
2
Testing for Tumor-Specific Genetic Markers
Until recently, cancer diagnosis depended mainly on study of the morphology of tumor cells at the light and, to a lesser extent, at the electron microscopic level. While still of primary importance in diagnosis and likely to remain so for the foreseeable future, morphological examination has several limitations. Most importantly, morphologic evaluation is subjective and may be prone to observer variability. Additionally, morphologic evaluation is relatively insensitive, allowing detection of tumor cells only if their frequency is greater than 1-10% in most circumstances. In contrast, methods using tumor-specific genetic markers offer the potential of a more objective means of diagnosing malignancy with far greater sensitivity than is possible based on morphology alone. Furthermore, genetic analyses of biopsy tissues may provide certain kinds of information about the prognosis and predicted biologic behavior of tumors that is not available from morphologic examination.
Because of the various inherent advantages of genetic methods of diagnosis, we believe that molecular analysis of biopsy tissues will revolutionize the way in which physicians diagnose and treat many tumors. As with the results of any other test, physicians will be able to utilize this information best only when the methodology, indications, and limitations of each test are understood. With these considerations in mind, the remainder of this report describes tests and associated genetic markers that are currently available to assess genetic changes in cancer, offers examples of tumors to
which application of these tests now appears most informative, and finally contains several recommendations concerning the ways in which the medical community should prepare itself for the widespread implementation of these tests in clinical practice. (Table 2-1 provides an overview of the status of genetic testing for cancer.)
Many of the methods now used to detect tumor markers identify changes in DNA structure or sequence directly. However, because genetic information encoded in DNA is transcribed into RNA and finally translated into protein, tests that assess RNA sequence or quantity, or protein structure, function, or quantity, are also informative in various situations. The molecular lesions associated with different kinds of tumors are diverse, necessitating a pragmatic approach suited to the individual type of tumor in question.
Microscopic Detection of Alterations in DNA Structure
Until recently, detection of chromosomal rearrangements has been dependent on cytogenetic analysis. Briefly, fresh tumor cells are disaggregated and grown for short periods of time in cell culture. Growing cells are then poisoned with a chemical that arrests the cells in metaphase, a stage in cell division (mitosis) during which DNA is condensed into readily recognizable chromosomes. The cells are then swollen in a hypotonic solution, fixed, and dropped onto glass slides. The slides are pretreated to induce banding and are stained with a dye that binds to the chromosomes. The chromosomes are analyzed under the microscope. Sets of chromosomes dispersed from single cells are then photographed, and each chromosome is cut from the photograph and assembled in an orderly portrait, or karyotype, showing the total chromosomal content of an individual cell.
To interpret cytogenetic data, a working understanding of the structural components of chromosomes and the nomenclature used to describe them is required. The normal diploid human cell contains 46 chromosomes, composed of 22 pairs of autosomes (numbered 1–22) and 2 sex chromosomes (two X chromosomes in the female, one X and one Y chromosome in the male). Each chromosome has a constriction called a centromere that divides the chromosome into two arms; the long arm is designated ''q'' and the short arm "p." The arms of each chromosome contain a characteristic pattern of light and dark bands revealed by staining the chromosomes with a variety of dyes, and each band is identified by a number.
The karyotype of an individual cell is reported as a list describing the number of chromosomes, the sex chromosomes present, and any observed abnormalities. For example, a normal karyotype for a female is 46,XX; for a male 46,XY. Abnormalities of chromosome number are referred to as aneuploidy and most commonly include the presence of an additional copy
Table 2-1 Overview of the Status of Genetic Testing for Cancer
|
|
Molecular |
Role in |
Means of Detection |
||||
|
Disease |
Target |
Dx |
Px |
Tx |
Established |
Ready |
Future |
|
HEMATOLOGIC CANCER |
|||||||
|
CML |
BCR-ABL |
C |
|
F |
Cyto, SBlot, PCR |
ISH |
?Immuno |
|
CLL |
Trisomy 12 |
|
C |
|
Cyto |
ISH |
|
|
ALL |
|||||||
|
B or T |
AGRs |
|
|
F |
SBlot |
|
PCR, RNase protection, denaturing gels |
|
B |
BCR-ABL |
|
C |
F |
As in CML |
|
|
|
|
t(1;19),(5;14), (8;14) (2;8),(8;22) |
|
C |
F |
Cyto |
PCR, ISH |
|
|
|
other trans. |
|
|
F |
Cyto |
|
SBlot, ISH, ?PCR |
|
T |
TCL5 rearr. |
|
|
|
SBlot |
PCR |
|
|
|
t(14q 11), |
|
|
F |
Cyto |
PCR |
ISH |
|
|
other trans. |
|
|
|
Cyto |
|
SBlot, ISH, ?PCR |
|
AML |
|||||||
|
M2 |
t(8;21) |
|
C |
F |
Cyto |
|
ISH, SBlot, PCR |
|
M3 |
t(15;17) |
C |
|
C |
Cyto |
SBlot |
ISH, PCR |
|
M4Eo |
inv(16) or t(16;16) |
C |
C |
F |
Cyto |
|
SBlot, ISH |
|
AML with baso. |
t(6;9) |
|
C |
F |
Cyto |
|
ISH, SBlot |
|
AML with high plts. |
inv(3) or t(3;3) |
|
C |
F |
Cyto |
|
SBlot, ISH, ?PCR |
|
AML with-7 or-5 |
del 5 or 7 |
|
C |
F |
Cyto |
ISH |
|
|
Lymphoma |
|||||||
|
All cases |
AGRs |
C |
|
F |
Immuno, SBlot |
|
PCR, RNase protection, denaturing gels |
|
Follicular |
t(14;18) |
C |
|
F |
Cyto, SBlot, PCR |
|
ISH |
|
Burkitt's |
t(8;14), t(8;22) t(2;8) |
C |
|
F |
Cyto |
SBlot, PCR |
ISH |
|
|
Molecular |
Role in |
Means of Detection |
||||
|
Disease |
Target |
Dx |
Px |
Tx |
Established |
Ready |
Future |
|
SOLID TUMORS |
|||||||
|
Sarcomas |
|||||||
|
Synovial sarcoma |
t(X;18) |
C |
|
|
Cyto ?ISH, |
|
?PCR, ?SBlot |
|
Myxoid liposarcoma |
t(12;16) |
C |
|
|
Cyto |
|
" |
|
Extraskeletal myxoid chondrosarcoma |
t(9;22) |
C |
|
|
Cyto |
|
" |
|
Ewing's sarcoma/ PNET |
t(11;22) |
C |
|
|
Cyto |
|
" |
|
Alveolar rhabdomyosarcoma |
t(2;13), t(1;13) |
C |
|
|
Cyto |
|
" |
|
Neuroblastoma |
del(1p) |
C |
C |
F |
Cyto, SBlot |
|
ISH |
|
|
MYCN amp, |
C |
C |
F |
SBlot |
|
ISH |
|
Breast cancer |
ERBB2 amp. |
|
C |
F |
Immuno, SBlot |
|
|
|
Ovarian cancer |
ERBB2 amp. |
|
F |
F |
Immuno, SBlot |
|
|
|
KEY: Dx: diagnostic marker |
|||||||
|
Px: prognostic marker |
PCR: polymerase chain reaction |
||||||
|
Tx: used to monitor response to therapy, or detection modifies induction therapy |
ISH: in situ hybridization Immuno: immunohistochemistry trans.: translocations |
||||||
|
C: current |
rearr.: rearrangement |
||||||
|
F: future |
baso.: basophilia |
||||||
|
Ready: available, needs to be evaluated |
plts.: platelets |
||||||
|
Cyto: cytogenetics |
AGR: antigen receptor gene rearrangement |
||||||
|
SBlot: Southern blotting |
amp.: amplification |
||||||
of a chromosome (trisomy) or the loss of a chromosome (monosomy). The gain of a chromosome is denoted in the karyotype by a plus sign; for example, (47,XY,+21) indicates the presence of an extra copy of chromosome 21. Loss of a chromosome is indicated by a minus sign [e.g., (45,XX,-7)]; deletion of a part of a chromosome is denoted by del followed by the deleted portion of the chromosome [e.g., del(7q)]. In some tumors, the entire haploid complement of chromosomes is present in one or two additional copies—conditions called triploidy and tetraploidy, respectively. In other instances, the number of chromosomes is normal, but structural rearrangements involving one or more chromosomes are present. One type of rearrangement involving a single chromosome is an inversion, an event recognized by an inverted sequence of chromosome bands. Inversions are reported with the symbol inv, for example, inv(16). Rearrangements that produce the fusion of two or more fragments of different chromosomes are called translocations and are indicated by listing the involved chromosomes. For example, t(9;22)(q34;q11) symbolizes a translocation occurring between chromosomes 9 and 22 at bands 9q34 and 22q11, and identifies the so-called Philadelphia chromosome seen in the blood cells of patients with chronic myelogenous leukemia (Figure 1-1). Occasionally, duplication of a chromosome arm occurs with loss of the other arm, creating an isochromosome, designated by the abbreviation i; for example, i(17q) refers to a chromosome composed of long arms of chromosome 17 fused at the centromere. Other abnormalities seen in tumor cells include double minute chromosomes (dmin), small fragments of chromosomes that are present in numerous copies and contain amplifications of specific genes. In some circumstances, amplification of genes occurs within a chromosome, generating a visible local expansion of chromosomal material, referred to as a homogeneously staining region, or hsr.
Traditional cytogenetic study of tumors has been of fundamental importance in identifying pathogenic molecular events in an increasing variety of cancers. However, it has a number of practical limitations. It can be applied only when fresh, viable tissue is available and therefore cannot be used on archival tissue in paraffin blocks—the form in which most tissue is currently stored in pathology departments. Even when fresh tissue is available, samples may be too small for reliable growth or may not grow well enough in culture to yield sufficient numbers of metaphase cells for evaluation. Finally, cytogenetic studies can only identify mutations that produce structural changes large enough to be seen with the light microscope.
In Situ Hybridization
These difficulties can be circumvented to some degree by a new approach to cytogenetic analysis called in situ hybridization (ISH) (Plate 1).
This procedure, like all hybridization procedures, is based on the complementarity of nucleic acids. The two strands of double-stranded DNA can be dissociated by heat or various chemical treatments and incubated under proper conditions of temperature, pH, and ionic strength with single-stranded fragments of DNA, often termed probes, prepared by recombinant DNA techniques. If the nucleotide sequence of the probe is complementary to a sequence in one of the two strands of the dissociated target DNA, it can bind to that sequence, a process termed hybridization. Techniques have been developed that permit hybridization of specific DNA sequences in metaphase chromosomes or intact nuclei by incubating preparations with tagged probes complementary to the sequences of interest. In principle this technique allows analysis of a single cell for the presence of any known DNA sequence.
Previously, the probes were labelled with radioisotopes, such as 3H or 35S, and binding was detected with autoradiography. While sensitive, this method is cumbersome and often requires exposure times of several weeks or longer before results can be analyzed, and the resultant signal is not well localized to the bound probe. More recently, a derivative method called fluorescence in situ hybridization (FISH) has solved these difficulties (1–3). This method is sensitive and specific, and yields a discrete, well-localized signal that can be detected in real time with a fluorescence microscope. Briefly, DNA within metaphase chromosomes or interphase nuclei is denatured and then hybridized to a probe conjugated with a hapten—a covalently bound chemical side group that can be recognized by specific antibodies. After being washed, the preparation is incubated with anti-hapten antibody bearing a fluorescent tag. By using probes with different hapten groups and anti-hapten antibodies with different fluorescent tags, two or more sequences can be studied simultaneously in the same cell. By substituting enzymes for the fluorochrome on the anti-hapten reagents, colorimetric staining techniques can be developed that will permit analysis by light microscopy. Methods for the direct attachment of fluorochromes to probes will also be available in the near future and are likely to result in much simpler protocols.
The kinds of probes used will vary depending on the clinical setting and the differential diagnosis. Probes derived from repetitive centromeric sequences that recognize specific chromosomes can be used to detect trisomies or monosomies. Whole chromosomes can be stained (or "painted") with the collection of probes distributed along the full length of the chromosome. Finally, probes specific for individual genes can be used to detect alterations in gene structure or number, in either metaphase chromosomes or intact nuclei. Aberrations detected by such probes include deletions, inversions, translocations, and gene amplifications.
Deletions of a gene or chromosome are seen as the loss of hybridiza-
tion signal from the cell. This seems simple, but several potential difficulties must be considered. One is purely technical: Not every target sequence can be stained, which results in a finite false-positive rate. In the specific case of studies performed on nuclei, additional false-positives result from both copies of the target sequence overlying one another by chance. For probes directed at individual genes, the probability of this occurring is about 1%; with centromeric probes, which recognize a larger target, the frequency increases to about 3 to 8%. It may be possible to alleviate this problem by using special confocal microscopes capable of "optical" sectioning, thereby allowing resolution of vertically superimposed hybridization targets, but this is currently a relatively slow procedure.
Detection of trisomy is straightforward with this technique, particularly because false-positive signals leading to pseudotrisomy rarely constitute a significant problem. Gene amplification in the form of double minute chromosomes or homogeneous staining regions is relatively easy to identify in metaphase cells and interphase nuclei.
Rearrangements of particular sequences, such as a translocation or inversion, can be detected with probes targeted to DNA at the breakpoints. Depending on the location of target sites of the probe(s) being employed, these types of events bring together two sequences that are normally far apart or that separate two normally closely spaced signals. Appropriate choice of hybridization probes can potentially reduce the false-negative and false-positive detection frequencies to well below 1%.
Gene rearrangements that produce no change in chromosome banding patterns, and are therefore missed by karyotype analyses, are usually detectable by using in situ hybridization. Conversely, complex rearrangements resulting in so-called marker chromosomes can be analyzed by using the probes for whole chromosomes. Components of marker chromosomes may be difficult to analyze by standard cytogenetics but can be identified relatively unambiguously by in situ hybridization. Simultaneous multicolor hybridizations will increase the efficiency of this type of analysis in the near future. These probes are also useful for detecting all fragments of a particular chromosome, no matter how they may be rearranged and distributed within the genome.
Clinical applications of in situ hybridization will develop rapidly in the near future. Labeled probes for specific chromosome centromeres are now available commercially, and probes for whole chromosomes will soon be as well. When probes are detectable by colorimetric staining, widely available conventional transmitting light microscopy will be suitable for analysis. The increased flexibility and perhaps sensitivity in spatial resolution of fluorescence microscopy will make it more common outside academic centers in time. Computer-assisted microscopes for analysis of complex multiprobe
hybridizations are now being developed in a number of research laboratories and commercial enterprises, and these instruments may eventually have a significant impact on the application of in situ hybridization.
The term in situ hybridization has also been used to describe a somewhat different technique in which probes are used for hybridization to RNA within the cytoplasm of intact cells on microscope slides. This procedure is sometimes useful for analyzing gene expression at the RNA level and may provide information about cell differentiation, for instance, in characterizing the tissue origin of various tumors. The method has also been used to identify viral transcripts or genomes in infected tumor cells (see below). However, with some exceptions, such as those involving detection of a rapidly secreted protein, immunohistochernistry offers a good alternative to in situ hybridization. Furthermore, in situ hybridization to cells or tissue sections on glass slides can be technically difficult and is subject to a high degree of variability in nonexpert hands. Consequently, its application for routine diagnostic purposes has been restricted.
Ploidy Analysis
Two other strategies that provide certain kinds of information about the state of chromosomes in tumor cells involve flow cytometry and image analysis. Application of these techniques is based on the correlation of higher or lower DNA content in cancer tissues, due either to aneuploidy or to increased DNA synthesis and cell division, compared to nonmalignant tissues. For flow cytometry, cells or cell nuclei in suspension are stained with dyes that bind quantitatively to DNA. The stained cells are passed through an instrument called a flow cytometer, which measures the intensity of staining cell by cell. Comparable information can be obtained in tissue sections by using special image analysis microscopes. Both flow cytometry and image analysis measure the relative DNA content of the cells. Populations of tumor cells within the suspension or tissue frequently show a well-defined and distinctive level of DNA content, different from that associated with normal cells. The ratio of this value to that of normal cells is called the DNA index and is an overall estimate of tumor ploidy. However, no information about specific chromosomal abnormalities in the tumor is obtained.
Another assessment that is possible by using these techniques involves numbers of dividing cells. Cells synthesizing DNA have higher DNA content. The portion of cells showing this higher level of DNA is referred to as the S phase fraction. The DNA index and the S phase fraction have been shown to have prognostic value in several different forms of cancer. In addition, quantitative analysis of DNA by flow cytometry or image analysis has the advantage of being applicable to fixed, archival material.
Techniques for Detecting Changes in DNA Structure at the Submicroscopic Level
Ultimately, all of the genetic changes associated with cancer involve changes in the DNA sequence. The most direct way to assess this is through direct nucleotide sequence analysis of DNA. However, at present, DNA sequencing, although greatly simplified in recent years, is still relatively cumbersome and labor intensive, and thus is currently of limited clinical use.
Southern Blot Hybridization
Other means of indirectly detecting changes in DNA sequence are available, however. One of these involves the use of restriction enzymes—proteins that are isolated from a wide variety of bacteria. These enzymes recognize and cut DNA at sites defined by specific sequences, usually four to eight base pairs in length. Treatment of DNA with a restriction enzyme results in a set of DNA fragments of varying size, which can be separated by electrophoresis on agarose gels. Individual restriction fragments within the complex array of differently sized fragments resolved by the gel can be identified by using a technique called Southern blot hybridization (4) (Figure 2-2). The double-stranded DNA fragments within the gel are denatured into
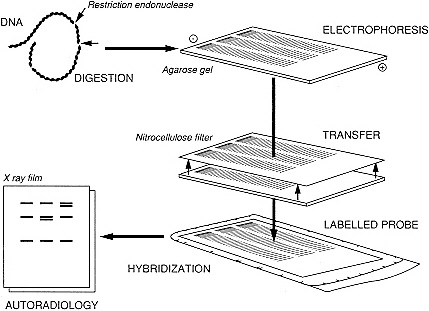
Figure 2-2 Southern blot hybridization. See text for explanation.
single strands by soaking the gel in a basic solution and then transferring the single strands of DNA to a nitrocellulose or nylon membrane, where the DNA fragments become permanently immobilized. Radiolabelled single-stranded probes complementary to the DNA sequence under study are then incubated in solution with the membrane to permit the nucleotide sequences in the probe to find the complementary sequences on the membrane. The membrane is rinsed, dried, and placed against X-ray film for autoradiography. The result is an autoradiogram displaying a band or series of bands whose positions depend on the sizes of the restriction fragments containing the relevant DNA. The sizes of the restriction fragments, in turn, reflect the relative positions of restriction sites in and around the DNA being probed. The feature of this type of test that is important for clinical testing is that rearrangements within or surrounding sequences homologous to the probe produce changes in the size of the fragments and therefore in the pattern of bands.
Southern blot hybridization, perhaps more than any other technique, has opened up the field of molecular diagnosis. Nevertheless, this technique has several limitations. In addition to requiring cloned hybridization probes, the Southern blotting technique usually works best when one knows the target sequence and has some idea where rearrangements within the sequence are likely to occur, so that restriction enzymes and probes with optimal sensitivity can be chosen. Southern blotting detects only a subset of alterations involving the sequence of interest; point mutations will be missed unless a restriction site is destroyed or created. Deletions that completely excise all sequences homologous to a probe may often go unrecognized. By and large, DNA must be extracted from fresh tissue, frozen tissue, or tissue fixed in ethanol, and for maximal sensitivity, radiolabelled probes have thus far been essential. The technique is relatively slow, requiring about one week from receipt of the specimen under the best of circumstances. Despite these disadvantages, Southern blotting has proved invaluable in recent years for screening tissues for various forms of DNA rearrangements, including translocations, deletions, antigen receptor gene rearrangements, and gene amplifications. Furthermore, chemiluminescent probes just now becoming available may eliminate the need for radioisotopes and improve turnaround time by decreasing the duration of blot exposure. Automated technology is also under development.
Southern blotting can also provide useful information about genes whose sequences and precise identity are unknown. This sort of indirect analysis requires only the availability of probes that recognize sequences located near the presumptive gene of interest on the chromosome. The method relies on normal variation in the sequence of DNA in human populations, some of which involves sites recognized by restriction enzymes. A restriction site present in more than 1% and less than 99% of the chromosomes in a population is said to be polymorphic. If DNA from multiple individuals
is cut with an enzyme that recognizes a polymorphic site, two patterns of bands are obtained on Southern blots, one for the chromosome homologue from each parent. The variation is referred to as a restriction fragment length polymorphism (RFLP).
RFLP sites themselves are usually phenotypically silent, but because they may be closely linked physically to genes that cause disease and tend to be co-inherited with these genes, they can serve as markers for inheritance of the disease-causing gene. If, for instance, an RFLP is found to lie near a gene responsible for the familial tendency to a certain form of cancer, the RFLP may be used to track that gene in that family, although the gene itself has not been identified. The use of RFLPs to identify cancer predisposition has many limitations, however. First, sufficient affected and unaffected family members must be available in order to identify the specific RFLP that segregates with the affected phenotype or cancer. Second, the RFLP must be sufficiently polymorphic or variable so that there is a good probability of heterozygosity in key individuals, thus allowing identification of the distinct RFLP inherited from each parent. Third, the RFLP must be closely linked to or within the gene of interest, to reduce the probability of meiotic recombination and separation of the RFLP from that gene. This probability can be reduced by the use of multiple RFLPs flanking both sides of the gene under study. Ultimately, these problems are surmounted only by identification of the affected gene, which allows direct testing for the mutant allele.
RFLP analysis has also been used to identify regions of the genome likely to contain tumor suppressor genes. This is done by comparing the Southern blot obtained from tumor DNA with that obtained from DNA of normal tissue from the same individual. As mentioned earlier, the function of tumor suppressor genes in tumors may be diminished or abolished by acquired mutations. This is frequently accomplished by mutation of one allele, together with deletion of the chromosome carrying the normal allele. In some cases the chromosome carrying the mutant allele is duplicated. Whether or not duplication occurs, only the chromosome carrying the mutant allele remains in the tumor cells; that is, the cells are no longer heterozygous for the mutant allele but have become homozygous. This so-called loss of heterozygosity (LOH) may be detected by the disappearance of RFLPs linked to the deleted chromosome. Certain tumors are recurrently associated with LOH of certain chromosomes or chromosome regions, and it is often assumed that tumor suppressor genes reside within these regions.
Pulsed-Field Gel Electrophoresis
Pulsed-field gel electrophoresis (PFGE) is a method that is closely related to conventional restriction enzyme analysis and is useful for detecting
changes in DNA over large distances (5). This method entails cutting DNA with special restriction enzymes that recognize rare sequences. By using these rare cutters, large restriction fragments are generated that, because of their size (up to several million base pairs), require a special electrophoretic system for separation. In this system, the electric field is applied from either side of the gel in alternating pulses at an angle to the overall direction of migration of the DNA. For reasons that are not well understood, this procedure permits separation of large DNA fragments, which ordinarily tend to aggregate at the top of standard gels. By combining PFGE with Southern blotting, DNA far away from sequences complementary to a specific probe can be investigated relatively quickly. The technique, however, demands intact, unsheared DNA for analysis, preferably from viable cells, and special extraction procedures must be used. Overall, PFGE is labor intensive and technically difficult. It seems likely to be confined to the research laboratory for the foreseeable future.
Polymerase Chain Reaction
Another technique that can be applied to detection of changes in DNA sequence, which has assumed enormous importance in recent years, is the polymerase chain reaction (PCR)(6,7) (Figure 2-3). This technique permits in vitro production of many copies of a defined DNA sequence up to several kilobases long, provided DNA sequences flanking this region on both sides are known. The technique requires the enzyme DNA polymerase, a DNA template such as total cellular DNA, the four nucleotide triphosphates from which DNA is synthesized, a buffered solution containing appropriate ions, and two so-called oligonucleotide primers.
DNA polymerase purified from bacteria is used to copy the DNA during the PCR. An essential feature of this enzyme is that polymerase cannot by itself begin to copy the template at simply any point. Rather, it carries out synthesis by adding nucleotides in a template-directed manner to the end of single-stranded DNA fragments previously hybridized to the template strands. These priming fragments in the PCR usually consist of two short oligonucleotides approximately 15 to 40 nucleotides in length. They are synthesized chemically to complement the known sequences lying on either side of the region to be copied. The two primers are hybridized to opposite strands of the DNA, with their 3' ends (the ends extended by DNA polymerase) oriented to face each other.
DNA is synthesized in repetitive cycles (usually totaling between 20 and 60). At the start of the first PCR cycle, the ingredients for the reaction are mixed together, heated to separate the two strands of the template DNA, cooled to permit the primers to bind to their complementary sites, and then brought to a temperature at which DNA synthesis can occur. At the end of
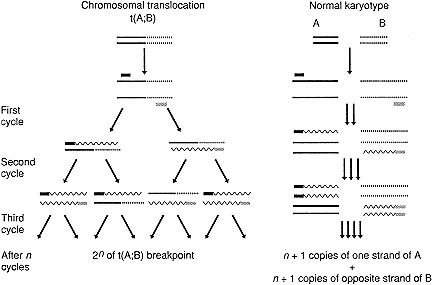
Figure 2-3 The figure illustrates major principles of the polymerase chain reaction for amplification of DNA, contrasting successful amplification of DNA across a chromosomal translocation breakpoint (right) to an unsuccessful attempt to amplify the same DNA from cells lacking the translocation (left). Chromosomal DNA of chromosome A is represented by a solid line; DNA from chromosome B by a dashed line. The two oligonucleotide primers are represented as solid and cross-hatched small rectangles. DNA synthesized during the amplification reaction is represented by a wavy line. The length of these extended strands of newly synthesized DNA is heterogeneous after the first cycle of synthesis, but in fragments containing two partners, the size of the fragments is homogeneous and corresponds to the distance between the primer-related sequences in the original template DNA.
this step, DNA corresponding to the region between the primers is doubled. The cycle is repeated, and the DNA for the region doubles again because DNA synthesized during the earlier cycle becomes template for the next cycle. DNA for the specified region keeps doubling in this way for as many cycles as are desired (sometimes requiring replenishing of reagents for longer reactions). Products accumulated over the cycles consist of complementary fragments of DNA with lengths corresponding to the distance between the sites of hybridization of the two primers in the template DNA. These fragments can be detected rapidly and easily as a band of appropriate size after gel electrophoresis. PCR amplification of DNA has been greatly simplified by the use of heat-resistant DNA polymerase found in Thermus aquaticus, bacteria native to hot springs, and through the development of program-
mable instruments to carry out the repeated thermal cycling in a precisely controlled manner.
Under various circumstances, PCR can detect insertions, deletions, translocations, point mutations, and even gene amplification. It works best on fresh tissue, but can be applied to fixed tissue and tissue that has been embedded in paraffin.
Its advantage and disadvantage are one and the same: its extreme sensitivity. The technique can detect as few as 1 tumor cell in 105 or 106 normal cells, and may prove to be the technique of choice in assessing the effectiveness of cytoreductive therapy. Its principal drawback lies in the fact that contamination of reagents with even one molecule of previously synthesized PCR product can result in a false-positive signal. Use of positive displacement pipets, UV irradiation of reagents, and substitution of deoxyuracil triphosphate (dUTP) for deoxythymidine triphosphate (dTTP), coupled with pretreatment of the reaction mixture with uracil N-glycosylase, an enzyme that degrades uracil residues, may decrease the risk of contamination but does not eliminate it. For these reasons, scrupulous attention to quality control is essential if PCR is to be clinically useful.
Techniques for Detection of Point Mutations in DNA
Several techniques have been described for rapid screening of DNA fragments for point mutations without requiring complete sequence analysis of the fragment. One approach to this problem already touched on is analysis of DNA by cleavage with restriction endonucleases. Since deviation from the recognition sequence for an endonuclease by as little as a single base pair prevents cleavage at that site, failure to cleave a sequence (or, conversely, creation of new cleavage sites at sequences normally differing from restriction sites by one base pair) can be used to identify point mutations. However, this approach has the obvious disadvantage of screening a relatively small amount of DNA with each enzyme used.
A more general approach for detection of point mutations has involved the technique of RNase protection (8). In this technique, a radiolabelled RNA probe for a given sequence is hybridized to target DNA (or RNA), followed by digestion of the hybrid with RNase A and/or RNase T1. Mismatched, single-stranded nucleotides are digested by the RNase, thereby cleaving the probe. Alteration in the probe can be assayed by gel electrophoresis. In many but not all situations, this technique is sufficiently sensitive to detect single nucleotide differences between the probe and the target nucleic acid. Although construction of RNA probes for DNA sequences has been simplified by PCR with primers that incorporate promoters for in vitro transcription of the fragment into RNA (9), RNase protection still requires this extra step compared to other techniques, and some care because of the instability of RNA relative to DNA.
An alternative technique, denaturing gradient gel electrophoresis (DGGE), exploits changes in the local stability of DNA duplexes as a function of nucleotide sequence and the reduced mobility of partially single-stranded DNA in electrophoretic gels (10). In the most frequently used version of this technique, fragments of double-stranded DNA (such as those resulting from PCR amplification of a DNA sequence) are analyzed in gels containing an increasing gradient of chemical denaturants (urea and formaldehyde), which tend to force the two strands of DNA to separate or ''denature.'' Strands of the DNA seem to separate stepwise over stretches of sequence referred to as domains, and the denaturant concentration at which each domain denatures is highly dependent on the sequence within that domain and is affected by even single base pair changes. Because any single-strandedness in the fragment greatly reduces the mobility of that fragment in the gel, a band of partially denatured DNA fragments forms roughly at the position in the gel where the first domain begins to denature. DNA fragments with altered nucleotide sequences can often be distinguished as bands in positions different from those created by fragments with wild type sequences. An advantage of this technique is that DNA can be viewed in the gel under UV light after staining with ethidium bromide, and therefore it does not require radioactivity.
Another, simpler technique relies on the principle of so-called single-strand conformational polymorphism (SSCP) (11, 12). Single-stranded DNA, generated simply by heating double-stranded DNA until the strands separate, will spontaneously anneal to itself, forming a complex tangle of double-stranded stems and single-stranded loops. Single base pair differences can apparently often produce sufficient changes in the overall three-dimensional structure of these tangles that a mutant fragment will migrate differently from a wild type fragment in a polyacrylamide gel. In practice, the technique works with fragments of DNA up to about 500 base pairs in size, but the precise conditions for gel electrophoresis (mostly concerning proper temperature) must be rigorously established for each fragment to be tested.
A final technique for detecting point mutations utilizes preferential modification of mismatched thymidine or cytosine nucleotides in a heteroduplex of probe and target DNA with osmium tetroxide or hydroxylamine, followed by cleavage at the modified nucleotide with piperidine (13,14). If a probe radiolabelled at one end is used, the technique has the advantage of offering a way to rapidly determine the position of the mismatch by measuring the size of the fragment resulting from piperidine cleavage. On the negative side, the chemicals involved in this technique are toxic and potentially explosive.
Techniques Directed at Analysis of RNA
It is possible to study RNA by Northern blot hybridization—a method analogous to that described above for Southern blot analysis. The RNA is
separated on a gel, transferred to a membrane, and analyzed by hybridization with specific probes. In this way, changes in the levels of RNA or in RNA structure due to gene rearrangement can be detected. In practice, however, this technique is of restricted clinical use, because of the relative instability of RNA.
The polymerase chain reaction provides a means to partially circumvent the problem of RNA instability. RNA prepared from tumor can be used as a template for the synthesis of a complementary strand of copy DNA (cDNA) by using an enzyme called reverse transcriptase. The polymerase chain reaction is then carried out as described above. Because of the great sensitivity of this technique, degradation of RNA (and false-negative results) are less of a problem than with Northern blotting; indeed, positive signals have been generated from aspirate smears stored for several years at room temperature. Again, the principal danger lies in the generation of false-positive results due to contamination of reagents with PCR products. False-negative results can be avoided by using controls that amplify any of a variety of ubiquitous genes.
Techniques for Analyzing Changes at the Protein Level
All of the techniques used in clinical testing involve antibodies directed against the protein of interest. In one form of analysis called the Western blot, proteins isolated from tumor are separated by gel electrophoresis, transferred to a membrane, and identified by incubating the membrane with specific antibody. This technique has been used extensively in research laboratories, but has found limited clinical application.
Immunohistochemistry
A method of generally greater utility than Western blotting is performed by incubating a specific antibody directly with tissue sections or cells in suspension. Antibody binding to cells in tissue sections, a technique referred to as immunohistochemistry, can be assessed by using antibody conjugated to fluorescent molecules or with anti-antibodies conjugated to enzymes capable of depositing a colored product at the site of binding. Antibody binding to cells in suspension is usually assessed by using fluorescent antibody and analyzing the fraction of stained cells in a flow cytometer. Although analysis at the level of protein might initially seem somewhat removed from underlying changes in gene structure, in certain respects it represents the best available technique for analyzing genetic changes associated with cancer. Both immunohistochemistry and flow cytometry permit correlation of protein antigens with other cytological features. Immunohistochemistry also permits simultaneous assessment of many morphological characteristics routinely analyzed by light microscopy. Although most ap-
plicable to frozen tissue, immunohistochemistry can often be applied to paraffin-embedded tissue. Moreover, the technique is generally available at present in most pathology departments and can be used on archival material as new antibody reagents are developed, provided the antibody recognizes antigens that are not destroyed by paraffin embedding.
Immunohistochemistry is frequently helpful in determining the lineage of tumors and can be used to evaluate the clonality of many B lymphocyte proliferations, as well as to generate useful information regarding the malignant or benign nature of T cell proliferations (15,16). As such, it is particularly important in the diagnosis of lymphomas and leukemias, with several caveats: (1) aberrant expression of myeloid antigens on lymphoid neoplasms or vice versa—a phenomenon variously referred to as lineage infidelity (17), heterogeneity (18), or promiscuity (19,20)—is seen in a significant minority of acute leukemias, making precise assignment of lineage difficult in this subset of cases; and (2) direct assessment of clonality in T cell proliferations with immunohistochemical reagents is not feasible at present.
Immunohistochemistry can also be valuable for determining the tissue of origin of solid tumors and can provide prognostic information in certain cases. The principal limitation at present is that most genetic changes associated with oncogenesis cannot be assayed by using this technology.

















