
1
Setting the Stage
The prospect of identifying all of our genes raises the hope of understanding and finding ways to alleviate the diseases that afflict humankind. Some diseases are caused by mutations of a single gene; identifying the gene often may be helpful in learning its normal function, and understanding function can greatly accelerate development of therapies to prevent manifestations of these diseases, or even arrest or reverse their course. Conventional therapies, such as drugs or special diets, can compensate for the genetic aberration. Other therapies, based on identification and cloning of the normal gene, can supply the product of the normal gene. Gene therapy might substitute or add a normal gene for the defective one, inactivate a defective gene, or even repair it.
Most single-gene diseases are rare, but some of these rare diseases have more common analogues. Learning more about the basic defect in rare, single-gene forms of hyperlipidemic heart disease, Alzheimer disease, and some cancers, may teach us more about the nature of the defect in the common forms, and might also help us design effective treatments.
Knowing which genes cause or contribute to the occurrence of disease also permits scientists to develop tests to detect people who will suffer the disease, before they experience symptoms, so they can benefit from early intervention (if it is available). Tests for many single-gene disorders already exist, and some of them are described in this report. Tests for detecting genetic predispositions to more common, complex disorders are under development. And even when treatments have not yet been developed, genetic tests can nevertheless help people decide about conceiving or bearing children. In addition to the benefits to be derived from genetic testing, a number of concerns have been raised about the use
of genetic testing. One such concern is that those who are poor or live in rural areas will not have access to testing. The development and widespread use of genetic tests before safe and effective treatments are available have raised fears about the uses of genetic testing—that genetic tests will be imposed while other approaches to alleviating human suffering are neglected. Their use also raises issues about discrimination and privacy—that people found to possess certain genetic characteristics will lose opportunities for employment, insurance, and education. Finally, genetic testing raises worries about inequities and intolerance—that not everyone will share equitably in the benefits of genetic testing, that some will be stigmatized, and that the beauty of human diversity will be denigrated due to a narrowed definition of what is acceptable.
This is not the first time the National Academy of Sciences (NAS) has addressed issues related to genetic testing. Since Genetic Screening: Programs, Principles and Research was published by the academy in 1975 (NAS, 1975), the technology underlying genetic testing has been revolutionized, greatly expanding the number of diseases for which genetic testing will be possible. This has brought new urgency to many of the issues raised in the 1975 report and raised some additional issues as well. In this chapter, we briefly review the technological changes and their implications for assessing genetic risks. We revisit the first NAS report, and consider its applicability today.
RECOMBINANT DNA TECHNOLOGY, GENE MAPPING, AND IDENTIFICATION OF DISEASE-RELATED GENES
During the 1970s, researchers discovered that human DNA (as well as DNA from other organisms) could be cut reproducibly into small segments, each of which could then be rapidly reproduced (cloned) by inserting it into a microorganism. Cloned segments can then be prepared in sufficient quantity to serve as "probes" to find the longer piece of human DNA from which the segment has been cut. Further advances facilitate determination of the chromosome on which the segment of DNA resides.
The availability of DNA probes made it possible to search for individual variation in DNA. Different kinds of DNA variation have been found. Probes can detect such DNA sequence variation among individuals (polymorphisms), and the inheritance of chromosome segments containing polymorphisms can be easily traced from parents to offspring (Botstein et al., 1980). Using DNA sequence variations as "markers," researchers began constructing maps defining their order and spacing along chromosomes (e.g., Donis-Keller et al., 1987). An important application of this genetic mapping technology was the localization at specific chromosomal sites of genes responsible for inherited disorders.
If, for instance, within a large family the relatives who had a genetic disease were found to have one form of a polymorphism significantly more often than blood relatives who did not have the disease, the disease-causing gene (still uni-
dentified) was probably "linked" to that polymorphism. Different families might have polymorphisms on the same chromosome and very close to the marker, or might have different forms of the same polymorphism, but if each of them had an inherited disease that originated from the same gene, they would be "linked" to that same marker. Since the chromosome location of the marker was known (or could be easily determined), the chromosome location of the disease gene was immediately evident. Then the search for the gene itself could begin. There can be a very long gap between gene localization—finding the general location of the gene on the chromosome—and gene identification and isolation. The Huntington disease gene was the first autosomal gene to be localized using new DNA marker techniques (Gusella et al., 1983). Ten years later, it was found (Huntington's Disease Research Collaborative Group, 1993). Many marker genes including those responsible for cystic fibrosis (CF) (Rommens et al., 1989; Collins, 1992), neurofibromatosis (Collins et al., 1989), and a form of colon cancer (Fearon and Vogelstein, 1990) have been isolated using this approach.
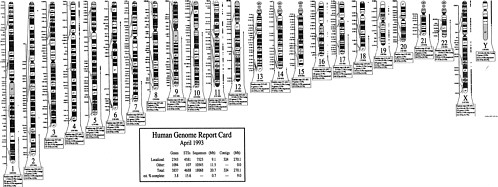
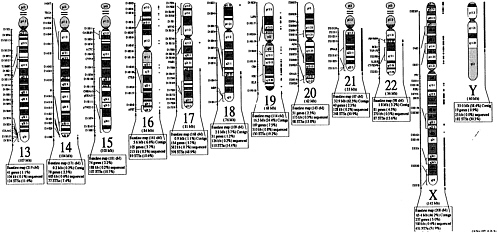
The international effort to map the human genome is accelerating the development of new markers and the construction of detailed linkage maps of the human genome. Disease-causing genes are also being discovered at an accelerating pace (NIH-CEPH Collaborative Mapping Group, 1992; Weissenbach et al., 1992). New technological innovation in mapping and sequencing will lead to the cloning of disease-causing genes and the discovery of alterations in those genes (mutations) that lead to the occurrence of specific disease. As of July 15, 1992, 3,836 polymorphic markers had been localized on specific regions of chromosomes, and 611 disease-related genes had been mapped (Donis-Keller et al., 1987, 1992; see Figure 1-1).
As a result of these discoveries, it is still a very difficult but technically more straightforward matter to localize and identify disease-causing genes for singlegene diseases—those that follow the rules of inheritance established by Gregor Mendel in 1865—provided that families in which the disease occurs are willing to participate in linkage studies and are of sufficient size to study. As the Human Genome Project progresses, genes will be found by means other than their association with a specific disease in such families. Theoretically, there could be as many diseases as there are genes (50,000 to 100,000 human genes by current estimates), although some genes are so essential to embryonic and fetal development that defects in them will result in spontaneous abortion rather than postnatal disease. It is possible that some functioning genes are so nonessential that defects in them will not result in any impairment. As is the case with most single-gene diseases, many of those waiting to be discovered will be rare. There is considerable conservation of gene sequences across species. This allows scientists to find homologous genes in mice, fruit flies, or primates, and to create artificial manifestations in the DNA to mimic the mutation in the human gene. Animal models carrying the actual human gene will permit the study of the normal and abnormal function of the gene in the development of new therapies.
When diseases that are suspected to be gene-influenced do not follow usual Mendelian patterns of inheritance, more than one gene, or a variety of environmental factors to which susceptible individuals are exposed, play a role. This is the case for many common diseases, including several birth defects, some forms of heart disease, hypertension, cancer, diabetes, mental illness, and infectious disorders. Evidence supporting the promise of linkage mapping for common diseases has been found in families in which the disease is inherited as a single-gene dominant disorder. For example, two different genes, each by itself responsible for Alzheimer disease have been mapped (Goate et al., 1991; Schellenberg et al., 1992). In another case, a gene thought to be responsible for only a rare form of colon cancer was later found to be implicated in the more common sporadic form of the disease (Fearon and Vogelstein, 1990). Success of the linkage approach in complex disorders has been tempered, however, by several failures. Earlier reports of linkage in manic-depressive illness in the Old-Order Amish could not be confirmed when new individuals in the family became ill (Egeland and Kidd, 1989); reports of linkage on the X chromosome previously reported for manic depression were recently retracted (Baron et al., 1992).
For disorders with complex etiologies, such as coronary artery disease, identification of a genetic defect in a rare, single-gene form may provide invaluable clues to causation and approaches to therapy in general. This has been demonstrated for familial hypercholesterolemia (Goldstein and Brown, 1989). It is possible, however, that mutations in the gene that is involved in the rare, single-gene forms of other common, complex disorders will not play a role in the complex forms. Moreover, it will be very difficult to use linkage studies to establish the role of genes that are neither necessary nor sufficient to cause the disease (Greenberg, 1993).
Implications of Recombinant DNA Technology for Genetic Testing
Prior to the discovery of recombinant DNA techniques, the determination of a person's risk of harboring genes that could lead to disease in that person, or in her or his offspring, was limited to those diseases for which a clinical diagnosis could be made or for which tests were available to detect an altered gene product (an enzyme or other protein) or a metabolite that accumulated in a readily accessible tissue such as blood, urine, skin (by biopsy), mucosa of the mouth, or hair. In the late 1960s, it proved possible to detect some altered gene products in cells obtained from the amniotic fluid by midtrimester amniocentesis, thus making prenatal diagnosis possible (Fuchs, 1966; Steele and Breg, 1966; Jacobson and Barter, 1967; Hahneman and Mohr, 1968; Nadler, 1968). However, effective treatment was not available for most prenatally diagnosable disorders. In the absence of effective treatment, prenatal diagnosis had to be done at a gestational age at which the mother had the option of aborting the affected fetus safely and legally. The number of diseases that could be diagnosed prenatally increased markedly when it became possible to localize a disease-related gene by linkage studies and
then identify the gene. Based on the same techniques that made localization and identification possible, tests could be developed whose purpose was to show whether mutations capable of causing a specific disease, or conferring susceptibility to its occurrence, were present or absent. Since all of a person's genes are present in all nucleated cells from the moment of conception, the requirements of having an accessible tissue or an active gene (resulting in the appearance of a gene product) were eliminated. For any disease-related gene that has been localized, linkage studies can be done in families in which the disease has already occurred to determine whether other relatives are likely to be affected. For any disease for which the disease-causing gene and, within it, disease-causing mutations have been identified, direct tests for the presence of those mutations can be done on anyone: having a family history is not a prerequisite; entire populations can be screened for the presence of specific mutations.
The development of genetic tests to assess individual risks of diseases followed quickly on the heels of the discovery of disease-related genes and mutations. For most diseases, testing has been confined to families in which the disease has already occurred, even when direct tests for mutations have been developed. This has greatly increased the opportunity for people in these families to learn their risks of disease or of having children with the disease. In the case of sickle cell anemia and thalassemia (autosomal recessive disorders for which carrier screening was possible by "gene product" techniques), direct tests of mutation have permitted safe and reliable prenatal diagnosis. As the Human Genome Project and other research lead to the discovery of more disease-related genes, the scope of testing will expand further. Tests for genetic predispositions to multifactorial disorders will also be possible, and eventually technologies will be available to simultaneously test for hundreds of different disease-causing mutations, either in the same or in different genes.
Other Recent Advances and Their Implications for Genetic Testing
A number of other techniques have been developed recently that also expand the opportunities for genetic testing. The most far-reaching is the polymerase chain reaction (PCR), a technique that permits the rapid amplification (reproduction) of predetermined segments of DNA. By starting with the DNA from as little as one cell, a sufficient amount of material can be replicated within hours to allow the amplified segment to be detected easily. This has greatly simplified genetic testing.
New techniques have also been developed for prenatal diagnosis. Chorionic villus sampling permits prenatal diagnosis as early as 9 weeks of gestation, compared to amniocentesis, which until recently was seldom performed before 16 weeks. Earlier amniocentesis is currently being investigated.
Very recently, successful prenatal diagnosis has been reported before the embryo is implanted in the uterus. The technique, known as preimplantation
diagnosis, involves removing cells at the blastocyst stage and using PCR to amplify the segments of DNA (one from each parent) containing the part of the disease-related gene that contains the disease-causing mutation of interest. If the disease-causing mutation is absent, the embryo(s) is then implanted in the woman's uterus (Simpson and Carson, 1992). Prefertilization determination of whether an ovum contains a specific disease-causing mutation has also been accomplished by polar body analysis (Verlinsky and Pergament, 1984; Verlinsky and Kuliev, 1992). If the ovum does not contain such a mutation, in vitro fertilization is carried out, and the resulting embryo is implanted in the woman's uterus; this technique also involves the use of PCR. These techniques are still considered experimental and are discussed further in Chapter 2.
Recombinant DNA technology has also added new techniques for detecting changes in the number of chromosomes present in a fetus's cells (aneuploidy). The technique involves fluorescent in situ hybridization (FISH) of chromosomespecific probes with cells obtained prenatally (Klinger, 1992). It is far less time-consuming and labor-intensive than the earlier technique of karyotyping, which nevertheless is capable of detecting a much wider range of disorders than FISH.
Empiric observations have revealed that when a woman is carrying a fetus with an extra chromosome (i.e., trisomy), she is likely to have altered concentrations of alpha-fetoprotein (AFP), chorionic gonadotropin, and estradiol in her blood (so-called triple-marker testing for increased risk of abnormalities in the fetus) (Haddow et al., 1992). This has permitted the development of maternal blood tests for these substances as initial screens of pregnant women for increased risk of such fetal abnormalities. Confirmatory tests are necessary for actual diagnosis.
New techniques for prenatal diagnosis are also being evaluated involving fetal cells that are usually present in and can be isolated from the maternal circulation in the first trimester of pregnancy. These techniques are still highly experimental (Bianchi et al., 1991; Elias et al., 1992). Although this technique is noninvasive of the fetus, the scientific, ethical, legal, and social issues associated with other forms of prenatal diagnosis are still present (see Chapters 2 and 4).
Many of the techniques discussed thus far involve gene changes (mutations) that are—with rare exceptions (i.e., mosaicism)—usually present in all cells of the body, having occurred as germline mutations and inherited from one or both parents. On the other hand, there is now evidence that as cells become cancerous they undergo a number of nongermline, noninherited, somatic cell mutations in different genes (Fearon and Vogelstein, 1990). Preliminary work suggests that the presence of these somatic cell mutations may be detected (via PCR) by analysis of fluids in which mutated cells might be shed (e.g., fecal matter in the case of colon cancer), which would serve as an early warning of the development of malignancy (Sidransky et al., 1992).
Advances in ultrasound have resulted in its use to detect structural changes in the fetus, some of which may be associated with genetic or chromosomal abnormalities. Although not a genetic test in itself, ultrasonography is used as an ad-
junct to help detect structural abnormalities. In some countries, ultrasonography is performed on every pregnant woman, and its use is increasingly common in the United States as well. It may be the most frequently used prenatal diagnostic test. The general consensus is that ultrasound should not be used routinely in every pregnancy (NIH, 1984; ACOG, 1988), but that consensus may not reflect actual obstetrical practice today. Recent reviews in the literature have identified some factors affecting reliability in identification of fetal anomalies through ultrasound (Filly et al., 1987; Goldstein et al., 1989); others have reported variability in quality by center and practitioner and error rates of more than 10 percent (Levi et al., 1991; Lancet, 1992). The committee heard testimony that raised its concern about the reliability of interpretation of prenatal ultrasound images and about the consequences of decisions based on such interpretation (see Chapter 2).
Limitations of Genetic Testing
Genetic tests are seldom perfect predictors of clinical risk. No biochemical screening test is sensitive enough to detect all cases, and even current DNA methods cannot detect all possible mutations that cause a specific disease. For example, more than 300 mutations in the CF transmembrane regulator gene have been found that cause CF; most of them are extremely rare. Current technology permits the simultaneous detection of approximately 20 of the most common ones, accounting for about 85 to 90 percent of mutations in whites but a much smaller proportion of mutations in nonwhites (Cutting et al., 1992). Moreover, the frequency of the different mutations for CF can vary within subpopulations; what applies to a northern European population does not necessarily apply to Italian or Jewish populations. The 0.4 to 0.6 percent of normal individuals who carry undetectable mutations will not know—even after testing—that they are at risk, if they mate with another carrier, of having a child with CF. Similar allelic diversity occurs in many other disorders. Thus it becomes important to distinguish between analytic sensitivity, the ability of a test to detect the various mutations it was designed to detect, and clinical sensitivity, the ability of the test to detect all patients who will get, or who have, the disease.
A way could be found around the less-than-perfect sensitivity of current DNA tests in the presence of allelic diversity. The multiple mutations that result in a specific disease all impair the normal expression of the gene. Consequently, a test that measures normal gene expression could tell, indirectly, whether any mutation capable of altering expression is present. Tests of altered gene product antedated DNA-based tests, but they could only be performed when evidence of gene expression could be found in readily accessible tissues. Recent work indicates that minute amounts of mRNA for proteins that are detectable only in specialized tissues are present in accessible tissues such as white blood cells and cultured chorionic villus cells (Chelly et al., 1989; Sarkar and Commer, 1989). The protein could be translated from the mRNA and its structure and function assayed.
Even when a test can detect a mutation capable of causing a single-gene disease, the test may not be able to predict with certainty whether disease symptoms will appear or when they will appear. In such cases of incomplete penetrance, the specific mutation may be a necessary but not a sufficient condition for the disease to become manifest; other conditions must be present as well. These may be mutations at other gene loci or environmental factors; some or all of these other factors may be unknown. As a result, genetic test information on predispositions to a disorder has a potential for falsely labeling persons as being at risk for the disorder.
Many factors may modify the severity of single-gene diseases: the disease can have variable expressivity. The degree of severity (or extent of expressivity) cannot usually be predicted by a genetic test, even a test for a specific mutation; however, some specific mutations tend to be associated with specific levels of severity of the disorder.
Aside from problems that arise as a result of allelic diversity, incomplete penetrance, and variable expressivity, tests for genes implicated in single-gene disorders have other limitations. The accuracy of linkage tests will be impaired when an incorrect diagnosis is made in a relative or when the social father is not, in fact, the biological father. Linkage studies often have some uncertainty attached to them because the DNA marker linked to a disease gene can be separated by recombination. Such recombination events become more frequent as the physical distance between the marker and the disease gene increases. Tests will also be imperfect predictors if the laboratories performing them do not do so correctly and accurately.
Problems of penetrance and expressivity become even greater in testing for complex disorders in which multiple factors, of which the gene being tested is only one, contribute to the causation of the disease. In some of these disorders, a gene at a different locus (or genes at more than one locus) than the one being tested could contribute to causation. This is a form of genetic heterogeneity.
For some disorders, treatments of proven effectiveness will be available, but these treatments could be harmful to people who do not have the genetic disease (such as those in which a test was falsely positive). For many diseases, however, no definitive therapy will be possible when testing becomes available, although identification of the underlying genetic defect is likely to accelerate the discovery of future treatments. The duration of the lag period between testing and treatment capabilities will vary from disease to disease. For some disorders, the lag time will be so long that (1) individuals confronted with having to decide whether to be tested will not have any prospect of effective treatment to benefit them, and (2) they could not postpone having children long enough to see whether a treatment to benefit a prenatally diagnosed fetus will be discovered. Under such circumstances, nonmedical benefits and harms, as well as ethical considerations, dominate the decision about whether testing should be undertaken, both for individuals and for society.
Because of the imperfect nature of genetic tests and the implications of both true positive and false positive test results, as well as false negative results, the understanding of those who offer tests and of the recipients themselves is crucial to appropriate use of genetic testing. It was these issues that prompted the first NAS report on genetic screening.
LESSONS FROM THE PAST
From 1972 to 1975, a committee convened by the National Research Council (NRC) considered problems related to genetic testing. At that time, the most frequent use of genetic tests was in screening programs. That committee defined genetic screening ''as a search in a population for persons possessing certain genotypes that (1) are already associated with disease or predispose to disease, (2) may lead to disease in their descendants, or (3) produce other variations not known to be associated with disease" (NAS, 1975, p. 9). A report on screening had been published by Wilson and Jungner (1968), but genetic aspects were not emphasized in that report. Thus, the NAS committee set out to gather ideas and information with the intention of producing a review of current practices and recommendations for the future. That NAS report focused on two disorders for which screening programs had recently been developed: the testing of newborns for phenylketonuria (PKU), which started in the late 1950s; and screening for sickle cell anemia and carriers, which started in 1971. The 1975 committee quickly perceived that much of the genetic testing then being done was undertaken prematurely and was haphazard or ill-informed. The Tay-Sachs carrier screening program, which started in the Jewish community in Baltimore in 1971, suggested that screening could be organized rationally (Kaback and Zeiger, 1972). The report also considered screening for Tay-Sachs disease and thalassemia. A brief description of early screening programs follows.
Phenylketonuria
Screening for PKU began with the testing of infants' urine for a metabolite of phenylalanine, an amino acid that accumulated in PKU. Within a few years it became apparent that as many as half of infants with PKU were missed by the urine test (Medical Research Council, 1968). It was replaced by a test for phenylalanine in the blood. This test could be performed on a spot of blood obtained from a heel prick before the infant left the hospital nursery. After a short field trial, this test was made mandatory by most states. Some state laws gave parents a right to refuse testing, primarily on religious grounds. In most states, however, screening was done without informing the parents about the test or their right to refuse.
Mandatory screening was based on the belief that restricting the phenylalanine in the diet of infants with PKU would prevent the mental retardation that was
almost invariably associated with the disorder. Scientists had proven that the low-phenylalanine diet promptly lowered serum phenylalanine levels in infants with PKU. However, at the time mass screening began, there was no conclusive evidence that the diet prevented mental retardation. Because of the claims of benefit, it was impossible to mount a randomized controlled trial to determine the effectiveness of the diet. (In retrospect, 14 children detected by screening, and meeting rigorous criteria for PKU, would have been needed in a two-year trial to demonstrate the effectiveness of the diet; Holtzman, 1977.) Only after 10 years did a collaborative trial (in which all infants were treated with the special diet, but to varying degrees) provide convincing evidence of an association between early institution of the low-phenylalanine diet and maintenance of good dietary control on the one hand, and intellectual performance on the other (Committee on Genetics, 1982; N. Holtzman et al., 1986).
In the meantime, it also became apparent that not all infants with elevated phenylalanine had classical PKU. Some infants had variant forms of PKU that resulted in seizures, retardation, and death even with dietary restriction of phenylalanine; fortunately, these variants are very rare (Scriver et al., 1989). In other infants, the elevation of phenylalanine was transient or, if it persisted, was mild and not associated with mental retardation. A few infants who did not actually have PKU died or suffered irreversible damage when they were started—without proper monitoring—on the low-phenylalanine diet, resulting in depletion of their supply of phenylalanine, an essential amino acid (Holtzman, 1970).
A 1970 survey of health department and PKU clinics indicated that 5 to 10 percent of infants with PKU were being missed by screening and were being diagnosed because of mental retardation. Although it is not altogether clear what accounts for some infants being missed, two factors contribute significantly: (1) the inherent lack of sensitivity of the screening test, particularly when it is performed the first day after birth, and (2) human errors in the performance of the test, which include errors in specimen collection, laboratory procedure, or follow-up, such as failure to notify parents or the infant's physician (C. Holtzman et al., 1986). The current practice to reduce the length of stay of newborns in order to reduce hospital costs may well contribute to an increasing rate of false negatives with the PKU screening test.
Thus, screening for PKU had been undertaken widely before either the validity of the test, the quality of the laboratories performing it, or the efficacy or safety of the treatment had been firmly established. As a result, some infants suffered irreparable harm; however, many more infants benefited from early PKU detection and appropriate therapy.
Sickle Cell Anemia and Trait
A more serious set of errors was made in the establishment of sickle cell screening programs in the African-American community in the early 1970s. Some
of these screening programs were established without adequate consultation and education of the affected communities. A test then in common use failed to distinguish individuals with the disease from those who were only carriers. No treatment for sickle cell anemia as effective as the low-phenylalanine diet for PKU was available or on the horizon. The only benefit of early detection for infants with the disease was to ensure that children would receive medical care promptly when they became sick, yet improved access to medical care was not usually part of the programs. Nor was prenatal diagnosis available at that time; the only way parents could avoid having affected children was not to have children at all or to avoid mating with another carrier.
Since 8 percent of African-Americans are carriers of the sickle cell trait, many carriers were detected in screening programs. However, knowledge of having the sickle cell trait had no medical benefit since carriers are healthy. Detection of the trait was appropriate only for reproductive purposes since there is a 1 in 4 risk of having an infant with sickle cell anemia if both parents carry the trait. However, confusion about the significance of carrying the common sickle cell trait (about 1 in 12 African-Americans) and the rare sickle cell anemia (with a frequency of about 1 in 600) often led to stigmatization and discrimination.
In the early 1970s, seven states passed mandatory laws requiring sickle cell screening, while ten others had voluntary laws. Some laws called for newborn screening. Others erroneously regarded sickle cell anemia as an infectious disease and called for screening before a child could enter school. Others required it as a condition of obtaining a marriage license. In at least one state law, as well as in the National Sickle Cell Anemia Control Act of 1972, it was evident that legislators had confused the frequency of the trait (about 1 in 12 African-Americans) with the frequency of the disease of about 1 in 600 (Reilly, 1977). The national act required programs supported with federal funds to be voluntary, not mandatory. Nevertheless, even in the late 1970s, some screening in at least one state was done without informed consent (Farfel and Holtzman, 1984).
The experiences in the sickle cell screening programs of the early 1970s reinforce the need for education before screening and for counseling after screening, and require that participants be assured of the confidentiality of their test results (see Chapter 8). Enthusiasm turned to suspicion as many African-Americans concluded that the intent was to eradicate the sickle cell gene by preventing carriers from reproducing—thereby reducing the birth rate in the black community. These genetic testing programs were perceived by some as genocidal in intent.
Considerable time, effort, and money have been required to overcome these early mistakes. To promote research and develop high-quality programs of screening and counseling, the National Heart, Lung, and Blood Institute provided funding in the 1970s under the national act to establish 10 centers for sickle cell disease research and 25 clinics to carry out education, screening, and counseling in high-risk populations. Under these auspices, protocols for more effective education, screening, and counseling were developed and the criteria for appropriate
management of sickle cell anemia were developed. Early studies by the Centers for Disease Control (CDC) found that the laboratories providing tests often made technical errors and were sometimes doing primary screening with a test that did not distinguish sickle cell trait from the disease. Federal support through the CDC proficiency testing and assistance program helped laboratories to improve the quality of testing. Similarly, federal support for community-based sickle cell programs included education of providers and consumers, improving the understanding of both groups. It was not until 1978 that sickle cell anemia could be diagnosed in amniotic fluid cells obtained by amniocentesis (Kan and Dozy, 1978). Prior to that time, diagnosis was possible on fetal blood, although obtaining it carried much greater risks to the fetus than amniocentesis (Hollenberg et al., 1971). And not until 1986, as the result of a collaborative nationwide randomized controlled trial, was it established that penicillin prophylaxis reduced infant and childhood mortality from sickle cell anemia (Gaston et al., 1986), thereby providing a therapeutic rationale for screening newborns for sickle cell anemia. Although 42 states now screen newborns for sickle cell disease (CORN, 1992), not all infants with the disease receive treatment.
Tay-Sachs Disease and Trait
That community-based programs could be effectively mounted was demonstrated by the development of Tay-Sachs carrier screening. Beginning in Baltimore, this program spread rapidly to Jewish communities across the country and around the world. Unlike sickle cell anemia, whose manifestations and severity vary considerably, the predominant form of Tay-Sachs disease in Jews of eastern European (Ashkenazi) origin was uniformly fatal in early childhood. The basic defect in this form of the disease—the absence of an enzyme—and the ability to identify carriers had been established shortly before screening was contemplated. Prenatal testing was also available—it was possible to detect fetuses with the disorder by assaying amniotic fluid cells for the missing enzyme. This meant that couples in which both partners were found to be carriers could avoid the birth of any child with this uniformly fatal disease without having to forgo childbearing.
Before Tay-Sachs disease screening started, intense discussions of the goals and methods, as well as means of publicizing the program, were held with rabbis, physicians in the community, and other community and religious leaders. Pilot studies were conducted to determine the validity and reliability of the automated serum enzyme assay to be used for screening, and a more definitive confirmatory test (leukocyte assay) was established. Organizers decided that screening would be most relevant to reproductive decision making and, therefore, would be targeted to young married couples; it was later expanded to include unmarried individuals 18 years and older. Screening was held in community sites—primarily community centers, religious schools, and synagogues—medical geneticists and genetic counselors were present, along with community volunteers, to provide on-
|
BOX 1-1 Tay-Sachs Disease: Chevra Dor Yeshorim Program Preventing Tay-Sachs disease by avoiding reproduction between carriers was the option selected by Hasidic Jews, originally in New York City (Chevra Dor Yeshorim Program) (Merz, 1987). This community has a high risk of Tay-Sachs disease and also opposes abortion and contraception. The program is based on the community practice of arranged marriages. When a woman turns 18, or a man,, a blood sample is tested anonymously for Tay-Sachs disease. The laboratory assigns a code number, which it retains (without names) with the results; the laboratory gives the code number but not the result to the person tested. When a marriage is planned, a matchmaker queries the registry by giving the code numbers of the two prospective partners. A couple considering marriage can also query the registry. Only if both partners are carriers will they be told that they should each seek another partner. This system was designed with a relatively high degree of anonymity to avoid possible stigmatization of carriers (Merz, 1987). Originating in New York, the program has spread to communities elsewhere in the United States, Canada, Europe, and Israel. This approach was designed for the particular religious beliefs and customs of ultra-Orthodox Jews. |
site education and respond to questions. Results were mailed personally but, in addition, those with positive tests were usually informed by telephone. Couples in which both partners were found to be carriers were routinely referred for genetic counseling with a genetic counselor. Informed consent was obtained before all testing. The results were kept confidential and were made available only to the individuals tested by use of a self-addressed envelope filled out at testing.
Couples found by screening to be at risk for having a child with Tay-Sachs disease almost always decided to proceed with prenatal diagnostic testing and to terminate affected pregnancies when diagnosed (President's Commission, 1983). By 1992, more than 2,400 pregnancies at risk for Tay-Sachs disease were tested in programs throughout the world; nearly 500 fetuses were found to be affected by the fatal disorder, and virtually all of these pregnancies were terminated (Kaback, 1992). Of particular significance, more than 1,800 healthy offspring have been born to these at-risk couples; many of these children might never have been conceived or brought to term without genetic testing. Since 1971, screening in the Ashkenazi Jewish populations has led to more than a 90 percent reduction in Tay-Sachs disease in that population in the United States and Canada. By contrast, it is the committee's impression that the incidence of sickle cell anemia among African-Americans has not decreased since screening began.
We still have more to learn about education and counseling for a population. Even in the relatively well-educated population being screened for Tay-Sachs, there was occasional confusion about the meaning and implications of test results (Childs et al., 1976; Zeesman et al., 1984; Kaback, 1993). Nevertheless, this
continues to be a highly effective genetic carrier detection effort in which more than a million people have been screened to date. The program has wide acceptance, with little evidence of personal, family, or community stigmatization of individuals found to be carriers or of couples at risk of Tay-Sachs disease in their offspring.
Thalassemia
Thalassemia screening began in 1976, based substantially on the Tay-Sachs disease model (early planning with the relevant communities, laboratory quality control, and prescreening and posttest education and counseling). With the availability of prenatal testing, thalassemia carrier screening has been organized in a number of Mediterranean countries and in communities in other countries with populations that are at high risk for thalassemia. In some of these countries, the frequency of thalassemia, and particularly the need for blood transfusions in patients with the disorder, was so high that a sizable fraction of the population suffered from the disorder and a significant fraction of health resources was consumed in management of the disorder. In Sardinia (Cao et al., 1981, 1989), Cyprus (Angastiniotis, 1990), and among Cypriots in London (Modell et al., 1980), after thalassemia screening, couples were offered prenatal diagnosis and if found to be carrying a fetus with thalassemia, the majority decided to abort; this has resulted in a 10-fold reduction in infants born with this disease. In Sardinia, for example, the incidence of thalassemia has declined from 1 in 250 births to one in 1,200 births since 1974, and because of the educational program and the follow-up screening of families, the majority of persons are now being screened before pregnancy (Cao et al., 1989). This model has also been adapted to high-risk populations in Canada and in the United States (Fisher et al., 1981; Rowley et al., 1984; Scriver et al., 1984).
Important lessons emerged from early screening programs. In PKU, screening was made mandatory at a time when neither the benefits of the therapy for PKU nor the sensitivity of the test had been clearly established. In sickle cell, screening was started hastily without a clear effort to distinguish screening for carriers from screening for disease, as well as without adequate education of the public. When problems such as these were analyzed and worked out in advance, as in Tay-Sachs disease and thalassemia screening, genetic screening programs were developed in a more appropriate way, and implementation raised fewer scientific, ethical, legal, and social issues.
UPDATING THE FINDINGS OF THE 1975 NAS COMMITTEE
The 1975 NAS committee concluded that it would be useful to elaborate some fundamental principles and rules of procedure for genetic testing. In reviewing these 1975 principles, the current Institute of Medicine (IOM) committee
asked whether these principles and rules are still valid for the 1990s and beyond; whether changing conditions necessitate new principles; and whether specific actions are needed to ensure implementation of the principles. To a large extent, these questions guide the discussion in the chapters that follow. As mentioned earlier, the 1975 committee focused on population-based screening, although most of its principles apply equally to other forms of genetic testing.
Aims of Testing and Screening
The aims of testing and screening are multiple. The first is management (i.e., a search for people with treatable genetic disorders that could prove dangerous to their health if left untreated). A second is providing reproductive options to people with high probability of having children with severe, untreatable disease, for whom counseling could be helpful and prenatal diagnosis or abortion of interest, with Tay-Sachs disease as the prototypical example. A third is enumeration of cases (e.g., for some public health purpose such as determining the incidence and prevalence of congenital anomaly). Finally, research is an aim which might (1) involve the testing of hypotheses relating to human physiology or evolution, (2) serve to enumerate the incidence of other diseases, or (3) investigate the feasibility and value of new methods or tests.
(summarized from NAS, 1975)
The aims of screening remain the same, but the potential scope of screening has expanded remarkably as a result of the ability to test at the DNA level, and new aims have been identified. In screening for "management" of disease, newborns may now be screened for up to 11 disorders, depending on the policy of the state in which they are born. However, the treatment for some of these disorders may not always restore normal function (e.g., some cases of galactosemia and maple syrup urine disease), and, in some instances, may prolong life that many would view as of poor quality. In addition, screening of people of various ages for genetic predispositions to disease is on the verge of becoming a reality. If cholesterol screening is viewed as a search for genetic disease (since hypercholesterolemia often has a genetic basis), such screening is already here. As more is learned about the role of somatic cell mutations in the chain of events leading to malignant neoplasms, it is becoming possible to screen individuals for certain precancerous mutations (e.g., p53). All of these types of screening offer benefit through permitting the individual to reduce the risk of severe disease by taking medications, undergoing periodic monitoring, or altering behavior or life-style (see Chapter 2).
Opportunities have also expanded in screening for reproductive options. Maternal serum alpha-fetoprotein (MSAFP) is now widely used to screen pregnant women of all ages to determine if they are at higher risk of carrying a fetus
with a neural tube or other serious birth defect, including increased risk of chromosomal abnormalities. Amniocentesis remains the principal technique for prenatal diagnosis, although chorionic villus sampling can be performed earlier in pregnancy (with a somewhat increased risk to the fetus). To increase a couple's reproductive options as early in pregnancy as possible, new technologies are being devised rapidly. Recently, highly experimental genetic tests have become possible prior to fertilization of the ovum and prior to implantation of the fertilized ovum, although not without the stress, cost, and other issues associated with in vitro fertilization. Work is also in progress in genetic testing of fetal cells isolated from the maternal blood-stream early in pregnancy (Bianchi et al., 1991; Elias et al., 1992).
Multiplex testing—using technologies that will concurrently test for dozens if not hundreds of disease-related mutations—was not considered in 1975 except to the extent that each test would have to satisfy the committee's criteria for screening. The sheer number of tests that could be performed at one time on a single sample raises serious issues of informed consent (see Chapter 8).
Genetic testing is also already being used for nonmedical purposes, such as forensic identification (OTA, 1990a; NAS, 1992). The U.S. Armed Forces Institute of Pathology is now collecting and storing blood samples from every member of the Armed Forces, and the Federal Bureau of Investigation has developed a national DNA profile data bank system for the identification of criminals. At least 17 states maintain DNA samples for criminal identification, and some states (see Virginia, 1990) require a sample for DNA identification as a condition of parole. Such nonmedical uses of genetic information may lead to breaches of privacy and confidentiality (see Chapter 8).
It has been conjectured that genetic testing may be able to predict an individual's intellectual capabilities and aptitudes. The committee doubts that—in the foreseeable future—genetic testing will permit an accurate assessment of a child's specific physical or mental capabilities; these traits are far too complex (in their genetic and environmental interactions) to enable a genetic test to predict them accurately. Consequently, any such use of testing is likely to result in mislabeling of individuals and will result in considerable harm.
Of much greater immediate concern is the use of genetic test results—intended for medical diagnosis or management—for purposes that may not be in the best interest of the person being tested. One such use is specifically to determine insurability or employability. Although this practice is not now widespread (OTA, 1990b, 1992), there is a growing danger that the results of genetic testing, obtained by an individual primarily for purposes of disease management or for reproductive options, may be used by an insurer or employer to deny health or life insurance or employment (see Chapters 7 and 8).
The 1975 committee indicated research purposes as another aim of screening. This report does not deal with such uses, except for investigations, including pilot
programs, undertaken to assess the value of screening for management or the provision of reproductive options.
Criteria for Testing
Tests should be of benefit to the individual tested, and if offered to anyone they should be offered to everyone. Singling out of subpopulation groups for genetic testing raises the potential for stigmatizing such groups. The tests should be reliable and accurate, being reasonably specific and highly sensitive. Results should be communicated quickly and under circumstances that take into account the implications of the news and its impact on the feelings of those receiving it. Standards should be uniform throughout the Nation. Mechanisms to ensure consistency and continuous evaluation should be instituted. Centralization of screening laboratories is desirable to maintain standards. Screening should not be undertaken in the absence of pilot studies or facilities for follow-up.
(summarized from NAS, 1975)
The committee finds itself in strong agreement with these criteria, especially the principle that tests should be of benefit to the individual being tested. This principle has several applications. In newborn screening, it means that testing should be undertaken only to benefit the newborn. In screening for disease management, the interventions that follow the test should benefit those screened, and not be used against them. The committee was concerned that some uses of genetic test information may represent harmful and unwarranted intrusions on individual privacy. For example, in adoption cases, information from genetic screening programs should not be used in a detrimental way to determine suitability as adoptive parents or as a potential adopted child. Obviously, the use of genetic information, as well as other medical information, in adoption is a complex subject, and requires further study (see Chapter 9).
The committee also agrees that there should be equal access to testing, with the further condition that equal access should be provided for people at approximately equal risk of having a genetic disorder. If there are wide discrepancies in risks to subsegments of the population, there would be no need to screen those at lower risk; the committee believes that the mere availability of a genetic test should not mean that it should necessarily be offered to everyone. Such a policy might be difficult to implement, but—if a low-risk group can be delineated—the committee does not believe that it would be necessary to offer them testing. This will help to hold down the costs per case averted at a time when neither the states nor the federal government is as likely to finance an expansion of genetic testing or screening as they were for PKU and sickle cell anemia. Equal access may also depend on reforms in our health insurance system that will either pay for, or reim-
burse for, appropriate genetic tests and follow-up services regardless of an individual's ability to pay (see Chapters 7 and 8).
Quality of Testing
With the expansion of genetic testing, concern about its reliability and validity is greater today than it was in 1975. There are several reasons. First, the proliferation of laboratories and new tests may outpace the establishment of criteria for judging quality and even for deciding when the validity of the test warrants its use clinically. Second, defining what constitutes reasonable specificity and sensitivity is difficult and can be decided only when evidence is available on the performance characteristics of the tests under consideration, and criteria are likely to vary with different types of tests depending on the ratio of benefits to risks. Third, this proliferation of testing sites makes it more difficult to reach all sites with external quality assessment programs, and increases the chance of errors in testing. Fourth, greater concerns surround genetic testing performed to provide reproductive options, given the potential consequences of errors.
Like the earlier committee, we view high laboratory quality and demonstrated safety, quality control, and effectiveness of tests as critical principles with which there can be no compromises. Federal legislation is already in place both for assessing the characteristics of new tests before they are marketed and for assessing the quality of laboratories that are providing clinical services. However, these are not being fully applied to genetic testing. Although centralization of testing facilities could help to maintain standards of testing, that centralization may not be possible as long as companies compete with each other, and in some instances with academic centers, for the testing business. However, centralization is important and attainable for tests for rare disorders, which are less lucrative financially (see Chapter 3).
Conflicts of Interest
The proliferation of tests and extension of testing into commercial laboratories have led to new issues in conflicts of interests. A key new issue is the potential for conflict of interest on the part of those who are in a position to recommend tests. Such individuals, including geneticists and primary care physicians, may have a business or financial interest in laboratories or companies that provide testing and/or hold patents on genes from which royalties are generated through the testing process. The committee believes that all such holdings should be publicly disclosed and strongly discouraged. The American Medical Association recently voted overwhelmingly that it is unethical in most cases for a physician to send a patient for tests to a clinic owned by the physician (AMA, 1992). The committee believes that the same disclosure requirement should apply to genetics
and laboratory personnel who have a financial interest in a laboratory providing genetic tests.
Pilot Studies
This committee also agrees that pilot studies should precede routine testing, and it would emphasize the principle that pilot studies must have prespecified objectives, clear methods, defined end points, and outside evaluation. In the years since the 1975 report, this principle has not always been followed rigorously. In newborn screening, for example, ''pilot studies" imperceptibly became part of routine testing (Holtzman, 1991). Even though pilot studies cannot replicate all aspects of the situation that would ensue if screening were to become routine, pilot programs can be very useful in helping to establish a standard of care. This is especially true as genetic screening becomes incorporated into routine medical practice, which the committee believes will happen increasingly. Medical followup will also become far less of a problem than it has been in past screening programs (organized on a state public health or community-wide basis), except for people without a regular source of medical care (see Chapter 7). However, ensuring adequate patient education and genetic counseling will pose new challenges (see Chapter 4).
Auspices and Settings
The sponsorship of state and local government is essential to provide guidance, facilities, and follow-up. The 1975 study suggested that states or other communities institute commissions with authority to recommend new tests and programs, to monitor those in place, and to ensure standards for education, counseling, follow-up, treatment, and test procedures, thereby mitigating public concern about the many ethical issues that arise.
(summarized from NAS, 1975)
It is in this area that the situation is changing greatly from the time of the earlier recommendations. As already indicated, neither state nor federal government is likely to organize screening, although state health agencies may require private physicians to offer such tests (e.g., the MSAFP screening program in California), and state insurance laws may ensure that charges for genetic tests are reimbursed. But today the concern is no longer that of ensuring that new genetic screening tests will be done, but rather that pretest education and counseling, the offering of tests, their performance, and the counseling and medical interventions that follow positive test results, will be done appropriately. As testing technology becomes more simplified, walk-in testing (e.g., at shopping malls), mail-order kits, and home test kits become real possibilities; because of the importance of
education and counseling and current deficiencies in public understanding, the committee believes that testing at nonmedical sites is inappropriate at this time.
There are not now, nor in the future are there likely to be, sufficient numbers of medical geneticists and individuals trained primarily as genetic counselors to explain genetic tests and test results to all who might want them. Thus, it seems likely that much of genetic testing will be incorporated into the mainstream of medical care and provided particularly by primary care practitioners (family physicians, internists, obstetrician-gynecologists, and pediatricians). More problematic is the lack of time and inclination to provide counseling among primary care practitioners. The committee notes that, in many instances, primary care physicians, by virtue of having continuing contact with their patients, are better suited in several respects to provide counseling than specialists who are likely to first encounter patients only after positive test results have been obtained. Physicians' knowledge of genetics currently has serious deficiencies, and more attention to genetics and genetic testing is needed in medical education (see Chapter 6). Genetic counselors can help in the training of physicians, and in the training of nurses and social workers, who in turn can assist primary care physicians in genetic education and counseling (see Chapters 4 and 6).
Standards of Care
If testing is to be provided primarily by primary care physicians, how will these providers know when to adopt genetic tests? One protection will be afforded by ensuring that new tests and the laboratories performing them have been rigorously examined before they are approved by the appropriate federal agencies (see pilot studies above, Chapters 3 and 9). The committee believes, moreover, that in considering genetic tests, agencies must also consider problems of test interpretation, so that physicians receiving the results have a thorough and understandable explanation of their major implications and limitations. In addition, those providing reimbursement for testing should also require evidence of their safety and validity before reimbursement is provided (see Chapter 7). Of paramount importance is the role of professional organizations, such as the American Society of Human Genetics, American College of Medical Genetics, the American Academy of Pediatrics (AAP), and the American College of Obstetricians and Gynecologists, that can establish expert review panels to collect and review the evidence for deciding whether or not specific genetic tests should be designated "standard of care" and under what conditions. These panels should be guided by the principles enumerated in this chapter and by the criteria for specific types of testing outlined in Chapter 2. Objective, professional input is particularly important as physicians are increasingly bombarded by advertisements and other promotional material from laboratories providing tests or from companies selling various products.
The committee recognizes that successful litigation against physicians for failing to provide prenatal tests (Holtzman, 1989; Andrews, 1992) probably accel-
erated the adoption of some genetic tests and may induce physicians to adopt tests whose limitations are unclear to them. The committee would hope that prospective standard setting—and making physicians aware of such standards—will reduce the chance of poor practices and, consequently, of liability suits.
The committee believes that the standard is for offering the test, not actually providing it, and that no genetic test should be done without the consent of the persons being tested or, in the case of newborns, the consent of their parents, as discussed below and in depth in Chapter 8.
The committee strongly supports continued attention to scientific, ethical, legal, and social issues in genetics at all levels. The committee sees a particular need for advisory bodies—with grass roots consumer representatives—to guide state health departments or legislatures on such issues as deciding when tests should be added to state-run screening programs and to ensure that the offering, testing, and associated education and counseling are always conducted in accord with the principles suggested in this report. The committee also sees the need for continuing national oversight for the evaluation of existing and new genetic tests, and of pilot projects for the use of such tests, to help states decide what tests to adopt, to advise federal agencies with responsibilities related to genetic testing, and to provide broad policy advice on genetic testing (see Chapter 9 for the committee's recommendations).
Age for Testing
The optimal age for testing depends on the aims of the test. If the test is performed for disease management, the time to test is sometime before the age at which treatment must be started in order to be effective. If the test is performed for reproductive counseling, the time to test is when reproduction is being considered.
(summarized from NAS, 1975)
The committee agrees with this principle: there is little point, and possibly some harm, to testing at an age earlier than necessary to prevent irreversible damage. For instance, at the moment, there is considerable controversy about screening children for hypercholesterolemia. It is not clear that screening per se, or even lowering of cholesterol in children, is without harmful effects (Holtzman, 1992). Nor is it clear that lowering cholesterol in childhood confers any additional benefit of reducing the risk of future coronary artery disease over lowering cholesterol in early adulthood. However, getting accustomed to a prudent diet relatively early may be an advantage.
The committee rejects the assertion that the timing of screening should be determined primarily by when people come for health care and much prefers reform in the health care system to improve access so that many more people will come for care at optimal times. Because many women who contemplate pregnan-
cy will not come for care until they are pregnant, some have argued that there is little point in providing carrier screening until pregnancy. The committee rejects this notion. First of all, some women will come for care too late in pregnancy to obtain any benefit from carrier testing in that pregnancy (see Chapter 2). Second, by delaying screening until pregnancy, women are deprived of options other than prenatal diagnosis and abortion. Nevertheless, when testing must first be done in pregnancy, it should be done in accord with the principles and criteria stated in this report.
Education of the Public
Screening programs cannot fulfill their aims unless the public is aware of the purpose of the test, the disease it is intended to detect, its availability, its benefits to individuals, and its limitations.
(summarized from NAS, 1975)
The principle of having an informed public takes on added importance as the scope of genetic testing increases. A recent survey (March of Dimes, 1992) shows that whereas members of the public have strong opinions about both genetic testing and gene therapy, they report little knowledge or understanding of either. Even when sufficient information is provided when tests are being offered, people with little familiarity with genetics will find it difficult to understand all of the ramifications. Informed decision making will be helped by improved teaching of genetics, probability, human variation, and ethical issues, beginning in the elementary grades, and increased attention to the issues in genetic testing in the media, so people will be better prepared to make informed decisions (see Chapter 5).
Ethical Issues
People should know that their presence at the place of testing and the results of testing will be held in confidence. With this knowledge, people should be free to decide whether or not they want to be tested or screened. The only possible exception is newborn screening leading to effective treatment "if it were found that nonmandatory screening leaves many babies unscreened because of parental noncooperation or physicians' ignorance or oversight" (NAS, 1975, p. 191). People asking for tests should understand clearly that the test will be done only for the intended purposes and that there is no element of discrimination or of eugenic purpose.
(summarized from NAS, 1975)
The expansion of testing heightens the importance of these ethical principles and the need to address others. In fact, much of this report is an exposition of
what is needed for the principle of autonomy, in particular, to be taken seriously. The chapters on genetic counseling (see Chapter 4) and public education (see Chapter 5) emphasize the need for effective education before testing, which in turn depends on the client's ability to understand as much as possible about the basic concepts of genetics and probability (see Chapter 4). The instrument for ensuring the principle of autonomy is informed consent: whether or not a person should be tested is for him or her to decide after giving explicit consent based on adequate information. This is especially important in the case of reproductive choices and in prenatal diagnosis, where the growing range of technological options associated with genetic testing is placing increasing pressures on women to make difficult and unprecedented decisions (see Chapters 2, 4, and 9). Individuals also need to be assured that the results of a test will not be disclosed to others without their explicit permission, although there may be rare occasions when such disclosures can be justified (see Chapter 8).
Allocation of Resources
It is becoming common in health care to make decisions regarding programmatic support and reimbursement based on cost-benefit determinations. The committee explored cost-benefit and cost-effectiveness issues to a limited degree. There are troubling implications of cost-benefit analysis of genetic testing at this stage in the development of such testing, especially when effective treatment exists for relatively few diagnosable disorders. The implication of such analysis—that if an economic benefit were to be established, testing should be encouraged—violates the principle of patient autonomy, with particularly grave consequences in testing for reproductive options.
Most—but certainly not all—people who undergo prenatal testing have chosen to avoid having children with very severe disorders (especially those that cause extreme retardation or early death) without any degree of directiveness. However, some degree of coercion would probably be needed to gain widespread testing for carriers of defects that are less severe, but entail high costs (perhaps because survival is longer). It will, moreover, be difficult for people to agree on what is severe. If directiveness is accepted for any form of testing for which a safe, effective, and widely accepted treatment is not available, this compromises a critically held tenet: patient autonomy. The decision to support or reimburse genetic tests should be based on how well they meet the criteria and principles stated here and in Chapters 2 and 8, not on economic considerations. The committee believes that cost-effectiveness analyses may be appropriate when safe, effective, and widely accepted treatment is available, but additional research is needed on how to apply these techniques appropriately to genetic testing issues (see Chapter 9).
Recognition of Human Diversity and Respect and Tolerance for People with Disabilities
The committee heard powerful and moving testimony from individuals with disabilities who believe that genetic testing and the consequent increase in abortion promote the premise that individuals with disabilities (whether tests exist for early detection or not) have less intrinsic worth. They pointed out that many people with disabilities lead full and productive lives and that society's negative view of disabilities is sometimes of greater harm to them than the disabilities themselves (Waxman, 1992). Some people with disabilities would resist any expansion of genetic testing. The committee recognizes the weight of these concerns and urges broad public education to dispel myths about people with disabilities—genetic or otherwise—and to reduce barriers to their participation in society (see Chapter 5). The committee rejects the notion of restricting any expansion of genetic testing based on the concerns of persons with disabilities that the technologies are inherently harmful. Such a restriction would undermine individual autonomy as much as the view that people should be urged or forced to use genetic tests and abortion to prevent disabilities. Nevertheless, the concerns of persons with disabilities are critical ones for our society. The committee is concerned that society may be moving closer to adopting the view that people should be urged or forced to use genetic tests and abortion to prevent disabilities; steps must be taken to counteract this tendency by decreasing pressures to test and increasing education and understanding of disabilities. If testing becomes widespread, efforts to urge people to undergo genetic testing might engender a greater intolerance for persons with disabilities—even though most disabilities are not the result of genetic causes. Avoiding the social pitfalls of intolerance based on genetic testing will require continuing vigilance.
REFERENCES
American College of Obstetrics and Gynecology (ACOG). 1988. Ultrasound in Pregnancy (Technical Bulletin No. 116), Washington, D.C.
American Medical Association (AMA). 1992. Resolution of the AMA House of Delegates on Physician Ownership and Self-Referral. December 12, 1992.
Andrews, L. 1992. Torts and the double helix: Malpractice liability for failure to warn of genetic risks. 29 Houston Law Review 149.
Angastiniotis, M. 1990. Cyprus: Thalassemia programme. Lancet 336:119-120.
Baron, M., et al. 1992. Diminished support for linkage between manic depressive illness and X-chromosome markers in three Israeli pedigrees. Nature Genetics 3:49-55.
Bianchi, D., et al. 1991. Fetal cells in maternal blood: Prospects for non-invasive prenatal diagnosis. Paper presented at the International Congress of Human Genetics, Washington, D.C.
Botstein, D., et al. 1980. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. American Journal of Human Genetics 32:314-331.
Cao, A., et al. 1981. Prevention of homozygous beta-thalassemia by carrier screening and prenatal diagnosis in Sardinia. American Journal of Human Genetics 33:592-605.
Cao, A., et al. 1989. The prevention of thalassemia in Sardinia. Clinical Genetics 36:277-285.
Chelly, J., et al. 1989. Illegitimate transcription: Transcription of any gene in any cell type. Proceedings of the National Academy of Sciences USA 86:2617-2621.
Childs, B., et al. 1976. Tay-Sachs screening: Motives for participating and knowledge of genetics and probability. American Journal of Human Genetics 28:537-549.
Clow, C., and Scriver, C. 1977. The adolescent copes with genetic screening: A study of Tay-Sachs screening among high school students. In Kaback, M. (ed.) Tay-Sachs Disease: Screening and Prevention. New York: Liss.
Collins, F. 1992. Cystic fibrosis: Molecular biology and therapeutic implications. Science 256(5058):774-779.
Collins, F., et al. 1989. Genetic and physical maps come into focus. American Journal of Human Genetics 44(1): 1-5.
Committee on Genetics. 1982. New issues in newborn screening for phenylketonuria and congenital hypothyroidism. Pediatrics 69:104-106.
Council of Regional Networks for Genetic Services (CORN). 1992. Newborn Screening Report: 1990 (Final Report, February 1992) . New York: CORN.
Cutting, G., et al. 1992. Analysis of four diverse population groups indicates that a subset of cystic fibrosis mutations occur in common among Caucasians. American Journal of Human Genetics 50(6):1 185-1194.
Donis-Keller, H., et al. 1987. A genetic linkage map of the human genome. Cell 51:319-337.
Donis-Keller, H., et al. 1992. A genetic linkage map of the human genome. Science 258:67-86.
Egeland, J., and Kidd, K. 1989. Re-evaluation of the linkage relationship between chromosome 11 p loci and the gene for bipolar affective disorder in the Old Order Amish. Nature 342(6247):238-243.
Elias, S., et al. 1992. First trimester prenatal diagnosis of trisomy 21 in fetal cells from maternal blood. Lancet 340:1033.
Farfel, M., and Holtzman, N. 1984. Education, consent, and counseling in sickle cell screening programs: Report of a survey. American Journal of Public Health 74:373-375.
Fearon E., and Vogelstein B. 1990. A genetic model of colorectal tumorigenesis . Cell 61:759-767.
Filly, R., et al. 1987. Fetal spine morphology and maturation during the second trimester. Journal of Ultrasound in Medicine 6(11):631-636.
Fisher, L., et al. 1981. Genetic counseling for beta-thalassemia trait following health screening in a health maintenance organization: Comparison of programmed and conventional counseling. American Journal of Human Genetics 33:987-994.
Fuchs, F. 1966. Genetic information from amniotic fluid constituents. Clinical Obstetrics and Gynecology 9:565.
Gaston, M., et al. 1986. Prophylaxis with oral penicillin in children with sickle cell anemia. New England Journal of Medicine 314:1593-1599.
Goate, A., et al. 1991. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 348:704-706.
Goldstein, J., and Brown, M. 1989. Familial hypercholesterolemia. Pp. 1215-1245 in Scriver, C., et al. (eds.) The Metabolic Basis of Inherited Disease (6th edition). New York: McGraw Hill.
Goldstein, M. et al. 1977. Health behavior and genetic screening for Tay-Sachs disease: A prospective study . Journal of Social Sciences and Medicine 11:515-520.
Goldstein, R., et al. 1989. Sonography of anencephaly: Pitfalls in early diagnosis. Journal of Clinical Ultrasound 17(6):397-402.
Greenberg, D. 1993. Linkage analysis of "necessary" disease loci versus "susceptibility" loci. American Journal of Human Genetics 52:135-143.
Gusella, J., et al. 1983. A polymorphic DNA marker genetically linked to Huntington's disease. Nature 306:234-238.
Haddow, J., et al. 1992. Prenatal screening for Down's syndrome with use of maternal serum markers. New England Journal of Medicine 327:588-593.
Hahneman, N., and Mohr, J. 1968. Genetic diagnosis in the embryo by means of biopsy from extraembryonic membrane. Bulletin of the European Society of Human Genetics 2:23-29.
Hampton, M., et al. 1974. Sickle cell "non-disease." American Journal of Diseases of Children 128:58-61.
Hollenberg, M., et al. 1971. Synthesis of adult hemoglobin by reticulocytes from the human fetus at midtrimester: Possible applications to prenatal detection of sickle cell anemia and other disorders. Science 171:689-702.
Holtzman, C., et al. 1986. PKU newborn screening in descriptive epidemiology of missed cases of phenylketonuria and congenital hypothyroidism. Pediatrics 78(4):553-558.
Holtzman, N. 1970. Dietary treatment of inborn errors of metabolism. Annual Review of Medicine 21:335-356.
Holtzman, N. 1977. Anatomy of a trial. Pediatrics. 60:932-934.
Holtzman, N. 1989. Medical professional liability in screening for genetic disorders and birth defects. In Rostow, V., and Bulger, R. (eds.) Medical Professional Liability and the Delivery of Obstetrical Care. Vol. II: An Interdisciplinary Review. Washington, D.C., National Academy Press.
Holtzman, N. 1991. What drives neonatal screening programs? New England Journal of Medicine 325:802-804.
Holtzman, N. 1992. The great god cholesterol. Pediatrics 89(4):686-687.
Holtzman, N., et al. 1986. Effect of age at loss of dietary control on infant performance and behavior of children with phenylketonuria. New England Journal of Medicine 314:593-598.
Huntington Disease Collaborative Research Group. 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971-983.
Jacobson, C., and Barter, R. 1967. Intrauterine diagnosis and management of genetic defects. American Journal of Obstetrics and Gynecology 99:769.
Kaback, M. 1992. In Scriver, C., et al. (ed.) The Metabolic Basis of Inherited Disorders. New York: Liss.
Kaback, M., and Leonard, C. 1971. Control studies in the antenatal diagnosis of human genetic-metabolic disorders. In Harris, M. (ed.) Early Diagnosis of Human Genetic Defects: Scientific and Ethical Considerations. Fogarty International Center Proceedings 6:169-178.
Kaback, M., and Zeiger, R. 1972. Practical and ethical issues in an adult genetic screening program: The John F. Kennedy Institute Tay-Sachs Program. In Hilton, B. (ed.) Ethical Issues in Human Genetics. New York: Plenum Press.
Kaback, M., et al. 1986. Tay-Sachs disease: A model for the control of recessive genetic disorders. Pp. 248-262 in Motulsky, A., and Lenz W. (eds.) Birth Defects. Amsterdam: Excerpta Medica.
Kan, Y., and Dozy, A. 1978. Antenatal diagnosis of sickle cell by DNA analysis of amniotic fluid cells. Lancet 2:910-912.
Klinger, K. 1992 (published in 1994). New clinical and laboratory procedures: Fluorescence in situ hybridization. In Fullarton, J. (ed.) Proceedings of the Committee on Assessing Genetic Risks. Washington, D.C.: National Academy Press.
Lancet. 1992. Screening for fetal malformations (editorial). 340:1006-1007.
Levi, S., et al. 1991. Sensitivity and specificity of routine antenatal screening for congenital anomalies by ultrasound. Ultrasound in Obstetrics Gynecology 1:102-110.
March of Dimes Birth Defects Foundation News Release. 1992. White Plains, N.Y. September 29.
Massarik, F., et al. 1977. Community-based genetic education, communication channels and knowledge of Tay-Sachs disease. Pp. 353-366 in Kaback, M. (ed.) Tay-Sachs Disease: Screening and Prevention: Progress in Clinical and Biological Research. Vol. 18. New York: Alan Liss.
Medical Research Council. 1968. Present status of different mass screening procedures for phenylketonuria. British Medical Journal 4:7-13.
Meryash, D., et al. 1981. Prospective study of early neonatal screening for phenylketonuria. New England Journal of Medicine 304:294.
Merz, B. 1987. Matchmaking scheme solves Tay-Sachs problem. Journal of the American Medical Association 258:2636-2639.
Modell, B., et al. 1980. Effect of introducing antenatal diagnosis on reproductive behavior of families at risk for thalassemia major. British Medical Journal 280:1347-1350.
Nadler, H. 1968. Antenatal detection of hereditary disorders. Pediatrics 42:912.
National Academy of Sciences (NAS). 1972. Sickle Cell Disease in the Armed Forces. Washington, D.C.: NAS.
National Academy of Sciences (NAS). 1975. Genetic Screening: Programs, Principles and Research. Washington, D.C.: NAS.
National Academy of Sciences/National Research Council (NAS). 1992. DNA Technology in Forensic Science. Washington, D.C.: National Academy Press.
National Institutes of Health Centre d'Etude du Polymorphisme Humain (NIH/CEPH) Collaborative Mapping Group. 1992. A comprehensive genetic linkage map of the human genome. Science 258:67-86; 148-162.
National Institutes of Health (NIH) Consensus Conference. 1984. Diagnostic Ultrasound Imaging in Pregnancy. Public Health Service. U.S. Department of Health and Human Services. NIH Publication No. 84-667. Washington, D.C.: U.S. Government Printing Office.
Office of Technology Assessment (OTA). 1990a. Genetic Witness: Forensic Uses of DNA Tests. OTA-BA-438. U.S. Congress. Washington, D.C.: U.S. Government Printing Office.
Office of Technology Assessment (OTA). 1990b. Genetic Screening in the Workplace. OTA-BA-456. U.S. Congress. Washington, D.C.: U.S. Government Printing Office.
Office of Technology Assessment (OTA). 1992. Cystic Fibrosis and DNA Tests: Implications of Carrier Screening. OTA-BA-532. U.S. Congress. Washington, D.C.: U.S. Government Printing Office.
President's Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research. 1983. Screening and Counseling for Genetic Conditions. Washington, D.C.: U.S. Government Printing Office.
Reilly, P. 1977. Genetics, Law and Social Policy. Cambridge, Mass: Harvard University Press.
Rommens, J., et al. 1989. Identification of the cystic fibrosis gene: Chromosome walking and jumping. Science 245:1059.
Rothman, B. 1992 (published in 1994). Early prenatal diagnosis: Unsolved problems. In Fullarton, J. (ed.) Proceedings of the Committee on Assessing Genetic Risks. Washington, D.C.: National Academy Press.
Rowley, P., et al. 1984. Screening and genetic counseling for beta-thalassemia trait in a population unselected for interest: Comparison of three counseling methods. American Journal of Human Genetics 36:677-689.
Sarkar, G., and Commer, S. 1989. Access to a messenger RNA sequence or its protein product is not limited by tissue or species specificity. Science 244:331-334.
Schellenberg, G., et al. 1992. Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science 258:668-671.
Scriver, C., et al. 1984. Beta-thalassemia disease prevention: Genetic medicine applied. American Journal of Human Genetics 36:1024-1038.
Scriver, C. 1989. Phenylketonuria. In Scriver, C., et al. (ed.) The Metabolic Basis of Inherited Disease (6th ed.). New York: McGraw Hill.
Sidransky, D., et al., 1992. Identification of ras oncogene mutations in the stool of patients with curable colorectal tumors. Science 256:102-105.
Simpson, J., and Carson, S. 1992. Preimplantation genetic diagnosis (editorial). New England Journal of Medicine 327(13):951-953.
Stamatoyannopoulos, G. 1974. Problems of screening and counseling in the hemoglobinopathies. Pp. 268-276 in Motulsky, A., and Lenz, W. (eds.) Birth Defects. Amsterdam: Excerpta Medica.
Steele, M., and Breg, W. 1966. Chromosome analysis of human amniotic fluid cells. Lancet 1:383.
Verlinsky, Y., and Kuliev, 1. (eds.) 1992. Pre-Implantation Diagnosis of Genetic Diseases. New York: Alan Liss.
Verlinsky, Y., and Pergament, E. 1984. Preimplantation diagnosis by embryonic biopsy. American Journal of Human Genetics 36:199S.
Virginia Code Annotated. 1990. Section 19.2-310.6.
Waxman, B. 1992 (published in 1994). Human Genome Program: A disability perspective. In Fullarton, J. (ed.) Proceedings of the Committee on Assessing Genetic Risks. Washington, D.C.: National Academy Press.
Weissenbach, J., et al. 1992. A second-generation linkage map of the human genome. Nature 359:794-801.
Wilson, J., and Jungner, Y. 1968. Principles and practice of mass screening for disease. Bol. Oficina Sanitaria de Panama 65(4):281-293.
Zeesman, S., et al. 1984. A private view of heterozygosity: Eight-year follow-up study on carriers of the Tay-Sachs gene detected by high school screening in Montreal. American Journal of Human Genetics 18:769-778.





























