
B12
Antiprogestogens: Perspectives from a Global Research Program
P.F.A. VAN LOOK, M.D., Ph.D. and H. VON HERTZEN, M.D., D.D.S.
Special Programme of Research, Development and Research Training in Human Reproduction World Health Organization, Geneva
ABSTRACT
Because of their unique ability to block the action of progesterone at the cellular level through binding to the progesterone receptor, antiprogestogens are proving to be one of the most significant developments in endocrinology in recent years. In this paper we assess the current status—and make some suggestions on future directions—in the area of antiprogestogen research from the perspective of an international, public sector research program that supports basic, animal, and clinical studies with these compounds in some 16 countries worldwide. Five aspects in particular are reviewed, namely: the nature and number of currently available antiprogestational compounds, the (in)adequacy of the animal models used, the mechanisms of action, the possible clinical uses, and the prospects of antiprogestogens. The focus of our discussion is on the potential applications in fertility regulation since this is the area in which most of the research to date has been conducted. The results of this research leave little doubt that it is no longer a matter of ''the antiprogesterones are coming" but that the "antiprogestogens have come to stay." The time has come for scientists, policymakers, and the general public to acknowledge this fact.
INTRODUCTION
Studies by, among others, Fraenkel (1903), Loeb (1907), and Bouin and Ancel (1909a,b) at the beginning of this century strongly suggested that the corpus luteum was an endocrine organ and intimately involved
in the establishment and maintenance of pregnancy. Convincing proof of this concept was furnished in 1929 when Corner and Allen demonstrated that injections of corpus luteum extracts into rabbits, ovariectomized shortly after mating, resulted in the development of a progestational condition similar to that observed in early pregnancy. Attempts to isolate the active principle from these extracts were successful five years later, in 1934, when four laboratories announced independently the isolation of the pure corpus luteum hormone, progesterone (Allen and Wintersteiner, 1934; Butenandt and Westphal, 1934; Hartmann and Wettstein, 1934; Slotta et al., 1934).
Like other steroid hormones, progesterone exerts its effects through interaction with intracellular receptors that, after binding the hormone, undergo a "transformation" or "activation" process. Although the molecular details of this transformation have not been established unequivocally, the process apparently imparts to the transformed receptors the ability to interact with nuclear components with high affinity. Modification of the expression of the hormonally regulated genes is thought to be the result of this interaction between activated receptors and specific nuclear acceptor sites (for review see, for example, Savouret et al., 1990).
Progesterone receptors are found primarily, albeit not exclusively, in the organs of the female reproductive tract and in the pituitary and hypothalamus, in keeping with the hormone's central role in female reproductive physiology. Other normal tissues in which receptors have been demonstrated include, inter alia, the cerebral cortex, thymus, and muscle cells of uterine arteries (Savouret et al., 1990), and more recently, endothelial cells of decidual blood vessels (Wang et al., 1992). Progesterone receptors have also been detected in some benign (e.g., meningioma) and malignant tumors (e.g., breast, kidney, prostate, and endometrial cancer), which is why progesterone antagonists have been proposed for the treatment of some of these conditions.
Recognition of the indispensability of progesterone for pregnancy establishment and maintenance in most, if not all, mammalian species including the human, led to the search for compounds with antiprogestational activity (i.e., substances that would prevent the biological effects of progesterone on its target organs). Given the multifaceted role of progesterone in the reproductive process, such antiprogestational compounds could be expected to have a number of potential applications in human fertility regulation, including termination of pregnancy, induction of missed menses, post-coital contraception, and possibly also as once-a-month drugs and ovulation inhibitors.
The Special Programme of Research, Development and Research Training in Human Reproduction (the Special Programme) was established in 1972 by the World Health Organization to coordinate, promote,
conduct, and evaluate international research in human reproduction. Recognizing the potential of safe and effective antiprogestational drugs in fertility regulation, the Special Programme started to provide support to this area of research very soon after its inception and has continued to do so in a growing manner to the present day. Work supported in the 1970s included some of the studies by Baulieu and his colleagues on the cyclical changes of estrogen and progesterone receptors in the human endometrium (Bayard et al., 1978) and the demonstration that the C-11 position of progesterone was critical in the hormone's interaction with its receptor (Holmes et al., 1981). As described below, most of the antiprogestogens reproted to date have a bulky substituent in this C-11 position of the steroid molecule.
Research on antiprogestogens, in particular mifepristone, was started by the Special Programme in 1983, shortly after results from the first clinical trial with the compound had been published (Herrmann et al., 1982). The outcome of the Special Programme's first study—a dose-finding study in which mifepristone was given alone for termination of early pregnancy (Kovacs et al., 1984)—was somewhat unexpected. The compound's efficacy in inducing complete abortion was lower than anticipated, a finding later confirmed by numerous other investigators. Subsequent work, supported by the Special Programme, led to the discovery that mifepristone increases the sensitivity of the uterus to prostaglandins and that sequential treatment with the antiprogestogen and a low dose of a prostaglandin analogue such as sulprostone could terminate early pregnancy in about 95 percent of women (Bygdeman and Swahn, 1985). Since then, the research of the Special Programme has been focused on examining various combination regimens of mifepristone and different prostaglandin analogues, and on determining the lowest effective doses. Concomitantly, studies have been initiated on other possible uses of mifepristone in fertility regulation, including ripening of the cervix, induction of missed menses, prevention of implantation, and post-coital contraception. In addition, support is being given to research aimed at finding new, and possibly more potent and "pure," antiprogestogens.
The purpose of this paper is to assess the current status—and make some suggestions on future directions—in the area of antiprogestogen research from the perspective of an international, public sector research program that supports such work in about 16 countries worldwide (Figure B12.1), which together conducted a total of 32 research projects during 1992 involving mifepristone and other antiprogestogens (Figure B12.2). Five aspects in particular are reviewed, namely: the nature and number of currently available compounds, the (in)adequacy of animal models used, the mechanisms of action, the possible clinical uses, and the prospects of antiprogestogens. Because of the mandate of the Special
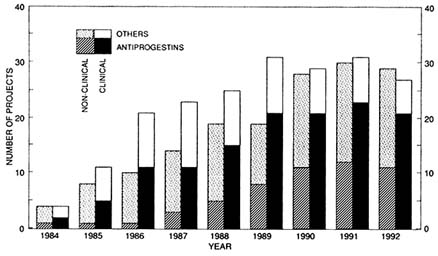
FIGURE B12.2 Number of ongoing, nonclinical, and clinical research projects on antiprogestogens and other postovulatory methods of fertility regulation supported during the period 1984–1992 by the Special Programme of Research, Development and Research Training in Human Reproduction at the World Health Organization.
Programme, which is focused on human reproduction research, and also because most of the antiprogestogen research conducted to date has been in this area, our comments relate predominantly to the possible applications of these compounds in fertility regulation. Potential uses outside this field are addressed in other papers of this report. Greater detail about antiprogestogens in general and about the Special Programme's research with these compounds in particular can be found in our earlier reviews (Van Look and Bygdeman, 1989; Puri and Van Look, 1991; Van Look and von Hertzen, 1992a,b, 1993).
COMPOUNDS
Compounds with antiprogesterone activity can be grouped into two main categories: (1) steroid enzyme inhibitors that prevent the biosynthesis of progesterone at the level of the steroid-producing cells, and (2) progesterone-receptor blockers (designated as "antiprogestogens" in this paper) that interfere with the binding of progesterone to its cellular receptor in target tissues.
Progesterone Synthesis Inhibitors
Progesterone biosynthesis in steroid-producing tissues proceeds by reactions involving removal of a C6 fragment from cholesterol to form
pregnenolone, which in turn is converted into progesterone by the Δ5-3β-hydroxysteroid dehydrogenase enzyme complex (3β-HSD) (Dorfman, 1973). Several synthetic progestogens have been shown to be capable of inhibiting ovarian 3β-HSD activity (Shinada et al., 1978), but clinical exploitation of this "antiprogestational" effect has been prevented by the intrinsic progestational activity of these compounds. Of more clinical interest is the series of steroidal 3β-HSD inhibitors that includes azastene, trilostane, and epostane. Cyanoketone also belongs to this series, but this compound is unsuitable for fertility regulation because it causes irreversible inhibition of 3β-HSD (Goldman, 1967).
Although azastene, trilostane, and epostane are competitive inhibitors of 3β-HSD, the compounds differ with regard to their relative potency in vivo as inhibitors of adrenal or ovarian/placental steroidogenesis. Azastene and epostane appear to be preferential inhibitors of ovarian and placental steroidogenesis rather than of adrenal hormone production, at least in primates (Schane et al., 1978; Creange et al., 1981). Epostane is the more potent of these two compounds; hence virtually all of the studies in primates, including the human, have been done with this inhibitor. A detailed review of these studies has been published (Van Look and Bygdeman, 1989).
As one might expect, the clinical effects of epostane administration are not unlike those resulting from treatment with a progesterone-receptor blocker such as mifepristone. Repeated dosing during the midluteal phase of the cycle causes premature menstruation in the majority of subjects, whereas daily treatment from the beginning of the cycle has been shown to modulate or inhibit follicular development and ovulation, depending on the dose used (Rannevik et al., 1988).
When given to pregnant women over a period of several days, the drug induces uterine contractions and increased myometrial reactivity to prostaglandins. In the two largest efficacy trials conducted (Birgerson et al., 1987; Crooij et al., 1988), a seven-day course with epostane (four times 200 mg/day) resulted in 89 (84 percent) complete abortions among 106 women with amenorrhea of less than 56 days. The limited experience that is available suggests that, as with mifepristone, the complete abortion rate can be increased significantly by complementing the epostane treatment with a prostaglandin analogue in a sequential regimen (Webster et al., 1985). Like mifepristone, administration of epostane during the second trimester of pregnancy augments uterine sensitivity to exogenous prostaglandins and significantly shortens the time needed to induce abortion with extra-amniotically administered prostaglandin E2 (Selinger et al., 1987).
Epostane and related molecules were synthesized by the pharmaceutical company Sterling-Winthrop. When this firm was taken over by the Eastman-Kodak company, epostane was no longer made available to
clinical researchers, and further development of the compound was discontinued. Although epostane appears to be a preferential inhibitor of the ovarian and placental 3ß-HSD enzyme, this compound—along with others like it—is probably less suitable for chronic administration than the more specific progesterone-receptor blockers because of possible inhibitory effects on adrenal corticosteroid synthesis. For occasional short-term use, however, such as in the termination of early pregnancy, a potent 3ß-HSD enzyme inhibitor could be a potential alternative to an antiprogestogen. Also, combining a 3ß-HSD inhibitor with a progesterone-receptor blocker may result in a powerful synergistic effect, as has been demonstrated in the pregnant guinea pig (Elger et al., 1988).
In view of the foregoing it would seem worthwhile to pursue the development of epostane or related compounds as a possible substitute for—or complement to—progesterone-receptor blockers. An active search for 3ß-HSD inhibitors that are more potent than epostane or azastene is currently in progress in the People's Republic of China.
Progesterone-Receptor Blockers
As described by Ulmann et al. (1990), progesterone-receptor blockers were discovered by chance rather than by design. Following the invention of a method of synthesizing 11ß-substituted steroids (Bélanger et al., 1981; Teutsch et al., 1988) and the observation that some of these 11ß-substituted compounds behaved as glucocorticoid antagonists, a formal research project for the development of glucocorticoid antagonists was launched at Roussel-Uclaf toward the end of 1979. Mifepristone (RU 38486, later shortened to RU 486) was produced a few months later, in April 1980 and, when tested for in vitro binding to the five classes of steroid receptors, was found to possess high affinity not only for the glucocorticoid receptor but also for the progesterone receptor, and low affinity for the androgen receptor (Philibert, 1984). The ability of antiprogestogens to interact with both the progesterone and the glucocorticoid receptors is probably related to the more than 50 percent homology in amino acid composition that exists between these two receptors in their C-terminal end (i.e., the region thought to be involved in steroid binding) (Misrahi et al., 1987). Subsequent tests in various animal models soon revealed that mifepristone possessed both antiglucocorticoid and antiprogestational activities. Thus, the scientists at Roussel-Uclaf had inadvertently managed to produce the progesterone antagonist long awaited by investigators and clinicians interested in birth control.
Following the initial discovery, several hundred compounds with antiprogesterone activity were synthesized, not only at Roussel-Uclaf (Teutsch, 1984, 1985), but also by other pharmaceutical companies
including Schering AG (Wiechert and Neef, 1987) and Organon (Kloosterboer et al., 1988) and by private institutions (Cook et al., 1990). Other companies such as Upjohn, ICI, and Jenapharm also have been granted patents describing antiprogestational drugs but, as far as we are aware, have not published any biological data on these compounds in the scientific literature. Most of the molecules shown to possess antiprogestational activity have a bulky substituent at C-11, usually a dimethylaminophenyl group as found in mifepristone, lilopristone (ZK 98 734), and onapristone (ZK 98 299) (Figure B12.3). In some of the Organon compounds (e.g., ORG 31167 and ORG 31343) the dimethylaminophenyl group is situated at C-18 (Kloosterboer et al., 1988).
From the published literature it appears that only a few of the many compounds synthesized to date have been evaluated to any significant extent in biological screening models and, to our knowledge, only three of them have been given to humans, namely, mifepristone, lilopristone (which has been used in a dose-finding study for termination of early pregnancy; Swahn et al., 1993), and onapristone (which is about to enter Phase II studies). The available preclinical data suggest that there can be marked differences between antiprogestogens in their antiprogestational and antiglucocorticoid potencies in vitro and in vivo, even for compounds that are very comparable in chemical structure. For example, lilopristone, which differs from mifepristone only in the structure of the 17a side chain (Figure B12.3), is reported as having a much reduced antiglucocorticoid activity (Table B12.1). The two Organon compounds referred to above also appear to possess reduced antiglucocorticoid activity (Kloosterboer et al., 1988). Since no "pure" antiprogestogens have been described to date, it is unclear whether the ancillary antiglucocorticoid activity present in currently available antiprogestogens has a modulatory influence—either positive or negative—on the antiprogestational potency. In the case of lilopristone, the more favorable antiprogestational/antiglucocorticoid potency ratio found in animal studies did not make the compound more potent in the termination of early pregnancy in the human compared to mifepristone (Swahn et al., 1993).
The results obtained to date with mifepristone leave little doubt that antiprogestogens will make a significant impact on fertility regulation in obstetrics and gynecology and, possibly also, in other branches of medicine. Data from animal work also suggest that antiprogestogens may differ among themselves in terms of the relative effects they have on different target tissues, but this requires confirmation in the human. Current research on antiprogestogens suffers from the fact that all the compounds that have been studied to any significant extent possess antiglucocorticoid activity, and there is an urgent need therefore to produce molecules in which these two activities—the antiprogestational and the antiglucocorticoid—have been separated. The potential advan-
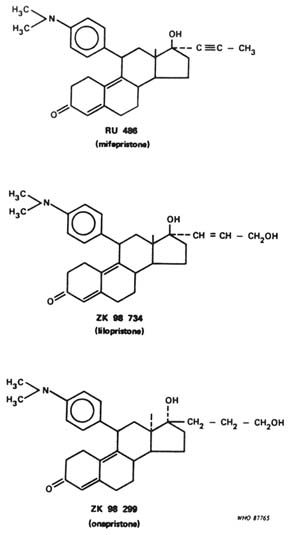
FIGURE B12.3 Chemical structures of the three best-known antiprogestogens.
tages of having pure, and preferably also more potent, antiglucocorticoids and antiprogestogens are manifold.
Availability of selective glucocorticoid- and progesterone-receptor blockers will provide basic researchers with powerful tools to study the effects of selective receptor blockage at the cellular level and thus provide further insights into the mechanism of action of these compounds. From a clinical point of view, pure antiprogestogens that are
TABLE B12.1 Antiprogestational and Antiglucocorticoid Activities of RU 486, ZK 98 734, and ZK 98 299
|
Parameter |
RU 486 |
ZK 98 734 |
ZK 98 299 |
|
Relative binding affinity (R 5020 = 100%) for progesterone-receptors using rabbit uterine cytosol (%) |
68 |
72 |
19 |
|
Antiglucocorticoid activity (%)a |
100 |
4 |
5 |
|
Abortifacient activity |
|
|
|
|
Rats (days 5–7 post coitum) |
|
|
|
|
3 mg/day |
4/4b |
4/4 |
4/4 |
|
1 mg/day |
2/4 |
4/4 |
4/4 |
|
0.3 mg/day |
0/4 |
1/4 |
0/4 |
|
Guinea pigs (days 43-44 post coitum) |
|
|
|
|
30 mg/day |
4/9 |
6/6 |
7/9 |
|
10 mg/day |
3/9 |
5/7 |
5/9 |
|
1 mg/day |
1/7 |
5/8 |
0/9 |
|
a Based upon reversal of dexamethasone-induced tyrosine aminotransferase activity in cultured rat hepatoma cells. b The abortion figures indicate the number of animals aborting versus the number of animals treated. Compounds were administered by the subcutaneous route in an oil base once daily on days 5–7 post coitum in rats and on days 43– 44 in guinea pigs. SOURCE: Based on Puri and Van Look (1991). |
|||
devoid of antiglucocorticoid activity are almost a sine qua non if they are going to be used for long periods of time, such as in the treatment of breast cancer or endometriosis, or as a daily contraceptive pill. On the other hand, pure antiglucocorticoids need to be developed that have no effect on the menstrual cycle and early pregnancy, and hence could also be used in countries where a compound with dual antiglucocorticoid and antiprogestational activities may not be made available becuase of, for example, restrictive abortion legislation. Attempts are under way to produce more selective molecules by using information from structure-activity relationships of existing compounds (Philibert et al., 1989) and from computer modeling studies of the stereochemical complementarity of steroid hormones and cavities between base pairs in DNA (Hendry and Mahesh, 1992).
ANIMAL MODELS
Identification of potential antiprogestogens generally involves, in the first instance, determination of the in vitro binding affinities of the newly synthesized compounds for the progesterone receptor and receptors of other steroid hormones. Compounds showing high binding affinity for the progesterone receptor are then assessed in appropriate animal models to determine if they are agonists or antagonists. The models used for this purpose and for evaluating the full hormonal and
antihormonal profile of new compounds have been described on several occasions (see, for example, Philibert et al., 1985) and are not discussed here. In general, the animal models used have been fairly reliable in predicting the effects of mifepristone in the human in spite of some major differences between humans and even their closest relatives, the nonhuman primates, in important areas such as the pharmacokinetics of the compound and the type of placentation.
Receptor Binding
Antiprogestogens have been demonstrated to bind to progesterone-receptor preparations from a variety of species including rat, rabbit, calf, marmoset, bonnet monkey, and human (for review see Van Look and Bygdeman, 1989). In the chicken, where progesterone has a different role than in mammals, mifepristone does not bind to the oviductal progesterone receptor (Groyer et al., 1985) but to an immunologically distinct macromolecule, possibly ovalbumin, that does not bind progesterone (Moudgil et al., 1986; Eliezer et al., 1987). However, the chicken glucocorticoid receptor binds mifepristone very well (Groyer et al., 1985). Absence of competition by mifepristone for binding of progesterone to its uterine cytosol receptor has also been reported for the hamster (Okulicz, 1987) and the tammar wallaby (Fletcher and Blandon, quoted in Baulieu, 1989).
In the case of the chicken, Benhamou et al. (1992) have shown that the absence of mifepristone binding is due to the presence of cysteine at position 575 in the hormone binding domain of the progesterone receptor. Substitution of this cysteine by glycine—but not by methionine or leucine—generated a mutated receptor that bound the antiprogestogen. In fact, all receptors that bind mifepristone, including those for glucocorticoids and androgens, have a glycine residue at the corresponding position. The progesterone receptor in the hamster, like that in the chicken, has a cysteine. Substitution of the corresponding glycine residue at position 722 by cysteine in the human progesterone receptor abrogated the binding of mifepristone but not that of a progesterone agonist, whereas the same substitution in the glucocorticoid receptor resulted in a loss of binding of both dexamethasone and mifepristone. Interestingly, the studies of Benhamou and colleagues also showed that antagonism is not per se an intrinsic property of an antihormone, because one human progesterone-receptor antagonist acted as an agonist when tested with a mutated receptor.
About 1 percent of women given mifepristone alone for induction of abortion do not respond to the antagonistic action of the antiprogestogen (Baulieu, 1989); vaginal bleeding is not induced in these subjects and the pregnancies continue, seemingly unaffected. Since these
women can be assumed to have normal responsiveness to progesterone as evidenced by their ability to conceive, it would be of interest to examine whether in these cases a mutation at position 722 of the progesterone receptor might be responsible for the lack of responsiveness to mifepristone. Further research in this area may also contribute to the designing of more potent and more selective compounds—antagonists as well as agonists—through a better understanding of the factors that determine the nature of the response when a ligand interacts with a receptor.
Plasma Protein Binding
One important aspect that needs to be kept in mind when evaluating animal data is the fact that, in contrast to the human, no animal species appears to have a plasma protein with binding affinity for antiprogestogens. In the human, on the other hand, mifepristone is transported in the circulation bound to orosomucoid, an a1-acid glycoprotein that acts as a high-affinity, limited-capacity binding protein (Philibert et al., 1986). Orosomucoid becomes saturated at mifepristone concentrations exceeding 2.5 µM (Heikinheimo et al., 1987); drug in excess of this concentration is probably bound with low affinity to albumin, and hence is available for metabolism and extravasation into tissues (Lähteenmäki et al., 1987). The presence of a plasma carrier protein in the human is the likely explanation for the divergent pharmacokinetic behavior of mifepristone between the human and other mammalian species (Deraedt et al., 1985). For example, plasma clearance of the compound in man (0.02 liter/hour per kilogram of body weight) is much lower than in rats and monkeys (3.0 and 1.5 liter/hour per kilogram of body weight, respectively). Administration of orosomucoid to rats reduces the clearance rate (Tremblay et al., 1989).
Lilopristone also exhibits binding to a serum protein that is not displaced by cortisol, dihydrotestosterone, or progesterone, but only by high concentrations of lilopristone or mifepristone (unpublished data of Heubner and Pollow, quoted by Henderson, 1987), suggesting that this antiprogestogen also is bound to orosomucoid. In contrast, onapristone does not appear to bind to this plasma protein (K. Chwalisz, personal communication, November 4, 1992) and, consequently, has a much shorter plasma elimination half-life (2–4 hours) compared to mifepristone (20–24 hours). It is unclear whether the absence of binding of onapristone to orosomucoid is due to this compound's configurational inversion at positions C-13 and C-17 relative to natural steroids (Figure B12.3).
The availability of compounds that do not bind to a plasma carrier protein and hence have a relatively short half-life could represent an
advantage in some situations; for example, in the use of an antiprogestogen as a late luteal, once-a-month contraceptive where a ''spillover" effect into the next cycle is to be avoided. To develop such compounds, further research into those molecular characteristics of antiprogestogens that determine their binding to carrier protein(s) may prove useful.
Pregnant Guinea Pig Model
One animal model that has been employed extensively, particularly by Elger and his colleagues at Schering AG, for the study of the abortifacient potency and mechanism of action of antiprogestogens is the guinea pig in advanced stage of pregnancy (i.e., around day 43–44 post coitum) (Table B12.1). The reasons for selecting this species and stage of pregnancy as a model of pregnancy and uterine motor function in the human have been described by Elger et al. (1987). Studies in this model and similar research in the human (Bygdeman and Swahn, 1985) led to the development of the sequential treatment regimen of mifepristone followed by prostaglandin, now used clinically in France, Great Britain, and Sweden as a nonsurgical method for early pregnancy termination (for review see, for example, Van Look and von Hertzen, 1992a). Subsequently, the same group of workers also reported a marked synergism between antiprogestogens and epostane, and between antiprogestogens and tamoxifen in inducing abortion in the pregnant guinea pig model (Elger et al., 1988). From these observations they postulated that the "estrogen background" may exert an inhibitory influence on the onset of uterine contractions in antiprogestogen-treated animals.
In order to examine if a similar effect exists in the human, a study was undertaken with the support of the Special Programme in five hospitals in Beijing, China [Wu, Clinical study of mifepristone in combination with tamoxifen and 15-methyl-prostaglandin F2a methyl ester in the termination of early pregnancy (provisional title), in preparation]. A total of 990 women with amenorrhea of up to 49 days were given a single dose of 200 mg of mifepristone followed, 72 hours later, by a vaginal suppository of 1 mg of dl-15-methylprostaglandin F2a methyl ester. In addition, half the women received tamoxifen (20 mg twice a day for two days starting at the time of mifepristone intake), and the other half received placebo tablets. The results of this trial did not confirm the synergistic effect of tamoxifen observed in the guinea pig. In fact, the complete abortion rate in the tamoxifen group (91.5 percent) tended to be lower than in the placebo controls (93.4 percent), and the interval between prostaglandin administration and expulsion of the amniotic sac was significantly (P < .02) longer in women given the antiestrogen. These results point to the need for further evaluation of the guinea pig
model, particularly for studying synergism and antagonism for different compounds given in combination with antiprogestogens. It would be of interest, therefore, to examine in the human the efficacy of other combination regimens with synergistic activity in the guinea pig, particularly the combination of mifepristone with a 3ß-HSD enzyme blocker.
MECHANISMS OF ACTION
The manner in which antiprogestogens exert their pharmacological actions has not been fully elucidated. To date most of the research has focused on the effects on the pregnant uterus, and relatively little work has been done to explain actions such as, for example, the noncompetitive antiestrogenism or the inhibition of pituitary gonadotropin secretion. Work supported by the Special Programme has been concentrated primarily on the changes induced by antiprogestogens in three areas, namely, prostaglandin metabolism, myometrial gap junctions, and progesterone- and estrogen-receptor concentrations in decidua and trophoblast (for review see Van Look and von Hertzen, 1993).
Prostaglandin Metabolism
Initial studies established that mifepristone administration resulted in an increase in the sensitivity of the uterus to exogenous prostaglandins followed by the onset of spontaneous uterine contractility, which reached a maximum level about 36–48 hours after the start of therapy (Bygdeman and Swahn, 1985; Swahn and Bygdeman, 1988). Subsequent work using the pregnant guinea pig as the animal model showed that the antiprogestogen-induced increase in myometrial sensitivity to exogenous prostaglandins appears to be due to a reduction in the catabolism rather than to a stimulation of the synthesis of prostaglandins (Brooks et al., 1990; Kelly and Bukman, 1990). Very recent experiments have demonstrated that the same applies to the human (Cheng et al., 1993). In particular, it was shown that the concentration of prostaglandin dehydrogenase, the enzyme that catalyses the first step of prostaglandin metabolism, was significantly reduced in decidual tissue obtained during surgical abortion in women treated 12, 24, or 36 hours earlier with a single dose of 200 mg of mifepristone. The activity of the enzyme was also lower in the chorionic villi of mifepristone-treated women than in controls, but this reduction was not significant. Immunohistochemistry, using a monoclonal antibody specific for prostaglandin dehydrogenase, confirmed the results of the functional assays of enzyme activity. In addition, the investigators were able to confirm immunohistochemically that the reduction in prostaglandin dehydrogenase activity was accom-
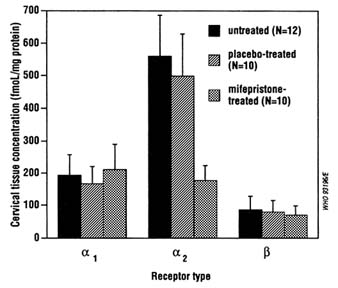
FIGURE B12.4 Effect of mifepristone on adrenergic receptors in cervical tissue specimens of early pregnant women.
SOURCE: Based on Kovacs and Falkay (1993).
panied by an increase in the concentration of prostaglandin E, particularly in perivascular cells, and a decrease in the concentration of prostaglandin E metabolites in these cells.
The exact significance of the elevated prostaglandin concentration in perivascular cells after mifepristone treatment is not clear, but the importance of this reaction may lie in the regulatory role of these cells in controlling ingress of leukocytes. To examine this possibility, studies are ongoing to quantify the effect of mifepristone treatment on the number of cells in the decidua that express the leukocyte common antigen, CD 45. It is of interest to note in this context that a marked leukocyte infiltration has been reported in an electron microscopy study of guinea pig cervical tissue after administration of onapristone (Hegele-Hartung et al., 1989).
The results described above on the effects of antiprogestogens on prostaglandin metabolism suggest that compounds capable of inhibiting prostaglandin catabolism such as inhibitors of the dehydrogenase enzyme could have abortifacient activity similar to antiprogestogens. Further research is required to examine this possibility. Additional research is also needed on the role played by adrenergic receptors in mediating the actions of antiprogestogens on the uterus. In both the rabbit (Falkay, 1990) and the human (Figure B12.4; Kovacs and Falkay, 1993), mifepristone treatment is associated with a marked reduction of
the a2-adrenoreceptor concentration in cervical tissue, but the functional significance of this change remains to be determined.
Myometrial Gap Junctions
There is now overwhelming evidence that gap junctions between myometrial cells play a key role in enabling the myometrium to behave as a functional syncytium by providing metabolic and electrical coupling between the individual cells (for review see, for example, Garfield et al., 1988). It seemed of interest, therefore, to evaluate the effect of antiprogestogens on the presence, distribution, and functional characteristics of gap junctions. To this end, immunocytochemical and immunoblot studies were carried out using a specific antibody to the gap junction protein, connexin 43. In addition, the degree of cell-to-cell coupling between the myometrial cells was assessed by measuring electrical input resistance with intracellular glass microelectrodes and by monitoring the spread of an intracellularly injected dye to neighboring cells.
The results of these studies, carried out by Garfield and his group in myometrial tissues of pregnant guinea pigs, rats, pigs, and humans (see Van Look and von Hertzen, 1993, for references) confirmed that gap junctions (connexin 43 content) in the myometrium and intercellular coupling of myometrial cells increase significantly in preterm labor induced by antiprogestogen treatment. Thus, compounds that affect the formation and/or function of gap junctions may prove useful to replace or complement antiprogestogens for induction of abortion and labor, and research aimed at finding such agents may prove rewarding.
Estrogen and Progesterone Receptors
The Special Programme has supported research on the distribution and characteristics of estrogen and progesterone receptors in decidua and trophoblast following mifepristone treatment in early pregnancy. These studies have shown that progesterone-receptor levels in cytosol of mifepristone-exposed decidua were less than half those found in placebo-treated controls; nuclear binding sites for progesterone, on the other hand, did not change (Shi et al., 1992). Estrogen-receptor levels in both nucleus and cytosol of decidual samples taken 12 hours after a single dose of 200 mg of mifepristone were about twice the concentration found in placebo-treated controls. Other workers (Zaytseva et al., 1993) have also reported increases in estrogen-receptor concentrations at 36 hours after three doses of 25 mg of mifepristone given at 12-hour intervals and after a single dose of 600 mg. In addition, Shi et al. (1992) found a marked increase in the dissociation constants of the progester-
one binding sites in both cytosol and nucleus following mifepristone treatment, an unexpected finding that is being studied further.
Immunohistochemical staining of mifepristone-exposed decidua revealed that antiprogestogen treatment resulted in increased staining for both progesterone and estrogen receptors in specimens presumed to represent decidua parietalis because of the absence of invading cytotrophoblast cells. The increased staining was most noticeable in the decidual vessels and stromal cells, and weak to absent in glandular epithelial cells. In specimens with invading cytotrophoblast cells (decidua capsularis), immunostaining of progesterone and estrogen-receptors was weaker than in decidua parietalis and not influenced by mifepristone treatment (Shi et al., 1993a). The finding of a seemingly lower progesterone-receptor content in decidua capsularis may explain the observation (Wu et al., 1990) that prolactin production and morphological decidualization in this part of the decidua are not affected by mifepristone treatment, in contrast to the decidua parietalis. Steroid binding assays on villous cytosol failed to demonstrate the presence of a specific progesterone-binding component, and immunostaining for progesterone receptor was weak in both villous and extravillous trophoblasts (Shi et al., 1993b).
The above results indicate that in addition to its ability to compete with progesterone for the cellular hormone receptor, mifepristone may influence, either directly or indirectly, the concentration and affinity of progesterone binding sites in the decidua. These effects are most marked in the decidua parietalis, which suggests that this tissue, and particularly its blood vessels, may be the primary target site of antiprogestogens.
Increases in estrogen-receptor concentration following mifepristone treatment have also been observed in the decidua and myometrium during antiprogestogen-induced premature delivery in rhesus monkeys (Haluska et al., 1990) and in estrogen-treated, ovariectomized rhesus monkeys (Wolf et al., 1989; Neulen et al., 1990). In this latter model, mifepristone has been demonstrated to antagonize the mitogenic effects of estrogen on the endometrium, which has led to the concept that mifepristone possesses a functional, noncompetitive antiestrogenic effect. A similar antiestrogenic action has been observed in the case of onapristone (K. Chwalisz, personal communication, November 4, 1992). It is at present entirely unclear through which mechanism antiprogestogens exert this antiestrogenic action. Equally unclear is the role, if any, that this functional antiestrogenism may play in the effects of antiprogestogens on endometriosis (Kettel et al., 1991), uterine leiomyomas (Murphy et al., 1993), and possibly, breast cancer (Romieu et al., 1987). In fact, there is no convincing evidence as yet that antiprogestogens possess antiestrogenic activity in the human, although the obser-
TABLE B12.2 Suggested Indications of Areas in Which Antiprogestogens May Be of Clinical Value
|
1. |
Antiprogestational Effect |
|
|
|
Gynecology |
|
|
|
• |
Cervical ripening (e.g., prior to curettage or vacuum aspiration) |
|
|
• |
Endometriosis (?) |
|
|
• |
Uterine leiomyomas (?) |
|
|
• |
Breast cancer (?) |
|
|
• |
Endometrial cancer (?) |
|
|
• |
Ectopic pregnancya |
|
|
• |
Premenstrual tensiona |
|
|
Obstetrics |
|
|
|
• |
Therapeutic pregnancy termination in second or third trimester (therapeutic abortion, intrauterine fetal death, compromised pregnancies) |
|
|
• |
Cervical ripening/induction of labor at term |
|
|
Fertility Regulation |
|
|
|
Other |
|
|
|
• |
Meningioma (?) |
|
2. |
Antiglucocorticoid Effect |
|
|
|
• |
Hypercortisolism (selected cases) |
|
|
• |
Depression (selected forms) (?) |
|
|
• |
Ocular hypertension (?) |
|
|
• |
Wound healing (?) |
|
a Currently available data suggest little or no use of antiprogestogens in this condition. |
||
vation that mifepristone (50 mg daily for three months) administered in an attempt to induce regression of uterine leiomyomas was associated with an initial rise in the plasma level of luteinizing hormone and clinical menopausal symptoms (hot flashes) in the face of follicular phase levels of estrogen, supports a possible functional antiestrogenic effect in the human too.
Further research is also needed to unravel the mechanism by which continuous mifepristone administration acts to induce chronic anovulation. As shown by the recent work of Ledger et al. (1992) and of Croxatto et al. (1993), daily mifepristone doses of 10, 5, and 2 mg, but not of 1 mg, are capable of suppressing the final stages of follicular maturation and ovulation, suggesting the possible use of antiprogestogens in oral contraception.
POSSIBLE USES
Since its discovery in early 1980, mifepristone has been suggested as of potential use in a wide range of conditions but, as shown in Table B12.2, the clinical evaluation of the compound's therapeutic potential in many of these indications has been either slow or nonexistent for a variety of reasons and, consequently, many question marks remain.
As described in other papers in this volume and in earlier reviews
(e.g., Puri and Van Look, 1991), the gynecological and obstetrical applications in which antiprogestogens have been shown of value include ripening of the pregnant cervix prior to pregnancy termination, sensitization of the uterus to prostaglandins in second-trimester abortion, and induction of labor in cases of intrauterine fetal death and in compromised or normal pregnancies. Available data suggest that antiprogestogens have no place in the conservative treatment of ectopic pregnancy (see Puri and Van Look, 1991, for references) or in the treatment of premenstrual tension (Schmidt et al., 1991).
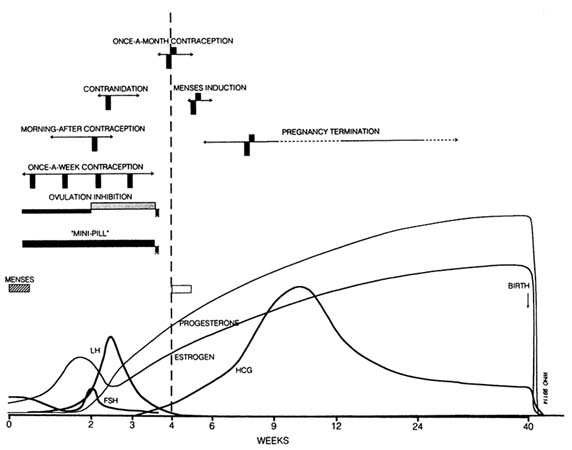
Potential uses of antiprogestogens in fertility regulation (Figure B12.5) are multiple and have been detailed in other papers in this report and earlier reviews (e.g., Van Look and von Hertzen, 1992a). To date, most of the research has focused on the use of mifepristone in combination with a prostaglandin analogue (represented by the hatched vertical bars in Figure B12.5) for pregnancy termination. Such a sequential mifepristone-prostaglandin regimen has been shown effective also for menses induction (see Baird, Appendix B4) and can be expected to be an efficacious once-a-month contraceptive. Mifepristone alone, without adjuvant prostaglandin, has shown promising results as an anti-implantation agent and in emergency contraception (see Baird, Appendix B4). Other potential uses include once-a-week contraception, ovulation inhibition (in a sequential regimen with a progestogen), and as a daily "mini-pill."
Confirmed and suggested applications of the antiglucocorticoid property of currently studied antiprogestogens include the palliative treatment of hypercortisolism due to Cushing's syndrome, certain forms of depression and of glaucoma, and in wound healing. However, as discussed earlier, it would be preferable if compounds were developed that were pure glucocorticoid antagonists devoid of antiprogestational activity.
The potential therapeutic range of antiprogestogens is wide, but as indicated above, much research still needs to be undertaken before the true value of these compounds can be determined.
PROSPECTS
Because of their unique ability to block the action of progesterone at the cellular level through binding to the progesterone receptor, antiprogestogens are proving to be one of the most significant developments in endocrinology in recent years. To date, clinical research has been dominated very much by studies on the termination of pregnancy, the indication for which the drug has been registered in four countries so far (France, China, Great Britain, and Sweden). However, as evidenced by the papers in this report and our brief review, antiprogesto-
gens are not merely "abortion pills"; they have a variety of other potential applications in several fields, including fertility regulation. Indeed, the ability to prevent pregnancy may well turn out to be a much more significant characteristic of these compounds than their abortifacient activity. Moreover, because of their antiprogestational properties and their apparent functional antiestrogenism, antiprogestogens are likely to have several therapeutic uses in the fields of obstetrics and gynecology and of reproductive health care.
In 1985, Healy and Fraser warned the medical community through the editorial pages of the British Medical Journal that "the antiprogesterones are coming." Since then, these compounds have been the subject of intense basic and clinical research, and the results obtained have left little doubt that "antiprogestogens have come to stay" (Van Look and von Hertzen, 1992a). The time has come for scientists, policymakers, and the general public to acknowledge this fact.
ACKNOWLEDGMENT
We are grateful to Ms. C. Anderson for typing the manuscript.
REFERENCES
Allen, W.M., and Wintersteiner, O. Crystalline progestin. Science 80:190–191, 1934.
Baulieu, E.-E. Contragestion and other clinical applications of RU 486, an antiprogesterone at the receptor. Science 245:1351–1357, 1989.
Bayard, F., Damilano, S., Robel, P., et al. Cytoplasmic and nuclear estradiol and progesterone receptors in human endometrium. Journal of Clinical Endocrinology and Metabolism 46:635–648, 1978.
Bélanger, A., Philibert, D., and Teutsch, G. Regio and stereospecific synthesis of 11ß-substituted 19-norsteroids: Influence of 11ß-substitution on progesterone receptor affinity. Steroids 37:361–383, 1981.
Benhamou, B., Garcia, T., Lerouge, T., et al. A single amino acid that determines the sensitivity of progesterone receptors to RU 486. Science 255:206–209, 1992.
Birgerson, L. Ölund, A., Odlind, V., et al. Termination of early human pregnancy with epostane. Contraception 35:111–120, 1987.
Bouin, P., and Ancel, P. Sur la fonction du corps jaune. Action du corps jaune vrai sur l'utérus (deuxième note préliminaire). Comptes Rendus hebdomadaires des Séances et Mémoires de la Société de Biologie et de ses Filiales 66:505–507, 1909a.
Bouin, P., and Ancel, P. Sur la fonction du corps jaune (quatrième note préliminaire). Démonstration expérimentale de l'action du corps jaune sur l'utérus et la glande mammaire. Comptes Rendus hebdomadaires des Séances et Mémoires de la Société de Biologie et de ses Filiales 66:689–690, 1909b.
Brooks, J., Holland, P., and Kelly, R. Comparison of antiprogestin stimulation of uterine prostaglandin synthesis in vitro. Prostaglandins, Leukotrienes and Essential Fatty Acids 40:191–197, 1990.
Butenandt, A., and Westphal, U. Zur Isolierung und Charakterisierung des Corpusluteum-Hormons. Berichte Deutsche chemische Gesellschaft 67:1440–1442, 1934.
Bygdeman, M., and Swahn, M.L. Progesterone receptor blockage. Effect on uterine contractility and early pregnancy. Contraception 32:45–51, 1985.
Cheng, L., Kelly, R.W., Thong, K.J., et al. The effects of mifepristone (RU 486) on prostaglandin dehydrogenase in decidual and chorionic tissue in early pregnancy. Human Reproduction 8:705–709, 1993.
Cook, C.E., Lee, Y.W., Wani, M.C., et al. Hormonal properties of 11ß-(4-N,N-dimethyl-aminophenyl)-19-norpregna-4,9-diene-3, 20-diones. Journal of Steroid Biochemistry 36 (Supplement):68S, 1990.
Corner, G.W., and Allen, W.M. Physiology of the corpus luteum. II. Production of a special uterine reaction (progestational proliferation) by extracts of the corpus luteum. American Journal of Physiology 88:326–339, 1929.
Creange, J.E., Anzalone, A.J., Potts, G.O., et al. WIN 32,729, a new, potent interceptive agent in rats and rhesus monkeys. Contraception 24:289–299, 1981.
Crooij, M.J., de Nooyer, C.C.A., Rao, B.R., et al. Termination of early pregnancy by the 3ß-hydroxysteroid dehydrogenase inhibitor epostane. New England Journal of Medicine 319:813–817, 1988.
Croxatto, H.B., Salvatierra, A.M., Croxatto, H.D., et al. Effects of continuous treatment with low dose mifepristone throughout one menstrual cycle. Human Reproduction 8:201–207, 1993.
Deraedt, R., Bonnat, C., Busigny, M., et al. Pharmacokinetics of RU 486. In E.E. Baulieu and S.J. Segal (eds.) The Antiprogestin Steroid RU 486 and Human Fertility Control. New York: Plenum Press, 1985.
Dorfman, R.I. Biosynthesis of progestogens. In Handbook of Physiology . Section 7: Endocrinology. Vol. II: Female Reproductive System. Part 1. Greep, R.O., ed. Washington, D.C.: American Physiological Society, 1973.
Elger, W., Shi, Q.S., Fähnrich, M., et al. The mechanism of action of new antiprogestins. In Fertility Regulation Today and Tomorrow. Diczfalusy, E. and Bygdeman, M., eds. New York: Raven Press, 1987.
Elger, W., Neef, G., Ottow, E., et al. Studies on interactions of antiprogestins with prostaglandins and sex hormone-related agents at the myometrial level in pregnant guinea pigs. In Hormone Antagonists for Fertility Regulation. Puri, C.P. and Van Look, P.F.A., eds. Bombay: Indian Society for the Study of Reproduction and Fertility, 1988.
Eliezer, N., Hurd, C.B., and Moudgil, V.K. Immunologically distinct binding molecules for progesterone and RU 38486 in the chick oviduct cytosol. Biochimica et Biophysica Acta 929:34–39, 1987.
Falkay, G. Effects of antiprogestogen (RU 486) treatment on myometrial and cervical alpha and beta-adrenoreceptors in pregnant rabbits. Human Reproduction 5:924–927, 1990.
Fraenkel, L. Die Funktion des Corpus luteum. Archiv für Gynäkologie 68:438–545, 1903.
Garfield, R.E., Blennerhassett, M.G., and Miller, S.M. Control of myometrial contractility: Role and regulation of gap junctions. Oxford Reviews of Reproductive Biology 10:436–490, 1988.
Goldman, A.S. Stoichiometric inhibition of various 3ß-hydroxysteroid dehydrogenases by a substrate analogue. Journal of Clinical Endocrinology and Metabolism 27:325–332, 1967.
Groyer, A., Le Bouc, Y., Joab, I., et al. Chick oviduct glucocorticosteroid receptor. Specific binding of the synthetic steroid RU 486 and immunological studies with antibodies to chick oviduct progesterone receptor. European Journal of Biochemistry 149:445–451, 1985.
Haluska, G.J., West, N.B., Novy, M.J., et al. Uterine estrogen receptors are increased by RU 486 in late pregnant rhesus macaques but not after spontaneous labor. Journal of Clinical Endocrinology and Metabolism 70:181–186, 1990.
Hartmann, M. and Wettstein, A. Ein krystallisiertes Hormon aus Corpus luteum. Helvetica Chimica Acta 17:878–882, 1934.
Healy, D.L. and Fraser, H.M. The antiprogesterones are coming: Menses induction, abortion, and labour? British Medical Journal 290:580–581, 1985.
Hegele-Hartung, C., Chwalisz, K., Beier, H.M., et al. Ripening of the uterine cervix of the guinea pig after treatment with the progesterone antagonist onapristone (ZK 98.299): An electron microscopic study. Human Reproduction 4:369–377, 1989.
Heikinheimo, O., Lähteenmäki, P.L.A., Koivunen, E., et al. Metabolism and serum binding of RU 486 in women after various single doses. Human Reproduction 2:379–385, 1987.
Henderson, D. Antiprogestational and antiglucocorticoid activities of some novel 11ß-aryl substituted steroids. Pp. 106–131 in Pharmacology and Clinical Uses of Inhibitors of Hormone Secretion and Action. Furr, B.J.A., and Wakeling, A.E., eds. London: Baillière Tindall, 1987.
Hendry, L.B., and Mahesh, V.B. Stereochemical complementarity of progesterone, RU 486 and cavities between base pairs in partially unwound double stranded DNA assessed by computer modeling and energy calculations. Journal of Steroid Biochemistry and Molecular Biology 41:647–651, 1992.
Herrmann, W., Wyss, R., Riondel, A., et al. Effect d'un stéroide anti-progestérone chez la femme: Interruption du cycle menstruel et de la grossesse au début. Comptes Rendus de l'Académie des Sciences (Paris) 294:933–938, 1982.
Holmes, S.D., Van, N.T., Stevens, S., et al. Affinity labelling of the human uterine progesterone receptor with 21-, 16 alpha- and 11 alpha-bromoacetoxyprogesterone. Endocrinology 109:670–672, 1981.
Kelly, R.W., and Bukman, A. Antiprogestagenic inhibition of uterine prostaglandin inactivation: A permissive mechanism for uterine stimulation. Journal of Steroid Biochemistry and Molecular Biology 37:97–101, 1990.
Kettel, L.M., Murphy, A.A., Mortola, J.F., et al. Endocrine responses to long-term administration of the antiprogesterone RU 486 in patients with pelvic endometriosis. Fertility and Sterility 56:402–407, 1991.
Kloosterboer, H.J., Deckers, G.H.J., van der Heuvel, M.J., et al. Screening of antiprogestagens by receptor studies and bioassays. Journal of Steroid Biochemistry 31:567–571, 1988.
Kovacs, L., and Falkay, G. Changes in adrenergic receptors in the pregnant human uterine cervix following mifepristone or placebo treatment in the first trimester. Human Reproduction 8:119–121, 1993.
Kovacs, L., Sas, M., Resch, B.A., et al. Termination of very early pregnancy by RU 486—An antiprogestational compound. Contraception 29:399–410, 1984.
Lähteenmäki, P., Heikinheimo, O., Croxatto, H., et al. Pharmacokinetics and metabolism of RU 486. Journal of Steroid Biochemistry 27:859–863, 1987.
Ledger, W.L., Sweeting, V.M., Hillier, H., et al. Inhibition of ovulation by low-dose mifepristone (RU 486). Human Reproduction 7:945–950, 1992.
Loeb, L. Über die experimentelle Erzeugung von Knoten von Deciduagewebe in dem Uterus des Meerschweinchens nach stattgefundener Copulation. Zentralblatt für Allgemeine Pathologie und Pathologische Anatomie 18:563–565, 1907.
Misrahi, M., Atger, M., d'Auriol, L., et al. Complete amino acid sequence of the human progesterone receptor deduced from cloned cDNA. Biochemical and Biophysical Research Communications 143:740–748, 1987.
Moudgil, V.K., Lombardo, G., Hurd, C., et al. Evidence for separate binding sites for progesterone and RU 486 in the chick oviduct. Biochemica et Biophysica Acta 889:192–199, 1986.
Murphy, A.A., Kettel, L.M., Morales, A.J., et al. Regression of uterine leiomyomata in response to the antiprogesterone RU 486. Journal of Clinical Endocrinology and Metabolism 76:513–517, 1993.
Neulen, J., Williams, R.F., and Hodgen, G.D. RU 486 (mifepristone): Induction of dose dependent elevations of estradiol receptor in endometrium from ovariectomized monkeys. Journal of Clinical Endocrinology and Metabolism 71:1074–1075, 1990.
Okulicz, W.C. Effect of the antiprogestin RU 486 on progesterone inhibition of occupied nuclear estrogen receptor in the uterus. Journal of Steroid Biochemistry 28:117–122, 1987.
Philibert, D. RU 38386: An original multifaceted antihormone in vivo. In Adrenal Steroid Antagonism. Agarwal, M.K., ed. Berlin: Walter de Gruyter, 1984.
Philibert, D., Moguilewsky, M., Mary, I., et al. Pharmacological profile of RU 486 in animals. In The Antiprogestin Steroid RU 486 and Human Fertility Control. Baulieu, E.E. and Segal, S.J., eds. New York: Plenum Press, 1985.
Philibert, D., Moguilewsky, M., Bonnat, C., et al. Influence of human alpha 1-acid glycoprotein (AAG) on pharmacokinetics and biological activity of RU 486. In Abstracts of the 68th Meeting of the Endocrine Society. Bethesda, Md.: The Endocrine Society, 1986.
Philibert, D., Hardy, M., Gaillard-Moguilewsky, M., et al. New analogues of mifepristone with more dissociated antiprogesterone activities . Journal of Steroid Biochemistry 34:413–417, 1989.
Puri, C.P., and Van Look, P.F.A. Newly developed competitive progesterone antagonists for fertility control. Antihormones in Health and Disease, Frontiers of Hormone Research 19:127–167, 1991.
Rannevik, G., Doeberl, A., and Valentin, L. Epostane in nonpregnant females: Effects on progesterone, 17α-hydroxy-progesterone, and 17β-estradiol of two dose levels given for one month. Fertility and Sterility 50:893–902, 1988.
Romieu, G., Maudelonde, T., Ulmann, A., et al. The antiprogestin RU 486 in advanced breast cancer: Preliminary clinical trial. Bulletin du Cancer (Paris) 74:455–461, 1987.
Savouret, J.F., Misrahi, M., and Milgrom, E. Molecular action of progesterone. International Journal of Biochemistry 22:579–594, 1990.
Schane, H.P., Creange, J.E., Anzalone, A.J., et al. Interceptive activity of azastene in rhesus monkeys. Fertility and Sterility 30:343–347, 1978.
Schmidt, P.J., Nieman, L.K., Grover, G.N., et al. Lack of effect of induced menses on symptoms in women with premenstrual syndrome. New England Journal of Medicine 324:1174–1179, 1991.
Selinger, M., MacKenzie, I.Z., Gillmer, M.D., et al. Progesterone inhibition in midtrimester termination of pregnancy: Physiological and clinical effects. British Journal of Obstetrics and Gynaecology 94:1218–1222, 1987.
Shi, W.-L., Wang, J.-D., Xu, L.-K., et al. Early changes of affinity and binding sites of progesterone and oestrogen receptors in decidua exposed to RU 486. Human Reproduction 7:934–939, 1992.
Shi, W.-L., Wang, J.-D., Fu, Y., et al. Estrogen and progesterone receptors in human decidua after RU 486 treatment. Fertility and Sterility, 1993a (in press).
Shi, W.-L., Wang, J.-D., Fu, Y., et al. The effect of RU 486 on progesterone receptor in trophoblast population of human early pregnancy. Human Reproduction, 1993b (in press).
Shinada, T. Yokota, Y., and Igarashi, M. Inhibitory effect of various gestagens upon the pregnenolone 3β-ol-dehydrogenase-Δ5-4-isomerase system in human corpora lutea of menstrual cycles. Fertility and Sterility 29:84–87, 1978.
Slotta, K.H., Ruschig, H., and Fels, E. Reindarstellung der Hormone aus dem Corpusluteum. Berichte Deutsche chemische Gesellschaft 67:1270–1273, 1934.
Swahn, M.L., and Bygdeman, M. The effect of the antiprogestin RU 486 on uterine contractility and sensitivity to prostaglandin and oxytocin. British Journal of Obstetrics and Gynaecology 95:126–134, 1988.
Swahn, M.L., Kovacs, L., Aedo, A.R., et al. Termination of early pregnancy with ZK
98.734: A dose-finding, pharmacokinetic study. Human Reproduction, 1993 (manuscript submitted).
Teutsch, G. 11ß-Substituted 19-norsteroids: At the crossroads between hormone agonists and antagonists. In Adrenal Steroid Antagonism. Agarwal, M.K., ed. Berlin: Walter de Gruyter and Co., 1984.
Teutsch, G. Analogues of RU 486 for the mapping of the progestin receptor: Synthetic and structural aspects. In The Antiprogestin Steroid RU 486 and Human Fertility Control. Baulieu, E.E. and Segal, S.J., eds. New York: Plenum Press, 1985.
Teutsch, G., Ojasoo, T., and Raynaud, J.P. 11ß-Substituted steroids: An original pathway to antihormones. Journal of Steroid Biochemistry 31:549–565, 1988.
Tremblay, D., Busigny, M., Bonnat, C., et al. Experimental demonstration in the rat on the role played by human 1 glycoprotein (HAAG) in the nonlinearity of RU 486 pharmacokinetics in women. In Alpha1-Acid Glycoprotein: Genetics, Biochemistry, Physiological Functions, and Pharmacology. New York: Alan R. Liss, 1989.
Ulmann, A., Teutsch, G., and Philibert, D. RU 486. Scientific American 262(6):42–48, 1990.
Van Look, P.F., and Bygdeman, M. Antiprogestational steroids: A new dimension in human fertility regulation. Oxford Reviews of Reproductive Biology 11:2–60, 1989.
Van Look, P.F., and von Hertzen, H. Antiprogestins in fertility regulation. Magyar Nöorvosok Lapja 55:215–222, 1992a.
Van Look, P.F., and von Hertzen, H. Post-ovulatory methods for fertility regulation. In Annual Technical Report 1991. Geneva: World Health Organization, 1992b.
Van Look, P.F., and von Hertzen, H. Post-ovulatory methods for fertility regulation. In Annual Technical Report 1992. Geneva: World Health Organization, 1993.
Wang, J.-D., Fu, Y., Shi, W.-L., et al. Immunohistochemical localization of progesterone receptor in human decidua of early pregnancy. Human Reproduction 7:123–127, 1992.
Webster, M.A., Phipps, S.L., and Gillmer, M.D.G. Interruption of first trimester human pregnancy following epostane therapy. Effect of prostaglandin E2 pessaries. British Journal of Obstetrics and Gynaecology 92:963–968, 1985.
Wiechert, R., and Neef, G. Synthesis of antiprogestational steroids. Journal of Steroid Biochemistry 27:851–858, 1987.
Wolf, J.P., Hsiu, J.F., Anderson, T.L., et al. Noncompetitive antiestrogenic effect of RU 486 in blocking the estrogen-stimulated luteinizing hormone surge and proliferative action of estradiol on endometrium in castrate monkeys. Fertility and Sterility 52:1055–1060, 1989.
Wu, W.X., Glasier, A., Norman, J., et al. The effects of the antiprogestin mifepristone, in vivo, and progesterone in vitro on prolactin production by the human decidua in early pregnancy. Human Reproduction 5:627–631, 1990.
Zaytseva, T.S., Goncharova, V.N., Morozova, M.S., et al. The effect of RU 486 on progesterone and estrogen receptor concentration in human decidua of early pregnancy. Human Reproduction, 1993 (in press).

























