
FOLD, SPINDLE, AND REGULATE
How Proteins Work
by David Holzman
Protein molecules are the building blocks of living systems. Cartilage, skin, hair, nails, and eyeballs are formed from the same basic substance. So are the shell of a snail, the wings of a beetle, feathers, the horn of a rhinoceros, and most everything else that makes up the bodies of creatures. In addition, the molecular machines that perform, or catalyze the building, tearing down, and maintenance work that keeps all organisms functioning are virtually all made of protein. There are literally hundreds of thousands of molecular tools and machines in a human body.
Not surprisingly, then, an understanding of how proteins work could lead to a host of useful applications. The symptoms of genetic disease are caused by defects in proteins. For example, sickle cell anemia results from a defect in the blood-transporting hemoglobin molecule. "If we could manipulate proteins, we could fix genetically defective proteins," says Tom Alber, an enthusiastic young molecular biologist who was then at the University of Utah and who presented his research
at the 1992 "Frontiers of Science" symposium.
But this, said Alber, now at the University of California, Berkeley, is only the beginning of the benefits that could accrue. "We could design proteins that are useful to people but don't necessarily occur in nature." For example, pharmaceuticals that require no refrigeration could bring the benefits of modern medicine to the world's isolated and impoverished populations. Novel catalysts could be designed to mediate chemical reactions that are not found in biological systems, reactions that might be useful in the food or pharmaceutical industries.
THE IMPORTANCE OF THREE-DIMENSIONAL STRUCTURE
Although evolution has been building with proteins for 600 million years, scientists have only begun to understand the rudiments of how these substances work, and years or even decades may elapse before engineers will be able to systematically design proteins for specific tasks.
Three-dimensional structure determines function, Alber explained. Enzymes—the catalysts of living substances—fit their substrates as precisely as keys fit locks. For example, it is shape that allows the different enzymes that reside in the gastrointestinal tract to catalyze the disassembly of food molecules into the building blocks that the body uses for nourishment. But there are different enzymes to break down different types of protein and still others to dismantle starch into its constituent sugar molecules.
And it is shape that prevents the starch-digesting enzymes from allowing us to eat grass or bark. Cellulose, the woody material that prevents humans from grazing with the cattle, and from dining with termites on old, dead trees, is composed of chains of sugar molecules, just like starch. The difference between a two-by-four and potatoes or pasta is largely the result of slight differences in the way the sugar molecules are strung together. These slight differences render cellulose indigestible by starch-cleaving enzymes.
Like enzymes, cells of the immune system that adhere to invading microbes also fit their targets with the specificity of lock and key. Recognition is so precise that there are different antibodies for each disease.
"The problem [is] where does the structure come from?" says Alber. "Our work is aimed at understanding the architectural principles that
determine the shapes of proteins. We're starting to understand some of the principles that govern shape.''
ANATOMY OF PROTEINS
To illustrate the magnitude of the challenge, Alber projected a diagram of the cellular catalyst ATCase, a large protein consisting of roughly 30,000 atoms. Like many proteins, its convoluted topology is roughly as complex as a tangle of telephone wire, yet it has the structural grace of a suspension bridge or a bicycle wheel.
The structure of proteins is maintained by chemical bonds that form within proteins. Sometimes these are covalent bonds, the powerful forces that bind atoms together to form molecules, but more often they are the much weaker forces such as electrostatic charges that cause some atoms or molecules to associate with one another in solution. Then there are the water-hating side chains that also attract one another and—surprise—repel water. These are also referred to by scientists as "hydrophobic" or just plain "greasy." The difference between covalent bonds and the other forces is analogous to the difference between the strength of the ties of family and friendship.
Both the covalent bonds and the various weaker forces are properties of the building blocks of proteins, small molecules called amino acids. Proteins are chains of amino acids, which are strung together sort of like poppet beads. But instead of uniform inert beads, there are 20 different kinds of amino acids that occur naturally as well as some synthetic ones. The various forces within proteins are properties of the side chains of amino acids.
It is possible to melt the structure of ATCase by adding urea to the solution containing the protein. Melting overcomes the forces between the chemical groups, destroying ATCase's structure, so that the protein chain now flops around like boiling spaghetti. But remove the urea and the chemical groups pull the protein back into its proper shape.
THE SEARCH FOR STRUCTURE: X-RAY CRYSTALLOGRAPHY AND NUCLEAR MAGNETIC RESONANCE
Christian B. Anfinsen was the first to try this experiment, on the enzyme ribonuclease, in the late 1950s. That the protein regained its shape and its natural enzymatic activity in minutes suggested that all the information that directs folding is contained in the amino acid sequence and not in templates, such as the DNA template that directs the replication
of DNA and the transcription of RNA from DNA. For this work, Anfinsen received the Nobel prize.
Theoretically, then, it should be possible to predict a protein's structure from its amino acid sequence. In fact, to researchers who study protein folding, this is the holy grail, and hundreds are currently attempting to discover the rules that would make this possible. But so far, such efforts have proven frustrating, to put it mildly. This is not surprising.
For a protein composed of just 100 amino acids, a rather small protein, there may be as many as a google (10100) alternative structures that the protein could form, more structures than the number of atoms in the universe, Alber explained. It is amazing, he said, that the protein itself finds the right confirmation, "spontaneously, in a few milliseconds," even though a random search through all possible structures "would take longer than the age of the universe."
To understand how a protein folds, it is necessary to know that protein's three-dimensional structure. Until recently, piecing structure together was such a difficult process—impossible in some cases, taking years in others—that this goal was a dream. Now new developments in protein imaging technologies, x-ray crystallography, and nuclear magnetic resonance (NMR) have enabled researchers to piece structures together within a year, where formerly it could take longer than a decade. Recombinant DNA techniques have made it possible to perform experiments that are beginning to yield some rules of protein folding. And with this information coming in, the dream of predicting three-dimensional structure from amino acid sequence is becoming a tantalizing possibility on the distant horizon of the field.
X-ray crystallography is an ancient technology by scientific standards, dating back to the first half of the century. Rosalind Franklin's crystallographic work was vital to cracking the structure of DNA, and many observers believe that for this contribution she should have shared the Nobel prize with James Watson and Francis Crick. This alleged slight may have had to do with prevailing attitudes about gender.
Crystallography is often the only way to figure out the molecular structure of a protein. The reason is simple. At best, crystallography provides a picture of the entire protein. It accomplishes this by revealing the location of electron densities throughout the protein. These correspond to chemical groups on amino acids.
Nonetheless, the technique frequently does not succeed so marvelously. There are built-in errors of about 5 percent. Furthermore, usually parts of the protein crystallize poorly or not at all, so that the corresponding electron densities leave no signature in the diffraction pattern.
Because of these limitations, structures derived from x-ray diffraction patterns are prone to error, and results are considered uncertain unless researchers know the sequence, which they can use to verify a structure.
But recent advances have made the technique much easier to use and more successful. New technologies have reduced the time it takes to collect a set of x-ray diffraction data from as much as a month to less than a day; they have greatly increased accuracy as well. No longer must machines distinguish approximately among shades of diffraction spots on photographic paper. Electronic film can literally count photons. Whereas photographic film provided a dynamic range of accuracy of 3 to 5, the modern devices are accurate over a dynamic range of 100,000.
Compared to x-ray crystallography, NMR spectroscopy has a few advantages and a variety of disadvantages. The big advantage is that to use NMR it is unnecessary to crystallize protein, which is an arduous and often impossible undertaking. Moreover, portions of proteins that do not crystallize are often important for activity, and, not only can NMR detect these, but it can follow their movements. On the down side, the largest proteins that NMR can decipher are an order of magnitude smaller than for crystallography.
Like x-ray crystallography, NMR has undergone dramatic advances in the last 5 years or so. These advances have made the technique easier to use and more powerful. The size of the proteins that NMR can study has increased from 80 amino acids to 150 to 200 residues.
NMR detects atoms by magnetizing them. Of atoms that occur naturally in amino acids, only hydrogens can be magnetized. It is possible to set up NMR so that it detects atoms in pairs, because one atom can magnetize another provided the two are within several chemical bond lengths of each other. When one hydrogen is a member of more than one pair, researchers can begin to get spatial information.
Though hydrogens are the most abundant atoms in protein, they are not structural elements in its backbone. If a spinal column represents this backbone, and each vertebra represents an amino acid, hydrogens are analogous to appendages to the vertebrae that do not contribute to the connections between them. For reasons related to this, it is difficult for researchers using hydrogen-based NMR to "walk"—in the parlance of that technique—down the backbone from one amino acid to the next. Furthermore, the differences in the NMR signals of hydrogen are so small that the number of individual hydrogens that can be distinguished in a single protein is small.
However, in the mid to late 1980s, researchers began inserting
isotopes of carbon and nitrogen into proteins. These isotopes made NMR more powerful because they are structural atoms in protein, which made it easy to walk the length of the chain. In addition, since the isotopes transferred their magnetic spin to other atoms more efficiently, it became possible to magnetize triplets, even quadruplets of atoms, resulting in obtaining bigger chunks of information at once. A final advantage is that the spectra of the isotopes are about 20 times wider than the spectrum for hydrogen.
THE STRUCTURE OF PROTEIN IS RESILIENT
Today, researchers are using both techniques for structural analysis of several dozen proteins. T4 lysozyme, a 164 amino acid protein that viruses use to break open bacterial cell walls, is one of the most thoroughly studied. It was the subject of Alber's postdoctoral research under Brian Matthews of the University of Oregon. (Lysozyme is a powerful antibacterial and is present in tears to protect the eyes.)
Matthews first turned his attention to lysozyme in the early 1970s. His colleague, the late George Streisinger, had created about 100 versions of the protein, using a traditional genetic technique that introduces random mutations. "We thought it would be a wonderful opportunity to take advantage of that genetic information," says Matthews, to find out how the mutations affected three-dimensional structure and function.
There was an intrinsic advantage to the use of lysozyme for the study of structure. An important characteristic of proteins is their stability. Heat a protein and eventually the structure falls apart as the weaker noncovalent bonds are breached. The measure of stability is the temperature at which the structure melts. Some proteins are hard to work with because they coagulate in the melted state, like an egg white, losing the ability to reform their proper structure—but not T4 lysozyme.
From 1974 to 1979, Matthews spent most of his time identifying interesting mutants, purifying the proteins, and analyzing their three-dimensional structure with the help of his colleague Rick Dahlquist, an expert on NMR. At that time, "There was a perception that protein structures were very delicately poised between folding and unfolding … and that a single mutation might tip the balance," says Matthews. But he doubted this. He had noted that, while the hemoglobins of horses and humans are structurally similar, they differ in amino acid sequence by nearly 50 percent. Still, he says, "what you don't know in looking at these naturally occurring variants is whether the 50 percent that are
different are different for functional reasons or just because they don't matter."
But his late colleague's mutant lysozymes reinforced his doubts. Although a few of the mutations destroyed the protein's structure, most did virtually nothing to change it. Nonetheless, Matthews knew that it would have been dangerous to generalize from his results since Streisinger's techniques had created mutations in but a small fraction of lysozyme's 164 amino acid positions. "For large parts of the protein there was no information at all."
The advent of recombinant DNA made it possible to extend this research to the rest of the protein. One could now substitute any amino acid at any position and crank out copious copies of the mutant. By the mid-1980s, lysozyme had been cloned, and Matthews' studies would soon take off on a wave of technical progress. When Alber came to Matthews's laboratory in 1982, Matthews had studied five mutants. By the time he left in 1987, 50 had been characterized. The total has now reached 500. And as the experimental record grew, it continued to support the hypothesis that most single mutations do not change the structure of proteins.
In one of his first experiments to take advantage of recombinant DNA, in 1984, Alber took one of Streisinger's mutants, which had an amino acid substitution at position 157, and created a series of additional mutants by inserting virtually every other amino acid into that position, in turn. He performed the first x-ray crystallography that had ever been done on any of these variants, and the surprising result was that all were very similar to one another. The stability of each mutant was then measured by heating it until it melted. The mutants were all quite stable.
To further test these ideas, Matthews's postdoctoral student Xue-Jun Zhang systematically substituted the amino acid alanine at each position in the protein, one by one. Alanine is one of the simplest of all the amino acids and lacks the reactive chemical groups that interact with those on other amino acids. In subsequent experiments, Zhang substituted 10 alanines at 10 consecutive positions at once, repeating the process all over the protein. The structure of lysozyme was impervious to these changes at all but a few sites.
The experiments with alanine, and extensive substitutions with other amino acids, such as those at position 157, led to two general conclusions. First, "Probably less than 50 percent of the amino acids in the protein are critical to structure," says Matthews. And different proteins can have the same basic three-dimensional structures despite large differences in amino acid sequence.
To Alber, this makes perfect sense. "Just as a house is held together by nails, glue, screws, beams, and other binders, many forces give a protein its shape: van der Waals forces, hydrogen bonds, disulfide bonds, and electrostatic attractions and repulsions." But as few single fasteners or frame elements are critical to the integrity of a house, so most amino acids can be changed without destroying a protein.
The second result was that the most destructive substitutions almost always occurred in the core of the protein, while those on the surface were predominantly benign. The logic of this is that amino acids in the core, packed together as tightly as they are, are well placed to interact with each other, while those on the surface are not. (Surface amino acids might well interact with other substances in the surrounding solution or the solution itself.) "We now believe that for folding it is the core that is critical," says Matthews.
DESIGNING SUPERSTABLE PROTEINS
If that were true, Matthews speculated at the time of these earlier experiments, it should be possible to increase a protein's stability by creating additional bonds or stronger bonds in the core. Since the strongest structural bridges in proteins are the covalent disulfide bridges, Matthews and his postdoctoral researcher, Masazumi Matsumura, created disulfide bonds in the core of lysozyme. This meant substituting cysteine, the sulfur-containing amino acid, in place of other amino acids on either side of a fold in the protein.
Matsumura, now at Scripps Research Institute, built the bridges. Not all of them worked. "In many cases, the introduction of these bridges actually makes the protein less stable," says Matthews. The reason for this is that the geometry of these bridges needs to be very precise, more so than generally is possible. Otherwise, "you distort the protein, and that makes it less stable."
Nonetheless, the scientists discovered one approach to covalent bridge building that worked well. Certain parts of the protein are naturally flexible. Here, precision was unnecessary. The flexing prevented the bridges from distorting the rest of the protein; yet a disulfide bridge could increase stability typically by 5 to 10°C. Three bridges in one protein raised its melting temperature by 25 degrees.
Matthews' group also discovered that noncovalent bonds could more easily be used to increase stability since the force of their attraction was not great enough to cause the destabilizing distortion.
Tying pieces of protein together with opposite charges could also
increase stability, the researchers discovered, performing the experiment in an alpha helix, which is a common structural element of many proteins, including lysozyme. (Screws, slinkies, and stairways that wind around to save space are all examples of helices, as is DNA.)
One end of an alpha helix is positively charged, while the other end is negatively charged. By placing an opposite charge one turn of the helix from one of the end charges, the researchers found that they could raise the melting temperature of lysozyme by about 2°C. Several similar substitutions added further stability.
THE LEUCINE ZIPPER AND GENE REGULATION
From Oregon, Alber went to the University of Utah, where he was soon caught up in studies of the leucine zipper, collaborating with Peter Kim of the Whitehead Institute and several other researchers.
Leucine zippers are alpha helices that direct gene regulation. Gene regulation is the process that determines when the body will produce each protein. For example, eat a donut and the gastrointestinal tract must crank out the enzymes that digest starch. Then, as sugar enters the bloodstream, the pancreas must begin to manufacture insulin. Leucine zippers manage the regulatory process.
The work on leucine zippers ultimately would support Matthews's findings that, although most amino acids exert little influence on shape, that of core amino acids can be profound. Alber got caught up in the leucine zipper work as Kim et al. were piecing together the structure.
But it was Steven McKnight, of the Carnegie Institution of Washington
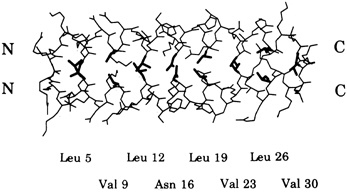
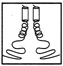
FIGURE 10.1 Side chains (thick lines) make contacts between the helices in a schematic drawing of a leucine zipper.
in Baltimore, who actually discovered the leucine zipper, in 1987, around the time that Alber departed for Utah. McKnight recounted his discovery in the April 1991 issue of Scientific American.
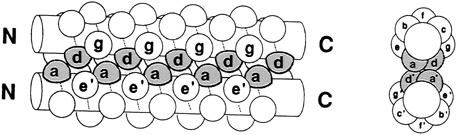
Leucine zippers are alpha helices, which are named for the fact that the amino acid leucine appears in the helix at intervals of seven amino acids, or slightly less than two turns of the screw. Moreover, the leucines, and another, similarly hydrophobic amino acid, which also appears at intervals of seven, form a zipper-like structure that binds two alpha helices together (see Figures 10.1 and 10.2). The pairs are called dimers. (The nomenclature here is counterintuitive. A leucine zipper is only one alpha helix, comprising only half of the zipper-like structure that binds two helices together into the dimer. To make things more confusing, scientists usually refer to both single and paired leucine helices as leucine zippers. However, in this account, leucine zipper refers only to the unpaired alpha helix; dimer or leucine zipper dimer refers to the pairs.)
More than 80 leucine zippers have been discovered during the past 4 years, and about 100 are known. Most are very similar to one another. Each leucine zipper is attached to a regulatory protein. Pairing brings the regulatory proteins together, which is necessary in order for them to turn genes on or off. Free ends of each helix then wrap part way around the DNA, fitting nicely along the groove of the DNA helix, thereby placing the regulatory proteins in the proper position to control expression of the gene(s) (see Figure 10.3).
It is the structure of the helices that determines which regulatory
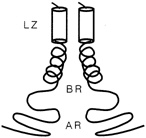
FIGURE 10.3 Schematic drawing of a leucine zipper protein. The leucine zipper (LZ) is the pair of cylinders. The amino acid tails include the basic region (BR), which binds DNA, and the activation region (AR), which turns genes on.
proteins come together. Slight differences in the amino acid sequence of the helices result in different pairings.
Different combinations of regulatory proteins regulate different genes in different ways. Some leucine zipper dimers turn genes on; others turn them off. For example, when the leucine zippers bind the regulatory protein jun to itself, the resulting complexes turn on many different genes. But the regulatory protein fos sunders the jun/jun pairs, and then binds with jun, turning off those same genes.
Some leucine zipper dimers control just a few genes, while others control huge batteries of genes. The GCN4 leucine dimer, which Kim et al. studied extensively, "is like a switch hooked up to 30 lights," says Alber. "You flick one switch, and all the lights go on."
"Gene regulatory proteins," McKnight recounted, "were discovered as an outgrowth of research into the structure of DNA and, later, into the organization of genes." His discovery of the leucine zipper grew out of his efforts to learn more about how DNA binding proteins—of which the leucine zipper dimer is one class—activate genes. The first such proteins had only recently been identified.
The big clue that the leucine zipper was important came when McKnight and his colleagues sequenced a regulatory protein that goes by the evocative acronym C/EBP and discovered via computer search that a segment of 60 amino acids was very similar to segments of the regulatory proteins fos and myc. (Fos and myc are proto-oncogenes, genes that normally play important roles in the body but that contribute to the development of cancer when they undergo certain mutations. More on that later.)
Then, McKnight set out to model the structure of the leucine zipper. "We knew that parts of many proteins fold into an alpha helix, a kind of coil," McKnight wrote. "We therefore began by considering whether the segments relating C/EBP, fos and myc might adopt that structure."
A frequent characteristic of alpha helices is that the two sides of a helix have different chemical properties. (Imagine the slinky with lines running the length of it, on opposite sides.) One side might be soluble in water, or hydrophilic, while the other side might be hydrophobic—and never the twain shall mix. The amino acids on each side of the helix determine these properties. And so it was with the C/EBP leucine zipper. The leucines, which are extremely oil soluble, were "aligned in a plane along the length of the helix, forming a prominent ridge," McKnight wrote. He had originally proposed that the hypothetical leucine ridges bound the helices together with each facing in opposite directions, the leucines forming interlocking teeth, exactly like the teeth of a zipper.
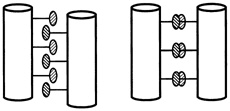
FIGURE 10.4 The old and new models of the leucine zipper. The side chains do not interdigitate (left), but come together side by side (right).
Compelling as it was, the model of two cylindrical helices side by side, fastened together with a molecular zipper proved somewhat inaccurate. Instead, the leucine zipper is a structure well known to experts in protein folding, called a coiled coil, which Linus Pauling and Francis Crick had first proposed to be the structure of the protein keratin, independently of each other, nearly 40 years earlier. Rather than lying side by side, the two helices of a coiled coil wind around each other in a super helix, like a pair of slinkies wrapped around each other or like the twining of rope.
Instead of one zipper running the length of the two helices, the structure of the binding mechanism is more like a series of very short zippers forming individual connections between the two helices (see Figure 10.4).
THE LEUCINE ZIPPER IS A COILED COIL
It was a freshly minted graduate of Smith college, who was working as a technician in Peter Kim's lab at the Massachusetts Institute of Technology, who spotted McKnight's mistakes. Her insight ultimately led to her Ph.D.
"McKnight's paper came out in June, 1988," says Alber. "The week it came out, [Erin] O'Shea read it. Peter and I had come back from a meeting, and Erin ran up to us in the hall, gave him the papers, and said, 'Read these by tomorrow, the peptide is on the synthesizer'." O'Shea, a sure-footed young woman of 26, is more modest about her achievement, explaining that she was quite familiar with coiled coils. "I had majored in biochemistry, and my advisor worked on coiled coils." So it was not surprising that she could spot a distorted one.
"It was evident from what I read that McKnight's [model] couldn't be correct," says O'Shea. "It takes 3.6 amino acids to make one full turn of a protein helix," she explains. "If the helical repeat were 3.5, the leucines, spaced every seventh amino acid, would fall on top of each other in the coil," as McKnight had described it. "One-tenth of a helix doesn't sound like much, but it is enough to make the ridge of leucines spiral around the helix, instead of lying flat.''
To hear Alber tell it, Kim and O'Shea already had a great circumstantial case. But if circumstantial evidence is considered to be a weak basis for conviction in a court of law, in science it almost never passes muster. So the scientists would approach the problem from many perspectives over the next 4 years in order to nail down the structure.
One of the Kim lab researchers' first steps was to synthesize the leucine zipper—minus the DNA binding part—on a protein-synthesizing machine and see if it would fold. At that time, the dogma had been that small fractions of proteins, such as the leucine zipper, could not fold by themselves. But Kim had been challenging this dogma and had already shown that a small piece of a protein called trypsin inhibitor could fold. Now the leucine zipper folded into its normal helical form, and Kim had another example.
Next, Kim and O'Shea synthesized the leucine zippers of fos and jun, and demonstrated that these disembodied sequences could also fold into helices that could then form dimers. This, says Alber, "set the stage for all the subsequent discoveries, because it gave people confidence that what was being studied was relevant for [the] regulation of genes."
Using a relatively crude but simple imaging technique called circular dichromism, O'Shea was able to detect the helices in the leucine zipper. But she was unable to determine that they coiled around each other, let alone view any of the molecular details of the structures. That proof should have had to come from the x-ray crystal structure that O'Shea would finish later, but serendipity provided a quicker path to proof.
A CANINE PINCUSHION POINTS THE WAY
One day in Utah, one of Alber's dogs, a chocolate labrador retriever, returned from the adjacent foothills to the lab with his nose looking like a pincushion. Marley had tangled with a porcupine. Quills must be removed with extreme care, because they are designed to tear the tissues during extraction. This gave Alber plenty of time to think. Quills are made from keratin, as are fingernails, the scutes of turtle shell, hair, and other common proteins. Of all of them, quills make the cleanest x-ray diffractions because the fibers are especially well lined up. A clear image of the diffraction pattern of a quill had been published in the British journal Nature in 1943, but no one had derived the structure from the pattern.
In 1949 Pauling had proposed that some proteins would form alpha helices. He knew how amino acids fit together, and he could take a protein full of them and manipulate the topology in his mind, and now he and Crick were in a race to find an actual protein that had this structure.
Each figured out what kind of diffraction pattern a helix would produce, and at roughly the same time both men deduced that the helix could explain the diffraction pattern of keratin if two helices were coiled around each other, like the fibers in a rope.
(The political story, says Alber, is that although Pauling, an American, submitted his paper to Nature first, toward the end of 1952, the British-born Crick's paper was published first, in 1952, while Pauling's was held until 1953.)
Anyway, as he was extracting quills from his dog's nose, Alber realized that he now had a means to quickly compare the diffraction patterns of the leucine zipper and keratin. "We had known from looking at the leucine zipper," says Alber, "that it was qualitatively similar to the published pattern of keratin." However, the nearly half-century-old x-ray provided no detail on the molecular level. But once they had a clear diffraction pattern for the quill, they could see that the diffraction patterns of that and the leucine zipper, which O'Shea had already obtained, were very similar to one another.
Besides proving that the leucine zipper is a coiled coil, the comparison of diffraction patterns showed that the structure of the zipper is similar in detail to that of keratin. But at the time, no one had ever deciphered the fine structure of keratin from the diffraction pattern. NMR and O'Shea's work on crystallography would fill in those details.
At that time, says Alber, "We only knew that some part of the peptide was helical, from the circular dichromism and from the original x-ray pattern." "The NMR showed exactly which residues were in the helix." NMR also proved O'Shea's hypothesis that the two helices did not zip together quite as McKnight had described. Lawrence McIntosh and Terry Oas performed the NMR in the winter of 1989, "practically overnight," says Alber. Ironically, it was a property of NMR that is usually a limitation that made the work go so quickly.
When two or more hydrogens lie in identical chemical environments, only one shows up on the NMR spectrum. This is because hydrogens in identical chemical environments produce the same peak in the same place on the NMR spectrum. (The forces that are relevant in NMR to an atom's chemical environment are only those that are located within a couple of chemical bonds from the atom. See Box beginning on p. 304.)
McKnight's zipper hypothesis required the leucine zippers to face in opposite directions; otherwise, the chemistry of the binding would not work. Although the leucine zippers that McIntosh and Oas were studying were identical to one another, had they faced in opposite directions,
|
Deciphering Structure: The Methodology of Molecular Modeling Of all imaging techniques, x-ray crystallography can usually provide the most complete picture of a protein's structure. In a sense, crystallography takes a photograph of the entire protein (though this can be clear or blurry depending on how much information is generated in the form of x-ray photographs). It does so by revealing the density and location in space of a protein's electron clouds. This provides information on where the different atoms of a protein might be located, because different atoms have different electron densities. The technique's biggest limitation is that the protein must be crystallized. "You might get a crystal in a day, or a week, and sometimes you never get a crystal at all," says Alber. In other words, you might spend months walking down a long road only to find yourself at a dead end in the middle of a desert. "It's a process of trial and error to find the right conditions to make a crystal," says Alber. Whether you get a crystal or not is very sensitive to conditions of temperature, pH, salt concentration, the buffer salt you use, and how much protein you have. So the basic strategy is to get close, and then you vary each of the conditions in turn, and all the sudden you have a crystal. "By chance, with the GCN4 leucine zipper we tried conditions that were close enough that we knew we were on the right track," says Alber. "We changed each little variable in the conditions in turn, and we discovered that the crystallization was incredibly sensitive to all the conditions. In that sense it was very unusual. Many proteins can crystallize over a range of 2 pH units or more, and some over 7. In this one you have to be within a couple of tenths of a pH unit. We were very lucky to hit close enough to begin with." They had their crystal within 3 months. Sometimes parts of the protein crystallize poorly or not at all, so that electron clouds of those atoms do not contribute to the diffraction pattern. Another limitation is the need for sequence data. It is not an absolute necessity, but it helps, in order to be able to verify the structure once it has been derived. The problem is that the electron density data are prone to errors of up to 5 percent. These errors inevitably crop up in the counting of literally billions of photons, as well as from background radiation, which is hard to accurately subtract from the picture, and from some of the mathematical equations used to generate the positions of the electron clouds, which are |
|
approximations, says Alber. For example, when Alber and O'Shea deciphered the structure of the leucine zipper, "We could only see one-half of it clearly," says Alber. "But the half that we could see was enough to bootstrap the rest." The first step, once a protein has been crystallized, is to take x-ray pictures of the crystal. Often thousands of x-rays are taken, from every possible angle encircling the crystal. Until recently, the x-ray patterns were recorded on ordinary film, and the machine judging of how strong the x-ray diffraction had been by the shade of spots on the film was imprecise. Now, photographic film has been replaced by electronic film that can literally count photons, greatly increasing accuracy. In fact, it is almost ironic how sophisticated the equipment for deciphering crystals has become, compared to the intellectual process of piecing together the structure. Using the diffraction data, a powerful computer maps the electron densities in space and displays them on a TV screen. The computer allows the crystallographer to turn the map around in order to peer at the pattern from every angle. If the crystallographer has a sequence, it can be generated on the screen as well, and, by twirling a set of dials, the crystallographer can manipulate each atom in three dimensions, among the electron clouds. Without a sequence, the crystallographer simply tries fitting different atoms or chemical groups among the electron clouds. Poor Watson and Crick had to manipulate their models of DNA's structure in their heads and on paper, and then they had to build them. Sequence or none, there is method to the madness of deciphering structure. Due to the various built-in errors, the electron clouds are ambiguous indicators. This can leave the crystallographer in the position of a rat trying to run a three-dimensional maze. The most important work is to track the backbone of the protein. But researchers can get sidetracked onto side chains, particularly when two connect. In sum, crystallography is a time-consuming and exacting process of trial and error, so much so that there is a whole field dedicated to making model fitting methodical. Where x-ray crystallography fails, nuclear magnetic resonance (NMR) can sometimes succeed (and vice versa). Proteins that fail to crystallize may yield their structural secrets to NMR. The mobile fractions of proteins that cannot be crystallized can be viewed through the lens of NMR. Furthermore, NMR can reveal the different configurations of such proteins. Researchers are beginning to use NMR to track the pathways of folding proteins. And, finally, NMR can reveal interactions between charged sidechains that stabilize a protein. |
|
But where x-ray crystallography is like taking a photograph and getting the big picture, NMR is more like assembling a jigsaw puzzle—putting the structure together from tiny pieces of disembodied information. Not surprisingly, then, for NMR, having the sequence is like having a picture of the entire jigsaw. But recent advances have made NMR much easier, says Thomas James, a researcher at the University of California, San Francisco. These advances have also allowed researchers to use the technique to examine much bigger proteins: with 150 to 200 amino acids, up from about 80 amino residues. Prior to the advances, NMR was limited. It is sort of like a game of connect the dots. NMR detects atoms by magnetizing them. Magnetized atoms can magnetize other atoms if they are close enough together—within 5 angstroms, or the equivalent of a couple of chemical bonds. NMR can be set up to reveal the pairs and the distance between them. Stick a third atom into the mix, one that is within 5 angstroms of both of the others, and now one can deduce the angles between the hydrogens as well. Instead of one dimension, one is now working in two. In the same way, a fourth hydrogen within 5 angstroms of the other three can add the third dimension. But if the third hydrogen is more than 5 angstroms from, say, the second hydrogen, NMR cannot reveal the distance between them, or the shape of the triangle outlined by the three, unless some clue from another source is available. Another limitation of the old hydrogen-based NMR is that hydrogen atoms are deadends in the covalent structure of protein. Moreover, although hydrogens are attached to most of the atoms in this chain, certain carbons within the backbone of each amino acid lack hydrogens. The result is that the smallest hydrogen-hydrogen distance across the carbon gap via the backbone exceeds the 5-angstrom limit, forcing researchers to trace their way from one amino acid to the next through space, a more difficult technique. Yet another problem was that the NMR spectrum for hydrogen is narrow, which had much to do with placing the 80 amino acid limit on the size of proteins that could be studied. Imagine shrinking the FM dial to one-tenth its size. It might still be able to accommodate all the stations available in a small city like Bismarck, North Dakota, but forget about trying to squeeze in the airwaves of New York. |
|
This was the state of the art until the mid-1980s. Researchers had long realized that NMR could be much easier and more versatile if certain isotopes of carbon and nitrogen that can be magnetized were incorporated into protein. But no one had tried it until then. One advantage of the isotopes was that their NMR spectra are about 20 times wider than that of hydrogen. This contributed to increasing the size of proteins that the technique could study. Another was that the use of carbon-13 provided a new, more direct way to connect atoms along the length of the protein chain—through the bonds rather than through space. Still another advantage of isotopes is that it became possible to connect triplets and even quadruplets of atoms directly, instead of just pairs. Researchers could trace larger pieces of structure at once, speeding up solving the puzzle. In theory, it would have been possible to connect triplets under the old hydrogen-based system, because when hydrogen magnetizes a neighboring hydrogen the neighboring hydrogen can magnetize a third atom. In practice, hydrogen magnetizes its neighbor so inefficiently that only about 1 percent of the magnetic "spin" would get transferred to hydrogen #3, too little to show up on NMR. But the carbon and nitrogen isotopes transfer roughly 90 percent of the spin to their neighbors. NMR picks up additional information on structure from so-called torsional angles. All atoms have preferred bonding angles, and scientists could have used these to deduce the spacial confirmation of a protein. However, the forces that give the protein its shape bend the bonding angles in somewhat the way a load of cargo bends the springs of a car. Torsion angles between molecular bonds can be deduced from NMR spectra. Ultimately, all this information is processed by using computers, using several different methods to deduce a protein's structure. Since experimental error is inevitable, the structure is analyzed by a program called the distance geometric algorithm, which determines what parts of the protein's structure have been accurately defined, and what parts have been poorly defined. The data are delivered in the form of a "family of structures," which the researcher can compare to one another. Accuracy is determined by the degree of congruence among the structures in the family. But an area where the structures are different may not mean poor definition. Instead, it may mean that that part of the molecule is mobile and that the different structures correspond to different real confirmations of the molecule. |
the chemical environments for the hydrogens from each helix would have been slightly different, and so both sets of hydrogens would have appeared on the NMR spectrum. Instead, McIntosh and Oas saw spectral peaks corresponding to only one helix. The zipper hypothesis was becoming increasingly untenable.
X-RAY CRYSTALLOGRAPHY: FINDING THE MOLECULAR DETAILS
Only the high-resolution structure would divulge the location of all the atoms in the protein. This was a job for x-ray crystallography. The researchers began their attempts to crystalize the protein in November 1988. This and the rest of the preparatory work, through collection of x-ray data, took nearly a year. Then, at the end of 1989, O'Shea went to Alber's laboratory to take advantage of his skills and facilities.
It was amazing, says O'Shea, how quickly she generated the structure. Within 9 months she had confirmed her original model. Technological developments in data collection and software made this speed possible, in particular, programs written by Wayne Hendrickson. O'Shea had gotten stuck, but once she began using his program, which exploited the various symmetry relationships that exist in many proteins, she was able to finish the job in 2 months.
The solution of the crystal structure not only proved beyond a doubt that the leucine zipper is a coiled coil but was also a milestone in the study of protein structure. The list of suspected coiled coils had included many important and ubiquitous proteins, including muscle proteins; dynein, which forms molecular motors; intermediate filaments, which make up some of the girders that give cells their structural integrity; and oncogene products. But O'Shea had uncovered the first complete high-resolution structure of a coiled coil. This "made the models of myosin, keratin, and all these other coiled coils much more believable," says Alber.
THE COILED COIL: RULES OF FOLDING
Meanwhile, in 1991, Pehr Harbury, one of Peter Kim's graduate students, set out to determine how structure influences function in the leucine zipper.
Leucine and the other hydrophobic amino acids in the zipper are spaced as evenly as possible within an odd-numbered repeating sequence: Four positions from leucine to the other hydrophobe, and back to leucine again is three, and so on. Since there are 3.6 amino acids in
one turn of the helix, this places the other hydrophobic amino acids nearly in line with the leucines.
Harbury made certain changes in the sequence, substituting different hydrophobic amino acids at the positions of leucine and the other hydrophobe that forms the zipper mechanism. Then he asked how the amino acid substitutions changed the structure of the leucine zipper, the way it bonded, and its stability.
One set of substitutions barely altered the structure. But other changes caused the leucine zipper to form tetramers (quadruplets) or trimers (triplets) instead of dimers (see Figure 10.5). (To visualize the structure of these, imagine winding three and then four fibers around each other to form a rope). In the latter cases the main difference between the normally occurring hydrophobes and their replacements was shape. The substituting amino acids had side chains that branched at the first carbon atom; leucine does not.

FIGURE 10.5 Dimer and tetramer coiled coils viewed end-on. The amino acid side chains of the coils pack very tightly between the coils, and their shape,which can be changed by changing amino acids in certain positions within the helix, determines the number of coils in a structure.
It was surprising that a couple of substitutions could cause such drastic changes in bonding patterns, Alber said at the "Frontiers of Science" symposium, "because intuitively, you know that sequences that are similar generally form the same structure. That's a very big idea in protein structure. If you have two similar sequences, they have the same fold. This is an exception to that rule."
"These sequences are in fact 75 to 87 percent identical, and yet they form different arrangements," Alber continued. "Why is that? The only thing that we've changed in these sequences is the geometry of these [amino acids that connect the helices]. So it must be the packing of the interface that determines the number of strands. We've figured out how that can happen by determining by x-ray crystallography the three-dimensional structure of this tetrameric coil." (see Figure 10.6.)
The other reason the result seemed surprising is that researchers had

FIGURE 10.6 The different shapes of these three amino acids, in the a and d positions of the helices, influence whether coils group together as dimer, trimer or tetramer.
based their predictions on chemical forces and the size of amino acid side chains, ignoring their shapes, and for the most part this had worked. (But note that the result should not seem quite so surprising as all that because the amino acids causing the changes in structure are the interacting ones, which are comparable to those in the core of a protein.) But now Harbury had demonstrated that protein folding was still more complex than this. And in particular, says Alber, how the amino acids fit together in the core of the coiled coil determines the number of strands.
The rule of folding that the results suggest, says Alber, is that amino acids with side chains that branch at the first carbon do not fit the holes in the so-called knob-in-hole packing that leucines occupy in the leucine zipper dimer.
This, at any rate, is the current hypothesis. Harbury has since studied the sequences of many helices that come together as twins or triplets and has found that these rules generally hold (see Figure 10.7). The next step will be to design and synthesize idealized trimers and tetramers and see if they come together as predicted.
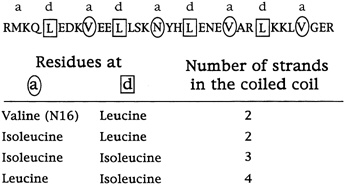
FIGURE 10.7 The specific amino acid changes in the a and d (circled and boxed) positions that determine the number of coils that come together.
THE ASPARAGINE RULE
Oddly enough, it might seem, among all the hydrophobic amino acids that bind leucine zippers in pairs, there is one water-loving amino acid that is found in virtually every leucine zipper helix: the amino acid asparagine. ''So it's a really important thing for function," says Alber. Since such an entity could only weaken the zipper, the researchers were mystified as to what its purpose might be. To answer the question, they substituted in turn four different amino acids for asparagine and then determined the structure and bonding strength variant. Chavela Carr, a graduate student in Kim's lab, studied the stabilities, and Russ Brown, a postdoctoral student with Alber in Utah, deciphered the structures. Two things happened. First, the new helices bonded so strongly that even boiling didn't sunder the zipper. Second, these asparagine-free leucine zippers formed both pairs and triplets, and Alber speculates that they may have formed skewed dimers as well (see Figure 10.8).
In hindsight, it was easy to deduce what the purpose of the destabilizing asparagine might be. Any biological switching device must be capable both of holding on to and letting go of its substrate. For example, hemoglobin must bind oxygen strongly enough to pull it from the lungs as blood cells pass through the alveolar tissue but weakly enough to release it throughout the rest of the body. Carbon monoxide molecules are poison because they adhere so tightly to blood cells that they rarely let go, thereby preempting oxygen. Similarly, a leucine zipper would be useless if it bound so tightly to one partner that it could never let go. Furthermore, too strong a propensity to bond would allow the helices to pair in an improper alignment. An improperly aligned leucine dimer would be unable to bind to the DNA because the DNA binding ends of the leucine zippers would be askew.
For biological switches to function, then, the strength of the binding must be finely tuned: Strong enough to come together but weak enough to do so without misaligning the molecules and weak enough to break apart when the time comes. In the leucine zipper, asparagine accomplishes this fine tuning.
Most coiled coils are not as regular as leucine zippers, and frequently another hydrophobic amino acid occupies the "leucine position." Harbury hopes to develop a set of rules for predicting the structures and bonding arrangements of this variety of coiled coils, and he is asking such questions as how many substitutions of leucine by isoleucine, one of the so-called beta-branching hydrophobes that fail to fit in the knob-in-hole
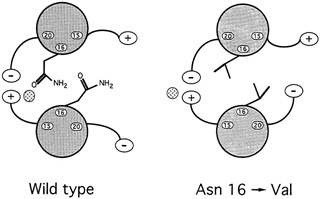
FIGURE 10.8 When experimenters replaced asparagine with valine, in the 16th amino acid position of the GCN4 leucine zipper, the result was a much stronger, more stable dimer than occurs naturally.
fastener, it takes for alpha helices to come together in trimers, instead of dimers. The results of experiments now under way will tell.
ANOTHER APPROACH IN THE QUEST FOR THE HOLY GRAIL
Despite the growing knowledge of the mechanics of lysozyme and leucine zippers, the Holy Grail of protein research, predicting structure from sequence, remains elusive. This was driven home to Harbury and his colleagues when, prior to synthesizing his experimental leucine zippers, he tried to predict what their structures would be. "Because the leucine zipper is such a tiny repetitive structure, Pehr could use effectively the most sophisticated methods for doing this calculation," says Alber. Nonetheless, "The calculation failed dramatically."
"We know the laws of chemical physics, so why can't we compute structure, given sequence?" David Eisenberg, professor of molecular biology at the University of California, Los Angeles, demands rhetorically. One cannot describe the folding of protein from first principles, he explains, because protein is too big and complex to be fitted into Schroedinger's equations, which are the mathematical expression of the chemical physics laws.
Another hypothetical approach would be to find the conformation with the greatest stability. Stabilities of different conformations can be calculated for small molecules, and, with certain exceptions, the conformation with the greatest stability is the correct one. The problem with this approach is that for proteins there are far too many potential conformations
to begin to do this, as Alber explained when he put the number at a google for ATCase. "It's not just that we don't have the computers to do that now," says Eisenberg. "We won't have them in 10 years."
Eisenberg has been taking the opposite approach to the problem of the relationship between sequence and structure. In his experiments he asks, as Stanford University engineering professor Eric Drexler did first in 1981: Given a specific structure, what amino acid sequences are compatible? But Eisenberg and his postdoctoral student Jim Bowie have simplified this approach in two ways.
First, they ask what known amino acid sequences are compatible with the structure. "That limits us to 50,000 sequences, whereas Ponder and Richards [researchers who also had previously tried the same approach] had looked at every conceivable sequence. Why should we care if there are sequences we don't know? Our job is to take information from the human genome and find the structure for each protein sequence. That limits the dimensionality of the search."
"The second major simplification is that instead of working with a three-dimensional structure, we have simplified that into a one-dimensional string which we can compare to amino acid sequences." ("String" is a computer term meaning things that have been strung out in one dimension.)
Eisenberg replaces three-dimensionality with the details of the chemical environment of each amino acid position. For example, "What are the amino acid side chains around that position, and do they prefer an apolar or a charged environment?" He has divided the chemical environment into 18 different environmental classes, and each amino acid in a protein is assigned to one of them. "We call the string of classes the three-dimensional profile. By looking at the environment, we are looking at the footprint, rather than the foot."
Eisenberg predicts the job will be manageable because he believes the number of types of protein folds is limited. "When we learn the structure of a new protein, it often has the structure of a known protein. But we can't necessarily predict that from the sequence, because the sequence may have diverged too much. This suggests that there may be a finite number of folds."
In early 1992 Cyrus Chotia, of the MRC Laboratory in Cambridge, England, estimated that there may be only 1000 to 1500 distinctive folds. "If that is true, then the job of the person studying protein structure will be to assign sequences to one of these folds," says Eisenberg. "That is what our method is aimed at."
Three-dimensional protein structures are being compiled in a data
base at Brookhaven National Laboratory, and sequences are stored at Georgetown University and the National Institutes of Health. There are about 1200 three-dimensional structures in the Brookhaven data base, and Georgetown and NIH have a combined total of about 60,000 different sequences. "We used those data bases in our research," says Eisenberg. "We started with the structure of lysozyme, and computed a three-dimensional profile, and we were able to find all the sequences in the sequence data base which are compatible with the lysozyme structure."
But so far this method has had its own limitations and failures. A coiled coil that Eisenberg had predicted should be a dimer turned out to be a trimer. And Alber says, "Our finding that the shape of an amino acid makes a difference to the structure suggests that something is missing from his calculations, because he doesn't consider shape at all."
"It's not that his method is wrong," Alber hastens to add. "It's just not fully developed." Will the two methods eventually merge somehow in a way that will generate general principles, much as theoretical physicists hope to combine the four forces into a grand unifying theory?
The best answer that either researcher could give was Alber's response that on occasion his predictions agree with Eisenberg's. For example, in the case of one coiled coil sequence, both methods correctly predicted a trimer. This weak response suggests that both methodologies remain embryonic.
APPLICATIONS: A CANCER DIAGNOSTIC
While a grand unified theory of protein folding is probably years in the future, the basic research is already yielding useful developments. So far, most of them involve research methods, but Alber and Ray White, of the University of Utah, are working on a diagnostic system for colon cancer.
White and Bert Vogelstein, of Johns Hopkins University, had discovered mutations in the so-called APC gene. That gene is present in many people who have colon cancer. The mutations truncate the APC protein but leave a coiled coil intact. One of White's first-year graduate student advisees at Utah who had been working with him brought Alber and White together. Now Alber is designing a coiled coil to be used in the diagnostic system.
The diagnostic would work as follows. Doctors would take cells from patients' colons, use laboratory techniques to separate the proteins by size, and label APC proteins using Alber's coiled coil—radioactively tagged—as a hook. These would then be analyzed to see if truncated APC proteins were present.
CONCLUSION
No doubt the quest for the Holy Grail will continue for years, if not decades. Researchers are only beginning to design novel proteins while sitting at the keyboard. The amino acid chain remains a Rosetta stone with the wealth of information on how proteins fold largely undeciphered. Nonetheless, the empirical data that researchers have collected are beginning to yield patterns that are providing valuable clues as well as insights into the mechanisms of gene regulation, which is one of the most fundamental processes in biology. The coiled coil, says Alber, "although simple, has been especially rich. On the one hand, we are finding out how protein-protein interactions control gene expression. On the other hand, we've described a simple structural motif in enough detail that we now think we know how to design this motif. This is a small step in the direction of being able to design proteins."
BIBLIOGRAPHY
Alber, T. 1992. Structure of the leucine zipper. Current Opinion in Genetics & Development 2:205–210.
Alber, T. 1993. How GCN4 binds DNA. Current Biology 3:182–184.
Brändén, C.-I., and T. A. Jones. 1990. Between objectivity and subjectivity. Nature 343:687–689.
Crick, F. H. C. 1952. Is keratin a coiled coil? Nature 170:882–884.
Crick, F. H. C. 1953. The packing of a-helices: simple coiled coils. Acta Crystallographica 6:689–697.
Harbury, P. B., T. Zhang, P. S. Kim, and T. Alber. 1993. A switch between two-, three- and four-stranded coiled coils revealed by mutants of the GCN4 leucine zipper. Science, in press.
Mattherw, B. W. 1993. Structural and genetic analysis of protein stability. Annual Review of Biochemistry 62:139–160.
McKnight, S. L. 1991. Molecular zippers in gene regulation. Scientific American 264(4):54–64.
Olson, A. J., and D. S. Goodsell. 1992. Visualizing biological molecules. Scientific American 267(5):76–81.
O'Shea, E. K., J. D. Klemm, P. S. Kim, and T. Alber. 1991. X-ray structure of the GCN4 leucine zipper, a two-stranded, parallel coiled coil. Science 254:539–544.
Pauling, L., and R. B. Corey. 1953. Compound helical configurations of polypeptide chains: structure of proteins of the a-keratin type. Nature 171:59–61.
Richards, F. M. 1991. The protein folding problem. Scientific American 264(1):54–63.
Wüthrich, K. 1989. Protein structure determination by nuclear magnetic resonance spectroscopy. Science 243:45–50.


























