
B1
1993: RU 486—A Decade on Today and Tomorrow
Étienne-Émile Baulieu, M.D., Ph.D.
Unité de Recherches sur les Communications Hormonales (INSERM U 33) and Faculté de Médecine Paris-Sud, Bicêtre Cedex, France
The development of RU 4861 (Figure B1.1), the first efficient antiprogestin, may be seen as a result both of the biomedical revolution of the last few decades and the efforts of the women's movement during the twentieth century to control their reproductive life. This conjunction was exemplified in the early 1950s when Margaret Sanger went to Gregory Pincus to discuss the possibility of developing a medical method to achieve "planned parenthood." The result of this meeting, which merged science (hormone research) and the cause des femmes, was the invention of the contraceptive pill (Pincus, 1965). "The pill" remains at least as important symbolically as it is useful practically. Scientifically, this development was based on the physiological concept that sex steroid hormones exert negative feedback control on ovulation. With progress in steroid chemistry, orally active compounds that mimicked the action of endogenous steroids were developed (Djerassi, 1970).
In the 1960s and 1970s, it became clear that the available contraceptive methods did not completely meet the needs of women and their families; nor would they alone have a sufficient demographic impact to
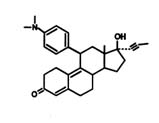
FIGURE B1.1 Mifepristone (RU 486).
limit the population explosion. During those decades, ideas for new methods of contraception emerged as biology became focused more on the cellular and molecular elements of regulation of the reproductive system. The hormone-responsive proteins of target cells in the reproductive tract (termed receptors) were discovered, while progesterone (P), designated as the hormone of gestation (pro gestare) by Corner (in 1932) (Corner, 1963), was now easy to quantitate by radioimmunoassays (Lieberman et al., 1959). The uterine progesterone receptor (PR) (Milgrom et al., 1970) and the synthesis and action of prostaglandins (PG) (Bergström et al., 1972) were described, while the role of progesterone in the establishment and maintenance of pregnancy in women was demonstrated (Csapo and Pulkkinen, 1977). As it became clear that progesterone is involved at all steps of the reproductive processes, antagonists of progesterone were actively sought. As early as 1975, the concept of a "midcycle" contraceptive, a method based on progesterone receptor down-regulation with an "antiprogesterone" ligand, was proposed (Baulieu, 1975). Now, in 1993, we have a number of efficient antiprogestins. Although induction of abortion has been the most immediate application of such compounds, other potential applications include delivery, contraception, and treatment of several hormone-dependent diseases.
When developing a procedure for the termination of pregnancy in women, it is important to be aware of both moral and physiological ideals, as well as psychological concerns. For centuries, abortion has been not only a morally difficult event for women, but also a physically painful and often dangerous procedure. A medical means for pregnancy termination should diminish this threat to women's health and, in turn, allow them to maintain their dignity. Furthermore, the distinction between abortion and contraception has lessened because the beginning of pregnancy is now understood, in physiological terms, to be a progression of steps. Hence, the term "contragestion" was proposed (Baulieu, 1985, 1989a, b) to clearly designate a method that can provoke pregnancy interruption (contra gestation) and operates as soon as possible after fertilization might have occurred, before the word abortion is appropriate (is an IUD considered an abortifacient?, see later discussion). This change in concept may be one of the most important outcomes of RU 486 development and usage.
ANTIHORMONES: THE 20 YEARS BEFORE RU 486
The aim of suppressing hormone activity is almost as old as the word hormone (wrm‘‘’ein: to excite) itself. If a hormone molecule is excitatory for the target cells, then suppression of its effects can be attained by (1) abolition of its production, (2) blockade of its transport from the gland that produces it to target organs, or (3) blockade of its action at the target cell. In the case of small, lipophilic steroids such as P, which act intracellularly, the latter possibility could mean prevention of its entry into potentially responsive cells.
The first of these possibilities, suppression of biosynthesis, seems feasible in humans for some situations. For example, enzymatic inhibitors such as Epostane (4,5-epoxy-17ß-hydroxy-4, 17a-dimethyl-3-oxo-5a-androstane-2-carbonitrile), which inhibits 3ß-hydroxysteroid dehydrogenase, have been tested with some success in abortion (Birgerson and Odlind, 1987; Crooij and Janssens, 1988). An approach that blocks the action of hormones using specific antihormone antibodies—such as antibodies that interact with P in the blood or in target organs (Wang et al., 1989)—does not seem easily applicable to humans. However, an antihormone that operates directly at the receptor level may act more rapidly and be more specific than an inhibitor of a key enzyme involved in the synthesis of many steroids. In fact, the center of hormone action and thus the best molecular target for antihormonal action is the receptor (R) protein molecule, a mandatory element for cellular responses to hormone.2
The image of a receptor portrayed as a lock whose key is the hormone and whose keyhole (in fact a "binding site") can be competitively occupied and consequently put out of order by a false key (an antihormone) has been popular for decades. Because steroids are rigid molecules of well-defined conformation, as the high-affinity binding site of the receptor should be, it seemed logical to expect that a breakthrough in the hormone antagonism field would occur first in the antisteroid field. Initially, steroid receptors were detected by the binding of a traceable (radiolabeled) hormone to tissue extracts. The first of these so-called "radioreceptor" experiments was performed with tritiated estradiol (the natural estrogen) and MER 25, an antiestrogen (Segal and Nelson, 1958) that competed efficiently for radioactive hormone uptake and retention in the uterus (Jensen and Jacobson, 1962). The structure of MER 25 (Figure B1.2) is not that of a steroid. It is a triphenylethylene stilbene derivative with two phenyl rings mimicking rings A and D of
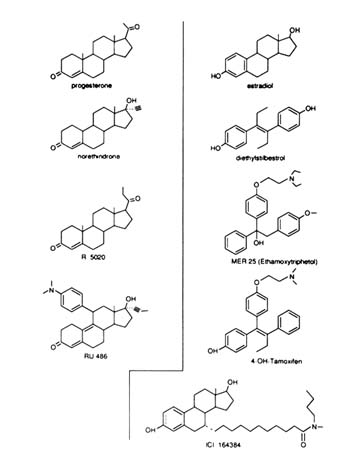
FIGURE B1.2 Structure of some progestins, estrogen agonists, and antagonists.
the steroids. X-ray crystallographic studies of the nonsteroidal estrogen diethylstilbestrol (DES) and estradiol (E) have delineated their similarity (Hospital et al., 1972). The third ring of triphenylethylene derivatives is perpendicular to the rest of the steroid-mimicking skeleton (Figures B1.2 and B1.3)—a fact that was of great importance. Given the high affinity that molecules such as E and DES show for the receptor, it was not surprising that triphenylethylene derivatives such as MER 25 and tamoxifen (Figure B1.2) had lower affinity than the agonists. However, the presence or absence of the third phenyl ring is not the critical factor for determining binding affinity since 4-hydroxytamoxifen, with an additional hydroxyl on the ring A equivalent of the tamoxifen molecule, mimicking the 3-hydroxyl group of estradiol, is a compound with high affinity for the receptor and has a resulting strong antiestrogenic effect ("pure" antagonist with no agonist activity in the chick) (Sutherland et al., 1977).
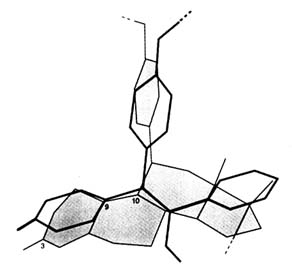
FIGURE B1.3 Superimposition of RU 486 and tamoxifen skeletons. Calculations from X-ray crystallographic data were made by Jean-Paul Mornon (Laboratoire de Crystallographie, CNRS URA 09 and Universités Paris VI and Paris VII).
I was very impressed by these last data concerning 4-hydroxytamoxifen because they contradicted the thinking of the time—all known antagonists had low affinity for their respective receptors: antiestrogens (e.g., tamoxifen), antiandrogens (e.g., cyproterone acetate, flutamid), antiglucocorticosteroids (e.g., P), and antialdosterone (e.g., spironolactone, P). Screening for antihormonal steroids tended to eliminate compounds demonstrating a high affinity for the receptor. In fact, there was no adequate theoretical reason to equate the quantitative notion of high affinity with the qualitative property of hormone antagonism. The latter was predictably due to specific conformational changes of receptor domain(s) that are involved in the transcription activation functions (TAF) of the receptor (see Figure B1.7 and later discussion), in particular in the ligand binding domain (LBD). In contrast, steroid binding affinity reflects interaction with the binding site, also located in the LBD, and is important only for kinetic quantitative aspects of the antihormone activity. I presented this scenario to Robert Bucourt who was head of chemistry at Roussel-Uclaf in the early 1970s.
Interestingly, Dr. Bucourt and his colleagues had collaborated with us to purify the estrogen receptor by affinity chromatography. Initially, this involved the screening of potential receptor ligands. Among the synthetic derivatives tested by Hélène Richard-Foy were estrogens with a long side chain grafted at the 7a-position. We selected one of them for receptor purification (Bucourt et al., 1978); however, Roussel did not test its biological activity. [About 10 years later, it was found to be an antagonist of estrogens by ICI researchers (Wakeling and Bowler, 1988).] It is important to note that the 7a-substitution on the steroid skeleton is somewhat symmetrical to an 11ß-substitution, consistent with the
structures of the triphenylethylene antiestrogens and of the 11ß-phenyl derivative compounds of the RU 486 series.
Also in the early 1970s, the Roussel chemists were working to improve the synthesis of new glucocorticosteroids and found a new way to produce 11ß-derivatives of steroids. They discovered that 5a, 10a-epoxides obtained by metachloroperbenzoic acid treatment of 5(10), 9(11)-estradienes are prone to nucleophilic opening with Grignard reagents (Nédélec and Gasc, 1970). In addition, either copper chloride-catalyzed Grignard reagents or lithium organocuprates efficiently gave the corresponding regio- and stereospecific 11ß-substituted 4,9-estradienes (Teutsch and Bélanger, 1979; Bélanger et al., 1981). Interestingly, the size of the substituent appears to largely determine agonistic or antagonistic activities.
Thus chemical research on the synthesis of glucocorticosteroids and biological studies of estrogens/antiestrogens converged when the RU 486 series of compounds was synthesized by Georges Teutsch and colleagues (Teutsch et al., 1988). The remarkable analogy of orientation of the third ring of tamoxifen and the fifth ring of RU 486 (approximately coplanar with the C-9 to C-11 bond, both perpendicular to the basic stilbene or steroid skeletons), is shown in Figure B1.3. Indeed, the 11ß-phenyl-N-dimethyl-substituted estradiol is a strong antiestrogen (unpublished result).
The rest of the RU 486 story, which has been presented in several publications (see footnote 1), continued with the observation of the antiglucocorticosteroid activity of RU 486, and thereafter the demonstration of its antiprogesterone property. The decision to test it for human abortion was made after the endocrinological and pharmacological studies performed by Daniel Philibert and colleagues. We proposed that the compound was active and probably safe, but the idea of using RU 486 in human beings was almost ''killed" by toxicologists who did not correctly interpret the signs of cortisol insufficiency when the product was given at very high doses in monkeys for several consecutive weeks. RU 486 was rescued by my insistence that it was just a beautiful (in vivo) demonstration of the antiglucocorticoid activity of the compound in primates (Baulieu, 1991c).
This compound became the subject of a political debate that is not relevant to this review. However, the scientific story is not complete, and should be pursued in order to improve and to extend the first discoveries.
CHEMISTRY: NOVEL MOLECULES
Almost all the potent antiprogestins and antiglucocorticosteroids so far described are 11ß-phenyl-substituted steroids. The exception is RU 43044 (Figure B1.4), a 17ß-substituted steroid: see later in the text. The
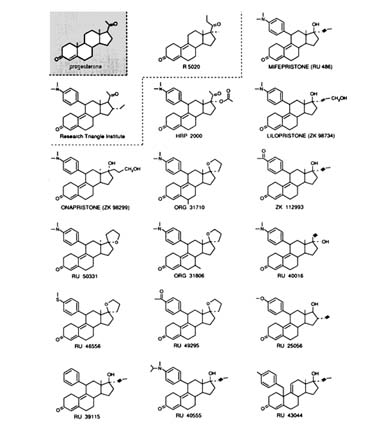
FIGURE B1.4 Structure of some currently available progestins and antiprogestins.
relatively long half-life of RU 486 in human beings (˜20 hours) seems to be due to its ability to bind to plasma orosomucoid (an a1-glycoprotein) (Moguilewski and Philibert, 1985; Grimaldi et al., 1992). This binding is not found in nonhuman primates or other animals. RU 40555 (see Figure B1.4 for structure of this and other compounds discussed in this section) does not bind to the orosomucoid and has a shorter half-life, which may be of interest for kinetic assessment of the hypothalamus-pituitary-adrenal axis in clinical endocrinology (Bertagna et al., 1984; Gaillard et al., 1984). However, the binding of RU 486 and lilopristone (ZK 98734) to orosomucoid may enhance the antisteroid activity since it protects the drug against metabolic inactivation and provides a reservoir system for sustained delivery to target cells.
Since the early studies with RU 486, chemists have tried to dissociate the two main antihormonal activities of the compound and have aimed, for obvious medical reasons, to obtain "pure" antiprogestin(s) and
"pure" antiglucocorticosteroid(s) that would not display any other endocrine effects.
At present, there is no published account of a pure antiprogestin compound. However, it is important to note that for abortion the antiglucocorticosteroid effect is apparently neither necessary nor even useful, and that a single dose of =600 mg of RU 486 does not create any medical problem related to corticosteroid insufficiency. RU 486 derivatives,* such as RU 46556 and RU 49295, are strong antiprogestins with limited antiglucocorticosteroid activity. ORG 31710 (more active) and ORG 31806 have less antiglucocorticosteroid activity than RU 486 (Mizutani et al., 1992). A 17a-acetoxy derivative such as HRP2000 (Research Triangle Institute, Cook et al., 1992), with a 17ß-progesterone side chain and a 11ß RU 486-like substituent, is both an antiprogestin and an antiglucocorticosteroid. Curiously, 17ß-acetyl, 16a-ethyl derivatives of 11ß-phenyl-substituted steroids are progestin agonists (Cook et al., 1992). The Schering group has synthesized lilopristone, with a 17ß side chain slightly different from that of RU 486; it has less antiglucocorticosteroid activity and higher binding to the androgen receptor. Another compound (ZK 112993) also has reduced antiglucocorticosteroid activity in the rat, due to an acetyl group on the 11ß-phenyl moiety. A significant change in the RU 486 structure was obtained by making onapristone (ZK 98 299); due to photochemical epimerization at C-13, inversion of the D ring and substitutions at the C-17 position occur (Elger et al., 1986; Neef et al., 1984). Onapristone does not bind to orosomucoid (contrary to RU 486), does not bind to the chicken (c) PR (like RU 486) (Nath et al., 1991), is an antiprogesterone (but less active than RU 486), and has weak antiglucocorticosteroid activity. Its mechanism currently is controversial (see later).
A pure antiglucocorticosteroid may be easier to use chronically in premenstrual women. One possibility is RU 40016, an RU 486-like compound with inversion of substituents at the C-17 position. Although not very active, it has relatively more antiglucocorticoid and fewer antiprogestin effects than RU 486. RU 43044 is chemically very different, since the additional phenyl substituent is in the 10ß position, and although this ring is partially superimposable spatially, with a phenyl group in 11ß, there is no binding to the PR, and the activity is purely antiglucocorticosteroid (but weaker than that of RU 486). The compound, perhaps because of its metabolism, has no activity in vivo in animals; however, its activity in situ may provide some clues for the synthesis of a series of locally active thereapeutic agents.
In conclusion, the 11ß-phenyl substitution is essential in determining the antagonistic properties of most antisteroids, while an 11ß-aliphatic
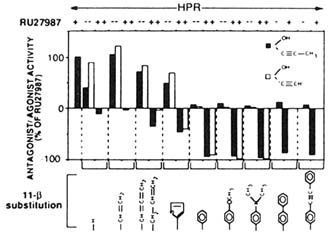
FIGURE B1.5 Agonists and antagonists of 11β-substituted steroids. Agonistic and antagonistic potential of two series of 11β-substituted steroids that differ from RU 486 only in their 11β-substitutions or their 11β- and 17α-substitutions. The chemical symbols in the top right corner of the upper panels illustrate the 17α-substitutions. Transcription activation was quantitated from normalized CAT assays in HeLa transiently-transfected cells with the MMTV-CAT reporter gene and the human progesterone-receptor (hPR) expression vector hPR1 in the presence of the various compounds. The agonistic potential of the hPR in the presence of 1 µM of these steroids alone (- RU 27987, at the top) is expressed as a positive value relative to the activation seen with 10 nM RU 27987 (arbitrarily assigned +100). Antagonistic potential was assayed by exposing transfected cells to 1 µM of a given steroid plus 10 nM RU 27987 (+ RU 27987 at the top) and is expressed as a negative value, with -100 indicating complete inhibition of RU 27987-induced transcription. The individual 11β-substitutions are depicted. RU 27987 is a 17α,21-dimethyl-3,20-dioxo-21-hydroxy-19-nor-pregna-4,9-diene progestin agonist. SOURCE: Garcia et al. (1992); © The Endocrine Society.
chain may result in agonistic derivatives (Figure B1.5). However, most steroidal structures do not carry an absolute intrinsic property of agonism or antagonism per se, as demonstrated by steroid binding differences between the PR of different species, changes of activity when mutating the receptor LBD, and activity differences in various target cells under different physiological states.
RU 486 and many corresponding compounds from Schering and Organon do not bind to cPR (Groyer et al., 1985), although they bind to the PR of humans (hPR) and most other mammals. The change of a cysteine (Cys) in the N-terminal region of the cPR LBD (Cys 575) to a glycine (Gly), as found in the hPR (cPR Cys 575 → Gly), permits the binding of RU 486 and antisteroid activity. Interestingly, RU 39115 (which is RU 486 minus N-dimethyl) is an antagonist of the hPR, but an agonist of the "humanized" chicken PR (cPR 575 → Gly). This indicates that the interaction of steroid and receptor is more complex than just binding
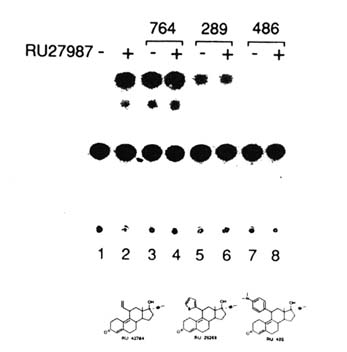
FIGURE B1.6 Agonists, antagonists, and mixed agonists-antagonists. Ligands for the hPR may generate three distinct types of TAF2-dependent transcriptional responses: they act as agonists with no antagonistic potential or as antagonists with no agonistic potential, or they may generate a mixed response, since they both activate and antagonize transcription activation. HeLa cells were transiently transfected with a reporter gene and a progesterone response element and exposed to the steroids in the absence or presence of RU 27987 (Figure B1.5). Steroids were used at 1 µM (- RU 27987; lanes 3, 5, 7); in cases where the antagonistic potential was analyzed, activation was achieved with 10 nM (RU 27987; lanes 4, 6, 8). Note that RU 28289 acts as both agonist and antagonist. SOURCE: Garcia et al. (1992); © The Endocrine Society.
ability, and may depend on the overall structure of the LBD and consequent modification of TAF2 function (see later). Systematic experiments indicate that depending on the nature and positioning of the 11ß-phenyl substituent, one may produce 11ß-substituted steroids with progestin agonistic, antagonistic, or mixed agonistic and antagonistic activities (with, as expected, no relationship to binding affinity) (Benhamou, 1992; Garcia et al., 1992) (Figure B1.6).
Indeed it is logical that the structure of the steroids and of the LBD combine ultimately to direct the conformation and, thus, the function of TAF2, therefore "deciding" if a compound will act as an agonist or an
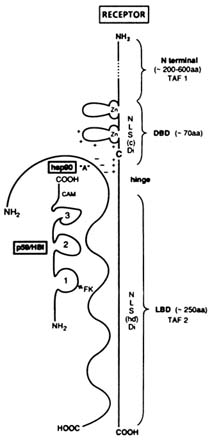
FIGURE B1.7 Schematic representation of a steroid hormone-receptor consensus structure in its "8S" heterooligomeric form. NOTE: aa = amino acid; NLS(c) = constitutive nuclear localization signal; NLS(hd) = hormone-dependent nuclear localization signal; Di = dimerization; FK = FK506; CAM = calmodulin; C = cysteine; + and - = conserved charged amino acids. Other abbreviations are as indicated in text.
antagonist. This is potentially important for cancer treatment, since the steroid-receptor mutations that are observed in certain tumors may radically change the properties of their receptors and the effectiveness of steroidal drugs.
CELLULAR AND MOLECULAR MECHANISMS OF ACTION OF ANTIPROGESTINS: THE RECEPTOR AT THE CENTER
The "consensus" anatomy of steroid receptors (Evans, 1988) and the concept of associated proteins (Lebeau et al., 1993) are illustrated in Figure B1.7. Shown in Table B1.1 and Figure B1.8 are several steps involved in the intracellular mechanism of steroid hormone and antihormone action.
Progesterone, cortisol, and their cognate synthetic agonists and antagonists seem to enter target cells freely and appear not to be
TABLE B1.1 Steps in the Intracellular Mechanisms of Steroid Hormone/Antihormone Actiona
significantly metabolized. They bind to receptors with high affinity (KD = 10 nM). A given ligand may bind to different receptors (for example, P and RU 486 bind to both PR and the glucocorticosteroid receptor [GR]), and a given receptor can bind multiple hormones (e.g., the GR binds both cortisol and P).
Upon binding of a ligand, transconformation of the receptor protein occurs. This molecular "reaction" is central to the mechanism of action of hormones and antihormones, and determines nuclear localization, binding to the hormone response element (HRE) of regulated genes, chromatin change, activation or inhibition of transcription, and possibly other activities.
The GR and PR, like other steroid hormone receptors, form heterooligomeric, non-DNA binding, "nontransformed" 8S complexes that include receptor-associated proteins (Baulieu et al., 1989; Lebeau et al., 1993). The most studied of these proteins are a heat shock protein of MW = 90,000 Da (hsp90), a "chaperone" protein (Baulieu and Catelli, 1989), and p59-HBI (a hsp90 binding immunophilin) (Callebaut et al., 1992) (Figures B1.7 and B1.8). Although their roles are not completely understood and remain controversial, certain relationships among these proteins are apparent.
The p59-HBI, a peptidyl-proline isomerase, also known as FKBP56 or 52, binds immunosuppressants such as FK506 and rapamycin. The binding of hsp90 by p59-HBI is not competitive with the binding of the immunosup-
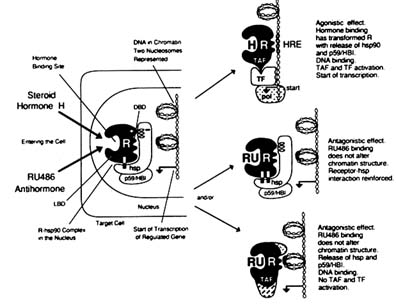
FIGURE B1.8 Cellular and molecular mechanisms of action of steroid hormone and antihormone. NOTE: hsp = heat shock protein, molecular mass ˜ 90,000 Da; pol = RNA polymerase II; TF = transcription factor. Other abbreviations are as indicated in text.
pressants, which are inhibitors of the isomerase activity. The possible effect of p59-HBI binding to hsp90 on receptor function is, at present, unknown. However, binding of p59-HBI to FK506 or rapamycin in vitro increases progestin and RU 486 binding to the rabbit uterus PR (Renoir et al., 1992) and immunosuppressants augment the response of transfected reporter gene to corticosteroids (Ning and Sanchez, 1993).
Hsp90 binds to the LBD apparently in a multipoint arrangement (Pratt et al., 1988; Cadepond et al., 1992), and also binds to a positively charged region at the C-terminal extremity of the DBD (Chambraud et al., 1990). This disposition obliterates the DNA binding to the DBD (Baulieu and Catelli, 1989), and hsp90-receptor complexes do not bind to DNA (Bourgeois et al., 1984).
In hsp90-containing 8S receptor complexes, there are two molecules of hsp90 (Radanyi et al., 1989) and one molecule of the PR, GR, or MR, or two molecules of the ER (Redeuilh et al., 1987a; Rexin et al., 1988; Rafestin-Oblin et al., 1989; Renoir et al., 1989, 1990).
Hsp90, in the role of chaperone, maintains the structure of the GR LBD in the appropriate ligand-binding conformation and seems to protect non-ligand-bound steroid receptors from chemical or enzymatic attack. It was also recently observed that hsp90 may competitively
interfere with the binding of the HRE of an estrogen-regulated gene to the ER, and thus could modulate the process of transcription (Redeuilh et al., unpublished). Agonist binding to the LBD modifies the structure of the latter in such a way that it favors the release of hsp90 from the receptor, and therefore allows binding of the receptor to the HRE and interaction with transcription factors (TFs) engaged in the transcription machinery. There may be a change of ligand affinity for the receptor even before dissociation of hsp90 (Redeuilh et al., 1987b). LBD modifications after binding of ligand are involved in TAF function, receptor dimerization, and probably chromatin changes.
Whether hsp90-receptor complexes are formed in the cytoplasm, the nucleus, or both, and whether hsp90 binding plays a role in the transfer and shuttling of the receptor between the two compartments, are not yet clear. Nuclear binding to the HRE and recycling of the receptor depend upon these movements.
The hormone-dependent homodimerization of receptors is important for their binding to the two halves of the (imperfect) palindromic HRE. The LBD and the DBD include domains for dimerization.
The progesterone response element (PRE) and the glucocorticosteroid response element (GRE) are enhancer sequences, as are other HREs for other steroid receptors. They are ligand activated following receptor binding and are most often physically situated in the 5'-promoter region of hormone-regulated genes. Schematically, HREs may be seen as allowing the receptor dimer to be placed in the appropriate position for interaction with other TFs, which bind to their specific DNA sequences and are involved in the function of RNA polymerase, the enzyme that is ultimately operational in gene transcription. There are proteins that appear functionally to link receptors and TFs, and are sometimes designated as transcription intermediary factors (TIFs). These factors are necessary when the HREs are situated far away, in molecular terms, from the transcription initiation site. Receptor transconformation following ligand binding determines the appropriate interaction of the receptor with TFs/TIFs. Receptor transconformation is also likely to be involved in inducing chromatin changes, which themselves eventually cooperate in hormone action by allowing or inhibiting TF/TIF function. The receptor domains involved in TAFs are described later.
RU 486 may interfere with several of the steps indicated in Table B1.1, which are discussed separately below.
Ligand Binding
RU 486 has high affinity for the hPR and the hGR (as 4-hydroxytamoxifen has for the hER), weak affinity for the human androgen receptor, and no affinity for the ER and the MR (thus being a useful
compound for the study of this last receptor in the presence of GR, since GR and MR share many high-affinity hormone ligands and GR is found in almost all cells). Kinetic experiments have shown differences between P and RU 486 binding to the PR (Skafar, 1991).
Receptor Transconformation
Receptor transconformation is a conformational change in the receptor that occurs after ligand binding. Transconformation may take place before dissociation of hsp90 from the 8S complexes (Allan, 1992a,b), as observed with the 8S ER (Redeuilh, 1987b), and thus precedes DNA binding and activation of the transcription function.
Selective, hormone-, or antihormone-related transconformation has been physically suggested by several observations:
-
Proteolytic enzymes do not have the same effect on the PR LBD when bound to progestin or RU 486 (Allan, 1992a).
-
An antibody such as mAb C262, raised against the last 14 C-terminal amino acids of the PR, binds to the RU 486-PR complex but not to the P-PR complex (Weigel, 1992).
-
The electrophoretic mobility of RU 486-PR bound to the PRE (gel shift experiments) is faster than that of P-PR bound to the same PRE (El-Ashri et al., 1989; Meyer et al., 1990). A difference in the receptor structure when bound to hormone versus antihormone has also been observed with antiestrogen-ER complexes (Sabbah et al., 1991).
-
In contrast to R 5020-PR (R 5020 is a synthetic progestin agonist of the 4S sedimentation coefficient), RU 486-PR forms 6S entities in salt-containing gradient centrifugation experiments (Mullick and Katzenellenbogen, 1986; Renoir et al., 1989). It is not clear whether this form is transconformed monomeric PR, homodimeric PR, or a heterodimer of one molecule of PR plus another protein, perhaps hsp70.
The lack of RU 486 binding by the cPR and the Cys 575 → Gly mutation that corrects this defect have been discussed above. Conversely, if the corresponding Gly of the hPR is transformed into cysteine, RU 486 no longer binds. In the hGR, if the corresponding Gly is transformed to Cys, the receptor does not bind dexamethasone or RU 486. Thus, exchanging cysteine and glycine may modify, in a complex manner, the overall LBD structure, particularly at the level of the pocket where it binds the 11β-substituent.
A truncated PR molecule (minus the 42 C-terminal amino acids) does not bind progestin agonists but does bind RU 486, which remarkably becomes an agonist (Vegeto et al., 1992). This, and other results mentioned above, suggest that there are both a common
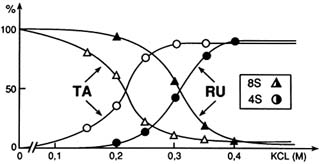
FIGURE B1.9 Glucocorticosteroid receptor (Δ0-417) 8S → 4S versus ligands. Stabilization of RU 486 by the heterooligomeric, non-DNA-binding form, of the glucocorticosteroid receptor in KCI-containing medium, as compared to the transformation effect of the agonist triamcinolone acetonide (TA). The receptor mutant Δ0-417 includes almost exclusively the DBD and the LBD. SOURCE: Segard-Maurel et al. (1992); reprinted with permission from Pergamon Press Ltd.
binding site for progestin and antiprogestin involving the C-terminal amino acids of the LBD, and another binding site for RU 486 (and other 11β-phenyl-substituted steroids) at the N-terminal extremity of the LBD. Moreover, from these results, it has been postulated that the C-terminal region of the receptor could act as an inhibitor of TAF1 in the absence of ligand or when bound to RU 486, with the negative function being released in the presence of an agonist (Vegeto et al., 1992).
Heat Shock Protein Binding
Transconformation of the LBD is probably involved in the modulation of hsp90 interaction with the receptor, and the in vitro stabilization of hsp90-containing 8S complexes after RU 486 binding has been demonstrated for RU 486-bound GR (Groyer et al., 1987) and PR (Renoir et al., 1989) (several other 11β-substituted steroids do the same) (Figure B1.9). In intact cellular systems, this stabilization has also been observed by several workers (Rajpert et al., 1987; Lefebvre et al., 1988; Segnitz and Gehring, 1990). Moreover, some experiments have indicated that RU 486-GR remains more cytoplasmic than agonist-GR (Ylikomi et al., 1992).
It should be acknowledged, however, that the situation is complex; nuclear retention of GR may vary depending on the phase of the cell cycle (Hsu et al., 1992). Also on this point, a number of studies in intact cells have strongly suggested that RU 486-receptor complexes move to the nucleus and then bind to HREs (see ''Binding to DNA"). The matter
is still controversial, and we cannot definitively state whether RU 486-receptor stabilization plays a significant role in antihormone action.
Receptor Dimerization
The extent of receptor dimerization frequently correlates with the specific DNA-binding activity of receptors. Indeed, RU 486 favors more dimerization of the PR than R 5020 (De Marzo, 1992). Due to the natural occurrence of distinct A and B subunits in the PR (PR-A and PR-B) and to the fact that the cPR does not bind RU 486, experiments have been done to analyze the potential heterodimerization of two PR subunits, one subunit binding the agonist R 5020 and the other subunit binding the antagonist RU 486 (Guiochon-Mantel et al., 1989; Meyer et al., 1990; De Marzo et al., 1992). In spite of different transconformations, R 5020-PR and RU 486-PR can form a heterodimer in solution, but not if the subunits bind to an artificially symmetrical palindromic PRE sequence (Meyer et al., 1990). If the PRE palindrome is imperfect, as it is naturally, formation of a heterodimer is possible (De Marzo et al., 1992). Therefore, dimerization is dependent not only on ligand-receptor interaction but also on receptor-DNA interaction. Heterodimerization including one RU 486-bound PR unit may be involved in the strong antihormonal activity of RU 486 via the negative effect of the RU 486 subunit on the heterodimer function.
Binding to DNA
RU 486, lilopristone, and ORG compounds all stimulate the binding of the PR to PREs (Bailly et al., 1986; Guiochon-Mantel et al., 1989; Turcotte et al., 1990; Mizutani et al., 1992). These results, in which the receptor is transformed and dissociated from hsp90, are consistent with the agonistic effects of RU 486 observed in some cell systems (Meyer et al., 1990) and some cell-free experiments with the GR (Schweizer-Groyer et al., 1988). The term "agonistic" implies that the receptor can bind to appropriate specific DNA. In the case of an antihormonal effect, indirect but suggestive evidence has been obtained by competition of a constitutively active (truncated) form of progesterone receptor with RU 486-PR complexes (Guiochon-Mantel et al., 1988).
When agonist-GR and RU 486-GR complexes bind to DNA, however, the results are kinetically different (Shauer et al., 1989). Since RU 486 binding to DNA is not followed by an effect on transcription, the RU 486-receptor complexes are unproductive, possibly due in part to a defect in chromatin structure and/or interaction with TF/TIF.
It has been suggested that the onaprisone-receptor complex does not have the ability to bind to HREs (Klein-Hitpass et al., 1991). This would
define a new type of antisteroid ("type one" for Klein-Hitpass, "type two" for Bocquel et al. [1993]). In competition experiments, onapristone inhibits the induction of DNA binding of PR by progestins and DNA-binding antiprogestins. This could be due to the lack of formation of a stable receptor dimer, but it has been recently disproved by Bocquel et al. (1993). In vitro, onapristone, in contrast to RU 486 (Schweizer-Groyer et al., 1988), may not display agonistic activity (Klein-Hitpass et al., 1991). Hyperphosphorylation of the receptor that is observed under agonist or RU 486 action is not observed using onapristone, again consistent with the hypothesis that a DNA-dependent protein kinase activity is involved (Bocquel et al., 1993).
However, the same proteolytic pattern of the PR was found with complexes involving so-called DNA binding and non-DNA-binding antiprogestins (Allan et al., 1992a). Moreover, these experimental differences between onapristone and other antiprogestins, particularly RU 486, may be ascribed to its low affinity for PR (Delabre et al., 1993).
Studies of Chromatin
In contrast to glucocorticosteroids, RU 486 does not modify chromatin structure, as assessed by DNase I experiments in the mouse mammary gland tumor virus (MMTV) and the liver tyrosine aminotransferase (TAT) systems (Véronique Marsaud and Hélène Richard-Foy, personal communication). The most likely explanation is that the chromatin modification induced by receptor transconformation after agonist binding does not occur after RU 486 binding. In addition, the binding of NF1, a nonspecific transcription factor, to its DNA site in the MMTV system does not take place after RU 486 binding as it does after the binding of an agonist.
Gene Transcription
Activation of gene transcription is regarded as the major mode of action of steroid hormones, mediated by two transcription activation functions of the receptor (Evans, 1988; Lees et al., 1989; Tora et al., 1989; Gronemeyer, 1991). TAF1 is situated in the N-terminal portion of the receptor molecule, is not hormone dependent, and can be regulated by cell-specific factors. TAF2 is hormone dependent, activated by agonist binding, and inhibited by antagonists, as demonstrated with tamoxifen derivatives for the ER (Webster et al., 1988) and RU 486 for the PR (Meyer et al., 1992). When TAF2 is inhibited, TAF1 may still operate differently depending on the cell type, and thus an antihormone may show some agonistic activity following binding of the receptor to DNA. This is true with RU 486 and theoretically should never be found with
non-DNA-binding antagonists (if they exist). The type of receptor may itself influence the result, with a probable role for the highly variable N-terminal domains. For instance, there are experiments in which RU 486 PR-B is agonistic and RU 486 PR-A is antagonistic toward the activation of a PRE-TK reporter gene (Meyer et al., 1990). TAF1 may also be controlled by an inactivation factor similar to that reported in yeast (McDonnell et al., 1992); if such an inactivation factor gene is suppressed, RU 486 becomes an agonist.
The number and positioning of the HREs, their varying structures in different genes stimulated (or repressed) by the same hormone, the variety of TFs/TIFs present along with receptors and HREs in different cells, and the other informational influences that reach the steroid-dependent transcription systems make the global network responding to steroids extremely diverse and difficult to interpret. An example of this is the modifying effect on antiprogestin action of another cAMP-dependent gene (Sartorius et al., 1993).
The expression of the receptor itself may change under the effect of its own ligand, as demonstrated for the down-regulation of the PR by progesterone (Milgrom et al., 1973). However, the effect of RU 486 on receptor down-regulation cannot be systematized, not being observed in certain cases (Sheridan et al., 1988) or being described in other instances (El-Ashry et al., 1989).
In conclusion, as would be predicted, everything depends on the complex formed by the ligand and the receptor, which is then transconformed after binding. It is important to note that genetic variation may abrogate the response to RU 486. Perhaps this is the case in the 1 percent of failures in abortion (see later), or in cancers if there are mutations such as those cited above. Hormone action involves a number of phenomena that are narrowly connected—at least temporally: release of hsp90 and other associated proteins, dimerization of the receptor, DNA binding and chromatin changes, and interaction with TFs/TIFs. The net result may be to increase or decrease gene transcription, and depends in part on specific HREs and TF(s). Antiprogestins are remarkable tools for dissecting complex cellular networks. In addition to more discoveries in cell biology, the study of these networks should lead to improved use of RU 486 and the development of novel molecules.
How lucky we were not to know of this complexity before testing RU 486, which worked so well on the basis of a "simple" hypothesis! (Herrmann et al., 1982.)
PHYSIOPHARMACOLOGICAL REPRODUCTIVE EFFECTS
The activity of RU 486 has been studied in many progesterone-responsive reproductive systems (Baulieu, 1989a; Brodgen et al., 1993) (Table B1.2). For
TABLE B1.2 Targets for Antiprogestins
example, studies in the uterus, endometrium, myometrium, and cervical tissue directly relate to the use of RU 486 in fertility control.
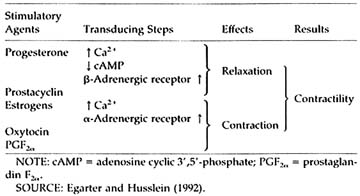
In the human myometrium (Table B1.3), RU 486 reverses the quiescence effect of progesterone, which depends on (poorly defined) effects on calcium cellular distribution and metabolism leading to decreased excitability; stimulates intercellular gap junction; and decreases ß2-adrenergic receptor synthesis (Vivat et al., 1992). In addition, prostaglandin PGF2a synthesis is increased and prostaglandin (PG) metabolism is decreased by RU 486. There is a decrease of prostacyclin release (Lobaccaro-Henri et al., 1992), and the myometrium sensitivity to the contractile effect of PGs of the F and E series is enhanced (Norman et al., 1991). In the rat, the increase of PG levels may not precede the stimulation of uterine contractility (Arkaravichien and Kendle, 1992).
Some hormonal components of RU 486 activity are difficult to classify. For instance, RU 486 and onapristone display an ER-independent antiestrogenic activity in the endometrium (Van Uem et al., 1989; Wolf et al., 1989a; Chwalisz et al., 1991; Shi et al., 1992), where they decrease the
TABLE B1.3 Interactions of Steroids/Prostaglandins/Catecholamines in the Human Myometrium
|
 |
glandular formation. This seems paradoxical since RU 486 and onapristone do not bind to the ER, and there is a PR-dependent increase of ER levels (indicated by both hormone binding and immunocytochemistry) (Haluska et al., 1990; Neulen et al., 1990).
In pregnancy, RU 486 inhibits the production of human chorionic gonadotropin (hCG) and prolactin in cultured syncytiotrophoblast cells and explants (Herrmann et al., 1985; Das and Catt, 1987). It decreases a marker of ER function, p29, in the placenta and the decidua (Rivera et al., 1991). RU 486 also increases the natural killer activity of lymphocytes (Hansen et al., 1992). In rats, it augments the interstitial collagenase activity of the cervix (Ikuta et al., 1991). RU 486 can inhibit the ovulation process (Loutradis et al., 1991) directly at the level of the ovaries, and may decrease gonadotropin-stimulated P production of granulosa cells (Parinaud et al., 1990).
Changes in the hypothalamus-pituitary-gonadal system have been reported. The PR is present in ß-endorphin neurons and dopamine neurons in the arcuate nucleus, but not in gonadotropin-releasing hormone (GnRH) neurons themselves, which are indirectly under P control (Yen, 1991). Luteinizing hormone (LH) is essentially depressed by RU 486 (Schaison et al., 1985; Garzo et al., 1988), and a decrease of follicle-stimulating hormone (FSH) may be explained by an increase of inhibin (Sanchez-Criado et al., 1992). Sexual behavior can be modified by the antiprogestin effect of RU 486 (Brown and Blaustein, 1986; Pleim, 1990).
In breast cancer cells, RU 486 inhibits P-induced transcriptional activity, which leads to the synthesis of fatty acid synthetase but, surprisingly, it stabilizes the mRNA of this enzyme (Chalbos et al., 1991).
Agonistic effects of RU 486 have been observed in vivo in the uterus (e.g., Gravanis et al., 1984) and are also responsible for 3T3 differentiation of preadipocytes (Rondinone et al., 1992), consistent with the
TABLE B1.4 Uses of RU 486
|
Contraception/Contragestion/Abortion |
|
5–9 weeks of amenorrhea |
|
Occasional luteal contragestion |
|
Once-a-month menses induction |
|
Emergency contraception |
|
Once-a-month anti-implantation |
|
"Endometrial" contraception |
|
Suppression of ovulation |
|
Medical Interruption of Pregnancy |
|
Second trimester |
|
Third trimester |
|
Labor Induction |
|
Medical Indications |
|
(Antiprogestin) |
|
Endometriosis |
|
Uterine fibroid |
|
Breast cancer |
|
Meningioma |
|
(Antiglucocorticosteroid) |
|
Endocrine test |
|
"Peripheral" hypercorticism |
|
Glaucoma |
|
Wound/burn healing |
observations made in vitro, as discussed above.
Thus, administration of RU 486 may produce effects in different directions, which have to be studied separately in each tissue. However, to provoke abortion or trigger labor, the effects on the decidua, endometrium, cervix, and even the trophoblast combine to make RU 486 a very efficient agent for the termination of pregnancy.
REPRODUCTIVE MEDICINE (TABLE B1.4)
Voluntary Early Pregnancy Interruption
On the basis of animal experiments, we expected RU 486 to meet the recognized need for an efficient medical means of early abortion, to be safer than a surgical technique, and to be relatively convenient and cheap (no anesthesia, no operating room). To develop a medical method of abortion was a must in terms of women's health and potentially a step toward more privacy for those having taken the difficult decision of pregnancy termination.
Early abortion offered the first opportunity to test RU 486 activity in
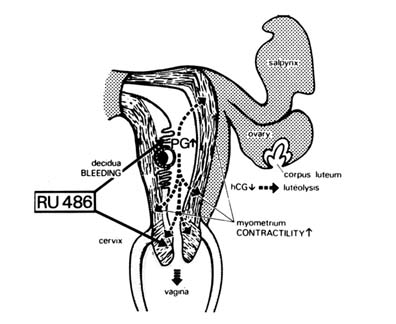
FIGURE B1.10 Physiopharmacological mechanism of action of RU 486 on the implanted blastocyst. Temporally, the antiprogesterone effect of RU 486 comes first, and then an increase in PG concentration and action, followed by a decrease in hCG-sustained corpus luteum function.
humans, and this was initially performed on a small group of women volunteers, under the direction of Walter Herrmann at the Hôpital Cantonal in Geneva in 1982. Since the postulated mechanism of action (Figure B1.10) suggested that a single dose should be sufficient, and since the compound was rapidly eliminated from the body, only rather short toxicological studies were necessary.
The results were so striking that RU 486 immediately got the nickname "abortion pill," despite the many other potential medical uses already predicted when the compound was announced (they progressively became reality). The first large-scale studies indicated approximately 80 percent success in pregnancies of £42 days of amenorrhea (Couzinet et al., 1986), and the compound was presented for registration to the French Ministry of Health. However, after the study by Bygdeman and Swahn (1985) (Table B1.5), the improved results obtained with the administration of a small dose of prostaglandin 48 hours after RU 486 administration (Baird et al., 1988; Dubois et al., 1988a) led to the approval (AMM: autorisation de mise sur le marché) in September 1988 of RU 486 plus prostaglandin for up to 49 days of amenorrhea pregnancies, in the context of the French abortion law. Sulprostone (a PGE2 analogue,
TABLE B1.5 Uterine Activity During Early Pregnancy in Control and RU 486-Treated Patients (Montevideo units; mean ± SEM)
|
|
Control |
36 Hours After 50 mg of RU 486 |
|
Mean uterine activity |
6 ± 4 |
222 ± 93 |
|
Sulprostone, 0.05 mga |
49 ± 24 |
711 ± 136 |
|
NOTE: SEM = Standard error on the mean. a Administered 0.5 hour after start of recording. SOURCE: Swahn and Bygdeman (1988). |
||
250 µg injected intramuscularly) was the prostaglandin used most. In Great Britain, the trials with gemeprost (a PGE1 analogue, 0.5 mg administered vaginally) were satisfactory when given up to 63 days of amenorrhea; registration (approval) was acquired there in July 1991, and in Sweden in 1992 with the same protocol. The largest study (Ulmann et al., 1992) indicated 95 percent complete efficacy, with 1 percent of ongoing pregnancies, emphasizing the obligation to evacuate the uterus instrumentally in case of failure. There were no particular bleeding problems—only approximately 1/1000 patients received a transfusion. However, three myocardial infarctions, including one fatal case (a medical mistake occurred when sulprostone was injected into a woman at great risk), were recorded after more than 60,000 cases. Sulprostone for intramuscular administration has since been withdrawn from the market in France.
Coincidentally, the death of a patient (having received intramuscular Sulprostone) was reported at the same time as the first trial of RU 486 plus orally active misoprostol (a PGE1 derivative) was published (Aubeny and Baulieu, 1991). We had hoped to use a safer prostaglandin (misoprostol has a record of millions of users for the prevention and treatment of gastrointestinal ulcers in individuals often at greater cardiovascular risk than normal pregnant women). It was also obvious that an orally effective, already available, cheap, and easy to store prostaglandin had the potential to be an important improvement, since it could allow a more convenient and private method of abortion. Most results (Peyron et al., 1993) have been obtained by using 600 mg of RU 486 and 400 µg (two tablets) of misoprostol 48 hours later. Four hours after misoprostol, approximately 70 percent of women aborted; if expulsion did not occur, a third misoprostol tablet (200 µg) was proposed to the patients. Greater than 98 percent efficacy was achieved according to the current trials for women with =49 days amenorrhea pregnancies (Table B1.6). Besides safety, this regimen was well tolerated—uterine cramps were minimized. Details and discussion of the method are found elsewhere (Peyron et al., 1993).
TABLE B1.6 Interruption of Early Pregnancy with RU 486 and One or Two Doses of Misoprostol
|
Treatment |
Total |
Number of Successesa (% of total) |
Number of Failuresb (% of total) |
|
|
Mifepristone, 600 mg |
385 |
|
|
|
|
Abortion before the time of administration of misoprostol (400 µg) |
385 |
21 (5.4%) |
|
|
|
Administration of first dose of misoprostol followed by abortion within 4 hours |
364 |
266 (69.1%) |
|
|
|
Later outcome |
98 |
|
|
|
|
Refuse more misoprostol |
27 |
26 (6.8%) |
Ongoing pregnancy |
1 (<1%) |
|
Second dose of misoprostol (200 µg) |
71 |
67 (17.4%) |
Partial retention |
2 (<1%) |
|
|
|
|
Synechiae with ongoing pregnancy |
1 (<1%) |
|
|
|
|
Ectopic pregnancy |
1 (<1%) |
|
Total |
|
380 (98.7%) |
|
5 (1.3%) |
|
a Success was defined as interruption of pregnancy and complete expulsion of the ovum. b Failures were defined as indicated. SOURCE: Peyron et al. (1993). |
||||
Currently in France, a woman suspecting an unwanted pregnancy sees a physician at a first visit, and after a delay for reflection she may return (second visit) to take the RU 486 pills. Two days later, she makes a third visit to the center to receive prostaglandin and then stays for four hours. A control (follow-up) visit (the fourth) should take place 10 to 15 days later. This method is currently not applicable to heavy smokers and women older than 35. All these precautions need to be reexamined, however, and most appear to be dispensable. In the future, it is hoped that a woman would consult her physician as early as possible in the case of missed menses and then receive, if this is her choice, RU 486 from a medically competent person who will have examined her. She will then take home the misoprostol pills for self-administration 48 hours later and return for a checkup in approximately two weeks.
Although RU 486 and misoprostol are safe drugs, pregnancy itself is a risk for women, no matter whether they wish to interrupt or continue it (e.g., ectopic pregnancy is not aborted by RU 486 and may be fatal if not treated surgically). Maintaining contact with a physician is mandatory, and there should be an appropriate permanent connection with a competent medical center in case of complications. Although it may be sufficient in the vast majority of cases for physicians (preferably gyne-
cologists) to see patients privately, it has been reported that many women prefer to be treated in a group at a medical center (Thong et al., 1992). Research should be conducted to define the best ways to administer the medications under specific conditions. It is certain that requirements for skilled personnel and sterile surgical facilities will be decreased (El-Refaey et al., 1992). The mechanism of action of prostaglandin at low doses indicates that the effect takes place only when progesterone activity has been much decreased by the antisteroid—after more than 24 hours. Whether a combination of RU 486 and prostaglandin to be administered simultaneously will become available is not predictable, since no technology for delayed prostaglandin release is in sight.
Application of this method in developing countries is necessarily more difficult, and local conditions must be considered, including the availability of medical facilities and personnel, cultural traditions (bleeding for several days may be a problem), and the social context. However, women have the right to obtain medical assistance in case of suspected pregnancy, to have the choice to decide to abort, and if so, also to have the choice of either a surgical or a medical method. Whether in developing or industrialized countries, we ought to offer a complete medical choice to women. Even the RU 486 plus misoprostol method may be imperfectly applied for a period of time in certain countries, but it can only be a definite improvement of the present situation. It has also been successfully used for missed abortions and anembryonic pregnancies (El-Refaey et al., 1992). Whether RU 486 plus misoprostol may be used to compensate for the lack of access to family planning and to health facilities is another question. Generally speaking, the best solution is to make available widely accepted and very efficient methods of contraception.
Pregnancy Interruption After Nine Weeks of Amenorrhea
The effects of progesterone, essentially on the decidua (implantation), the myometrium (calming effect), and LH secretion (depressed with lack of ovulation), are observed throughout the course of pregnancy; thus, it is not surprising that an antiprogesterone is potentially useful for pregnancy interruption and labor induction.
In France, voluntary pregnancy interruption is legally permitted until 12 weeks of amenorrhea. When women have passed beyond the current legal limit for RU 486 plus misoprostol treatment (seven weeks), vacuum aspiration is performed. This can be greatly facilitated by RU 486 taken 24 to 48 hours before the procedure—preoperative cervical preparation (ripening) (Henshaw and Templeton, 1991; Urquhart and Templeton, 1990). The cervical ripening may be due not to a change of prostaglandin metabolism in the cervix (Rådestad and Bygdeman, 1992), but to a
decrease of a2-adrenoreceptors (Kovacs and Falkay, 1993). A dose of 200–600 mg of RU 486 decreases the force required to dilate the cervix and has significantly fewer side effects than gemeprost (vaginal pessary). It also compares favorably with mechanical dilators such as Lamicel or Dilapan (Cohn and Stewart, 1991; Henshaw and Templeton, 1991; Gupta and Johnson, 1992; Thong and Baird, 1992).
In therapeutic second- and third-trimester abortions, RU 486 is most often used before the administration of prostaglandin, so that the dose of prostaglandins can be decreased, while pain and other side effects are reduced and expulsion is accelerated (Rodger and Baird, 1990). RU 486 also decreases the waiting time, and thus the pain and psychological suffering, in cases of a fetal demise (Cabrol et al., 1985).
Initiation of Labor
A decrease of progesterone activity occurs during parturition, but its precise role in successful delivery is unclear, particularly in primates (including humans) where it does not seem to be the primary event. In rats, RU 486 can synchronize delivery (Bosc et al., 1985), and in cattle (Li et al., 1991a,b) it is very efficient in facilitating parturition. In rhesus macaques near term, RU 486 provokes changes of prostaglandins and of the cervical status, but these modifications do not follow the same orderly sequence as those found during spontaneous delivery (Haluska et al., 1987; Wolf et al., 1993). It is not known if RU 486 increases gap junctions between myometrial cells in women as it does in rats (Garfield et al., 1987), but ß2-adrenoreceptor levels are unchanged in the myometrium (El Alj et al., 1989).
RU 486 has been tested in women at term who require labor induction for various medical indications such as post-term pregnancy and preeclampsia. When compared to placebo controls, the number of spontaneous deliveries is significantly increased; the amount of oxytocin, if required, is much lower; and the time to induce labor is shortened by RU 486 (Frydman et al., 1992). No undesirable incident, in mothers and newborns, was observed with the dose used (two times, 200 mg each), similar to observations made in monkey studies (Wolf, 1989b).
In summary, RU 486 appears to be safe for inducing labor when continuation of pregnancy is a risk for the fetus, the mother, or both. Systematic studies should now follow the development of babies born after RU 486 treatment, since it is known that in primates, RU 486 passes from mother to fetus during pregnancy (Wolf et al., 1988). The effects of neonatal and even embryonic (Wolf et al., 1990) exposure need to be assessed carefully (Weinstein et al., 1991). Until the absolute safety of antiprogestins is demonstrated in cases where there is a medical indication for labor induction, its use for convenience should be forbidden.
Contragestive and Contraceptive Methods Using RU 486
The previous considerations apply to pregnancy that has been demonstrated by missed menses and a positive pregnancy test, a situation clear to all women. Before this well-defined state, even if the biological steps are known, there is still confusion and ignorance as to when pregnancy begins. As a result, the vocabulary used to define the possible antihormonal interventions during the processes establishing pregnancy needs to be clarified.
If menses does not occur and a pregnancy test is positive, interruption is clearly defined as abortion. However, vacuum aspiration practiced very early, within approximately two weeks of menses delay, is called ''menses regulation" (e.g., officially in Bangladesh and Turkey) or menstrual induction, and can be considered as contragestive. On the other hand, any maneuver inhibiting fertilization is called contraception, for contra conception. The word conception is generally understood as fertilization; this is wrong etymologically, since concipire (Latin) means to retain (originally retain sperm and mother blood in the uterus to make the child).
Contraception is, therefore, commonly understood as a method to preclude fertilization, for instance, by suppression of ovulation or preventing sperm from reaching the ovum. However, physicians also designate as contraceptives methods that are applied before implantation is completed. They argue that a fertilized ovum that is not implanted after in vitro fertilization does not define a pregnancy. In fact, the available pregnancy tests are based on the measurement of human chorionic gonadotropin, produced by the embryonic chorion, which passes into the woman's blood and occurs only after implantation has been initiated. Intrauterine devices (IUDs) can be defined as "contraceptive" tools because they work, in part, as anti-implantation agents. The word post-coital contraception is also largely accepted and applies to a possibly fertilized ovum. Note also that the process of implantation is not instantaneous and takes several days during the last week of the fertile menstrual cycle, just before the time at which menses would occur. Coincidentally, the development of the embryo is characterized by the streak (a marker indicating that an individual embryo has been formed, and there is no further risk of twins), which should occur at approximately the same time—about 15 days after fertilization. Before that time, not only may genetic abnormalities or defects of implantation stop the process leading to pregnancy, but the very definition of a single potential person cannot be rigorously applied. In short, during the period between fertilization and the time at which menses should occur, interrupting methods are contragestive, differing from both abortion and contraception as defined above, and not hiding the fact that they oppose pregnancy.
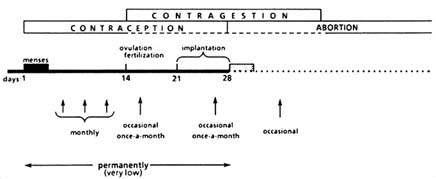
FIGURE B1.11 Fertility control with RU 486.
In summary, contragestion includes all treatments that operate over a period of approximately four weeks postfertilization. Such treatments include RU 486, other "morning-after" pills, early vacuum aspiration, and IUDs (as well as a potentially anti-hCG "vaccine").
According to the previous discussion, six methods for using RU 486 can be classified as contraceptive/contragestive terms (Figure B1.11):
Emergency Post-Coital Contraception (Contraceptive or Contragestive According to When it is Applied)
Recent studies have shown that RU 486 (single 600-mg dose) given to women after unprotected intercourse within the preceding 72 hours is highly efficient at preventing pregnancy (Glasier et al., 1992; Grimes, 1992; Webb et al., 1992), most likely acting as an anti-implantation agent. It is at least as effective as the high-dose combined estrogen and progestin preparations, and better tolerated by the women. Research needs to be pursued in order to determine the appropriate dose, and whether the administration can be repeated, how many times, and for how long, a possibility that appears rather remote because of probable changes in menstrual cyclicity.
Late Luteal Phase Administration (Occasional Use) (Contragestive)
The administration of 400 or 600 mg of RU 486, once or twice, two days before the expected menses, results in about a 20 percent failure in terms of initiating pregnancy. Since the probability of being pregnant is about 20 percent in normal couples, this leaves only 4 percent of women being pregnant (to be secondarily interrupted by other means) (Dubois et al., 1988b; Lähteenmäki et al., 1988; Grimes et al., 1992). The method is well tolerated and may be improved with the associated use of misoprostol.
Monthly Premenstrual, Late Luteal Phase Administration (Repeated Use) (Contragestive)
Giving RU 486 approximately two days before the expected day of menses over several months has not been successful (Van Santen and Haspels, 1987), the main reason being the irregularity of cycles, often due to the retardation of ovulation that is induced by the compound. In order to overcome this difficulty, a trial using a lower dose of RU 486 plus misoprostol, two days before and on the expected day of menses, has been undertaken. The prostaglandin may permit more rapid evacuation of the blastocyst and therefore a faster decline of hCG, which disturbs folliculogenesis during the next cycle (Aubény and Baulieu, work in progress).
Early Luteal Phase Administration (Once a Month) (Anti-Implantation)
Progesterone acts on the endometrium to prepare for implantation. This has been studied in detail in the ovariectomized rhesus monkey by analyzing the decidual transformation and the epithelial plaque formed in response to deciduogenic stimulus (Ghosh and Sengupta, 1992). An immunocytochemical study, assessing an endometrial secretory glycan (sialo-oligosaccharide), has shown that its production is P dependent (Graham et al., 1991). Moreover, experiments in rabbits have shown that endometrial receptivity and embryo implantation can be modified by antiprogestins (Hegele-Hartung et al., 1992), and RU 486 induces epithelium apoptosis (Rotello, 1992). Treatment with 200 mg of RU 486 was performed on women at the 2-day post-LH stage who had had unprotected intercourse at least once during the period three days before to one day after ovulation. Over 157 cycles, only one pregnancy occurred (Gemzell-Danielson et al., 1993). The main drawback of the method is the timing of the treatment. To use the method monthly, ovulation detection needs to become routine in the future. Doses of 10 mg of RU 486 administered on days 5 and 8 after the LH surge do not provoke hormonal change but do interrupt endometrial maturation with lowered PR levels (Greene et al., 1992).
"Endometrial Contraception" (Daily Delivery of Very Low Dose) (Contraceptive and Contragestive)
The provisional name "endometrial contraception" is given to the continuous exposure to a very low dose of RU 486. This may modify the genital tract in such a way that implantation and possibly fertilization do not occur, while ovulation and the pattern of estrogen and progesterone secretion are unchanged, and adrenal function is unmodified. Daily use in cycling guinea pig prevents implantation (Batista et al., 1991).
After administration of a low dose of RU 486 (12.5 mg to rhesus monkeys once per week), there was complete temporary infertility, but modifications of the cycle were very limited during three cycles of the experiment (Gary Hodgen, unpublished). In five women, 1 mg of RU 486 given daily for 30 days established a plasma concentration of ≥ 50 mg/ml; in one case, ovulation was suppressed (Croxatto et al., 1993). In a similar study, RU 486 (1 mg) given daily to nine women delayed ovulation and endometrial maturation, with a reduced peak of placental protein 14 (a glycoprotein marker for endometrium function) (Batista et al., 1992b).
Currently, studies are being organized using even lower daily doses of RU 486 given to women. An international comparative study will define the highest dose of RU 486 (actually very small) that can be administered chronically to women without perturbation of the cycle. Furthermore, this dose will be tested for contraceptive efficacy. Initially, these studies will be conducted using RU 486-containing pills administered daily. Secondarily, in case of success, we will move from pills to injectable microspheres that will allow the slow release of RU 486 for several months.
Ovulation Suppression (Daily Delivery of a Low Dose) (Contraceptive)
A number of observations demonstrate that P contributes to ovulation (Collins and Hodgen, 1986; Liu et al., 1987; Shoupe et al., 1987; Luukkainen et al., 1988; Danforth et al., 1989; Batista et al., 1992a). The administration of RU 486 during the follicular phase delays or suppresses ovulation. This may designate a new method of estrogen-free contraception. The main problem is to find an effective, well-tolerated dose. Using 5 mg of RU 486 per day may block ovulation without causing a change in adrenal function (Ledger et al., 1992; Croxatto et al., 1993). The sequential administration of RU 486 and progestin to maintain menstrual bleeding has been proposed (Croxatto and Salvatierra, 1991). We submit that this method with ovulation suppression will be more difficult to implement relative to continuous administration of a very low dose.
In conclusion, there is already hope, not to say certainty, for an occasional contraceptive/contragestive method, and "endometrial contraception" is a very appealing possibility. However, any contraceptive method should be studied carefully for a rather long period of time to delineate the possibility of side effects for the women and alteration of the embryo in case of failure (Wu, 1992).
Male Contraception
Progesterone increases calcium uptake by human sperm and favors the acrosomal reaction (Baldi et al., 1991; Blackmore et al., 1991; Parinaud et al., 1992; Uhler et al., 1992). There is likely to be a membrane
receptor mediating its action as in the progesterone-induced reinitiation of meiosis in Xenopus laevis oocytes (Blondeau and Baulieu, 1985). However, in sperm, as well as in oocytes, RU 486 may not act as an antiprogestin at the membrane-receptor level. A preliminary report (C.G.P. Puri, patent preview WO 9210194A1, 1992) of a contraceptive effect of RU 486 in monkeys with a decrease of sperm counts has not, to my current knowledge, yet been confirmed.
MEDICAL APPLICATIONS (TABLE B1.4)
Uterine Diseases
Endometriosis
RU 486 can suppress ovulation, and inhibit the mitotic action of estrogens in the monkey endometrium. These effects suggest that the compound may be useful for the treatment of endometriosis (Kettel et al., 1991). As expected, in women, RU 486 suppressed ovulation and menstrual cycle, and brought about an increase of LH (with augmented pulse amplitude but not frequency), adrenocorticotropic hormone (ACTH), and cortisol. Pain was decreased, but there was no decrease in the extent of the disease (corroborating observations in the rat by Tjaden et al., 1993); an effect on lipid peroxidation has also been described (Morales et al., 1992).
Fibroids
Murphy and coworkers (1993) have reported a 50 percent regression of uterine leiomyomas by daily administration of 50 mg of RU 486 for several weeks. This may be due principally to induced anovulation. Modulation of uterine growth factors and insulin-like growth factor (IGF)-binding proteins may also be involved (Murphy et al., 1993).
The treatment provokes an increase of LH, but not of FSH, and of dehydroepiandrosterone sulfate, androstenedione, testosterone, and cortisol, while estrogens, progesterone, sex steroid-binding plasma protein (SBP), thyroid-stimulating hormone (TSH), and prolactin (PRL) are unchanged, compared to original early follicular phase levels. In myometrial and leiomyomatous tissue, PR but not ER immunoreactivity is decreased. Amenorrhea and anovulation are constant. The significant decrease of PR levels may indicate a direct antiprogesterone effect; however, an alteration in ER function cannot be excluded, as discussed above.
The human papillomavirus (type 16, episomal) expression in ectocervical cells (cervical keratinocytes) is stimulated by glucocorticosteroids and progesterone (Mittal et al., 1993). Since RU 486 may inhibit this
induction in premalignant cervical lesions, do such data have application in human pathology?
Breast Cancer
Breast cancer in women is multifaceted and probably corresponds to several distinct pathophysiological and molecular processes (see recent review by Horwitz, 1992). Human carcinomas occur at different ages (before and after menopause); tumors in different animal species can be provoked by carcinogens and viruses; and various cultured cancer cells demonstrate different responses to progesterone. For example, in the classical experiments of Huggins and Yang (1962), progesterone was shown to be a promoter of DMBA-induced mammary tumors in the rat, while in contrast, a high dose of progestins is therapeutically useful in advanced cases of breast tumors in women (Pronzato et al., 1990).
RU 486 inhibits the growth of breast cancer cells in a PR-dependent manner (Bardon et al., 1985; Thomas and Monet, 1992). Interestingly, one may demonstrate (1) antiprogestin-, progesterone-, and PR-dependent effects of RU 486, lilopristone, and onapristone (Michna et al., 1992); and (2) hormone-independent, PR-dependent effects of the same "antiprogestins." In the latter cases, instead of simple degeneration and necrosis of tumor cells due to hormone deprivation, there may be terminal differentiation with a decrease in the number of cells in S-phase, an increase in cells in G0-G1, and evolution to apoptosis (Rotello et al., 1992). This has been observed in human tumor cells either cultured in vitro or transplanted into appropriate animals.
Clinically, only three studies concerning the effects of RU 486 and tumor development have been published (Maudelonde et al., 1987; Bakker et al., 1990; Klijn et al., 1990), but the percentage of objective remissions is promising enough to justify new trials. Trials are currently being performed in Canada and France. Indeed, clinical studies should concern pre- as well as postmenopausal women, and should analyze the mitotic pattern and the steroid and growth factor receptors under simultaneous or successive association of various antiprogestins with tamoxifen. Studies must also include compounds that may have less antiglucocorticosteroid activity, the association of antisteroid with antisteroidogenic drugs, and detailed molecular genetic description of PR mutations. We have seen that the latter may preclude hormone/antihormone action or even shift the response of some 11ß-phenyl derivatives from antagonistic to agonistic effects.
The hormonal biology of the breast and its tumors is different from that of normal and cancerous endometrium, including, therefore, the proliferative/antiproliferative potencies of progestins and antiprogestin; this difference justifies specific bioassays (Schneider et al., 1989; Michna
et al., 1991). Breast cancer studies may unveil original PR-dependent control mechanisms of the cell cycle—in particular, hormone-independent, receptor-dependent or -independent effects (as already suggested for tamoxifen). In other words, just as for antiestrogens (Bardon et al., 1987), there may be receptor-mediated antihormonal cytostatic activity, receptor-mediated cytotoxic activity (nonantihormonal), and nonspecific cytotoxic activity of RU 486 and parent drugs.
The growth of other cancers may be decreased by RU 486 treatment. It may act via the PR as in the case of androgen-insensitive R3327HI rat prostatic carcinoma (Mobbs and Johnson, 1991).
Meningioma
Meningiomas are common tumors, generally benign and slow growing, that can threaten brain function—or even life—if they are not surgically removed. More frequent in women, meningioma growth is accelerated during pregnancy. Most meningiomas contain PR (and often little or no ER), and it has been suggested that progesterone has either a permissive or a facilitating effect on their evolution, or possibly both (Magdalenat et al., 1982; Blankestein et al., 1983; Haak et al., 1990; Grunberg et al., 1991; Lamberts, 1992a,b,c). Administration of RU 486 over several months (usually 200 mg/day) has essentially been well tolerated; even the expected side effects reproducing the features of the familial syndrome of cortisol resistance have been observed—that is, relative glucocorticosteroid insufficiency in the presence of high blood cortisol with arterial pressure is maintained because aldosterone secretion is increased.
The treatment should be restricted to unresectable meningiomas. The spontaneous variety of evolution of these tumors makes a definitive evaluation of the beneficial effects very difficult, even though definite, sometimes spectacular improvement has been observed in about one-third of available reports. Instead of RU 486, an antiprogesterone without antiglucocorticosteroid activity would be welcomed. Conversely, there are gliomas whose growth is sensitive to glucocorticosteroid that may benefit from antiglucocorticosteroid compounds (Langeveld et al., 1992; Pinski et al., 1993).
ANTIGLUCOCORTICOSTEROID EFFECTS
While demonstrating antiglucocorticosteroid activity in vitro (Philibert et al., 1981; Jung-Testas and Baulieu, 1983), RU 486 is the first powerful antiglucocorticosteroid whose activity has also been demonstrated in vivo (Bertagna et al., 1984; Gaillard et al., 1984). As one might predict, given the elimination of the negative feedback control of cortisol on ACTH, RU 486 leads to increased ACTH and cortisol secretion. This
overcomes the hypocorticosteroid effect, which explains the good tolerance of RU 486 when it is administered briefly, as in abortion or emergency contraception.
There are several potential uses of RU 486 or antiglucocorticosteroid analogues. The long-term use of RU 486—for instance, to treat tumors—may be improved if a compound blocking steroid biosynthesis is given simultaneously. In breast cancer this could be an antiaromatase because production of estrogens increases when adrenal androgen hypersecretion occurs with RU 486 treatment.
An antiglucocorticosteroid with a short half-life should be useful for the kinetic studies of the hypothalamus-pituitary-adrenal axis, in particular to classify different types of depression (Ammar et al., 1986; Kling et al., 1989; Krishnan et al., 1992). In several instances, an acute increase of glucocorticosteroids, for instance, during the stress of aggressive conditions, might be antagonized by RU 486, which could therefore protect against immune depression. Conversely, RU 486 might be detrimental in the pharmacological manipulation of septic shock (Broukaert et al., 1992).
It is not impossible that RU 486 or another antiglucocorticosteroid deprived of antiprogesterone effect might be useful in the treatment of certain cases of psychosis or arterial hypertension, since these two pathological features are remarkably cured when present in Cushing's syndrome treated with RU 486 administration [here RU 486 has been a lifesaving drug (see review in Chrousos et al., 1989)]. However, this will concern only a small number of patients. Whether some chronic conditions involving hypercortisolism, such as certain forms of obesity, can benefit from RU 486 is still debatable (Okada et al., 1992). The use of RU 486 (or analogues) has been suggested in premenstrual syndrome, postmenopausal flushes, Alzheimer's disease, and AIDS, but there is currently no firm scientific background to justify trials.
More generally, any long-term treatment with an antiprogestin or an antiglucocorticosteroid should be carefully followed up. Chronic antiprogesterone activity may create an unopposed estrogenic state, counteract the osteogenic effect of P, or interfere with the activity of P in the central nervous system. Signs of adrenal insufficiency may also develop.
It is probably the local use of RU 486, or its derivatives with antiglucocorticosteroid activity, that will develop rapidly. Trials are on the way for treating glaucoma, and for accelerating or amplifying the slow healing of wounds and burns, particularly in stressed or aging patients (who have increased or normal cortisol and low adrenal androgen levels).
CONCLUSIONS
Much work remains to be done (Hodgen, 1991). I single out four approaches that I believe to be most important.
Use for Voluntary Pregnancy Interruption
The RU 486-plus-misoprostol combined method is ready to be used at large. It works, is safe, and is close to being as convenient as a medical method of abortion may be. Given the global demographic issue, the suffering of women, and related health problems, it must soon be made available in the United States—a key to further worldwide development. Studies must rapidly discern the best conditions for its distribution in parts of the world where there are obstacles, including developing countries. The early use of RU 486, as soon as a woman fears a pregnancy that she does not want, will help to defuse the abortion issue.
Contraception
Research should be conducted to define convenient and safe contraceptive methods with RU 486 or other antiprogestins. There are serious hopes, and it is now a matter of conducting systematic studies. However, it will take several years and a great deal of money. Significant success also will contribute to decreasing the practice of abortion as we know it.
Cancer
Nonreproductive medicine should investigate the regulatory properties of RU 486 and its derivatives in several diseases. The most cruel, breast cancer, should be first on the list of trials. Again, this may take time and money, but there already are clues that cannot be neglected.
Novel Antiprogestins: Biology and Chemistry
There is enough data to suggest that RU 486 is only the first in a series of new compounds with significant differences that could be medically exploitable (Table B1.7). Therefore, basic, novel, interactive chemical and biological research should be continued forcefully.
ACKNOWLEDGMENTS
I would like to acknowledge the editorial work of Rod Fiddes, Ph.D., and contributions to the manuscript by Françoise Boussac, Jean-Claude Lambert, Philippe Leclerc, Corinne Legris, Luc Outin, and Claude Secco. This work could not have been presented without the long-time collaboration of my INSERM colleagues and of the researchers at Roussel-Uclaf (Romainville, France).
TABLE B1.7 Objectives of Research for Novel Antiprogestins
|
No binding to orosomucoid (shorter half-life) |
|
Absolute specificity for progesterone receptor (no antiglucocorticosteroid activity) |
|
Novel cellular/molecular mechanisms |
|
No receptor dimerization |
|
No DNA binding |
|
Direct effect on cell membrane function |
|
Direct effect on enzymes |
|
Steroidogenesis |
|
PG synthesis |
|
Activity specific to central nervous system |
|
Activity exclusive of the central nervous system |
REFERENCES
Allan, G.F., Leng, X., Tsai, S.Y., et al. Hormone and antihormone induce distinct conformational changes which are central to steroid receptor activation. Journal of Biological Chemistry 267:19513–19520, 1992a.
Allan, G.F., Tsai, S.Y., Tsai, M-J., et al. Ligand-dependent conformational changes in the progesterone receptor are necessary for events that follow DNA binding. Proceedings of the National Academy of Sciences of the United States 89:11750-11754, 1992b.
Ammar, S., Allilaire, J.F., Lecrubier, Y., et al. Afternoon increase in plasma cortisol in depressed patients receiving an antiglucocorticosteroid in the morning. American Journal of Psychiatry 143:129–130, 1986.
Arkaravichien, W., and Kendle, K.E. Uterine contractile activity in rats induced by mifepristone (RU 486) in relation to changes in concentrations of prostaglandins E-2 and F-2a. Journal of Reproduction and Fertility 94:115–120, 1992.
Aubény, E., and Baulieu, E.E. Activité contragestive de l'association au RU 486 d'une prostaglandine active par voie orale. Comptes Rendus de l'Académie des Sciences (Paris) 312:539–545, 1991.
Avrech, O.M., Bukovsky, I., Golan, A., et al. Mifepristone (RU 486) alone or in combination with a prostaglandin analogue for termination of early pregnancy: A review. Fertility and Sterility 56:385–393, 1991.
Bailly, A., Le Page, C., Rauch, M., et al. Sequence-specific DNA binding of the progesterone receptor to the uteroglobin gene: Effects of hormone, antihormone and receptor phosphorylation. EMBO Journal 5:3235–3241, 1986.
Baird, D.T., Rodger, M., Cameron, I.T., et al. Prostaglandins and antiestrogens for the interruption of early pregnancy. Journal of Reproduction and Fertility 36:173–179, 1988.
Bakker, G.H., Setyono-Han, B., Portengen, H., et al. Treatment of breast cancer with different antiprogestins: Preclinical and clinical studies. Journal of Steroid Biochemistry and Molecular Biology 37:789–794, 1990.
Baldi, E., Casano, R., Falsetti, C., et al.Intracellular calcium accumulation and responsiveness to progesterone in capacitating human spermatozoa. Journal of Andrology 12:323–330, 1991.
Bardon, S., Vignon, F., Chalbos, D., et al. RU 486, a progestin and glucocorticoid antagonist, inhibits the growth of breast cancer cells via the progesterone receptor. Journal of Clinical Endocrinology and Metabolism 60:692–697, 1985.
Bardon, S., Vignon, F., Montcourrier, P., et al. Steroid receptor-mediated cytotoxicity of an antiestrogen and an antiprogestin in breast cancer cells. Cancer Research 47:1441–1448, 1987.
Batista, M.C., Bristow, T.L., Matthew's, J., et al. Daily administration of the progesterone antagonist RU 486 prevents implantation in the cycling guinea pig. American Journal of Obstetrics and Gynecology 165:82–86, 1991.
Batista, M.C., Cartledge, T.P., Zellmer, A.W., et al. Evidence for a critical role of progesterone in the regulation of the midcycle gonadotropin surge and ovulation. Journal of Clinical Endocrinology and Metabolism 74:565–570, 1992a.
Batista, M.C., Cartledge, T.P., Zellmer, A.W., et al. Delayed endometrial maturation included by daily administration of the antiprogestin RU 486: A potential new contraceptive strategy. American Journal of Obstetrics and Gynecology 167:60–65, 1992b.
Baulieu, E.E. Antiprogesterone effect and midcycle (periovulatory) contraception. European Journal of Obstetrics and Gynecology and Reproductive Biology 4:161–166, 1975.
Baulieu, E.E. RU 486: An antiprogestin steroid with contragestive activity in women. Pp. 1–25 in The Antiprogestin Steroid RU 486 and Human Fertility Control. Baulieu, E.E., and Segal, S.J., eds. New York: Plenum Press, 1985.
Baulieu, E.E. Contragestion and other clinical applications of RU 486, an antiprogesterone at the receptor . Science 245:1351–1357, 1989a.
Baulieu, E.E. RU 486 as an antiprogesterone steroid: From receptor to contragestion and beyond. Journal of the American Medical Association 262:1808–1814, 1989b.
Baulieu, E.E. RU 486 and the early nineties. Advances in Contraception 7:345–351, 1991a.
Baulieu, E.E. The antisteroid RU 486. Its cellular and molecular mode of action. Trends in Endocrinology and Metabolism 2:233–239, 1991b.
Baulieu, E.E. The Abortion Pill. New York: Simon & Schuster, 1991c.
Baulieu, E.E. Neurosteroids: A function of the brain. Pp. 63–73 in Neurosteroids and Brain Function. Costa, E., and Paul, S.M., eds. New York: Thieme Medical Publishers, 1991d.
Baulieu, E.E., and Catelli, M.G. Steroid hormone receptors and heat shock protein Mr 90,000 (hsp90): A functional interaction? Pp. 203–219 in Stress-Induced Proteins. Pardue, M.L., Feramisco, J.R., and Lindquist, S., eds. New York: Alan R. Liss, 1989.
Baulieu, E.E., and Segal, S.J. The Antiprogestin Steroid RU 486 and Human Fertility Control. New York and London: Plenum Press, 1985.
Baulieu, E.E., Godeau, F., Shorderet, M., et al. Steroid induced meiotic division in Xenopus laevis oocytes: Surface and calcium. Nature 275:593–598, 1978.
Baulieu, E.E., Binart, N., Cadepond, F., et al. Do receptor-associated nuclear proteins explain earliest steps of steroid hormone function? Pp. 301–318 in The Steroid/Thyroid Hormone Receptor Family and Gene Regulation. Carlstedt-Duke, J., Eriksson, H., and Gustafsson, J.A., eds. Basel: Birkhäuser Verlag, 1989.
Bélanger, A., Philibert, D., and Teutsch, G. Regio and stereospecific synthesis of 11ß-substituted 19-norsteroids: Influence of 11ß-substitution on progesterone receptor affinity. Steroids 37:361–383, 1981.
Benhamou, B., Garcia, T., Lerouge, T., et al. A single amino acid that determines the sensitivity of progesterone receptors to RU 486. Science 255:206–209, 1992.
Bergström, S., Diczfalusy, E., Borell, U., et al. Prostaglandins in fertility control. Science 175:1280–1287, 1972.
Bertagna, X., Bertagna, C., Luton, J.P., et al. The new steroid analog RU 486 inhibits glucocorticoid action in man. Journal of Clinical Endocrinology and Metabolism 59:25–38, 1984.
Birgerson, L., and Odlind, V. Early pregnancy termination with antiprogestins: A comparative clinical study of RU 486 given in two dose regimens and Epostane. Fertility and Sterility 48:565–570, 1987.
Blackmore, P.F., Neulen, J., Lattanzio, F., et al. Cell surface-binding sites for progesterone mediate calcium uptake in human sperm. Journal of Biological Chemistry 266:18655–18659, 1991.
Blankenstein, M.A., Blaauw, G., Lamberts, S.W.J., et al. Presence of progesterone receptors and absence of oestrogen receptors in human intracranial meningioma cytosols. European Journal of Cancer and Clinical Oncology 19:365–170, 1983.
Blondeau, J.P., and Baulieu, E.E. Progesterone-inhibited phosphorylation of an unique Mr 48,000 protein in the plasma membrane of Xenopus laevis oocytes. Journal of Biological Chemistry 260:3617–3625, 1985.
Bocquel, M-T., Ji, J., Ylikomi, T., et al. Type II antagonists impair the DNA binding of steroid hormone receptors without affecting dimerization. Journal of Steroid Biochemistry and Molecular Biology 45:205–215, 1993.
Bosc, M.J., Germain, G., Nicolle, A., et al. The use of antiprogesterone compound RU 486 to control timing of parturition in rats. Pp. 69–70 in The Antiprogestin Steroid RU 486 and Human Fertility Control. Baulieu, E.E., and Segal, S.J., eds. New York: Plenum Press, 1985.
Bourgeois, S., Mester, J., and Baulieu, E.E. DNA binding properties of glucocorticosteroid receptors bound to the steroid antagonist RU 486. EMBO Journal 3:751–755, 1984.
Brogden, R.N., Goa, K.L., and Faulds, D. Mifepristone: A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential. Drugs 45:384–409, 1993.
Brouckaert, P., Everaerdt, B., and Fiers, W. The glucocorticoid antagonist RU 38486 mimics interleukin-1 in its sensitization to the lethal and interleukin-6-including properties of tumor necrosis factor. European Journal of Immunology 22:981–986, 1992.
Brown, T.J., and Blaustein, J.D. Abbreviation of the period of sexual behavior in female guinea pigs by the progesterone antagonist RU 486. Brain Research 373:103–113, 1986.
Bucourt, R., Vignau, M., Torelli, V., et al. New biospecific adsorbents for the purification of estradiol receptor. Journal of Biological Chemistry 25:8221–8228, 1978.
Bygdeman, M., and Swahn, M.L. Progesterone receptor blockage. Effect on uterine contractility and early pregnancy. Contraception 32:45–51, 1985.
Cabrol, D., Bouvier d'Yvoire, M., Mermet, E., et al. Induction of labour with mifepristone after intrauterine fetal death. Lancet 8462:1019, 1985.
Cadepond, F., Gasc, J.M., Delahaye, F., et al. Hormonal regulation of the nuclear localization signals of the human glucocorticosteroid receptor. Experimental Cell Research 201:99–108, 1992.
Callebaut, I., Renoir, J.M., Lebeau, M.C., et al. An immunophilin that binds Mr 90,000 heat shock protein: Main structural features of a mammalian p59 protein. Proceedings of the National Academy of Sciences of the United States 89:6270–6274, 1992.
Chalbos, D., Galtier, F., Emiliani, S., et al. The anti-progestin RU 486 stabilizes the progestin-induced fatty acid synthetase mRNA but does not stimulate its transcription. Journal of Biological Chemistry 266:8220–8224, 1991.
Chambraud, B., Berry, M., Redeuilh, G., et al. Several regions of human estrogen receptor are involved in the formation of receptor hsp90 complexes. Journal of Biological Chemistry 265:20686–20691, 1990.
Chrousos, G.P., Laue, L., Nieman, L.K., et al. Clinical applications of RU 486, a prototype glucocorticoid and progestin antagonist. Pp. 273–284 in Adrenal and Hypertension: From Cloning to Clinic. Mantero, F., Scoggins, B.A., Takeda, R., et al., eds. New York: Raven Press, 1989.
Chwalisz, K., Hegele-Hartung, C., Fritzemeier, K.H., et al. Inhibition of the estradiol-mediated endometrial gland formation by the antigestagen onapristone in rabbits: Relationship to uterine estrogen receptors. Endocrinology 129:312–322, 1991.
Cohn, M., and Stewart, P. Pretreatment of the primigravid uterine cervix with mifepristone 30 h prior to termination of pregnancy: A double blind study. British Journal of Obstetrics and Gynaecology 98:778–782, 1991.
Collins, R.L., and Hodgen, G.D. Blockade of the spontaneous midcycle gonadotropin
surge in monkeys by RU 486. A progesterone antagonist? Journal of Clinical Endocrinology and Metabolism 63:1270–1276, 1986.
Cook, C.E., Wani, M.C., Lee, Y-W., et al. Reversal of activity profile in analogs of the antiprogestin RU 486: Effect of a 16a-substituent of progestational (agonist) activity. Life Science 52:155–162, 1992.
Cook, R.J., and Grimes, D.A. Antiprogestin drugs: Ethical, legal, and medical issues. Law, Medicine & Health Care 20:1992.
Corner, G.W. The Hormones in Human Reproduction. New York: Atheneum, 1963.
Couzinet, B., Le Strat, N., Ulmann, A., et al. Termination of early pregnancy by the progesterone antagonist RU 486 (mifepristone). New England Journal of Medicine 315:1565–1570, 1986.
Crooij, M.J., and Janssens, J. Termination of early pregnancy by the 3ß-hydroxysteroid dehydrogenase inhibitor epostane. New England Journal of Medicine 319:813–817, 1988.
Croxatto, H.B., and Salvatierra, A.M. Cyclic use of antigestagens for fertility control. Pp. 245–252 in Female Contraception and Male Fertility Regulation. Runnebaum, B., Rabe, T., and Kiesel, L., eds. Casterton, N.J.: Parthenon Publishing Group, 1991.
Croxatto, H.B., Salvatierra, A.M., Croxatto, H.D., et al. Effects of continuous treatment with low dose mifepristone throughout one menstrual cycle. Human Reproduction 8:201–207, 1993.
Csapo, A.I., and Pulkkinen, M.O. Control of human parturition. Pp. 159–188 in Progress in Perinatology. Kaminetzky, H.A., and Iffy, L., eds. Philadelphia: Stickley, 1977.
Danforth, D.R., Dubois, C., Ulmann, A., et al. Contraceptive potential of RU 486 by ovulation inhibition. III. Preliminary observations on once weekly oral administration. Contraception 40:195–200, 1989.
Das, C., and Catt, K.J. Antifertility actions of the progesterone antagonist RU 486 include direct inhibition of placental hormone secretion. Lancet ii:599–601, 1987.
Delabre, K., Guiochon-Mantel, A., and Milgrom, E. In vivo evidence against the existence of antiprogestins disrupting receptor binding to DNA. Pharmacology, 1993 (in press).
DeMarzo, A.M., Oñate, S.A., Nordeen, S.K., et al. Effects of the steroid antagonist RU 486 on dimerization of the human progesterone receptor. Biochemistry 31:10491–10501, 1992.
Djerassi, C. Birth control after 1984. Science 169:941–951, 1970.
Dubois, C., Ulmann, A., Aubeny, E., et al. Contragestion par le RU 486: Intérêt de l'associationà un dérivé prostaglandine. Comptes Rendus de l'Académie des Sciences de Paris 306:57–61, 1988a.
Dubois, C., Ulmann, A., and Baulieu, E.E. Contragestion with late luteal administration of RU 486 (mifepristone). Fertility and Sterility 50:593–596, 1988b.
Egarter, C.H., and Husslein, P. Biochemistry of myometrial contractility. Baillière's Clinical Obstetrics and Gynaecology 6:755–769, 1992.
El Alj, A., Breuiller, M., Jolivet, A., et al. ß2-adrenoceptor response in the rat uterus at the end of gestation and after induction of labor with RU 486. Canadian Journal of Physiology and Pharmacology 67:1051–1057, 1989.
El-Ashry, D., Oñate, S.A., Nordeen S.K., et al. Human progesterone receptor complexed with the antagonist RU 486 binds to hormone response elements in a structurally altered form. Molecular Endocrinology 3:1545–1558, 1989.
Elger, W., Beier, S., Chwalisz, K., et al. Studies on the mechanisms of action of progesterone antagonists. Journal of Steroid Biochemistry 25:835–845, 1986.
El-Refaey, H., Hinshaw, K., Henshaw, R., et al. Medical management of missed abortion and anembryonic pregnancy. British Medical Journal 305:1399, 1992.
Evans, R.M. The steroid and thyroid hormone receptor superfamily. Science 240:889–895, 1988.
Finidori-Lepicard, J., Shorderet-Slatkine, S., Hanoune, J., et al. Steroid hormone as
regulatory agent of adenylate cyclase. Inhibition by progesterone of the membrane bound enzyme in Xenopus laevis oocytes. Nature 292:255–256, 1981.
Gaillard, R.C., Riondel, A., Muller, A.F., et al. RU 486: A steroid with antiglucocorticosteroid activity that only disinhibits the human pituitary-adrenal system at a specific time of day. Proceedings of the National Academy of Sciences of the United States 81:3879–3882, 1984.
Garcia, T., Benhamou, B., Gofflo, D., et al. Switching agonistic, antagonistic, and mixed transcriptional responses to 11ß-substituted progestins by mutation of the progesterone receptor. Molecular Endocrinology 6:2071–2078, 1992.
Garfield, R.E., Gasc, J-M., and Baulieu, E.E. Effects of the antiprogesterone RU 486 on preterm birth in the rat. American Journal of Obstetrics and Gynecology 157:1281–1285, 1987.
Garzo, V.G., Liu, J., Ulmann, A., et al. Effects of an antiprogesterone (RU 486) on the hypothalamic-hypophyseal-ovarian-endometrial axis during the luteal phase of the menstrual cycle. Journal of Clinical Endocrinology and Metabolism 66:508–517, 1988.
Gemzell-Danielsson, K., Swahn, M.L., Svalander, P., et al. Early luteal phase treatment with RU 486 fertility regulation. Human Reproduction , 1993 (in press).
Ghosh, D., De, P., and Sengupta, J. Effect of RU 486 on the endometrial response to deciduogenic stimulus in ovariectomized rhesus monkeys treated with oestrogen and progesterone. Human Reproduction 7:1048–1060, 1992.
Glasier, A., Thong, K.J., Dewar, M., et al. Mifepristone (RU 486) compared with high-dose estrogen and progestogen for emergency post-coital contraception. New England Journal of Medicine 327:1041–1044, 1992.
Graham, R.A., Aplin, J.D., Li, T.-C., et al. The effects of the antiprogesterone RU 486 (mifepristone) on an endometrial secretory glycan: An immunocytochemical study. Fertility and Sterility 55:1132–1136, 1991.
Gravanis, A., Zorn, J.R., Tanguy, G., et al. The ''dysharmonic luteal phase" syndrome: Endometrial progesterone receptor and estradiol dehydrogenase. Fertility and Sterility 42:730–736, 1984.
Greene, K.E., Kettel, L.M., and Yen, S.S. Interruption of endometrial maturation without hormonal changes by an antiprogesterone during the first half of luteal phase of the menstrual cycle: A contraceptive potential. Fertility and Sterility 58:338–343, 1992.
Grimaldi, B., Barre, J., and Tillement, J-P. Les rôles respectifs des liaisons aux protéines sanguines et tissulaires de la mifépristone (RU 486) dans sa distribution quantitative dans l'organisme. Comptes Rendus de l'Académie des Sciences de Paris 315:93–99, 1992.
Grimes, D.A. Mifepristone (RU 486)—an abortifacient to prevent abortion? New England Journal of Medicine 327:1088–1089, 1992.
Grimes, D.A., Mishell, D.R., and David, H.P. A randomized clinical trial of Mifepristone (RU 486) for induction of delayed menses: Efficacy and acceptability. Contraception 46:1–10, 1992.
Gronemeyer, H. Transcription activation by estrogen and progesterone receptors. Annual Review of Genetics 25:89–123, 1991.
Groyer, A., Le Bouc, Y., Joab, I., et al. Chick oviduct glucocorticosteroid receptor. Specific binding of the synthetic steroid RU 486 and immunological studies with antibodies to chick oviduct progesterone receptor. European Journal of Biochemistry 149:445–451, 1985.
Groyer, A., Schweizer-Groyer, G., Cadepond, F., et al. Antiglucocorticosteroid effects suggest why steroid hormone is required for receptors to bind DNA in vivo but not in vitro. Nature 328:624–626, 1987.
Grunberg, S.M., Weiss, M.H., Spitz, I.M., et al. Treatment of unresectable meningiomas with the antiprogesterone agent mifepristone. Journal of Neurosurgery 74:861–866, 1991.
Guiochon-Mantel, A., Loosfelt, H., Ragot, et al. Receptors bound to antiprogestin form abortive complexes with hormone responsive elements. Nature 336:695–698, 1988.
Guiochon-Mantel, A., Loosfelt, H., Lescop, P., et al. Mechanisms of nuclear localization of the progesterone receptor: Evidence for interaction between monomers. Cell 57:1147–1154, 1989.
Gupta, J.K., and Johnson, N. Should we use prostaglandins, tents or progesterone antagonists for cervical ripening before first trimester abortion? Contraception 46:489–497, 1992.
Haak, H.R., De Keizer, R.J., Hagenouw-Taal, J.C., et al. Successful mifepristone treatment of recurrent, inoperable meningioma. Lancet 1:124, 1990.
Haluska, G.J., Stanczyk, F.Z., Cook, M.J., et al. Temporal changes in uterine activity and prostaglandin response to RU 486 in rhesus macaques in late gestation. American Journal of Obstetrics and Gynecology 157:1487–1495, 1987.
Haluska, G.J., West, N., Novy, M.J., et al. Uterine estrogen receptors are increased by RU 486 in late pregnant rhesus macaques but not after spontaneous labor. Journal of Clinical Endocrinology and Metabolism 70:181–186, 1990.
Hansen, K.A., Opsahl, M.S., Nieman, L.K., et al. Natural killer cell activity from pregnant subjects is modulated by RU 486. American Journal of Obstetrics and Gynecology 166:87–90, 1992.
Hegele-Hartung, C., Mootz, U., and Beirer, H.M. Luteal control of endometrial receptivity and its modification by progesterone antagonists. Endocrinology 131:2446–2460, 1992.
Henderson, D. Antiprogestational and antiglucocorticoid activities of some novel 11ß-aryl substituted steroids. Pp. 106–131 in Pharmacology and Clinical Uses of Inhibitors of Hormone Secretion and Action. Furr, B.J.A., and Wakeling, A.E., eds. London: Baillière Tindall, 1987.
Hendry, L.B., and Mahesh, V.B. Stereochemical complementarity of progesterone, RU 486 and cavities between base pairs in partially unwound double stranded DNA assessed by computer modeling and energy calculations. Journal of Steroid Biochemistry and Molecular Biology 41:647–651, 1992.
Henshaw, R.C., and Templeton A.A. Pre-operative cervical preparation before first trimester vacuum aspiration: A randomized controlled comparision between gemeprost and mifepristone (RU 486). British Journal of Obstetrics and Gynaecology 98:1025–1030, 1991.
Herrmann, W., Wyss, R., Riondel, A., et al. Effet d'un stéroide anti-progestérone chez la femme: Interruption du cycle menstruel et de la grossesse au début. Comptes Rendus de l'Académie des Sciences (Paris) 294:933–938, 1982.
Herrmann, W.L., Schindler, A.M., and Wyss, R. Effects of the antiprogesterone RU 486 in early pregnancy and during the menstrual cycle. Pp. 179–198 in The Antiprogestin Steroid RU 486 and Human Fertility Control. Baulieu, E.E., and Segal, S.J., eds. New York: Plenum Press, 1985.
Hodgen, G.D. Antiprogestins: The political chemistry of RU 486. Fertility and Sterility 56:394–395, 1991.
Horwitz, K.B. The molecular biology of RU 486. Is there a role for antiprogestins in the treatment of breast cancer? Endocrine Reviews 13:146–163, 1992.
Hospital, M., Busetta, B., Bucourt, R., et al. X-ray crystallography of estrogens and their binding to receptor sites. Molecular Pharmacology 8:438–445, 1972.
Hsu, S-C., Qi, M., and DeFranco, D.B. Cell cycle regulation of glucocorticoid receptor function. EMBO Journal 11:3457–3468, 1992.
Huggins, C., and Yang, N.C. Induction and extinction of mammary cancer. Science 137:257–361, 1962.
Ikuta, Y., Matsuura, K., Okamura, H., et al. Effects of RU 486 on the interstitial collagenase in the process of cervical ripening in the pregnant rat. Endocrinologia Japonica 38:491–496, 1991.
Jensen, E.V., and Jacobson, H.I. Basic guides to the mechanism of estrogen action. Pp.
387–414 in Recent Progress in Hormone Research. Vol. XVIII. Pincus, G., ed. New York and London: Academic Press, 1962.
Jung-Testas, I., and Baulieu, E.E. Inhibition of glucocorticosteroid action in cultured L-929 mouse fibroblasts by RU 486, a new anti-glucocorticosteroid of high affinity for the glucocorticosteroid receptor. Experimental Cell Research 147:177–182, 1983.
Kettel, L.M., Murphy, A.A., Mortala, J.F., et al. Endocrine responses to long-term administration of the antiprogesterone RU 486 in patients with pelvic endometriosis. Fertility and Sterility 56:402–407, 1991.
Klein-Hitpass, L., Cato A.C.B., Henderson, D., et al. Two types of antiprogestins identified by their differential action in transcriptionally active extracts from T47D cells. Nucleic Acids Research 19:1227–1234, 1991.
Klijn, J.G.M., Bakker, G.H., Setyono-Han, B., et al. Antiprogestin treatment of experimental and clinical breast cancer. Journal of Steroid Biochemistry 36:435, Abstract 117, 1990.
Kling, M.A., Whitfield, J.W., Brandt, H.A., et al. Effects of glucocorticoid antagonism with RU 486 on pituitary adrenal function in patients with major depression: Time-dependent enhancement of plasma ACTH secretion. Psychopharmacology Bulletin 25:466–472, 1989.
Kovács, L., and Falkay, G. Changes in adrenergic receptors in the pregnant human uterine cervix following mifepristone or placebo treatment in the first trimester. Human Reproduction 8:119–121, 1993.
Krishnan, K.R.R., Reed, B., Wilson, W.H., et al. RU 486 in depression. Progress in Neuro-Psyschopharmacology and Biological Psychiatry 16:913–920, 1992.
Lähteenmäki, P., Rapeli, T., Kääriäinen, M., et al. Late post-coital treatment against pregnancy with antiprogesterone RU 486. Fertility and Sterility 50:36–38, 1988.
Lambasts, SW., Kopek, J.W., and de Jong, F.H. Long-term treatment with RU 486 and glucocorticoid receptor resistance. Theoretical and therapeutic implications. Trends in Endocrinology and Metabolism 6:199–204, 1992a.
Lamberts, S.W., Koper, J.W., Reubi, J-C., et al. Endocrine aspects of the diagnosis and treatment of primary brain tumours. Clinical Endocrinology 37:1–10, 1992b.
Lamberts, S.W.J., Tanghe, H.L.J., Avezaat, C.J.J., et al. Mifepristone (RU 486) treatment of meningiomas. Journal of Neurology, Neurosurgery, and Psychiatry 55:486–490, 1992c.
Langeveld, C.H., van Waas, M.P., Stoof, J.C., et al. Implication of glucocorticoid receptors in the stimulation of human glioma cell proliferation by dexamethasone. Journal of Neuroscience Research 31:524–531, 1992.
Laue, L., Kawai, Udelsman, R., et al. Glucocorticoid antagonists: Pharmacological attributes of a prototype antiglucocorticoid (RU 486). Pp. 303–329 in Anti-inflammatory Steroid Action. Basic and Clinical Aspects. New York: Academic Press, 1989.
Lebeau, M.C., Binart, N., Cadepond, F., et al. Steroid receptor associated proteins: Heat shock protein 90 and p59 immunophilin. Meadow Brook Conference on Steroid Receptors in Health Disease. Moudgil, V. ed. Oakland University. Boston: Burkhaüser. 1993 (in press).
Ledger, W.L., Sweeting, V.M., Hillier, H., et al. Inhibition of ovulation by low-dose mifepristone (RU 486). Human Reproduction 7:945–950, 1992.
Lees, J.A., Fawell, S.E., and Parker, M.G. Identification of constitutive and steroid-dependent transactivation domains in the mouse oestrogen receptor. Journal of Steroid Biochemistry 34:33–39, 1989.
Lefebvre, P., Danze, P.M., Sablonnière, B., et al. Association of the glucocorticoid receptor binding subunit with the 90K nonsteroid-binding component is stabilized by both steroidal and nonsteroidal antiglucocorticoids in intact cells. Biochemistry 27:9186–9194, 1988.
Li, Y.F., Huang, C.J., Klindt, J., et al. Divergent effects of antiprogesterone, RU 486, on
progesterone, relaxin, and prolactin secretion in pregnant and hysterectomized pigs with aging corpora lutea. Endocrinology 129:2907–2914, 1991a.
Li, Y.F., Perezgrovas, R., Gazal, O.S., et al. Antiprogesterone, RU 486, facilitates parturition in cattle. Endocrinology 129:765–770, 1991b.
Liberian, S., Erlanger, B.F., Bemires, SM., et al. Steroid-protein conjugates: Their chemical, immunochemical, and endocrinological properties. Pp. 165–200 in Recent Progress in Hormone Research. Vol. XV. PINCs, G., ed. New York and London: Academic Press, 1959.
Lieu, C.H., Graze, G., Morris, S., et al. Disruption of follicular maturation and delay of ovulation after administration of the antiprogesterone RU 486. Journal of Clinical Endocrinology and Metabolism 65:1135–1140, 1987.
Lobaccaro-Henri, C., Saintot, M., Laffargue, F., et al. Effect of the progesterone antagonist-RU 486 on human myometrial spontaneous contractility and PGI2 release. Prostaglandins 44:443–455, 1992.
Loutradis, D., Bletsa, R., Aravantinos, L., et al. Preovulatory effects of the progesterone antagonist mifepristone (RU 486) in mice. Human Reproduction 6:1238–1240, 1991.
Luukkainen, T., Heikinheimo, O., Haukkamaa, M., et al. Inhibition of folliculogenesis and ovulation by the antiprogesterone RU 486. Fertility and Sterility 49:961–963, 1988.
Magdelenat, H., Pertuiset, B.F., Poisson, M., et al. Progestin and oestrogen receptors in meningiomas: Biochemical characterization, clinical and pathological correlations in 42 cases. Acta Neurochirurgica 64:199–213, 1982.
Mao, J., Regelson, W., and Kalimi, M. Molecular mechanism of RU 486 action: A review. Molecular and Cellular Biochemistry 109:1–8, 1992.
Maudelonde, T., Romieu, G., Ulmann, A., et al. First clinical trial on the use of the antiprogestin RU 486 in advanced breast cancer. Pp. 55–65 in Hormonal Manipulation of Cancer: Peptides, Growth Factors and New (Anti-)Steroidal Agents. Klijin, J.A.G., Paridaens, R., and Foekens, JA., eds. New York: Raven Press, 1987.
McDonnell, D.P., Vegeto, E., and O'Malley, B.W. Identification of a negative regulatory function for steroid receptors. Proceedings of the National Academy of Sciences of the United States 89:10563–10567, 1992.
Meyer, M.C., Pornon, A., Ji, J., et al. Agonistic and antagonistic activities of RU 486 on the functions of the human progesterone receptor. EMBO Journal 9:3923–3932, 1990.
Michna, H., Nishino, Y., Schneider, M.R., et al. A bioassay for the evaluation of antiproliferative potencies of progesterone antagonists. Journal of Steroid Biochemistry and Molecular Biology 38:359–365, 1991.
Michna, H., Nishino, Y., Neef, G., et al. Progesterone antagonists: Tumor-inhibiting potential and mechanism of action. Journal of Steroid Biochemistry and Molecular Biology 41:339–348, 1992.
Milgrom, E., Atger, M., and Baulieu, E.E. Progesterone in uterus plasma. IV—Progesterone receptor(s) in guinea pig uterus cytosol. Steroids 16:741–754, 1970.
Milgrom, E., Luu Thi, M., Atger, M., et al. Mechanisms regulating the concentration and the conformation of progesterone receptor(s) in the uterus. Journal of Biological Chemistry 248:6366–6374, 1973.
Mittal, R., Tsutsumi, K., Pater, A., et al. Human papillomavirus type 16 expression in cervical keratinocytes: Role of progesterone and glucocorticoid hormones. Obstetrics & Gynecology 81:5–12, 1993.
Mizutani, T., Bhakta, A., Kloosterboer, H.J., et al. Novel antiprogestins ORG 31806 and 31710: Interaction with mammalian progesterone receptor and DNA binding of antisteroid receptor complexes. Journal of Steroid Biochemistry and Molecular Biology 42:695–704, 1992.
Mobbs, B.G., and Johnson, I.E. Suppression of the growth of the androgen-insensitive
R3327 HI rat prostatic carcinoma by combined estrogen and antiprogestin treatment. Journal of Steroid Biochemistry and Molecular Biology 39:713–722, 1991.
Moguilewsky, M., and Philibert, D. Biochemical profile of RU 486. Pp. 87–97 in The Antiprogestin Steroid RU 486 and Human Fertility Control. Baulieu, E.E., and Segal, S.J., eds. New York: Plenum Press, 1985.
Morales, A.J., Murphy, A.A., and Parthansarathy, S. RU 486: An antioxidant. The American Fertility Society, p. S32, Abstract 0–071. 48th Annual Meeting, November 2–5, 1992.
Mulch, A., and Katzenellenbogen, BS. Antiprogestin-receptor complexes: Differences in the interaction of the antiprogestin RU38486 and the progestin R5020 with the progesterone receptor of human breast cancer cells. Biochemical and Biophysical Research Communications 135:90–97, 1986.
Murphy, A.A., Kettle, L.M., Morales, A.J., et al. Regression of uterine leiomyomata in response to the antiprogesterone RU 486. Journal of Clinical Endocrinology and Metabolism 76:513–517, 1993.
Nath, R., Bhakta, A., and Moudgil, V.K. ZK98299—A new antiprogesterone: Biochemical characterization of steroid binding parameters in the calf uterine cytosol. Archives of Biochemistry and Biophysics 292:303–310, 1991.
Nédélec, L., and Gasc, J.C. Alcoylation angulaire par condensation des organo-magnésiens sur les époxy-5,10 estrènes-9(11). Bulletin de la Société Chimique de France 2556–2564, 1970.
Neef, G. Antiprogestins. A new approach to contraception. Pp. 565–579 in Trends in Medicinal Chemistry. Mutschler, E., and Winterfeldt, E., eds. Weinheim, Germany: VCH, 1987.
Neef, G., Beier, S., Elger, W., et al. New steroids with antiprogestational and antiglucocorticoid activities. Steroids 44:349–372, 1984.
Neulen, J., Williams, R.F., and Hodgen, G.D. RU 486 (mifepristone): Induction of dose dependent elevations of estradiol receptor in endometrium from ovariectomized monkeys. Journal of Clinical Endocrinology and Metabolism 71:1074–1075, 1990.
Ning, Y.M., and Sanchez, E.R. Potentiation of glucocorticoid receptor-mediated gene expression by the immunophilin Logan's FK506 and Rapamycin. Journal of Biological Chemistry 268:6073–6076, 1993.
Norman, J.E., Wu, W.X., Kelly, R.W., et al. Effects of mifepristone in Vivo on decimal prostaglandin synthesis and metabolism. Contraception 44:89–98, 1991.
Okada, S., York, DA., and Bray, G.A. Mifepristone (RU 486), a blocker of type II glucocorticoid and progestin receptors, reverses a dietary form of obesity. American Physiological Society 262:R1106-R1110, 1992.
Parinaud, J., Perret, B., Ribbes, H., et al. Effects of RU 486 on progesterone secretion by human preovulatory granolas cells in culture. Journal of Clinical Endocrinology and Metabolism 70:1534–1537, 1990.
Parinaud, J., Label, B., and Vieitez, G. High progesterone concentrations induce acrosome reaction with a low cytotoxic effect. Fertility and Sterility 58:599–602, 1992.
Peyror, R., Aubény, E., Targosz, V., et al. Early termination of pregnancy with mifepristone (RU 486) and the orally active prostaglandin misoprostol. New England Journal of Medicine 328:1509–1513, 1993.
Philbert, D., Dearest, R., and Teutsch, G. RU 486 a potent antiglucocorticoid in Vivo. Program of the 8th International Congress of Pharmacology , Tokyo, Japan, Abstract 1463, 1981.
Philibert, D., Costerousse, G., Gailllard-Moguilewsky, M., et al. From RU 38486 towards dissociated antiglucocorticoid and antiprogesterone. Pp. 1–17 in Antihormones in Health and Disease. Vol. 19. Agarwal, MK., ed. Basel: Charger, 1991.
Pincus, G. The Control of Fertility. New York and London: Academic Press, 1965.
Pinks, J., Halimas, G., Shirahige, Y., et al. Inhibition of growth of the human malignant
gloom cell line (U87MG) by the steroid hormone antagonist RU 486. 1993 (submitted).
Pleim, E.T., Cailliau, P.J., Weinstein, M.A., et al. Facilitation of receptive behavior in estrogen-primed female rats by the anti-progestin, RU 486. Hormones and Behavior 24:301–310, 1990.
Prat, W.B., Jolly, D.J., Pratt, D.V., et al. A region in the steroid binding domain determines formation of the non-DNA binding, 9S glucocorticoid receptor complex. Journal of Biological Chemistry 263:267–273, 1988.
Pronzato, P., Brema, F., Amoroso, D., et al. Megestrol acetate: Phase II study of a single daily administration in advanced breast cancer. Breast Cancer Research Treatment 17:51–55, 1990.
Radanyi, C., Renoir, J.M., Sabbah, M., et al. Chick heat- shock protein of Mr 90,000, free or released from progesterone receptor, is in a dimeric form . Journal of Biological Chemistry 264:2568–2573, 1989.
Rådestad, A., and Bygdeman, M. Cervical softening with mifepristone (RU 486) after pretreatment with naproxen. A double-blind randomized study. Contraception 45:221–227, 1992.
Rafestin-Oblin, M.E., Couette, B., Radanyi, C., et al. Mineralocorticosteroid receptor of chick intestine: Oligomeric structure and transformation. Journal of Biological Chemistry 264:9304–9309, 1989.
Rajpert, E.J., Lemaigre, F.P., Eliard, P.H., et al. Glucocorticoid receptors bound to the antagonist RU 486 are not downregulated despite their capacity to interact in vitro with defined gene regions. Journal of Steroid Biochemistry 26:513–520, 1987.
Redeuilh, G., Moncharmont, B., Secco, C., et al. Subunit composition of the molybdate-stabilized "8–9S" non-transformed estradiol receptor purified from calf uterus. Journal of Biological Chemistry 262:6969–6975, 1987a.
Redeuilh, G., Secco, C., Mester, J., et al. Transformation of the 8–9S molybdate-stabilized estrogen receptor from low-affinity to high-affinity state without dissociation into subunits. Journal of Biological Chemistry 262:5530–5535, 1987b.
Renoir, J.M., Radanyi, C., and Baulieu, E.E. The antiprogesterone RU 486 stabilizes the heterooligomeric, non DNA-binding, 8S-form of the rabbit uterus cytosol progesterone receptor. Steroids 53:1–20, 1989.
Renoir, A.M., Radanyi, C., Jung-Testes, I., et al. The non-activated progesterone receptor is a nuclear heterooligomer. Journal of Biological Chemistry 265:14402–14406, 1990.
Renoir, A.M., Radanyi, C., and Bailee, EE. UN effete Des immunosuppresseurs FK506 et rapamycine sur le fonctionnement du receptor de la progesterone: La protein "p59-HI," UN careful de l'immunologie et de l'endocrinologie? Competes Rends de l'Académie Des Sciences de Paris 315:421–428, 1992.
Rexene, M., Bush, W., and Gearing, U. Chemical cross-linking of heteromeric glucocorticoid receptors. Biochemistry 27:5593–5601, 1988.
Rivera, J., Hill, N.C., Vernal, A.L., et al. P29, an estrogen receptor-associated protein, is down-regulated by mifepristone in first trimester human placenta and decidua. Human Reproduction 6:1338–1341, 1991.
Roger, M.O., and Bard, D.T. Pretreatment with mifepristone (RU 486) reduces interval between prostaglandin administration and expulsion in second trimester abortion. British Journal of Obstetrics and Gynecology 97:41–45, 1990.
Rondinone, C.M., Baker, M.E., and Redbird, D. Progestins stimulate the differentiation of 3T3-L1 preadipocytes. Journal of Steroid Biochemistry and Molecular Biology 42:795–802, 1992.
Rotello, R.J., Liberman, R.C., Lepoff, R.B., et al. Characterization of uterine epithelium apoptotic cell death kinetics and regulation by progesterone and RU 486. American Journal of Pathology 140:449–456, 1992.
Sabbah, M., Gouilleux, F., Sola, B., et al. Structural differences between the hormone and
antihormone estrogen receptor complexes bound to the hormone response element. Proceedings of the National Academy of Sciences of the United States 88:390–394, 1991.
Sánchez-Criado, J.E., Uilenbroek, J.Th.J., and de Jong, F.H. Antiprogesterone RU 486 increases serum immunoreactive inhibin levels and LH:FSH and testosterone:oestradiol ratios in cyclic rats. Journal of Endocrinology 134:51–57, 1992.
Sartorius, C.A., Tund, L., Takimoto, G.S., et al. Antagonist-occupied human progesterone receptors bound to DNA are functionally switched to transcriptional agonists by cAMP. Journal of Biological Chemistry 268:9262–9266, 1993.
Schaison, G., George, M., Lestrat, N., et al. Effects of the antiprogesterone steroid RU 486 during midulteal phase in normal women. Journal of Clinical Endocrinology and Metabolism 61:484–489, 1985.
Schneider, M.R., Michna, H., Nishino, Y., et al. Antitumor activity of the progesterone antagonists ZK 98.299 and RU38.486 in the hormone-dependent MXT mammary tumor model of the mouse and the DMBA- and the MNU-induced mammary tumor models of the rat. European Journal of Cancer and Clinical Oncology 25:691–701, 1989.
Schweizer-Groyer, G., Cadepond, F., Groyer, A., et al. Stimulation of specific transcription and DNA binding studies suggest that in vitro transformed RU 486-glucocorticosteroid receptor complexes display agonist activity. Journal of Steroid Biochemistry 30:291–294, 1988.
Segal, S.J., and Nelson, W.O. An orally active compound with anti-fertility effects in rats. Proceedings of the Society for Experimental Biology and Medicine 98:431–436, 1958.
Segard-Maurel, I., Jibard, N., Schweizer-Groyer, G., et al. Mutations in the "zinc fingers" or in the N-terminal region of the DNA binding domain of the human glucocorticosteroid receptor facilitate its salt-induced transformation, but do not modify hormone binding. Journal of Steroid Biochemistry and Molecular Biology 41:727–732, 1992.
Segnitz, B., and Gehring, U. Mechanism of action of a steroidal antiglucocorticoid in lymphoid cells. Journal of Biological Chemistry 265:2789–2796, 1990.
Shauer, M., Chalepakis, G., Willmann, T., et al. Binding of hormone accelerates the kinetics of glucocorticoid and progesterone receptor binding to DNA. Proceedings of the National Academy of Sciences of the United States 86:1123–1127, 1989.
Sheridan, P.L., Krett, N.L., Gordon, J.A., et al. Human progesterone receptor transformation and nuclear down-regulation are independent of phosphorylation. Molecular Endocrinology 2:1329–1342, 1988.
Shi, W.L., Wang, J.D., Xu, L.K., et al. Early changes of affinity and binding sites of progesterone and oestrogen receptors in decidua exposed to RU 486. Human Reproduction 7:934–939, 1992.
Shoupe, D., Mishell, D.R., Page, M.A., et al. Effects of an antiprogesterone RU 486 in normal women. II. Administration in the late follicular phase. American Journal of Obstetrics and Gynecology 157:1421–1426, 1987.
Skafar, D.F. Differences in the binding mechanism of RU 486 and progesterone to the progesterone receptor. Biochemistry 30:10829–10832, 1991.
Sutherland, R.L., Mester, J., and Baulieu, E.E. Tamoxifen is a potent "pure" antioestrogen in chick oviduct. Nature 267:434–435, 1977.
Swahn, M.L., and Bygdeman, M. The effect of the antiprogestin RU 486 on uterine contractility and sensitivity to prostaglandin and oxytocin. British Journal of Obstetrics and Gynaecology 95:126–134, 1988.
Teutsch, G., and Bélanger, A. Regio and stereospecific synthesis for 11ß-substituted 19-norsteroids. Tetrahedron Letters 22:2051–2054, 1979.
Teutsch, G., Ojasoo, T., and Raynaud, J.P. 11ß-substituted steroids, an original pathway to antihormones. Journal of Steroid Biochemistry 31:549–565, 1988.
Thomas, M., and Monet, J-D. Combined effects of RU 486 and Tamoxifen on the growth
and cell cycle phases of the MCF-7 cell line. Journal of Clinial Endocrinology and Metabolism 75:865–870, 1992.
Thong, K.J., and Baird, D.T. A study of gemeprost alone, Dilapan or mifepristone in combination with gemeprost for the termination of second trimester pregnancy. Contraception 46:11–17, 1992.
Thong, K.J., Dewar, M.H., and Baird, D.T. What do women want during medical abortion? Contraception 46:435–442, 1992
Tjaden, B., Galetto, D., Woodruff, J.D., et al. Time-related effects of RU 486 treatment in experimentally induced endometriosis in the rat. Fertility and Sterility 59:437–440, 1993.
Tora, L., White, J., Brou, C., et al. The human estrogen receptor has two independent non-acidic transcriptional activation functions. Cell 59:477–487, 1989.
Turcotte B., Meyer M.E., Bocquel M.T., et al. Repression of the a-fetoprotein gene promoter by progesterone and chimeric receptors in the presence of hormones and antihormones. Molecular and Cellular Biology 10:5002–5006, 1990.
Uhler, M.L., Leung, A., Chan, S.Y.W., et al. Direct effects of progesterone and antiprogesterone on human sperm hyperactivated motility and acrosome reaction. Fertility and Sterility 58:1191–1198, 1992.
Ulmann, A., Teutsch, G., and Philibert, D. RU 486. Scientific American 262:42–48, 1990.
Ulmann, A., Silvestre, L., Chemama, L., et al. Medical termination of early pregnancy with mifepristone (RU 486) followed by a prostaglandin analogue. Study in 16,369 women. Acta Obstetricia et Gynecologica Scandinavica (Umea) 71:278–283, 1992.
Urquhart, D.R., and Templeton, A.A. Mifepristone (RU 486) for cervical priming prior to surgically induced abortion in the late first trimester. Contraception 42:191–199, 1990.
Van Santen, M.R., and Haspels, A.A. Failure of mifepristone (RU 486) as a monthly contragestive, "Lunarette." Contraception 35:433–438, 1987.
van Uem, J.F., Hsiu, J.G., Chillik, C.F., et al. Contraceptive potential of RU 486 by ovulation inhibition: I. Pituitary versus ovarian action with blockade of estrogen-induced endometrial proliferation. Contraception 40:171–183, 1989.
Vegeto, E., Allan, G.F., Schrader, W.T., et al. The mechanism of RU 486 antagonism is dependent on the conformation of the carboxy-terminal tail of the human progesterone receptor. Cell 69:703–713, 1992.
Vivat, V., Cohen-Tannoudji, J., Revelli, J-P., et al. Progesterone transcriptionally regulates the ß2-adrenergic receptor gene in pregnant rat myometrium. Journal of Biological Chemistry 267:7975–7978, 1992.
Wakeling, A.E. Novel antiestrogens without partial agonist activity. Journal of Steroid Biochemistry 31:645–650, 1988.
Wang, W.M., Heap, R.B., and Taussig, M.J. Blocking of pregnancy in mice by immunization with anti-idiotype directed against monoclonal anti-progesterone antibody. Proceedings of the National Academy of Sciences of the United States 86:7098–7102, 1989.
Webb, A.M.C., Russell, J., and Elstein, M. Comparison of Yuzpe regimen, danazol, and mifepristone (RU 486) in oral post-coital contraception. British Medical Journal 305:927–931, 1992.
Webster, N.J.G., Green, S., Jin, J.R., et al. The hormone-binding domains of the estrogen and glucocorticoid receptor contain an inducible transcription activation function. Cell 54:199–207, 1988.
Weigel, N.L., Beck, C.A., Estes, P.A., et al. Ligands induce conformational changes in the carboxyl-terminus progesterone receptors which are detected by a site-directed anti-peptide monoclonal antibody. Molecular Endocrinology 6:1585–1597, 1992.
Weinstein, M.A., Pleim, E.R., and Barfield, R.J. Effects of neonatal exposure to the antiprogestin mifepristone, RU 486, on the sexual development of the rat. Pharmacology, Biochemistry and Behavior 41:69–74, 1991.
Wolf, J.P., Chillik, C.F., Itskovitz, J., et al. Transplacental passage of a progesterone
antagonist in monkeys. American Journal of Obstetrics and Gynecology 159:238–242, 1988.
Wolf, J.P., Hsiu, J.G., Anderson, T.L., et al. Noncompetitive antiestrogenic effect of RU 486 in blocking the estrogen-stimulated luteinizing hormone serge and the proliferative action of estradiol on endometrium in castrate monkeys. Fertility and Sterility 52:1055–1060, 1989a.
Wolf, J.P., Sinosich, M., Anderson, T.L., et al. Progesterone antagonist (RU 486) for cervical dilatation, labor induction, and delivery in monkeys: Effectiveness in combination with oxytocin. American Journal of Obstetrics and Gynecology 160:45–47, 1989b.
Wolf, J.P., Chillik, C.F., Dubois, C., et al. Tolerance of perinidatory primate embryos to RU 486 exposure in vitro and in vivo. Contraception 41:85–92, 1990.
Wolf, J.P., Simon, J., Itskovitz, J., et al. Progesterone antagonist RU 486 accommodates but does not induce labor and delivery in primates. Human Reproduction 8:759–763, 1993.
Wu, J.T. Failure of corticosterone (B) and progesterone (P) to reverse the inhibitory effect of RU 486 (RU) on the development of two-cell mouse embryos in vitro. Biology of Reproduction 46:77, Abstract 106, 1992.
Yen, S.S. The hypothalamic control of pituitary hormone secretion. Pp. 65–104 in Reproductive Endocrinology. 3rd Edition. Yen, S.S., and Jaffe, R.B., eds. Philadelphia: W.B. Saunders Company, 1991.
Ylikomi, T., Bocquel, M.T., Berry, M., et al. Cooperation of proto-signals for nuclear accumulation of estrogen and progesterone receptor. EMBO Journal 11:3681–3694, 1992.

















































