
7
Risks to Reproduction and Offspring
The preceding chapters have identified scientific, social, ethical, and legal issues that would need to be taken into account in implementing the committee's three fundamental principles of justice with regard to the inclusion of women in clinical studies (see Chapter 3). This chapter illustrates these issues, and potential mechanisms for resolving them, with regard to a particular subpopulation-subjects of reproductive age-in a particular category of clinical studies-clinical trials of drugs. Historically, concern for these risks has focused on women of reproductive potential, including pregnant and lactating women. Nevertheless, the possibility that certain drugs pose unique risks to the male reproductive system may also merit attention.
People of reproductive age get sick and take medications, and drugs intended for use by this population should therefore be tested in this population. Some of these drugs, however, have potential risks to reproduction or for the development of offspring. These risks give added importance to informed consent and contraceptive options. Risk assessment for reproductive and developmental toxicity is complicated by the high background rates of infertility and birth defects, as well as the difficulty of identifying the specific effects of the drug under investigation. Techniques such as animal studies, in vitro analysis, and surveillance for developmental effects, among others, can provide some information on potential hazards to humans.
Once the potential reproductive and developmental hazards of participation in a clinical study have been identified, investigators can attempt to design trials to minimize these hazards. In some cases, hazards may be
altogether avoidable; in others, it may be decided that hazards cannot be effectively minimized and that the trial should not proceed. The process of weighing potential risks against potential benefits is a complicated process with many players, including the investigator who proposes a study, the institutional review board (IRB) that assures that the protocol is consistent with human subjects regulations, and the potential study participants. This chapter describes some of the factors-such as toxicity data, subjects' understanding of risks, and contraception-that investigators must consider when they wish to study a drug in populations of reproductive age. It also describes the process of making risk-benefit assessments and the values that different people place on certain risks and benefits. The chapter concludes with the committee's recommendations for the conduct of research, including drug trials in persons of reproductive age and in lactating and pregnant women, and a discussion of the policy implications of these recommendations.
SCIENTIFIC ISSUES: RISK ASSESSMENT
For men and women of reproductive age, and for pregnant and lactating women, there are risks associated with taking experimental and nonexperimental drugs. In many cases, these risks are virtually impossible to detect before the drug is in widespread use because of the inherent limitations of animal studies and clinical trials. Nevertheless, concerns about reproductive and developmental toxicity do not override the need to improve the medical management of these populations. Our understanding of treatment options for all of these groups will only be advanced by their inclusion in clinical studies and by more systematic collection of empirical data on reproductive and developmental outcomes.
Identifying Reproductive and Developmental Toxicants
Reproductive and developmental toxicants are physical, chemical, and biological agents that produce a toxic effect in animals and/or humans. Reproductive toxicants alter fecundity, decreasing the ability or increasing the time needed to achieve pregnancy. Developmental toxicants alter the structure or function of offspring. Animal testing and clinical trials are the principal mechanisms for identifying drugs as either reproductive or developmental toxicants. The goal of these approaches is to determine the potential for reproductive and developmental toxicity prior to broader human exposure. In the absence of data from such experimentation, reports of adverse events in humans are also gathered in registries in an attempt to identify unrecognized toxicity. In addition, newborn infants are frequently
screened for birth defects so that drugs and other agents with developmental toxicity might be identified.
To determine whether or not a drug is a potential human reproductive or developmental toxicant requires two steps: (1) hazard identification and (2) hazard characterization. Characterization of the magnitude of the risk for adverse outcome requires two additional steps: (3) exposure assessment and (4) qualitative or quantitative risk characterization (NRC, 1983). Together, these four steps provide the framework for the assessment of risk to reproduction or offspring during a clinical trial.
Hazard Identification
The first step in risk assessment explores the question: Does the drug produce adverse effects on reproduction or offspring in animals or humans? Because data on the effects of drugs on human reproduction or pregnancy outcome are generally not available prior to a clinical trial, it is necessary to utilize data from animal studies or other studies, such as in vitro assays. Reproductive studies in animals are generally useful and valid for determining whether a drug represents a potential human reproductive or developmental toxicant (Wilson and Fraser, 1979; Shepard, 1983; Schardein, 1985; Jelovsek et al., 1989; Jelovsek et al., 1990). Laboratory animals and humans can differ in toxicokinetics, however, and the use of data from animals to determine health risks in humans must be assessed carefully (NRC, 1989).
Hazard Characterization
If a drug represents a potential hazard for reproduction or development, it is necessary to determine the dose-response relationship, site of action, and mechanisms through which the adverse effects are produced. Some drugs that appear to be reproductive or developmental toxicants in animals may not produce adverse effects in humans during a clinical trial. For example, compounds metabolized to toxicants in animals may produce different metabolites in humans, or substantially smaller amounts of the same metabolites, and therefore may not represent a hazard. The converse situation also occurs: a chemical may be non-toxic in the conventional animal assays, but be a human developmental toxicant. Fortunately, this is unusual-all known human teratogens are also teratogens in at least one animal species (Schardein, 1985). In addition, genetic differences among humans in their response to toxicants, such as that observed with hydantoin developmental toxicity (Phelan et al., 1982), may also modify risk to reproduction and development.
Exposure Definition
A critical step in characterizing risk is to define the exposure, including such variables as dose, duration, and timing of exposure with respect to reproduction or pregnancy. For example, exposures that occur before or after the development of a susceptible tissue or organ may carry no additional risk for fetal development. In the case of diethylstilbestrol (DES), the risk of vaginal adenosis is greater than 70 percent for female offspring exposed before the ninth week of gestation but less than 10 percent for exposure after the seventeenth week (see Appendix C). Similarly, in the sexually mature male, exposures to developmental toxicants that occur long before the development of the particular sperm cell that will fertilize an egg are unlikely to affect the development of the resulting offspring, due to the fact that sperm development occurs only in the few months prior to ejaculation.
Risk Characterization
Finally, after a reproductive or developmental hazard has been identified and characterized, and exposure has been defined, this information is analyzed using statistical models to characterize the risk to reproduction or pregnancy outcome. In this step it is important to define the amount and quality of the data available to characterize risk, including variability and uncertainty in the risk estimate. The risk characterization should also include the assessment of the background incidence of adverse reproductive or developmental outcomes.
Challenges to Identification of Reproductive and Developmental Toxicants
In order to isolate and evaluate observed drug effects, it is also necessary to consider the background incidence of adverse reproductive or developmental outcomes. This incidence is often higher than many potential study subjects would suppose. For example, about 15 percent of couples trying to conceive will not have done so after one year of trying (a common definition of infertility); 20 to 30 percent of recognized pregnancies end in miscarriage; 3 to 8 percent of all babies have birth defects; and 1 percent of liveborn children are born with severe mental retardation. Most of these adverse outcomes do not have recognized causes, and very few of those with known causes are the result of exposures to chemical, physical, or biologic agents.
With such a high background rate of many different adverse outcomes, identification of adverse effects imposed by a specific drug exposure can be
difficult. For example, consider an exposure that increases the incidence of limb reduction defects tenfold. If the baseline incidence of such defects is I in 10,000 (0.01 percent), the impact of an increase to 0.1 percent will be undetectable against a background incidence of total birth defects of about 5 percent. It is only when there is some idea of the kind of defect associated with the exposure that studies can be targeted at detecting an increase in incidence.
Determining whether a drug or other treatment is associated with an adverse reproductive or developmental effect also requires characterization of the endpoint of concern. Depending on the endpoint in question, sample sizes required to detect that endpoint can vary greatly (see Table 7-1). An uncommon endpoint such as a rare congenital malformation typically requires very large study populations: a malformation that occurs in only 1 of 10,000 births may not be detected in a trial involving 5,000 couples. When exposure produces a dramatic increase in the incidence of unusual abnormalities, however, it may not take very many cases before the association is recognized. This kind of recognition occurred, for example, with the birth defects associated with thalidomide and isotretinoin, with the cerebral-palsylike illness caused by methyl mercury, and with the severe testicular toxicity of the pesticide dibromochloropropane (DBCP).
Determining whether or not a drug is associated with a relatively common adverse outcome such as miscarriage may be defined with a much smaller number of subjects. Listed in Table 7-1 are some adverse reproductive and developmental outcomes of interest, the background rate of these adverse outcomes, and the sample size needed to determine a doubling of that outcome in a clinical trial.
Male-Mediated Developmental Toxicity
Although traditional concerns about developmental toxicity have focused on exposures (or treatment) of the female during pregnancy, scientists have long suspected that the male may also contribute to adverse pregnancy outcomes such as spontaneous abortion, stillbirth, impaired growth, and structural and functional abnormalities (Olshein and Mattison, in press). Most systematic studies of male-mediated developmental toxicity, however, have been rodent studies, and the implications are inconclusive for human males.
Scientists have postulated that the occurrence of developmental toxicity following exposure or treatment of the male may depend on a number of factors, including male reproductive status (fecundity), exposure (dose, duration), properties of the agent, pharmacokinetics (especially distribution to gonads and other endocrine organs), mechanism of action, stage of spermatogenesis affected, frequency and timing of intercourse with respect to
TABLE 7-1 Expected Frequency of Selected Endpoints of Reproductive or Developmental Failure and the Sample Size Needed to Detect a Doubling of that Endpoint in a Clinical Trial
|
Reproductive or Developmental Endpoint |
Denominator |
Frequency (%)a |
Sample Size of Treatment and Control Groups to Detect a Doubling of the Endpointb |
|
Azospermia |
Men |
1 |
3,300 Men |
|
Failure to conceive after one year of unprotected intercourse |
Couples |
10-15 (12) |
230 Couples |
|
Birth weight <2,500 g |
Live births |
5-15 (10) |
286 Live births |
|
Miscarriage |
Pregnancies |
10-20 (15) |
174 Pregnancies |
|
Chromosome aberration at miscarriage |
Miscarriages |
40-50 (45) |
25 Miscarriages |
|
Late fetal deaths (¬28 weeks) |
Late fetal deaths + live births |
1-4 (3) |
1,068 Late fetal deaths + live births |
|
Total birth defects at birth |
Live births |
2-3 (3) |
1,068 Live births |
|
Chromosome aberrations at birth |
Live births |
0.6 |
5,533 Live births |
|
Neural tube defects |
Late fetal deaths + live births |
0.005-1 (0.05) |
66,936 Late fetal deaths + live births |
|
Severe mental retardation |
Children to age 15 |
0.4 |
8,324 Children to age 15 |
|
a Where a range is given, the background rate used in the determination of sample size is shown in parentheses. b The sample size indicated is the size required of each population (i.e., both the study population and the control population will need to be at least as large as the sample size indicated) to detect a doubling in the rate of the indicated endpoint. In these calculations it is assumed that the control and study populations are the same size, and that the investigator wants a 90 percent chance to detect a statistically significant difference in the study and control populations at a p ≤ 0.05. The calculations were performed with the sample size estimation modules for an unmatched comparison of proportions as implemented in True Epistat 4.0, Epistat Services, Richardson, Texas (1991). The function for the sample size calculation for unpaired comparison of proportions is described on page 399 of Zar, 1984. |
|||
TABLE 7-2 Adverse Developmental Effects that May Occur Following Male Exposure
|
Reproductive Process |
Toxicological Effect of Male Exposure |
Endpoint Observed in Epidemiological Studies |
|
Conception |
Preimplantation loss |
Decreased fertility Increased time to pregnancy |
|
Implantation |
Postimplantation loss |
Increased spontaneous abortion |
|
Embryo development |
Failure/disruption of embryonic development |
Increased spontaneous abortion Increased fetal death Increased malformation Increased growth retardation Increased functional deficit Increased premature birth |
|
Fetal growth and development |
Failure/disruption of fetal growth and development |
Increased fetal death Increased malformation Increased growth retardation Increased premature birth Increased functional deficit |
exposure, and maternal reproductive characteristics (fecundity). In addition, the type of endpoint that is observed is likely to vary with the process affected in the male (see Table 7-2). Studies conducted over the past five decades have identified three potential mechanisms of male-mediated developmental toxicity:
- Genetic: damage to the genetic material contained in the sperm through the creation of a mutation or chromosomal abnormality.
- Epigenetic: damage to processes that control the expression of the paternal genes after fertilization.
- Transport of toxicant: transport of an agent through the ejaculate during postconception sexual intercourse and subsequent exposure of the conceptus, embryo, or fetus.
Although evidence is inconclusive concerning the role of the male in developmental toxicity, the possibility that he has a role provides reason enough for investigators to consider including discussion of developmental toxicity in the informed consent process for male subjects who may be exposed to developmental toxicants in the course of a clinical study. As discussed below, the provision of advice about contraceptive options may also be wise.
Evaluating Drugs for Use in Men and Women of Reproductive Age
Women of reproductive age (defined as 15 to 44 years of age) constitute a significant proportion of the population. Men of reproductive age constitute an even greater proportion, because the window of potential fertility is much larger for males. Not surprisingly, members of this large segment of the population experience a myriad of diseases requiring medical treatment, and drugs intended for use in this population should be tested in this population. As discussed in earlier chapters, federal policies and practices designed to protect women of reproductive potential from risks associated with experimental treatment have hindered the collection of information about drug effects in this subpopulation.
Investigators designing clinical studies need to be particularly concerned about the potential reproductive and developmental toxicity of the compounds they wish to study in this population. When a trial involves the exposure to a potential toxicant, issues of risk characterization, informed consent, and contraceptive options are paramount. Because people tend to overestimate the risks of harm to reproduction and development that are posed by drugs (Koren et al., 1989, 1990), enhanced subject education efforts may be required. Other characteristics of the population of reproductive age, such as use of hormonal contraceptives, must also be taken into account during trial design and recruitment. Discussed in greater detail below are the kinds of variables that investigators must consider when deciding how best to test new drugs in men and women of reproductive age.
Use of Toxicity Testing Data
If information is available on the reproductive and developmental effects of a given drug in animals, investigators must make several types of inferences in order to protect the reproductive and developmental health of their subjects (see Figure 7-1). First, are the animal data relevant to humans? Animal testing for developmental toxicants is generally relevant for human hazard identification (Jelovsek et al., 1989; Francis et al., 1990). While animal data may be less reliable as sources of information about human reproductive toxicity, these data have been used successfully to provide presumptive evidence of human reproductive toxicity (NRC, 1989).
Next, it is necessary to determine how to utilize the dose-response information. For example, investigators must consider whether characterization of a benchmark dose, and subsequent calculation of a reference dose, with safety or uncertainty factors, is adequate to protect human health (Barnes and Dourson, 1986; Gaylor, 1989; Kimmel, 1990; Gaylor, 1991). Alternative interpretations must also be considered: in some cases, it may be more appropriate to assume a linear, nonthreshold, low-dose relationship
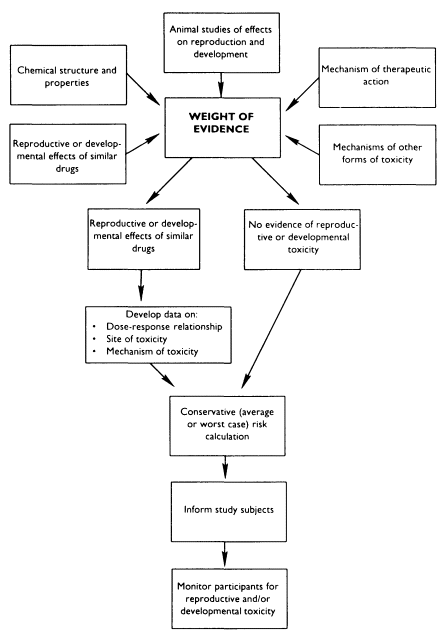
between the chemical and the reproductive or developmental effect (Meistrich and Brown, 1983; Rai and Van Ryzin, 1985; Kimmel and Gaylor, 1988; Meistrich, 1989a,b; Kimmel, 1990; Mattison, 1991; Meistrich and Mattison, in press).
Information from animal reproductive tests can be supplemented by other information. For example, data on the nonreproductive toxicity of an agent often provides clues about possible reproductive effects. If an agent is known to be toxic to the nervous system in adults, investigators testing this agent in pregnant animals know to examine the offspring for behavioral function as well as for gross birth defects. Investigators may also utilize what is known about the reproductive effects of certain agents to postulate the effects of related chemicals.
In the rare situation when there is no information on the reproductive and developmental effects of the drug being tested, other approaches may be taken to protect the health of the subject. For example, data from in vitro testing or structure-activity relationships may provide useful information for counseling subjects in a clinical trial. Recent data using expert systems to explore the relationship between chemical structure and developmental toxicity suggest that a substantial amount of useful information could be extracted from existing developmental toxicity data sets (Takihi et al., in press).
Finally, when available, human data on similar agents or classes of agents can be coupled with animal data to develop an overall assessment of an agent's potential for toxicity. In the case of fluoxetine, an antidepressant, the manufacturer collected several hundred cases of exposed pregnancies in which use of the drug was not associated with an increase in birth defects. This sort of evidence is reassuring, but by no means conclusive-an increase in birth defects of the kind and magnitude associated with, for example, valproic acid might not be detected with this number of patients. Arguments for the safety of fluoxetine for use in pregnant women are further strengthened, however, by the observation that rats and rabbits do not show an increase in developmental anomalies when exposed to the drug during pregnancy in standard protocols.
Subjects' Understanding of Developmental and Reproductive Risks
If men and women of reproductive potential are included in a trial in which they will be exposed to a potential reproductive or developmental toxicant, the potential risks must be characterized as accurately as possible so they can make an informed decision about whether or not to participate. If they decide to participate, they may also wish to consider measures to prevent pregnancy (see below). Finally, information about toxicity risk can help participants determine the likelihood that the baseline incidence of
adverse pregnancy outcome will have been increased by study participation, should a pregnancy occur during the trial.
In this regard, the use of reproductive and developmental toxicity information is similar to the use of information about toxicity to other organs and organ systems. Just as investigational agents can be evaluated in preclinical test phases for reproductive and developmental effects, they can be evaluated for carcinogenicity and for toxicity to liver, kidney, and bone marrow. Results from these evaluations are incorporated into the informed consent process, just as reproductive and developmental toxicity information is incorporated.
Contraception in Clinical Trials
When considering participation in clinical trials of agents with potential reproductive or developmental toxicity, men and women of reproductive age need to be made aware of the contraceptive options available to them as study participants and of what is known about the effectiveness of each method. Table 7-3 lists low and high reported failure rates for the range of contraceptive methods currently available in this country.
The investigator must be concerned with contraception for two major reasons. First, hormonal contraceptives, widely used by women in this country, may introduce complexities into the evaluation of drug effects (see Chapter 4). Second, demographic characteristics of a population can play a role in contraceptive use by that population, with implications for recruitment and the informed consent process.
An investigator may have difficulty assessing the independent effect of a drug in a woman who is using hormonal contraceptives, which can alter the pharmacokinetics (absorption, distribution, metabolism, and excretion) or pharmacodynamics (mechanism of action) of the drug under study. Accurate evaluation of drug effects can also be complicated by the need to clearly isolate the specific side effects of the hormonal contraceptive from those of the drug under study. Where scientific validity or contraceptive effectiveness may be compromised, investigators might consider excluding women using hormonal contraceptives, or recommending that an alternate, nonhormonal method of contraception be used.
There are important differences among population subgroups in the use of contraception, and contraceptive failure rates tend to vary according to age, education, race, ethnicity, marital status, and socioeconomic status. For example, with increasing age, contraceptive failure rates decline as a result of decreased frequency of intercourse and decreased ability to conceive. Non-use of contraceptives is higher among the unmarried, poor, and less educated—a phenomenon that may be associated with access to contraceptive services (Planned Parenthood Federation of America, 1991). Cultural
TABLE 7-3 Methods of Contraception
|
|
Failure Ratea |
|||
|
Category |
Method |
Mechanism of Action |
Low |
High |
|
Nonhormonal |
No method |
|
85.0 |
85.0 |
|
Spermicide alone |
Inactivation of sperm |
21.6 |
25.6 |
|
|
Sponge with spermicide |
Mechanical barrier to sperm; inactivation of sperm |
16.0 |
51.9 |
|
|
Withdrawal |
|
14.7 |
27.8 |
|
|
Periodic abstinence |
Avoidance of coitus during presumed fertile days |
13.8 |
19.2 |
|
|
Diaphragm or cervical cap with spermicide |
Mechanical barrier to sperm; inactivation of sperm |
12.0 |
38.9 |
|
|
Condom |
Mechanical barrier to sperm |
9.8 |
18.5 |
|
|
Intrauterine device, Copper T-380A |
Inhibition of sperm migration, fertilization, or ovum transport |
2.5 |
4.5 |
|
|
Hormonal |
Oral contraceptives |
|
3.8 |
8.7 |
|
Combined |
Suppression of ovulation, changes in cervical mucus and endometrium |
|
|
|
|
Progestin only |
Changes in cervical mucus and endometrium, possibly suppression of ovulation |
|
|
|
|
Intrauterine device Progesterone T |
Inhibition of sperm migration, fertilization, or ovum transport |
2.5 |
4.5 |
|
|
Medroxy-progesterone acetate (DepoProvera) |
Changes in cervical mucus and endometrium, suppression of ovulation |
<1 |
<1 |
|
|
Levonorgestrel subdermal implants (Norplant) |
Same as medroxy-progesterone acetate |
<1 |
<1 |
|
|
ª Percentage of accidental pregnancies during first year of use. ''Low" and "high" refer to rates among women in the United States who were more and less likely than average to use the method correctly and consistently (Harlap, 1991). |
||||
and religious norms may also affect some subjects' willingness to use certain forms of contraception. Awareness of these subgroup differences has important implications for both recruitment and informed consent procedures; investigators may wish to modify protocols according to the demographic characteristics of the population to be studied.
Evaluating Drugs for Use in Lactating Women
Investigators must be especially concerned about developmental toxicity when testing drugs in the subset of the population of reproductive age that is composed of lactating women. Exposure to drugs and chemicals can lead to the presence of these agents in their breast milk, creating concern for: (1) exposure of the infant to the agent and (2) impact of the agent on the quantity and quality of breast milk. Partly as a result of these concerns, lactating women are rarely recruited into trials of new drugs. Therefore, when lactating women require treatment for a medical condition such as pain, infection, depression, constipation, or vitamin deficiency, they often must take medications that have not been systematically evaluated in lactating women. To avoid risk by ceasing lactation during treatment is in many cases not advisable, considering lactation's important benefits (e.g., maternal-infant bonding; transmission to the infant of antibacterial and antiviral substances; enhanced nutrition, growth, and development of the infant).
Factors influencing the presence and amount of a drug in breast milk include maternal and mammary physiology and pharmacokinetics, chemical properties (e.g., lipid solubility, and protein binding), and infant feeding characteristics (frequency, duration, and amount). The impact, if any, of a drug on the child will depend on the amount of drug ingested, the pharmacokinetics of the drug (absorption, distribution, metabolism, and elimination), and the mechanism of action and toxicity of the drug. Not all drugs on the market have been fully characterized for their presence in breast milk and effect on the nursing infant, but some data are available to guide the practitioner; most drugs are compatible with breastfeeding (Briggs et al., 1986). Less well studied is the impact of drugs on milk production. Drugs suspected to alter milk production include dopaminergic agents, estrogencontaining oral contraceptives and other estrogens, antiestrogens, nicotine, prostaglandins, and the thiazide diuretics.
Investigators designing clinical studies in which lactating women may be recruited should carefully advise these women of the risks to the nursing child, including those of or cessation of lactation. Where possible, efforts should be made to characterize risks to the nursing infant based on known pharmacologic and toxicologic properties of an agent in other populations.
Evaluating Drugs for Use in Pregnant Women
Studies have shown that an average of 3.8 medications are used during each pregnancy (Heinonen et al., 1983) and that 75 percent of pregnant women use between 3 and 10 drugs while they are pregnant (Quirk, 1986). Medications used most commonly during pregnancy include analgesics, antipyretics, antimicrobials, antiemetics, diuretics, cough medications, and psychoactive agents (Quirk, 1986). Yet despite their frequent need for medical treatment, few clinical trials of new drugs include pregnant women. Thus, the initial use of treatments in pregnant women often involves therapies developed in men (and women) who are physiologically different (see Chapter 4).
For clinical conditions that are sufficiently common, controlled trials may be conducted in pregnant women several years after a drug has been put on the market (and several years after pregnant women have been taking the drug on what amounts to an experimental basis). This was true for antihypertensive medications, a number of which were only recently tested in controlled trials for use in pregnancy-induced hypertension. It is not uncommon for physicians to prescribe drugs for pregnant women on the basis of substantial anecdotal information about such use, but reliance on information is risky given the number of cases necessary to identify an association between a drug and an adverse effect.
The testing of therapies in pregnant women often depends on the initiative of independent investigators rather than on the marketing intentions of pharmaceutical manufacturers. It is unusual for a drug to be brought to market for the express purpose of treating pregnancy conditions or pregnant women. An exception is ritodrine, an agent used to treat preterm labor, and which has been marketed expressly for this indication. Ironically, many practitioners use terbutaline or magnesium sulfate to stop preterm labor, although they have not received FDA indications for this purpose. In general, the indications for use restrict the way the drug can be marketed but not how a physician uses the drug. These agents and others (e.g., indomethacin, sulindac, nifedipine) have been tested in controlled trials of preterm labor, although these trials were not part of the drug development efforts for these compounds.
The committee recommends that NIH strongly encourage and facilitate clinical research to advance the medical management of preexisting medical conditions in women who become pregnant (e.g., lupus), medical conditions of pregnancy (e.g., gestational diabetes) and, conditions that threaten the successful course of pregnancy (e.g., preterm labor).
While the inclusion of pregnant women in clinical studies introduces new complications and risks, the dearth of proven-safe treatment options for ill pregnant women carries its own set of complications and risks. If a drug is going to be used in pregnant women, then the availability of safety and effectiveness information applicable to that population is critical. Reliance upon adverse event reporting by clinicians is not in and of itself a sufficient basis upon which to assess the safety and effectiveness of drugs in pregnant women. Clinical trials, however, also have limitations. Clinical trials have limited power to detect some adverse effects due to the relatively small numbers of subjects included in clinical trials compared with the number of persons who may eventually use the drug under study. Adverse effects may not become evident until the drug is in widespread use. Therefore, systematic surveillance for developmental effects is essential to any plan to include pregnant women in clinical trials. Together, both methods will further our understanding of the medical management of the ill pregnant woman.
Surveillance for Developmental Effects
Surveillance for reproductive and developmental effects in the offspring is essential to our understanding of the safety of drug use during pregnancy. Such screening assumes that there are tools available to identify developmental effects and that these tools can be economically applied for the surveillance of a healthy population. Procedures may be as simple as a clinical evaluation of the newborn to determine if the child has a structural birth defect that can be identified on physical examination. One of the most critical steps in surveillance is the recording of screening results in a database so that they can be combined with other results for a more comprehensive analysis. Several programs currently exist to monitor populations for congenital malformations; these programs may provide a starting point for surveillance efforts related to pregnant women in clinical studies.
Monitoring of populations for congenital malformations began in the mid-1960s, and by the mid-1970s 7 countries had nationwide monitoring systems and 12 other countries had regional monitoring systems. In addition, the International Clearinghouse for Birth Defects Monitoring was created in the mid-1970s to collect, collate, analyze, and share information on local trends in birth defects identified by the various participating programs; at present there are 26 participating programs.
Monitoring systems use one of two major monitoring strategies: (I) monitoring of all malformations as reported or (2) monitoring of selected "sentinel" malformations, so-called because they are generally detected within the first week of life. Examples of sentinel malformations include anencephaly, spina bifida, hydrocephaly, orofacial clefts, gastrointestinal atresia, deformities of the extremities, Down's syndrome, and congenital hip dislo-
cation. Unfortunately, neither approach escapes the difficulty of ascertainment. Studies indicate that monitoring systems for congenital malformation experience substantial underreporting; in some cases, only one-third of infants with a given abnormality are identified by a monitoring system.
Most monitoring systems have established specific thresholds of malformation incidence that signal a significant increase in frequency (Holtzman and Khoury, 1986). These thresholds are characterized as excesses above an expected number (assumed to be a Poisson variable), excesses above a baseline rate determined from historical information, a decreasing time interval between consecutive births with the malformation (also determined from historical information), or changes in time-space clustering. As in all statistical analysis, it is important to avoid the erroneous assumption of causation when drugs are associated with birth defects. Given the large number of comparisons calculated for adverse developmental outcome, however, false positives are a continual problem.
As is the case with individual clinical trials, it is important to understand the capacity of a monitoring system to identify correctly a developmental toxicant, based on the number of births registered with the system. For example, cleft lip occurs in about I in every 1,000 births (incidence is 0.001). The ability to identify an exposure that increases the incidence of cleft lip varies with the size of the population monitored. If 75,000 births are monitored (half treated and half untreated), an increase of 1.3-fold over background could be identified; however, if 10,000 births are monitored (half treated and half untreated), the minimum increase that could be detected would be a 2.0-fold increase above background. In a related sense, the power of monitoring systems to detect adverse effects is also limited because few pregnant women will be exposed to a specific drug. While surveillance cannot guarantee detection of developmental toxicants, systematic collection of information about pregnancy outcomes in a wide range of situations, complemented with information gained through clinical trials that include pregnant women, provides an important element of protection for pregnant women and their offspring.
The committee recommends that a review be undertaken of existing birth defects monitoring programs to critically define what they are capable of doing and suggest improvements and reasonable expectations for their use.
Such a review would be of value in the development of reproductive and developmental screening systems and in further consideration of postmarketing surveillance for reproductive and developmental effects.
ETHICAL ISSUES: RISK-BENEFIT ANALYSIS
Assessing the potential risks and benefits of clinical research is not always an easy task. The very use of phrases such as "risk-benefit equation" and "balancing risks against benefits" has the misleading effect of making it seem that the process enjoys mathematical precision or scientific rigor. The task is made even more difficult because reasonable people disagree both in their evaluations of the magnitude of risks and benefits and how to weigh risks against potential benefits. Different people-be they medical scientists, patients, or healthy volunteers-may value the risks and benefits differently. They may consider some risks worth taking in relation to potential benefits, while other risks may be viewed as unacceptably high in relation to potential benefits.
Assessing risks and potential benefits has both a scientific component and a personal element that varies with the individual making the assessment. By "scientific" we mean that intersubjective agreement can be attained among scientists and researchers based on observations, previous studies, and clinical experience. For example, the identification of the risks and side effects of drugs and the probability of their occurrence is based on experience from earlier studies. Once a sufficiently large population has been studied, medical scientists should be able to agree on what risks might be expected, how likely they are to occur, and their impact on morbidity and mortality. The foregoing sections of this chapter illustrate the scientific dimension of assessing the risks of harm various substances are likely to produce. The same is true of benefits, when benefits are viewed in a relatively narrow, medical sense, best captured by the concept of efficacy: a drug does what it is designed to do-provide a cure, alleviate symptoms, produce a temporary remission, and so on.
But there also is an irreducibly personal element in risk-benefit assessments. By "personal" we mean the insertion of an individual's values, taken in the broadest sense, into the process of assessing the meaning of risks and benefits for one's life or the lives of others, as well as the weighing of risks against benefits. To take a common example, members of IRBs often disagree on how risky a particular procedure actually is.
A committee member who is a member of one IRB reports that heated disputes have arisen over how to characterize the level of risk of lumbar punctures in infants or demented elderly patients, insertion of urethral catheters in six-year-old boys, right-heart catheterization in cardiac patients, withdrawing medication from patients with mild-to-moderate hypertension, and withholding antipsychotic medication from patients with severe emotional disorders. Those who agree on the scientific facts concerning the magnitude and probability of side effects will bring different personal values and experiences to the question of whether the risks are acceptable.
Women's values can differ significantly from those of scientists (whether male or female) in assessing risk-benefit ratios. For example, women's health advocates tend to define the "safety" of contraceptive methods in terms different from those typically employed by biomedical scientists. According to one report:
Scientists' concern is to establish safety of methods according to specific measurable parameters. They assess toxicity, first in animals and then in carefully controlled studies in human volunteers. Subsequent studies address efficacy and short- to medium-term safety. . . Women's health advocates . . . give more priority to methods that have fewer side effects and that protect against sexually transmitted diseases and their consequences such as infertility. While scientists have tended to give priority to methods which minimize users' control, women's health advocates prefer methods controlled by the users. [World Health Organization, 1991:11.]
New policies to encourage the inclusion of women, and in particular women of childbearing age, in clinical studies evidence a greater acknowledgment of individual values and a respect of individual autonomy (see, for example, Merkatz et al., 1993). These policies will affect the responsibilities of IRBs and potential participants. The changes will be most evident in the communication of risks to participants and in IRB risk-benefit assessments.
As with any potential participant, a thorough discussion of the risks and potential benefits of participation is a prerequisite for an individual's ability to make an informed decision to enroll in a clinical study. For men and women of reproductive age, reproductive issues affect the type of information included in the informed consent process.
It will be the IRBs' obligation, as with all research involving presumptively competent adults, to continue to ensure that: (1) the selection of potential participants is fair; (2) the informed consent process is adequate; and (3) the risks to participants are outweighed by the potential benefits. This first duty-fair selection-is the subject of the committee's report and thus requires no additional comment. The other two duties will be discussed below.
Women (Not Pregnant or Lactating) and Men of Reproductive Age
Significant changes have occurred during the committee's tenure, in policies that govern the inclusion of women of childbearing potential in clinical studies, particularly studies of FDA-regulated products (see Chapter 6). FDA issued new guidelines permitting the participation of women of childbearing potential in the early phases of drug trials, and offered three reasons for this decision: (1) scientific gains in study design related to the early identification of gender differences in trials; (2) the ability to reduce
the risk of fetal exposure through protocol design; and (3) recent social changes indicating respect for women's autonomy and decisionmaking capacity in reproductive issues. NIH guidelines are currently under revision. These policy changes should have the effect of including more women of reproductive age in clinical studies, with implications for risk-benefit assessments.
In a study that poses risks to potential offspring, women who are not pregnant at the outset of the investigation may become pregnant while they are still participants. The committee believes that the informed consent process for these women should include information about contraception and the alternatives of voluntarily withdrawing from the study and terminating a pregnancy should conception take place. Similar discussions should be held with men who could father a child while participating in the study. As in all research involving human participants, every effort should be made to ensure that the consent decision is fully voluntary. An example of language for consent forms proposed by Moreno (1994) in his presentation to the committee appears below, as modified by the committee:
It is possible that your participation in this study may cause damage to children if you choose to have them. You have already been told what is known about this possibility, and you are encouraged to ask further questions. (Include as appropriate: We urge you or your partner not to become pregnant while you are part of this study.) You may want to discuss this with others before you agree to take part in this study. If you wish, we will arrange for a doctor, nurse or counselor who is not part of this study to discuss this possibility with you and anyone else you want to have present.
The committee recommends that investigators and IRBs not exclude persons of reproductive age from participation in clinical studies. In the case of women of reproductive age, the potential or prospect of becoming pregnant during the study may not be used as a justification for precluding or limiting participation. Risks to the reproductive system should be considered in the same manner as risks to other organ systems. Risks to possible offspring of both men and women who are not pregnant or lactating should not be considered in the risk-benefit calculation. It is the responsibility of investigators and IRBs to assure that the informed consent process include an adequate discussion of risks to reproduction and potential offspring, including, where appropriate, an adequate discussion of relevant considerations of birth control.
The committee recommends that the participant be permitted to select voluntarily the contraceptive method of his or her choice where there are no relevant study-dependent, scientific reasons to require
the exclusion of use of certain contraceptives (e.g., drug interaction).
The committee recommends that pregnancy termination options be discussed as part of the consent process in clinical studies that pose unknown or foreseeable risks to potential offspring.
Lactating Women
The possible transmission of drugs to nursing infants is a risk that must be considered when including lactating women in clinical studies. This additional consideration must be thoroughly discussed in the informed consent process.
The committee recommends that investigators and IRBs not exclude women who are lactating from participation in clinical studies. It is the responsibility of investigators and IRBs to ensure that the informed consent process includes, wherever appropriate, an advisory to potential participants that there may be special risks to their children if nursing mothers participate. No nursing mother should be permitted to agree to participate without first receiving additional information about these special risks.
Pregnant Women
As reflected in the recommendation presented earlier in this chapter, the committee wishes to encourage clinical research to advance the medical management of pregnant women who are or may become ill. In this context, the committee reviewed the current DHHS regulations concerning the involvement of pregnant women as research subjects. The committee's review was limited to situations in which the pregnant woman is the subject of the research (see Chapter 6). It did not include situations in which the fetus is the subject of the research (currently covered by the same regulation); fetal research was outside of the committee's charge.
The DHHS regulations begin with a presumption of exclusion-that is, "no pregnant woman may be a research subject" except under certain conditions. The regulations also require that IRBs ensure during their review of research protocols that the exclusionary standard enunciated in the regulations is met. In addition, the regulations classify pregnant women as a "vulnerable population" deserving of special protection. For the reasons discussed below, the committee concluded that the current regulatory scheme should be revised.
The committee acknowledges that the current regulations (45 C.F.R. 46
Subpart B) may reflect an inadvertent attribution of the vulnerability of the fetus (which obviously lacks autonomy) to the pregnant woman. Nonetheless, it is inappropriate for the regulations to retain a presumption of exclusion on the basis that pregnant women are a "vulnerable population" in need of special protection. In this context, "vulnerable" suggests that pregnant women are less autonomous or more easily exploited than other persons an inference that the committee has found no evidence to support. The labeling of pregnant women as a vulnerable population also might be viewed as suggesting that they cannot weigh the risks to a fetus or potential child in deciding whether to enroll in a clinical study; that pregnant women do not care sufficiently about the health or well-being of their future children to make sound decisions; and that the prevention of all potentially harmful outcomes of pregnancy is a goal that warrants governmental, regulatory, or other official intervention into the lives and free choices of women. The committee rejects these inferences as well. Removal of pregnant women from the regulatory category of "vulnerable" potential subjects would avoid any possible inference that pregnant women are less capable of making informed decisions by virtue of their pregnancy, than are other potential research participants.
For all potential research participants, risk-benefit assessment is a complex and difficult task. Nevertheless, it is no more difficult for pregnant women than it is for nonpregnant women or for men. Virtually all women desire healthy infants, even when their pregnancies are unplanned. While occasionally there may be pregnant women who are incapable of acting in the interests of their future children, it would be inappropriate to base a public policy on an atypical case, rather than a normative case.
There also is little public support for the proposition that the prevention of all potentially harmful outcomes of pregnancy is a goal that warrants governmental, regulatory, or other official intervention into the lives and free choices of women. Pregnant women may choose to work in stressful jobs, engage in recreational activities, drive automobiles, and do other things that could place their own or their fetuses' health or life in jeopardy.
The committee recommends that pregnant women be presumed to be eligible for participation in clinical studies. It is the responsibility of investigators and IRBs to ensure that pregnant women are provided with adequate information about the risks and benefits to themselves, their pregnancies and their potential offspring. Even when evidence concerning risks is unknown or ambiguous,1 the decision about acceptability of risk to the pregnancy or to offspring should be made by the woman as part of the informed consent process.
The committee was unanimous in the view that pregnant women should be presumed to be eligible for participation in clinical studies. The committee also unanimously endorsed the importance of recognizing, in public policy as well as in the deliberations of IRBs and investigators, that pregnant women should be treated as competent adults capable of making their own decisions about participation in research. It should be emphasized that the committee is not recommending that NIH impose an affirmative obligation on investigators to recruit pregnant women into every clinical study. What follows is further explication of the committee's intent with respect to the implementation of this recommendation.
Adequate Information
With respect to the obligation to ensure that pregnant women are provided with adequate information about the risks and benefits to their pregnancy and potential offspring, the committee recommends the following strengthened informed consent procedure. The disclosure statement of consent forms for all studies that pose a risk to pregnancy or potential offspring should include, highlighted in bold type, a statement such as: If you are pregnant or contemplating pregnancy, we urge you to consult your obstetrical care provider before deciding about participation in this study. Participation in this study may (does) pose a risk of (significant) harm to your pregnancy and/or your potential baby.
Investigators should ask all potential participants if they are pregnant as part of the initial screening phase of recruitment. If a woman is pregnant, her attention should be drawn to this bolded statement. This process should include a special disclosure statement that details in easily understood lay language what is known about the risks and potential benefits to her pregnancy and potential offspring, resulting from participation in the study. This statement should be reviewed with the pregnant woman, and she should be encouraged to consult with her obstetrical care provider before proceeding further in the consent process. It is important for a pregnant woman to have benefit of the advice of her obstetrical care provider in deciding whether to participate in a study. (In the case where the woman's own obstetrical care provider is the study investigator, the pregnant woman should be offered the opportunity to discuss her participation with a similarly qualified individual who is not associated with the study.) If the pregnant woman does not wish to consult with her obstetrical care provider, and even if she has had such a consultation, specific procedures should be instituted to ensure that she understands the relevant risks and benefits. For example, the potential participant could be asked to describe in her own words what the risks and benefits are. It should be clear that the pregnant woman understands
that no drug or other intervention can improve on normal pregnancy in a healthy woman. Alternatively, the pregnant woman could be asked to complete a knowledge test. Deficiencies in understanding discovered through either method should be addressed through continued discussion and education. Only after the woman demonstrates an adequate understanding should consent be solicited. These are procedures that are generally advocated to improve the quality and the meaningfulness of the informed consent process (Faden and Beauchamp, 1986; Appelbaum et al., 1987). They are particularly important when the stakes associated with participation are high, as is the case for pregnant women if participation entails significant risks to pregnancy or potential offspring.
Paternal Consent
It is appropriate for investigators to encourage a potential participant who is pregnant to discuss her participation in clinical studies and risks to potential offspring with the potential baby's father, but the committee rejects any requirement that the consent of the potential baby's father be a condition of the participation of a pregnant woman in research. The committee recognizes that the husbands of pregnant women, as well as future fathers who are not husbands, have an interest in the health of their children and that these men may have a deep emotional attachment toward their offspring prior to birth. Until a child is born, however, the future father can only protect the health of the potential child by controlling the decisions and actions of the woman. To give men the authority to veto the decisions of their wives or partners to participate in research grants men unacceptable power over women. It also would accord greater protection to fetuses than to children; only one parent's permission is required to enroll an infant or child in clinical research.
Scientific Criteria for Exclusion
The committee recognizes that, as in all clinical studies, there may be scientifically and medically valid reasons for excluding pregnant women from a particular study. A pregnant woman would be excluded if the medical condition of pregnancy disqualifies her as a subject in the same sense that anyone else, pregnant or nonpregnant, male or female, would be disqualified based on medical conditions that would interfere scientifically with the study. For example, a pregnant woman would be excluded from a study of hormone replacement or contraception. A pregnant woman also would be excluded from a study of weight loss, as would any person who, for example, was already very underweight; scientifically, it would not make sense to include either type of person in such a study. Similarly, a pregnant
woman would be excluded from a study when the condition of pregnancy places the woman in a risk category (because pregnancy increases the risk of harm to the woman) that would exclude others due to an unacceptable risk/benefit ratio.
Other Criteria for Exclusion
There was considerable discussion within the committee about whether there are any exceptional instances in which IRBs can be given the discretion to exclude pregnant women from participation for other than scientific reasons. Most committee members ultimately endorsed the following recommendation:
Investigators and IRBs may exclude pregnant women from participation only when the IRB finds, and records its finding in writing, that the following standard has been met: (1) there is no prospect of medical benefit to the pregnant woman, and (2) a risk of significant harm to potential offspring is known or can be plausibly inferred.
A finding that a risk of significant harm to potential offspring is ''known or can be plausibly inferred" may be based on evidence from animal studies, in vitro studies, structure-activity relationship data, or previous clinical experience.
Under this standard, IRBs may exclude pregnant women from the earliest phases of many drug trials, but most clinical studies would remain open to pregnant women. Committee members adopting this standard were motivated by a desire to be true to the underlying principle that pregnant women should be treated no differently than other presumptively competent adults in the context of IRB deliberations. In addition, these committee members were particularly concerned that if the exceptive case was not narrowly constructed, variation in interpretation could open the door to widespread exclusions of pregnant women.
A few members of the committee, however, were not able to endorse the above mentioned standard. They wished to reserve for the IRB the discretion to exclude pregnant women from participation not only when there is no prospect of medical benefit to the women but also when there is only potential for benefit to them that could be characterized as minimal or insignificant. The intent here is to allow the IRB more room for judgment about the appropriateness of exclusion. An example of a situation in which these members believed that IRBs should have the discretion to exclude pregnant women was that of a clinical trial of a medication thought to be helpful in the management of severe acne but known to cause malformations in offspring if taken during pregnancy. The standard endorsed by most
committee members would not permit a blanket exclusion of pregnant women from such a study, as it could not be claimed that there is no prospect of medical benefit to the pregnant participant.
The committee also struggled with how to accommodate within its support for the shift of the presumption to inclusion of pregnant women (from that of exclusion) a role for conscience and an individual investigator's moral commitments. It was agreed that, at a minimum, such a mechanism would require that the investigator provide the IRB with a written explanation of his or her concerns of conscience and that the IRB review any such requests in light of a presumption that favors the inclusion of pregnant women in clinical studies. It also would require the IRB to guard against any abuse of conscience claims, and, in particular, against circumstances in which a request for exemption on the basis of conscience is offered in lieu of other reasons not based in moral commitment. It is precisely because of the potential for abuse of a "conscience" exemption that the committee could not resolve whether or under what conditions such an exemption should be constructed. Appeals to conscience are in many respects unassailable; in some contexts, the force of such appeals has had a chilling effect on public policy.
Documentation and Monitoring of Exclusions
An IRB must record in writing both its reasons for permitting any exception to the general presumption of inclusion of pregnant women and the frequency with which it grants such exceptions. It is anticipated that IRBs would record such information in the minutes of their meetings and that the act of documentation would help the IRBs to properly implement the standard. Such record keeping also would provide a source of information should OPRR desire to evaluate the performance of an IRB on this issue.
Conclusion
The committee recognizes that its recommendation concerning the participation of pregnant women in clinical studies cannot ensure the prevention of a small, theoretical risk of harm to offspring. Pregnancy and the controversial moral and legal standing of the fetus or potential child raise unique considerations. We do not wish to dismiss or evade these important considerations. However, the committee was persuaded of the overriding value of ensuring that all women-pregnant or otherwise-be treated justly with respect to the opportunity to derive the benefits of research. The shifting of the presumption to one of inclusion of pregnant women in clinical studies from one of exclusion is an important step in that direction.
The committee believes that given the safeguards described above, holding IRBs and investigators to the presumption of including pregnant women in research is not a significant threat to the health of future generations. Except for studies specifically designed to investigate outcomes in pregnant women-which the committee strongly endorses-it is exceedingly unlikely that investigators will seek out pregnant women for recruitment. It should be emphasized that the committee is not recommending that NIH impose an affirmative obligation on investigators to recruit pregnant women into every clinical study. Moreover, the committee's conclusions are consistent with the position that women who are or who might become pregnant have a moral obligation to weigh risks to a future child when deciding whether to participate as research subjects. It is unlikely that pregnant women will seek admission into studies that pose a significant risk of harm to their offspring, unless there is some offsetting benefit to the health of the pregnant woman that in turn advances the interests of the potential child by its having a healthy mother. A policy of presuming that pregnant women are eligible to participate in clinical research, although introducing a possibility of harm to a potential child, is in fact likely to produce health dividends for mothers that will inure to their children. Although the committee is not indifferent to the risk of harm to even one potential child, the committee felt compelled to consider as primary the interests of all women in being treated justly and with dignity.
The committee recommends that OPRR revise and reissue Subpart B of the DHHS regulations for the Protection of Human Subjects, titled "Additional Protections Pertaining to Research, Development, and Related Activities Involving Fetuses, Pregnant Women, and Human In vitro Fertilization [45 C.F.R. 46, subpart B] in accordance with the committee's recommendations.
At least a technical amendment to Subpart A, sec. 46.111(a)(3), eliminating the reference to pregnant women as a "vulnerable population" will be required by this revision to Subpart B.
NOTE
|
|
presumption, since both procedures were subsequently determined to increase the risk of spontaneous abortion. IRBs and investigators may find it helpful to use this convention in discussing the level of risk with potential participants. |
REFERENCES
Appelbaum, P.S., Roth, L.H., Lidz, C.W., Benson, P., and Winslade, W. 1987. False hopes and best data: Consent to research and the therapeutic misconception. Hastings Center Report 17(April):20-24.
Barnes, D.G., and Dourson, M. 1986. Reference dose (RfD): Description and use in health risk assessments. Regulatory Toxicology Pharmacology 8:471-486.
Briggs, G.G., Freeman, R.K., and Yaffe, F.J. 1986. Drugs in Pregnancy and Lactation: A Reference Guide to Fetal and Neonatal Risk. 2nd ed. Baltimore, Md.: Williams & Wilkins.
DHEW (Department of Health, Education, and Welfare). 1979. DHEW Support of Fetoscopy. Report and Recommendations of the DHEW Secretary's Ethics Advisory Board, 23 February.
Faden, R.R., and Beauchamp, T.L. 1986. A History and Theory of Informed Consent. New York: Oxford University Press.
Fletcher, J.C., and Ryan, K.J. Federal regulations for fetal research: A case for reform. Law, Medicine and Health Care 15(3):126-138.
Francis, E.Z., Kimmel, C.A., and Rees, D.C. 1990. Workshop on the qualitative and quantitative comparability of human and animal developmental neurotoxicity: Summary and implications. Neurotoxicology and Teratology 12(3):285-292.
Gaylor, D.W. 1991. Comparison of the properties of reference doses based on the NOAEL and benchmark doses. Proceedings, Air and Waste Management Association 71: paper no. 91 173.6.
Gaylor, D.W. 1989. Quantitative risk analysis for quantal reproductive and developmental effects. Environmental Health Perspectives 79:243-246.
Harlap, S. 1991. Preventing pregnancy, protecting health. New York: Alan Guttmacher Institute.
Heinonen, O.P., Sloan, E.D., and Shapiro, S. 1983. Birth Defects and Drugs in Pregnancy. Boston: John Wright.
Holtzman, N.A., and Khoury, M.J. 1986. Monitoring for congenital malformations. Annual Review of Public Health 7:237-266.
Jelovsek, F.R., Mattison, D.R., and Chen, J.J. 1989. Prediction of risk for human developmental toxicity: How important are animal studies for hazard identification? Obstetrics and Gynecology 74:624-635.
Jelovsek, F.R., Mattison, D.R., and Young, J.F. 1990. Eliciting principles of hazard identification from experts. Teratology 42:521-533.
Kimmel, C.A. 1990. Quantitative approaches to human risk assessment for noncancer health effects. Neurotoxicology 11:189-198.
Kimmel, C.A., and Gaylor, D.W. 1988. Issues and qualitative and quantitative risk analysis for developmental toxicology. Risk Analysis 8:15-20.
Kline, J., Stein, Z., and Susser, M. 1989. Conception to Birth: Epidemiology of Prenatal Development. New York: Oxford University Press.
Koren, G., Bologna, M., and Pastyszak, A. 1990. The way women perceive teratogenic risk: The decision to terminate pregnancy. Pp. 373-381 in Maternal-Fetal Toxicology: A Clinician's Guide, G. Koren, ed. New York: Marcel Dekker.
Koren, G., Bologna, M., Long, D., Feldman, Y., and Shear, N.H. 1989. Perception of teratogenic risk by pregnant women exposed to drugs and chemicals during the first trimester. American Journal of Obstetrics and Gynecology 160:1190-1194.
Mattison, D.R. 1991. An overview of biological markers in reproductive and developmental toxicology: Concepts, definitions and use in risk assessment. Biomedical and Environmental Sciences 4:8-34.
Merkatz, R., Temple, R., Sobel, S., Feiden, K., Kessler, D., and the Working Group on Women in Clinical Trials. 1993. New England Journal of Medicine 329(4):292-296.
Meistrich, M. 1989a. Calculations of the incidence of infertility in human populations from sperm measures using the two-distribution model. Pp. 275-290 in Institute for Health Policy Analysis Forum on Science, Health, and Environmental Risk Assessment. Alan R. Liss .
Meistrich, M. 1989b. Interspecies comparison and quantitative extrapolation of toxicity to the human male reproductive system. In Toxicology of the Male and Female Reproductive Systems, P.K. Working, ed. Bristol, Penn.: Hemisphere.
Meistrich, M., and Brown, C.C. 1983. Estimation of the increased risk of human infertility from alterations in semen characteristics. Fertility and Sterility 40(2):220-230.
Meistrich, M., and Mattison, D.R. In press. Methods for quantitative assessment of reproductive risks. In: Assessing the Risks of Adverse Reproductive Outcomes, Monograph No. 4 of the Conte Institute for Environmental Health, D. Brenner and A. Bloom, eds. March of Dimes.
Moreno, J.D. 1994. Ethical issues related to the inclusion of women of childbearing potential in clinical trials. In: Women and Health Research: Ethical and Legal Issues of Including Women in Clinical Studies, Volume 2, A. Mastroianni, R. Faden, and D. Federman, eds. Washington, D.C.: National Academy Press.
NRC (National Research Council) 1983. Risk Assessment in the Federal Government: Managing the Process. Washington, D.C.: National Academy Press.
NRC. 1989. Biologic Markers in Reproductive Toxicology. Washington, D.C.: National Academy Press.
Olshein, A.R., and Mattison, D.R., eds. In press. Male-Mediated Developmental Toxicity. New York: Plenum.
Phelan, M.C., Pelloch, J.M., and Nance, W.E. 1982. Discordant expression of fetal hydantoin syndrome in heteropaternal twins. New England Journal of Medicine 307:99-101.
Planned Parenthood Federation of America. 1991. New Birth Control Conferences Report. Nine Conferences in Nine Cities: A Report, January 1990-January 1991. New York: Planned Parenthood Federation of America.
Quirk, J.G. 1986. Use and misuse of drugs in pregnancy. In: Drug and Chemical Action in Pregnancy: Pharmacologic and Toxicologic Principles, S. Fabro and A.R. Scialli, eds. New York: Marcel Dekker.
Rai, K., and Van Ryzin, J. 1985. A dose-response model for teratological experiments involving quantal responses. Biometrics 41:1-9.
Schardein, J.L. 1985. Chemically Induced Birth Defects. New York: Marcel Dekker.
Shepard, T.H. 1983. Catalog of Teratogenic Agents, 4th ed. Baltimore, Md.: Johns Hopkins University Press.
Sherwin, S. 1992. No Longer Patient. Philadelphia: Temple University Press.
Takihi, N., Rosenkranz, H.S., Klopman, G., and Mattison, D.R. In press. Structural determinants of developmental toxicity. Submitted to the Conference Proceedings for Workshop on Quantitative Methods in Developmental Toxicology, National Research Council, Washington, D.C.
Wilson, J.G., and Fraser, F.C., eds. 1979. Handbook of Teratology. New York: Plenum.
World Health Organization Special Programme of Research, Development and Research Training in Human Reproduction and International Women's Health Coalition. 1991. Creating Common Ground: Women's Perspectives on the Selection and Introduction of Fertility Regulation Technologies. Geneva: World Health Organization.
Zar, J.H. 1984. Biostatistical Analysis. Englewood Cliffs, N.J.: Prentice-Hall.




























