer is, in some respects, ironic because EBV is a ubiquitous virus and much more often responsible for benign infections. In fact, about 90% of all adult Americans show serological evidence of past infections, most of which are apparently subclinical. Infections acquired after childhood may be manifest as infectious mononucleosis, a benign self-limited proliferation of EBV-infected B cells. On the other hand, EBV DNA is found in most cases of endemic Burkitt's lymphoma (although only 5–20% of sporadic cases contain the genome) (244,245), and in most patients who develop B cell lymphoproliferative disorders in the setting of immunosuppression (246–249). Viral DNA is also detected in virtually all cases of nasopharyngeal carcinoma (245), a tumor worldwide in occurrence but found with greatly
increased incidence in southern China and in ethnic Chinese living elsewhere. Thymic lymphoepithelial carcinoma (250) and about 20% of Hodgkin's disease (251) have been reported to contain viral DNA, in the latter confined primarily to the Reed-Sternberg cell.
The mechanism by which EBV participates in malignant transformation of infected cells is not clear and may be complex. A variety of genes are active during latent infections, and several of these are candidates for mediators of transformation. However, attention has recently focused on a gene called LMPI, which apparently can induce increased expression of BCL2 and possibly other oncogenes, such as those of the RAS group (252).
HTLV I is a retrovirus related to human immunodeficiency virus I (HIV I), the virus that causes AIDS. Like HIV I, HTLV I infects T lymphocytes, some of which may eventually become transformed after many years to produce adult T cell leukemia (ATL) (253), an aggressive disease often associated with skin infiltrates and hypercalcemia. Viral infection has been shown to be endemic in the southern islands of Japan and in the Caribbean (254), both of which areas have an increased incidence of ATL. The virus is transmitted by exchange of body fluids much in the same manner as HIV I, a fact that has important implications for public health. Transforming activity of HTLV I seems to be located in the so-called tax gene of the virus (255,256). Products of this gene have the capacity to activate a set of cellular genes, including genes for the T cell growth factor IL-2, its receptor, and the oncogene FOS, which may explain the growth-promoting abilities of this virus (257).
HPV is a small DNA virus with a tropism for keratinocytes. Many different types have been identified. Most of these are found in benign warts and other skin disorders; however, three (HPV 16, 18, and 33) are detected in anogenital cancers, most importantly cervical cancer (258). Two genes, referred to as E6 and E7, have been implicated in the malignant transformation of cells by this virus. Both genes produce proteins that appear to interact with and inactivate the products of tumor suppressor genes. The E6 gene product seems to inactivate the TP53 protein, and the E7 gene product seems to inactivate the RB1 protein (259–261).
HBV is a single-stranded DNA virus that causes hepatitis B. Viral DNA is commonly also found in hepatocellular carcinoma, but its role in the pathogenesis of this disease has been unclear. Until recently, it was widely believed that cancers arose as a consequence of massive necrosis induced by HBV and regeneration of hepatocytes. However, new animal studies suggest that HBV—induced hepatocellular destruction need not precede tumor development and that the virus may be directly oncogenic (262–264). The gene HBx, which produces a protein able to activate transcription of a variety of viral and cellular genes, seems to represent the critical portion of the viral genome responsible for the oncogenic action of the virus (264,265).
From a diagnostic standpoint, genomes for each of these viruses are retained as DNA within replicating tumor cells and can be used both as markers for specific tumor type and as markers of clonality. Detection of the viral genomes can be performed by using a variety of techniques, including Southern blotting, polymerase chain reaction, in situ hybridization for RNA products, or immunological analysis for viral proteins. In those viruses that integrate their genomes into host DNA (HTLV I and HBV), the site of integration represents a clonal marker and can be assessed by Southern blotting with viral DNA probes and restriction enzymes that cut host DNA flanking the site of integration. Polymorphisms within the DNA of viral genomes, for instance, in EBV, can also be used to indicate clonal expansion of a population of cells bearing only one polymorphic form of the virus. However, any application of viral genomic markers for diagnosis must take into consideration that the presence of the virus does not indicate malignancy per se, since infections by the virus usually precede malignancy and/or are often present in patients with no evidence of cancer.




















