
1
Introduction
In response to the frequent claims that women's health has been less well served than men's health by clinical research practices and policies, the scientific community has begun to devote increased attention to what has now become a central public policy concern. It is contended that women are participants in clinical studies less frequently than men and that this affects the study of women's health in two central ways: (1) women's experiences and manifestations of health problems common to both men and women are not addressed; and, (2) health conditions specific to women (e.g., menopause) are not adequately investigated. In the past three years, both the National Institutes of Health (NIH) and Food and Drug Administration (FDA), two pivotal players in the biomedical research process, have issued new policies that are changing the ways women are studied by the research community. In June, Congress passed the NIH Revitalization Act of 1993 (P.L. 103-43), which mandates the increased inclusion of women and racial and ethnic groups in clinical studies. Several other bills concerning this issue are currently pending before Congress.
In order to responsibly and effectively respond to these concerns and initiatives, the research community must examine the available empirical evidence; determine the nature, scope, and severity of the problem; and then recommend action based on this assessment. A comprehensive analysis of the problems also requires the identification of criteria or standards for judging the proper balance of involvement of women and men, and their
interests, in research. This report attempts to identify and elaborate the factors that must be part of systematic examination of such criteria.
MANDATE AND SCOPE OF STUDY
At the request of the NIH Office of Research on Women's Health, the Institute of Medicine established a Committee on the Ethical and Legal Issues Relating to the Inclusion of Women in Clinical Studies. The committee's report is intended to provide guidance to NIH and to institutional review boards, scientists, and others who design and monitor clinical studies regarding appropriate policies for the admission of women to these studies.
This committee was asked to examine only the ethical and legal issues related to the participation of women as participants in clinical studies. In its attempt to execute this charge, however, the committee found it necessary to look at the accumulated scientific evidence and prevailing methods of clinical investigation in order to understand the principles behind the design of clinical studies and the relevance of biological and physiological differences between the sexes. Without this foundation of understanding, the committee believed it could not build reasoned ethical and legal arguments to support recommendations concerning future research policy.
STRUCTURE OF REPORT
This report was prompted by concerns that there has been insufficient attention in biomedical research to women's health conditions, and that studies of conditions affecting both genders may not be generalizable to women if they rely primarily on male participants. Chapter 2 provides a brief review of the history and current status of clinical research on women and attempts to gauge current levels of participation. The issues raised by these concerns are complex and controversial, involving considerations of ethics, science, and law, all of which may pose potential constraints on the participation by women in clinical studies.
First, there are compelling ethical arguments for the involvement of women and women's health interests in clinical research. Principles of justice require that all people and social groups be treated fairly. In the context of research, fair treatment requires that men and women be allowed to share equally in both the burdens and benefits of participation in research. Chapter 3 examines these ethical arguments and develops the basic principles that will guide the rest of the report.
Second, an overreliance upon male participants in clinical studies may also make for bad science. Chapter 4 examines some of the known biological, behavioral, and social differences between the genders. These differences are real and significant, and they can affect responses to drugs and
other treatments, creating the potential for differential health outcomes based on these documented differences. Consequently, it is not clear that results obtained from male-only studies can always be reliably and safely generalized to female populations. This uncertainty has practical ramifications for clinicians, who may not be able to recommend therapies and prescribe drugs to women when the only data available for decisions are derived from men.
Third, the social and historical context within which research occurs exerts powerful and often hidden influences on the scientific process. Chapter 5 examines the biases that permeate society and may distort the vision of the scientific community, leading to unjust treatment of gender, racial, and ethnic groups.
Fourth, there are concerns about the legal implications of including women in clinical studies—or of excluding them. For example, researchers focus on the potential for harm to the offspring of participants. At the same time, drug manufacturers may be subject to liability if a drug causes injury in a population on which it has not been tested. Chapter 6 examines these concerns in detail.
All these concerns are heightened with regard to the participation of women in early-stage drug trials and the risks those trials may pose to reproduction and fetal development. The drug development process is described in a later section of this introduction. Chapter 7 examines the scientific, social, ethical, and legal issues raised by these risks to reproduction and offspring.
Finally, based on full consideration of all these factors, the committee makes specific recommendations for the fair and effective inclusion of women and women's health interests in clinical research. While these recommendations address legal and ethical issues, they necessarily involve scientific and social considerations as well. Chapter 8 also provides direct, practical guidance to NIH with regard to implementing its recommendations.
DEFINITIONS
In the process of its deliberations, the committee endeavored to be careful with terminology that might be vague or inappropriately value laden. A few definitional clarifications and ''sensitive" words are discussed below.
Women in this report refers to all-females of all racial, ethnic, and socioeconomic backgrounds throughout the adult life cycle, including pregnant and lactating women. Unless otherwise specified, the report addresses the issues in relation to women as a class or to identified subgroups of women, and not women as individuals. Where appropriate, however, the report distinguishes between such class or subgroup interests and individual interests (for example, when discussing the risks and benefits of participating in a particular study).
Our report construes the term clinical studies broadly. The definition includes studies that require the involvement of human subjects, as well as studies that review records or data already in existence, but excludes research on animals. Clinical studies include randomized clinical trials of treatments and preventive interventions, as well as observational, behavioral, psychosocial, and medical outcomes studies. The committee acknowledges that some of the more complex ethical and legal issues arise in the context of clinical trials of drugs, and the report distinguishes such trials from other studies when appropriate (see below and Chapter 4).
Two imprecise terms have been widely employed to refer to the perceived lower rates of participation by women in clinical studies: underrepresentation and exclusion. The report endeavors to limit the use of these terms, which have multiple meanings and thus are often unclear. Underrepresentation, for example, has been claimed both: (1) when the proportion of female (or male) participants is less than the proportion of females (or males) affected by the disease under study and (2) when the study design provides inadequate statistical power to detect gender differences. Underrepresentation can also include studies or situations in which men and women are included in proportion to their numbers in the population that experiences the disease, and in sufficient numbers to support statistically valid subgroup analyses, but such analyses are not done.
In contrast, the term exclusion is generally understood to apply when women (or men) are explicitly barred from participation because of an express stipulation in the study protocol. In reported study findings, however, it is often difficult to distinguish between explicit exclusion and the failure or inability to recruit sufficient participants or conduct adequate analysis. And regardless of how the terms underrepresentation and exclusion are expressed or understood, they fail to capture broader considerations that are also critical to this report, such as the failure to adequately address women's (or men's) health issues in the overall research agenda or the failure to investigate outcomes or processes of particular interest to one gender when that gender is included in a study population.
It is also useful to distinguish between sex and gender. In general, the term sex refers to physiological and anatomical differences between men and women. Gender refers to the entire set of behaviors and social roles common to a particular sex in a given society. In the interest of consistency, the committee chose to use one term when referring to both men and women, selecting gender for use throughout this report.
THE DRUG DEVELOPMENT PROCESS
Much of the concern that led to this study has centered on drug trials, particularly the timing of the exposure of fertile women to experimental
drugs that may do harm to fertility, pregnancy, and fetal development. (See Box 1-1 for a description of the drug development and approval process.) There is usually a substantial body of knowledge about both the disease being treated and the drug being tested by the time a drug enters clinical investigation (see Figure 1-1). For many diseases, the natural history may be reasonably well understood—knowledge of the natural history of a disease is essential to an understanding of whether an intervention (whether pharmaceutical, surgical, or other procedure) is likely to have a beneficial impact. Political factors, however, may modify the typical process of drug development. For example, some drugs thought to be effective in treating human immunodeficiency virus (HIV) may have an accelerated timeline for testing in humans.
For diseases for which beneficial drug therapies exist, there is also likely to be some understanding of how the drugs act to modify the progression or etiology of the disease. This knowledge can be used to suggest modifications in the structure of new drugs, alterations in the treatment schedule (timing or dosing, for example, with meals) for existing drugs, or realization of the potential value of existing drugs not previously used in the treatment of the disease.
By the time a new drug is available for testing in humans, a substantial body of information has already been collected. For example, the chemical will have been evaluated for its ability to alter the etiology and progression of the disease from both in vivo and in vitro testing systems. At the same time, additional data are being gathered concerning the drug's most likely side effects and toxicities. Methods for collecting such data include a series of tests in animals, which may range from days to months in length and involve additional reproductive and offspring studies. Examples of endpoints of these animal testing requirements are displayed in Table 1-1. In vitro testing is also performed to evaluate the potential mechanisms of toxicity and side effects.
These data are then gathered and used to evaluate the potential efficacy of the drug, as well as its likely side effects and toxicity. This information is almost always available before any generalized clinical testing is done in humans. The two exceptions are the completion of the second-generation animal reproductive exposure studies, which generally are not completed until the middle of Phase II (primary efficacy and safety human tests), and chronic toxicity (predominantly conducted to determine carcinogenicity). These studies, which require two years of testing in two animal species, often are not started until Phase II and are not completed until shortly before the filing of a New Drug Application (NDA).
If, after all of these evaluation activities, the drug's potential for benefit in treating disease appears to be greater than its side effects and toxicity, a clinical trial program is developed. Over the course of a clinical trial, atten-
|
BOX 1-1 THE DRUG DEVELOPMENT AND APPROVAL PROCESS It takes 12 years on average for an experimental drug to travel from lab to medicine chest. Only five in 5,000 compounds that enter preclinical testing make it to human testing. One of these five tested in people is approved. 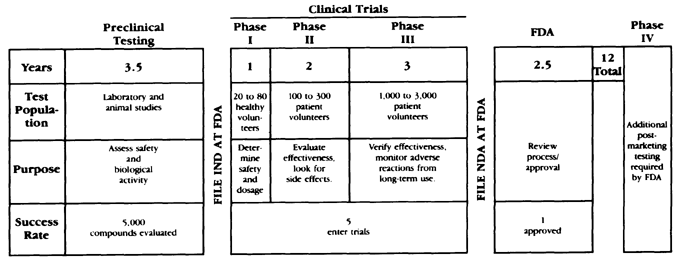 SOURCE: Wierenga and Eaton (1993). |
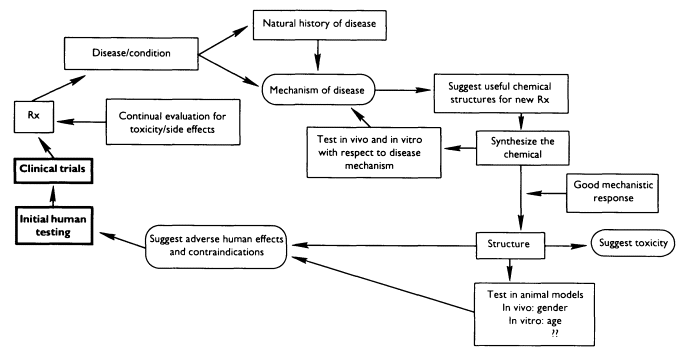
TABLE 1-1 Examples of End Points for Various Toxicity Studies
|
Study |
Endpoints |
|
Developmental toxicity |
Fetus: mortality, growth retardation, skeletal variations, gross external malformations, soft tissue/internal organ defects |
|
|
Female parent: general toxicity |
|
Reproductive toxicity |
Male parent: general toxicity, effects on fertility, reproductive organ changes |
|
|
Offspring: effects on viability, sex ratio, growth, behavior |
|
Carcinogenicity |
Tumor development and general toxicity |
|
Neurotoxicity |
Behavior, function, and motor activity deficits; microscopic nervous tissue changes |
|
Mutagenicity |
Heritable lesions leading to altered phenotypes |
|
SOURCE: EPA, 1984. |
|
tion is directed toward the identification of dosage range, toxicity, and side effects in treated individuals compared with individuals who receive a standard drug or a placebo, as well as the drug's benefit in treating the disease.
Because animal testing may not identify all types of human toxicity, initial testing of the drug in humans is generally performed under close observation in a series of trials (see Box 1-1). The initial clinical trials (Phase I) are conducted primarily to determine appropriate adult human dosage. Factors evaluated during these initial clinical trials include rate and extent of absorption by the routes chosen for administration. These tests generally can be conducted on a small number of healthy volunteers, although in some instances the testing may be conducted on individuals with the disease targeted for treatment. In Phase II clinical trials, the safety and efficacy of the drug is studied in a small number (on the order of 100) of individuals and the effectiveness of the drug in treating the disease (or diseases) is evaluated; the initial evaluation of toxicity, side effects, and contraindications is extended. It is not uncommon in practice for Phase I and Phase II to be combined.
If the effectiveness of the drug still appears promising and observed side effects and toxicity acceptable, it is studied in a larger number of patients (Phase III). During this phase, several thousand individuals with the appropriate diseases are studied during treatment to verify effectiveness, toxicity, and side effects. If a drug has progressed through all three initial phases of clinical trials and appears to have therapeutic benefit, all of the collected data are gathered, summarized, and included in the NDA, which is submitted to FDA. If, after review, FDA approves the NDA, the pharmaceutical company may choose to market the drug. Even after marketing begins,
however, additional information on the safety and efficacy of the drug will be gathered as physicians, the pharmaceutical industry, and FDA continue to monitor for toxicity, side effects, further benefits, and other indications. Data obtained are shared among health professionals through mailings or publications, and indeed may cause the modification of product data and use where deemed essential.
The formal postmarketing study of a drug may also take place in what is known as a Phase IV study. Phase IV studies are not mandated in FDA regulations but are discussed in FDA guidelines. A Phase IV study may be a condition of FDA approval to market a drug if the uncertainty about a drug's safety or efficacy does not warrant delaying its release on the market, or it may be initiated by a pharmaceutical company to further substantiate drug safety and efficacy and to support marketing claims (IOM, 1985). Phase IV studies may be controlled or observational.
REFERENCES
EPA (U.S. Environmental Protection Agency). 1984. Guidelines Subdivision F—Hazard Evaluation: Human and Domestic Animals, revised ed. Washington, D.C.: EPA.
IOM (Institute of Medicine). 1985. Assessing Medical Technologies. Washington, D.C.: National Academy Press.
Wierenga, D.E., and Eaton, C.R. 1993. The drug development and approval process. In: In Development: New Medicines for Older Americans. Washington, D.C.: Pharmaceutical Manufacturers Association.








