
2
Radiation Biology and Carcinogenesis
INTRODUCTION
The biologic effects of ![]() particles are at the core of our understanding of radon-induced lung cancer and risk models should appropriately reflect this understanding. Risk estimates for lung cancer related to radon in homes can be derived by at least two quite different approaches. In the so-called epidemiologic approach, lung-cancer data on underground miners are extrapolated from the high radon concentrations characteristic of mines to the much lower concentrations in homes. Differences in doses and dose rates can be addressed in laboratory experiments in radiobiology. Still, the data available for this purpose are limited and risk estimates are subject to major uncertainties.
particles are at the core of our understanding of radon-induced lung cancer and risk models should appropriately reflect this understanding. Risk estimates for lung cancer related to radon in homes can be derived by at least two quite different approaches. In the so-called epidemiologic approach, lung-cancer data on underground miners are extrapolated from the high radon concentrations characteristic of mines to the much lower concentrations in homes. Differences in doses and dose rates can be addressed in laboratory experiments in radiobiology. Still, the data available for this purpose are limited and risk estimates are subject to major uncertainties.
In the dosimetric approach, doses to the bronchial epithelium are estimated, and the long-term risks of lung cancer in Japanese atomic-bomb survivors are used to estimate lung-cancer risks from radon exposure. Two scaling factors are required:
-
A dose-rate correction, because the Japanese received a single
-
acute exposure, whereas radon exposure in homes occurs at low dose rates.
-
A factor that represents the change in the nature of the radiation, because the Japanese were exposed largely to sparsely ionizing gamma () rays, whereas radon progeny emit densely ionizing particles.
The dosimetric approach also is subject to major uncertainties, and neither approach is a priori better.
This section on the radiation biology of ![]() particles summarizes basic concepts in the field, focusing on how radiation biology can contribute to the assessment of radon risks, namely by using doserate corrections and the radiation quality factor for radon-progeny
particles summarizes basic concepts in the field, focusing on how radiation biology can contribute to the assessment of radon risks, namely by using doserate corrections and the radiation quality factor for radon-progeny ![]() particles. It also presents selected examples where recent information on mechanisms, oncogenes and tumor-suppressor genes, and possible biologic markers of
particles. It also presents selected examples where recent information on mechanisms, oncogenes and tumor-suppressor genes, and possible biologic markers of ![]() -particle exposures will be reviewed in depth in a Phase II study.
-particle exposures will be reviewed in depth in a Phase II study.
RANGE AND TRACK STRUCTURE OF  PARTICLES EMITTED BY RADON PROGENY
PARTICLES EMITTED BY RADON PROGENY
The decay series for radon involves the emission of two principal ![]() particles with energies of 6 and 7.7 MeV. The passage of
particles with energies of 6 and 7.7 MeV. The passage of ![]() particles through tissue produces essentially linear tracks that are dense columns of ionizations, which give rise to the locally high radiation dose deposited. The ranges of the two principal
particles through tissue produces essentially linear tracks that are dense columns of ionizations, which give rise to the locally high radiation dose deposited. The ranges of the two principal ![]() particles in tissue are 48 and 71
particles in tissue are 48 and 71 ![]() m. There is some debate over which respiratory epithelial cells are affected in the radiation induction of lung cancer by radon, but it is generally agreed that the target cells (whether basal or serous) are within the range of
m. There is some debate over which respiratory epithelial cells are affected in the radiation induction of lung cancer by radon, but it is generally agreed that the target cells (whether basal or serous) are within the range of ![]() particles depos-
particles depos-
ited on the surface of the bronchial epithelium, as illustrated in Figure 1. These target cells are likely to be close to the end of the ![]() -particle tracks where the density of ionization from the radiation along the tracks is high but changing rapidly (i.e., the radiation has a rapidly changing linear energy transfer, or LET). Figure 2 shows how dose decreases as lineal energy (yD), a quantity similar to LET, increases along the track of an a particle until near the end of its range, when yD declines sharply.
-particle tracks where the density of ionization from the radiation along the tracks is high but changing rapidly (i.e., the radiation has a rapidly changing linear energy transfer, or LET). Figure 2 shows how dose decreases as lineal energy (yD), a quantity similar to LET, increases along the track of an a particle until near the end of its range, when yD declines sharply.
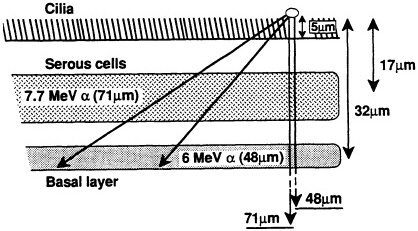
FIGURE 1. Ranges of two ![]() particles emitted by radon progeny deposited on lung surface and average depths of serous and basal cells in human lung, according to study of several hundred "normal" lung sections from Pathology Department at Columbia Presbyterian Medical Center. (Based on data collected by Charles Geard and David Brenner; reproduced from Hall, 1992, with permission of the publisher.)
particles emitted by radon progeny deposited on lung surface and average depths of serous and basal cells in human lung, according to study of several hundred "normal" lung sections from Pathology Department at Columbia Presbyterian Medical Center. (Based on data collected by Charles Geard and David Brenner; reproduced from Hall, 1992, with permission of the publisher.)
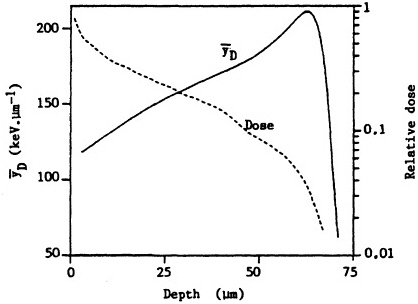
FIGURE 2. Change in dose and dose-averaged lineal energy yD (1-µm site size) with depth for ![]() particles simulating those emitted by radon progeny. (Reproduced from Brenner, 1990, with permission of the publisher.)
particles simulating those emitted by radon progeny. (Reproduced from Brenner, 1990, with permission of the publisher.)
RADIATION BIOLOGY OF -PARTICLE IRRADIATION
When tissues are exposed to ![]() particles, cells can be killed or sterilized, suffer mutations, or be transformed to a malignant state. Cellular models using these end points can address subjects-such as the slope of the dose-response relationship, the effects of dose
particles, cells can be killed or sterilized, suffer mutations, or be transformed to a malignant state. Cellular models using these end points can address subjects-such as the slope of the dose-response relationship, the effects of dose
rate, and the effects of radiation quality-that would be more difficult to address through animal experiments and impractical through human epidemiologic studies.
Cell Lethaity
A wide variety of rodent and human cell types have been irradiated with ![]() particles, and cell-survival curves have been generated (Barendsen et al., 1960; Thacker et al., 1982; Robertson et al., 1983; Hei et al., 1988b, 1993; Tsuboi et al., 1992). In all cases, as the dose increases, the fraction of cells surviving decreases approximately as an exponential function of dose. Figure 3 shows survival of mouse C3H10T1/2 cells irradiated in vitro. The shape of the survival curves implies single-hit kinetics-i.e., the cell is killed by a single
particles, and cell-survival curves have been generated (Barendsen et al., 1960; Thacker et al., 1982; Robertson et al., 1983; Hei et al., 1988b, 1993; Tsuboi et al., 1992). In all cases, as the dose increases, the fraction of cells surviving decreases approximately as an exponential function of dose. Figure 3 shows survival of mouse C3H10T1/2 cells irradiated in vitro. The shape of the survival curves implies single-hit kinetics-i.e., the cell is killed by a single ![]() particle- but at the mean lethal dose, the mean number of particles traversing the nucleus is often more than 1 and is reportedly as high as 13 (Lloyd et al., 1979). It is generally agreed that the high relative biologic effectiveness (RBE) of
particle- but at the mean lethal dose, the mean number of particles traversing the nucleus is often more than 1 and is reportedly as high as 13 (Lloyd et al., 1979). It is generally agreed that the high relative biologic effectiveness (RBE) of ![]() particles, compared with x rays or gamma rays, in killing cells is due to the
particles, compared with x rays or gamma rays, in killing cells is due to the ![]() particles' ability to induce irreparable DNA damage, inasmuch as these densely ionizing particles deposit a large amount of energy and efficiently induce localized multiple lesions (Ward, 1985; Goodhead, 1989).
particles' ability to induce irreparable DNA damage, inasmuch as these densely ionizing particles deposit a large amount of energy and efficiently induce localized multiple lesions (Ward, 1985; Goodhead, 1989).
Mutations in Cultured Cells
The mutagenic potential of ![]() particles with energies similar to those characteristic of radon progeny has been measured at several
particles with energies similar to those characteristic of radon progeny has been measured at several
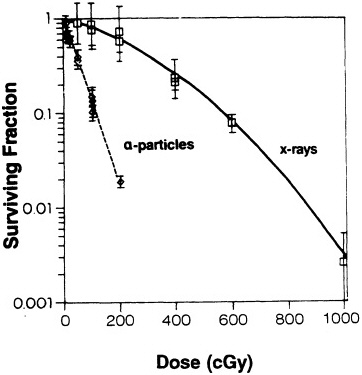
FIGURE 3. Survival of C3H10T1/2 cells irradiated with either x rays or ![]() particles. (Reproduced from Hall and Hei, 1985, with permission of the publisher.)
particles. (Reproduced from Hall and Hei, 1985, with permission of the publisher.)
geneti loci in a variety of human and rodent cell systems (Cox et al., 1977; Cox and Masson, 1979; Thacker, 1986; Hei et al.,1988c, 1994; Evans, 1991; Tsuboi et al., 1992; Jostes et al., 1994). In general, the mutagenicity of ![]() particles depends on both dose and LET. Figure 4 shows mutation induction at the HGPRT locus. As
particles depends on both dose and LET. Figure 4 shows mutation induction at the HGPRT locus. As
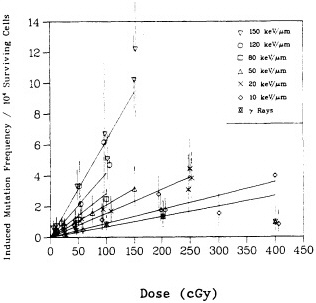
FIGURE 4. Mutation induction at HGPRT locus in primary human fibroblasts irradiated with gamma rays and charged particles of various LET. Mutations were expressed as number of mutants per 10,000 survivors. Data were analyzed according to linear quadratic response, and curves represent best fit to data by method of maximum likelihood. Bars represent 95 % confidence intervals. (Reproduced from Hei et al., 1988a, with permission of the publisher.)
shown in Figure 5, the RBE for mutation reaches a peak at an LET of about 100-200 keV/µm, similar to that for cell lethality, and the RBE at a given LET appears to be larger for mutational end points than for cell killing (Cox and Masson, 1979).
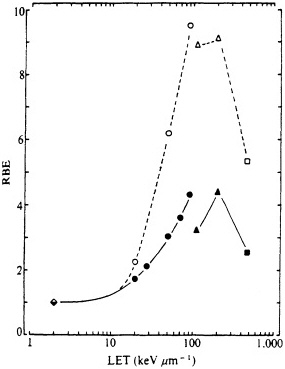
FIGURE 5. RBE-LET relationships for Chinese hamster V79 cells. Standard radiation was cobalt-60 ![]() rays, which was compared with helium, boron, and nitrogen ions (represented by circles, triangles, and squares respectively). Closed symbols represent cell lethality; open symbols, mutation at HGPRT locus. (Reproduced from Cox and Masson, 1979, with permission of the publishers.)
rays, which was compared with helium, boron, and nitrogen ions (represented by circles, triangles, and squares respectively). Closed symbols represent cell lethality; open symbols, mutation at HGPRT locus. (Reproduced from Cox and Masson, 1979, with permission of the publishers.)
Oncogenic Transformation In Vitro
Radiation has been used as both an initiator and a complete
carcinogen in rodent-cell models. In particular, a single exposure to either high- or low-LET radiation has been used successfully to transform a variety of rodent cells in culture (Borek and Hall, 1973; Miller and Hall, 1978; Elkind and Han, 1979; Lloyd et al., 1979; Balcer-Kubiczek and Harrison, 1983; Robertson et al., 1983; Watanabe et al., 1984: Hall and Hei, 1985, 1990; Yang et al., 1985; Hei et al., 1988a). Transformation induction by ![]() particles depends on both dose and LET (Yang et al., 1985, 1989; Hei et al., 1988a; Miller et al., 1993). A very large set of data has been generated by Miller and colleagues (Miller, personal communication) and is illustrated in Figure 6. The transformation incidence induced by
particles depends on both dose and LET (Yang et al., 1985, 1989; Hei et al., 1988a; Miller et al., 1993). A very large set of data has been generated by Miller and colleagues (Miller, personal communication) and is illustrated in Figure 6. The transformation incidence induced by ![]() particles can be modulated by various chemical and environmental agents, such as cigarette smoke, asbestos fibers, and 12-o-tetradecanoylphorbol-13-acetate (TPA) (Kennedy and Little, 1980; Han and Elkind, 1982; Hall et al., 1989; Piao and Hei, 1993). For example, Figure 7 shows that TPA substantially enhances the incidence of transformation resulting from low- or high-LET radiation. The
particles can be modulated by various chemical and environmental agents, such as cigarette smoke, asbestos fibers, and 12-o-tetradecanoylphorbol-13-acetate (TPA) (Kennedy and Little, 1980; Han and Elkind, 1982; Hall et al., 1989; Piao and Hei, 1993). For example, Figure 7 shows that TPA substantially enhances the incidence of transformation resulting from low- or high-LET radiation. The ![]() -particle-induced damage that results in oncogenic transformation is apparently not subject to repair during post exposure incubation of cells (Robertson et al., 1983).
-particle-induced damage that results in oncogenic transformation is apparently not subject to repair during post exposure incubation of cells (Robertson et al., 1983).
In contrast with rodent cells, which are readily transformable by ![]() particles, human cells in culture are refractory to malignant transformation by either radiation or chemicals. Also, unlike rodent cells, normal human cells in culture rarely undergo spontaneous transformation (Harris, 1987; Rhim, 1992). Most transformation studies with cells of human origin have involved either a retrovirus or a chemical carcinogen as the transforming agent (for review, see Rhim and Dritschilo, 1991). Of the few transformation studies involving ionizing radiation, essentially all used multiple high doses either to immortalize the cells (Namba et al., 1986) or to transform
particles, human cells in culture are refractory to malignant transformation by either radiation or chemicals. Also, unlike rodent cells, normal human cells in culture rarely undergo spontaneous transformation (Harris, 1987; Rhim, 1992). Most transformation studies with cells of human origin have involved either a retrovirus or a chemical carcinogen as the transforming agent (for review, see Rhim and Dritschilo, 1991). Of the few transformation studies involving ionizing radiation, essentially all used multiple high doses either to immortalize the cells (Namba et al., 1986) or to transform
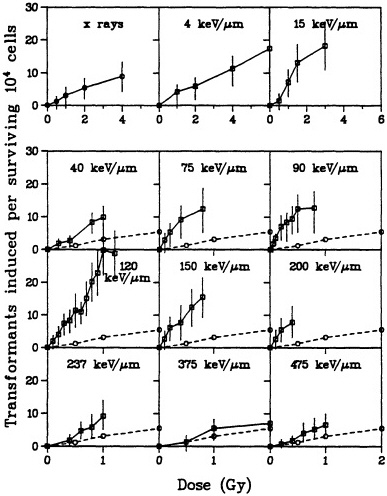
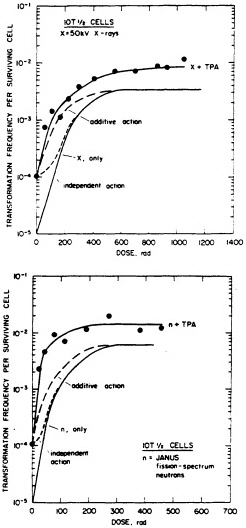
FIGURE 7. Transformation in C3H10T1/2 cells as a function of dose (neutrons and x rays) and TPA. (Reproduced from Han and Elkind, 1982, with permission of the publisher.)
the already immortalized cells after SV40 treatment (Thraves et al., 1990; Yang et al., 1991). Hei and colleagues (Hei et al., 1994) recently used human papillomavirus-immortalized human bronchial-
epithelial cells and showed for the first time that a single 30-cGy dose of ![]() particles can transform these cells.
particles can transform these cells.
Preliminary transformation data have also been reported on SV40-immortalized human keratinocytes irradiated with 10 cGy of ![]() particles (Martin et al., 1994) or fission neutrons (Thraves et al., 1993). No dose-response data on the transformation of cells of human origin are available.
particles (Martin et al., 1994) or fission neutrons (Thraves et al., 1993). No dose-response data on the transformation of cells of human origin are available.
Comparison of Effects In Vivo and In Vitro
An important step in the dosimetric approach to risk assessment is to convert exposure to radon progeny in working-level months (WLM) to absorbed dose in the bronchial epithelium. Production of micronuclei in ''in vivo" and "in vitro" exposure studies has been used as an index of the relationship between exposure and dose.
In a recent study reported by Khan et al. (in press), male rats were exposed to various numbers of WLM from radon progeny by inhalation. After sacrifice, the cells (deep-lung fibroblasts) were grown in culture and the incidence of micronuclei was determined.
In parallel experiments, lung fibroblasts in vitro were exposed to known radiation doses from radon progeny and the incidence of micronuclei was determined. Comparing ![]() -particle dose-response relationships in nonproliferating cells in vitro with the exposure response relationships in cells exposed by inhalation led to the estimate that a 1-WLM exposure in vivo caused the same amount of cytogenetic damage as 0.78 milligray (mGy) in vitro. Although these experiments used rat fibroblasts, not human epithelial cells, the results are of considerable interest. This direct comparison of
-particle dose-response relationships in nonproliferating cells in vitro with the exposure response relationships in cells exposed by inhalation led to the estimate that a 1-WLM exposure in vivo caused the same amount of cytogenetic damage as 0.78 milligray (mGy) in vitro. Although these experiments used rat fibroblasts, not human epithelial cells, the results are of considerable interest. This direct comparison of ![]() -particle exposure in vivo and in vitro must be viewed in the light of a comparison of uranium miners with atomic bomb survivors, though as noted, the latter had only minimal exposure to
-particle exposure in vivo and in vitro must be viewed in the light of a comparison of uranium miners with atomic bomb survivors, though as noted, the latter had only minimal exposure to ![]() particles.
particles.
Carcinogenesis in Laboratory Animals
The BEIR IV report summarized studies of experimental animals exposed to radon, including studies of the inverse dose-rate effect (National Research Council, 1988). Since the BEIR IV committee completed its work, additional analyses of data from experiments performed at Battelle, Pacific Northwest Laboratories, have taken account of competing risks and confirmed that, after exposures above 600 WLM, lung cancer in rats shows a significant inverse dose-rate effect. A dose-rate effect is termed inverse if higher dose rates produce less effect per unit dose than lower dose rates. In this case, exposures to the same total dose but spread over a longer period result in higher lung-cancer rates than acute exposures. Furthermore, the analyses suggest that the magnitude of the inverse dose-rate effect varies directly with total exposure and there is no
evidence of an effect at 320 WLM, the lowest exposure evaluated in these analyses (Gilbert, 1989; Gilbert and Cross, 1989).
However, a recent experiment conducted in France (Morlier et al., 1992) provides contrary evidence: that reduction in dose rate can reduce, rather than increase, the effect. The lung-cancer incidence in a group of rats that received a cumulative exposure of 25 WLM, over a period of 18 months, was similar to the incidence in control rats that were not exposed to radon progeny but significantly lower than the incidence observed in rats that received the same cumulative exposure over a period of 4-6 months. Thus, the influence of dose rate as assessed in experimental studies needs further investigation. However, the biophysical model described later (and illustrated in Figure 9 on page 36) appears to reconcile the apparent discrepancies.
Data from additional experiments conducted at Battelle, Pacific Northwest Laboratories, might also be available for consideration by a Phase II committee. These include results of experiments with lower total exposures and lower exposure rates than those used previously and results of initiation-promotion-initiation (IPI) experiments that address the combined effects of exposure to cigarette smoke and radon in various sequences. The IPI experiments show that cigarette-smoking promotes the radon-induced preneoplastic lesion adenomatosis. The duration of the smoke exposures, however, was insufficient to promote the induction of tumors, as occurred in earlier experiments conducted in France (F.T. Cross, 1992, personal communication).
RBE VERSUS LET
It was pointed out earlier that one of the most important ways in
which radiobiologic studies can affect radon risk estimates is to help to determine the quality factor (Q) for the densely ionizing ![]() particles emitted by radon progeny.
particles emitted by radon progeny.
Numerous past studies, with a variety of biologic systems from cell lethality to mutation to oncogenic transformation, have confirmed the general shape of the RBE-LET curve, namely, an increase of RBE with increasing LET up to a maximum at an LET of about 100-200 keV/µm, followed by a sharp decline at higher LET (as shown in Figure 5). Inasmuch as survival after ![]() -particle irradiation generally varies exponentially with dose (whereas the curves for survival after exposure to x rays or
-particle irradiation generally varies exponentially with dose (whereas the curves for survival after exposure to x rays or ![]() rays have a shape at low doses that is not exponential), RBE must vary inversely with dose; i.e., it is higher at lower doses and has its highest equivalent value (RBE) at the lowest doses, where the response to exposure to either kind of radiation is linear.
rays have a shape at low doses that is not exponential), RBE must vary inversely with dose; i.e., it is higher at lower doses and has its highest equivalent value (RBE) at the lowest doses, where the response to exposure to either kind of radiation is linear.
Another important subject is the dependence of RBE (at a given dose) on the type of end point. This is relevant to risk assessment using the dosimetric approach. In particular, in comparisons of high- and low-LET radiation, it appears to be a general finding that the RBEs are larger (at a given dose) for oncogenic transformation and for mutagenesis than for cell lethality (Borek, et al., 1978; Han and Elkind, 1979; Miller et al., 1989). Transformation incidence per surviving cell rises rapidly as a function of dose, but reaches a plateau at about 400 cGy of x rays or 100 cGy of ![]() particles. If, however, cell killing (which plays a larger part at higher doses) is factored in, the number of transformations per initial cell at risk rises with dose at first, reaches a peak, and then falls rapidly, paralleling the cell-survival curve (Han et al., 1980). The number of transformations per initial cell is probably a more relevant quantity for extrapolation to risk of carcinogenesis in a whole
particles. If, however, cell killing (which plays a larger part at higher doses) is factored in, the number of transformations per initial cell at risk rises with dose at first, reaches a peak, and then falls rapidly, paralleling the cell-survival curve (Han et al., 1980). The number of transformations per initial cell is probably a more relevant quantity for extrapolation to risk of carcinogenesis in a whole
organism. The peak transformation frequency moves to lower doses as the LET of the radiation increases and the peak occurs at about 300 cGy of x rays and 30 cGy of ![]() particles. The peak also reaches higher values for high-LET radiation: e.g., 30 cGy of
particles. The peak also reaches higher values for high-LET radiation: e.g., 30 cGy of ![]() particles results in a higher rate of transformations per initial cell at risk than is induced by any dose of x rays.
particles results in a higher rate of transformations per initial cell at risk than is induced by any dose of x rays.
Brenner (1990) has used the transformation incidence in C3H10T1/2 cells irradiated with ![]() particles of various LET in the track-segment mode to estimate the RBE as a function of range of
particles of various LET in the track-segment mode to estimate the RBE as a function of range of ![]() particles simulating those emitted by radon progeny (see Figure 8). The results should be compared with the quality factors derived from data on chromosomal aberrations in peripheral lymphocytes by the International Commission on Radiation Units and Measurements (ICRU). The "effective Q" depends heavily on the saturation characteristics at high LET of the biologic system used. The transformation data imply a much-reduced effectiveness of
particles simulating those emitted by radon progeny (see Figure 8). The results should be compared with the quality factors derived from data on chromosomal aberrations in peripheral lymphocytes by the International Commission on Radiation Units and Measurements (ICRU). The "effective Q" depends heavily on the saturation characteristics at high LET of the biologic system used. The transformation data imply a much-reduced effectiveness of ![]() particles near the end of their range and a lower overall effective Q over the range of the
particles near the end of their range and a lower overall effective Q over the range of the ![]() particles compared with those reported previously (International Commission on Radiation Units and Measurements, 1986).
particles compared with those reported previously (International Commission on Radiation Units and Measurements, 1986).
DOSE-RATE EFFECTS AND IMPLICATIONS FOR RISK ESTIMATES
For low-LET radiation, a unit of dose is usually more biologically effective at a high dose rate than at a low dose rate. This is sometimes expressed as a dose and dose-rate effectiveness factor (DDREF) with which high-dose-rate data are extrapolated to low dose rates. However, a considerable body of data shows that with
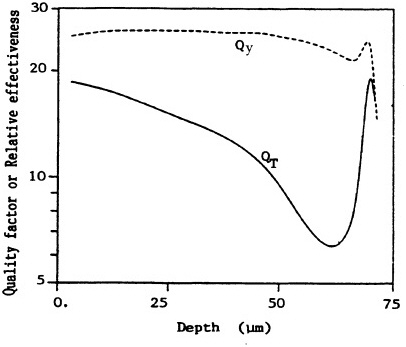
FIGURE 8. Variation in effectiveness (relative to 250-kVp x rays) as a function of depth, derived from the transformation data on C3H10T1/2 cells (QT) and from ICRU 40 (ICRU, 1986) function (Qy). Averaged over whole epithelium (10-72 µm deep), dose mean effectiveness is <QT> = 15 and <Qy> = 26. (Reproduced from Brenner, 1990, with permission of the publisher.)
oncogenic transformation in C3H10T1/2 cells as an end point, an inverse dose-rate effect is apparent for high-LET radiation such as fission-spectrum neutrons. The effect appears to be confined to particular radiobiologic end points; e.g., for clonogenic survival, the biologic effectiveness at medium or high LET is virtually independent of dose rate. It should be pointed out that published
data are equivocal; some investigators have found an inverse doserate effect, and some have not (Hill et al., 1984; Miller et al., 1988; Hieber et al., 1989; Bettega et al., 1992; Miller et al., 1993).
A mechanistic model has been proposed to reconcile the data that support the inverse dose-rate effect. It is based on the presence of a "window" of sensitivity to transformation at some point in the cell cycle (Rossi and Kellerer, 1986; Brenner and Hall, 1990; Elkind, 1991). Together, the model and the relevant data suggest that the magnitude of the inverse dose-rate effect varies in a complex fashion with dose, dose rate, and radiation quality. It can be argued on biophysical grounds that when the exposure rate and total exposure are so low that multiple traversals of target cells are rare, then the inverse dose-rate effect must disappear. That argument is relevant to the relationship of radon progeny exposure and lung cancer; epidemiologic studies have shown that lung-cancer risk from radon-progeny exposure depends on the exposure rate and increases as the exposure rate decreases. Figure 9 shows the result of calculations from the biophysical model referred to above-a surface that describes the enhancement in risk (relative to an acute exposure) due to dose protraction. As expected, the protraction effect increases as the exposure rate decreases, but decreases as the exposure itself decreases. The model implies that for domestic radon exposures, where an average of less than one particle traverses a given cell in the bronchial epithelium, protraction should have little or no effect on risk. However, in the miner studies, the higher exposure rates are more than compensated for by the higher exposures, the result being a significant protraction-related enhancement or a reduction due to shortening of exposure time.
The Phase II committee will need to select a model that controls for time since exposure and dose rate in an effort to determine that the dose-rate effect is really a cause-effect relationship. It should
be recognized that the risk of a point exposure is zero for a time after exposure, then moves gradually to a higher level, and remains elevated for as long as the subjects are followed. Thus 100 WLM 30 years ago has a bigger effect today than the same dose 10 years ago. More rapid accumulation of a fixed total dose may be correlated with a shorter time since initiation of exposure, so that an observed smaller effect is a result of shorter time since exposure rather than higher dose rate. Questions might arise how secular trends in exposure interact with secular trends in lung cancer. It is at least plausible that higher doses (and shorter times to a fixed total dose) tend to occur in early years, so that a larger part of the follow-up time was in years when rates were lower.
MECHANISMS OF CARCINOGENESIS
Recent years have seen an explosion in new information on the molecular genetics of cancer. In the context of radon risk estimates, it is a tantalizing possibility that the densely ionizing particles released by radon progeny produce characteristic genetic changes that can be recognized at the molecular level and would therefore constitute a "signature" or "footprint" of radon exposure. The possibility is being explored through the study of oncogenes and tumor-suppressor genes in tumors from uranium miners and in particle-induced tumors in experimental animals.
The identification and understanding of human oncogenes make it possible to understand why agents as diverse as retroviruses, ionizing radiation, and chemicals can result in tumors that are indistinguishable one from another (Bishop, 1983; Bishop and Varmus, 1984). Retroviruses insert a gene into the cell, and radiation and chemicals produce a mutation in a gene that is already in the cell. Today, about 100 oncogenes have been identified as
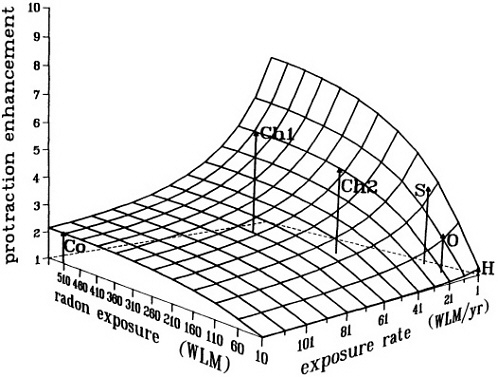
FIGURE 9. Calculated enhancement in risk (relative to an acute exposure) due to protraction, illustrating the interplay between radon exposure and exposure rate. This surface does not represent a dose-risk relation, but only the enhancement in risk over what would be expected from an acute exposure. Arrows indicate the positions in mean exposure/exposure rate space of various miner studies; calculated relative enhancements for the various miner cohorts, as shown by the heights of the arrows, do reflect the ordering of the relative risk estimated in these studies. Surface and points are from a preliminary calculation provided by D. Brenner, with a "sensitive window" of 20% of the total cell cycle, and with exposure-to-bronchial-dose conversion factors from NRC (1991). No fitting to experimental data was attempted in this exploratory calculation.
Co: Colorado miners (mean WLM; WLM/yr; calculated enhancement = 520; 120; 2.1, respectively).
Chl: Chinese tin miners (Lubin et al. 1990: mean WLM; WLM/yr; calculated enhancement = 507; 21; 5.3, respectively).
Ch2: Chinese tin miners (Xuan et al. 1993: mean WLM; WLM/yr; calculated enhancement = 275; 21; 4.4, respectively).
S: Swedish iron workers (mean WLM; WLM/yr; calculated enhancement = 120; 5; 3.9, respectively).
O: Ontario miners (mean WLM; WLM/yr; calculated enhancement = 10; 65; 2.4, respectively).
H: Typical domestic exposures.
associated with human cancer, of which more than 80% belong to the ras family. However, activated oncogenes are associated with only a small proportion (10-15 %) of human cancers and tend to be found more commonly in leukemias and lymphomas than in solid tumors. Oncogenes have been shown to be activated by point mutations, such as in ras (Bos, 1988); by deletions, such as infos; by reciprocal translocations, such as in myc (Dalla-Favera et al., 1983); and by gene amplification, such as in myc (Brodeur et al., 1984).
It was discovered in the 1970s that the tumorigenicity of tumor cells could be suppressed by hybridization with normal cells (Harris, 1971; Stanbridge, 1976). For example, the fusion of a normal human fibroblast with a HeLa cell suppressed the expression of the malignant phenotype of the HeLa cell. Those findings, which preceded the notion of oncogenes by several years, led to the hypothesis that normal cells contain a gene or genes that can suppress the neoplastic potential of tumor cells. The putative tumor suppressor genes have been mapped to specific chromosomes, such as chromosome 11 in the example quoted, by analyzing which chromosomes were lost from the hybrids that re-express tumorigenicity (Stanbridge, 1976). The importance of tumor-suppressor genes became evident in the work of Knudson (1971) with retinoblastoma, which occurs in both familial and sporadic forms. Knudson elaborated this two-hit hypothesis in the early 1970s, and by the mid-1970s, the location of the gene was identified on chromosome 13 (Cavenee et al., 1985). In the 1980s the Rb gene was cloned and sequenced (Lee et al., 1987). The Rb gene is associated in 100% of cases with retinoblastoma, and survivors of childhood retinoblastoma are also predisposed to bone cancers. Mutations of the Rb gene are also sometimes associated with various other tumors, such as small-cell lung cancer, bladder cancer, and mam-
mary cancer. An obvious mechanism for knocking out a suppressor gene is radiation-induced deletion. A tumor suppressor gene acts in a recessive way, so the deletion would have to occur in both chromosomes of a pair, an event of very low frequency. In practice, it is often found that the loss of a pair of suppressor genes occurs by the process of somatic homozygosity (Cavanee, 1989): one chromosome of a pair is lost, a deletion occurs in the other, and then the chromosome with the deletion is replicated. The cells in the tumor then have two chromosomes that originated from the same parent. That mechanism has been shown in retinoblastoma, small-cell lung cancer, and glioblastoma. Another possible mechanism is illustrated by familial heterozygosity in which subjects are at increased risk because they harbor one active and one inactive gene. In this case, the loss or mutation of the normal allele on chromosome 13 by nondisjunction, deletion, genetic recombination, or local mutation leads to production of cancer (Knudson, 1985). At least six tumor-suppressor genes and their locations and functions have been identified. The two most common and most intensively studied are the Rb gene and the p53 gene; both are involved in the arrest of cells in the G1 phase of the cell cycle and in tumor differentiation (DeCaprio et al., 1992).
MARKERS OF EXPOSURE
One of the workshops organized by the committee included a summary of studies of molecular markers of radon exposure. Mutations in the p53 tumor suppressor gene are common in many human cancers, including cancers of the colon, skin, breast, and lung. These mutations are not random, but clustered in the central portion of the gene, exons 5-9. Furthermore, the mutations caused
by exposure to tobacco smoke are characterized by G:C to T:A transversions in the coding strand, and lung cancers have ''hot spots" (regions of DNA with high mutation frequency) for these mutations. The mutation spectra of p53 and K-ras in lung cancers of uranium miners exposed to radon at high levels have been studied. The results in 19 miners, 18 of whom also smoked, showed no codon 12-13 mutations in K-ras (K-ras mutations are very common in lung cancers of cigarette smokers), but nine mutations in p53 were detected in seven of these patients. Twenty-two percent of the mutations were deletions. None of the mutations was a G:C to A:T transversion in the coding strand. Five missense mutations and one nonsense mutation were detected. A silent mutation was also detected. In addition to point mutations, two of the patients also had a small deletion in p53, but p53 deletions are not common in lung cancer. All these mutations were clustered between codons 146-161 and codons 195-208, which could be interpreted as "hot spots" for radon-induced mutations (Vähäkangas et al., 1992). In another study, 16 of 52 large-cell and squamous-cell cancers from uranium miners contained the same AGG to ATG transversion at codon 249, including cancers from three of five miners who had never smoked (Taylor et al., 1994).
Those data contrast with the findings in Japanese atomic-bomb survivors, who were exposed principally to low-LET radiation (Takeshima et al., 1993). Of 17 Japanese with adenocarcinomas, nine had been exposed to radiation, and eight were controls. There were four point mutations in p53 in the exposed and four in the nonexposed; i.e., the mutation was not characteristic of irradiation. A final conclusion must depend on a larger database, but preliminary data suggest that a signature of densely ionizing particles in p53 might exist, whereas it might not for low-LET radiation. However, in a recent study comparing the pathology of lung cancers
in the uranium miners and the atomic-bomb survivors, it was found that both had increased numbers of small cell carcinomas in proportion to the dose and few adenocarcinomas (Land et al., 1993).
SUMMARY AND RECOMMENDATIONS
Laboratory data provide a solid base of information that indicates that ![]() particles from radon progeny cause mutations in cultured cells, oncogenic transformation in cells in vitro, and tumors in experimental animals. The dose-response relationship appears to be linear down to the lowest doses at which an effect can be observed. The response is also very sensitive to the LET of the
particles from radon progeny cause mutations in cultured cells, oncogenic transformation in cells in vitro, and tumors in experimental animals. The dose-response relationship appears to be linear down to the lowest doses at which an effect can be observed. The response is also very sensitive to the LET of the ![]() particles and peaks at about 100-200 keV/µm. The range of end points and biologic systems studied has been considerably expanded since the BEIR IV report was published in 1988. Experiments with oncogenic transformation induced in cultured cells by high-LET radiation led to a biophysical model for the inverse dose-rate effect, whereby the biologic effectiveness of a given dose is increased if the dose is protracted. The biophysical model appears to be consistent with the dose-rate effect observed in studies of experimental animals and underground uranium miners and has implications for the extrapolation of data from radon exposures in mines to exposures in residences. That is an important development since the publication of BEIR IV and needs to be considered and evaluated by a Phase II (BEIR VI) committee.
particles and peaks at about 100-200 keV/µm. The range of end points and biologic systems studied has been considerably expanded since the BEIR IV report was published in 1988. Experiments with oncogenic transformation induced in cultured cells by high-LET radiation led to a biophysical model for the inverse dose-rate effect, whereby the biologic effectiveness of a given dose is increased if the dose is protracted. The biophysical model appears to be consistent with the dose-rate effect observed in studies of experimental animals and underground uranium miners and has implications for the extrapolation of data from radon exposures in mines to exposures in residences. That is an important development since the publication of BEIR IV and needs to be considered and evaluated by a Phase II (BEIR VI) committee.
The last few years have seen a dramatic increase in our understanding of the molecular genetics of cancer. Preliminary evidence indicates that it might be possible to identify molecular changes that are characteristic of cancers induced by ![]() particles.
particles.
As a result of recent radiation-biology studies in molecular
biology, transformation, and carcinogenesis, as well as some useful data from studies of neutron-radiation biology, the committee recommends that a Phase II Research Council committee
-
Evaluate biophysical models of the inverse dose-rate effect and their implications for risks associated with indoor radon exposure.
-
Compare risks and pathology of lung tumors induced by exposure to radon with risks and pathology of those induced by external exposures to low-LET radiation.
-
Examine in more detail the induction and repair of molecular changes resulting from -particle exposure.


























