
11
Rates and Patterns of Chloroplast DNA Evolution
Michael T. Clegg, Brandon S. Gaut, Gerald H. Learn, Jr., and Brian R. Morton
The chloroplast genome was almost certainly contributed to the eukaryotic cell through an endosymbiotic association with a cyanobacteria-like prokaryote. Moreover, present evidence suggests that the association that led to land plants occurred only once in evolution (Gray, 1993). The derivative plastid genome (cpDNA) is a relictual molecule of about 150 kbp that encodes roughly 100 genetic functions (reviews in Palmer, 1991; Clegg et al., 1991). This genome is the most widely studied plant genome with regard to both molecular organization and evolution. Complete sequences of six chloroplast genomes are now available, and these represent virtually the full range of plant diversity [an alga, Euglena (Hallick et al., 1993); a bryophyte, Marchantia (Ohyama et al., 1986); a conifer, black pine (Wakasugi et al., 1993); a dicot, tobacco (Shinozaki et al., 1986); a monocot, rice (Hiratsuka et al., 1989); and a parasitic dicot, Epifagus (Wolfe et al., 1992)]. With the possible exception of Euglena and Epifagus , the picture that has emerged is one of a relatively stable genome with marked conservation of gene content and a substantial conservation of structural organization. Mapping studies that span land plants and algae confirm the impression of strong conservation in gene content (Palmer, 1985).
Michael T. Clegg is professor of genetics and Gerald H. Learn, Jr. and Brian R. Morton are postdoctoral research associates in the Department of Botany and Plant Science at the University of California, Riverside. Brandon S. Gaut is assistant professor of genetics at Rutgers University, New Brunswick, New Jersey.
Because the photosynthetic machinery requires many more gene functions than are specified on the cpDNA of plants and algae, it is assumed that many gene functions were transferred to the eukaryotic nuclear genome. The strong conservation of cpDNA gene content cited above indicates that most transfers of gene function from the chloroplast to the nuclear genome occurred early following the primordial endosymbiotic event. Among land plants, gene content is almost perfectly conserved, although a few recent transfers of function from the chloroplast to the nuclear genome have been demonstrated (Downie and Palmer 1991; Gantt et al., 1991).
Conservation of gene content and a relatively slow rate of nucleotide substitution in protein-coding genes has made the chloroplast genome an ideal focus for studies of plant evolutionary history (reviewed in Clegg, 1993). These efforts have culminated in the past year with the publication of a volume of papers that presents a detailed molecular phylogeny for seed plants (Chase et al., 1993). This effort has involved many laboratories that have together determined the DNA sequence for more than 750 copies (by latest count) of the cpDNA gene rbcL encoding the large subunit of ribulose-1,5-bisphosphate carboxylase (RuBisCo). The sequence data span the full range of plant diversity from algae to flowering plants. This very large collection of gene sequence data has allowed the reconstruction of plant evolutionary history at a level of detail that is unprecedented in molecular systematics.
Accurate phylogenies are of more than academic interest. They provide an organizing framework for addressing a host of other important questions about biological change. For example, questions about the origins of major morphological adaptions must be placed in a phylogenetic context to reconstruct the precise sequence of genetic (and molecular) changes that give rise to novel structures. The mutational processes that subsume biological change can be revealed in great detail through a phylogenetic analysis. And, questions about the frequency and mode of horizontal transfer of mobile genetic elements can only be addressed in a phylogenetic context. One can make a very long list of biological problems that are illuminated by phylogenetic analyses.
Despite their obvious utility, many important questions remain about the accuracy of molecular phylogenies. All methods of phylogenetic reconstruction assume a fairly high degree of statistical regularity in the underlying mutational dynamics. Our goal in this article is to analyze the tempo and mode of cpDNA evolution in greater depth. We will show that below the surface impression of conservation there are a variety of mutational processes that often violate assumptions of rate constancy and site independence of mutational change. To facilitate an analysis of patterns of cpDNA mutational change, we divide the
genome into functional categories and we then study the pattern of mutational change within each functional category. The functional categories are (i) DNA regions that do not code for tRNA, ribosomal RNA (rRNA), or protein (referred to as ''noncoding DNA"); (ii) protein-coding genes; and (iii) chloroplast introns. The methodological approach is that of comparative sequence analysis, where complete DNA sequences from phylogenetically structured samples are analyzed to reveal the pattern of mutational change in evolution.
Evolution of Noncoding DNA Regions
The chloroplast genome is highly condensed compared with eukaryotic genomes; for example, only 32% of the rice genome is noncoding. Most of this noncoding DNA is found in very short segments separating functional genes. A number of recent studies have revealed complex patterns of mutational change in noncoding regions. The most widely studied noncoding region of the chloroplast genome is the region downstream of the rbcL gene in the grass family (Poaceae). This noncoding sequence is flanked by the genes rbcL and psaI (the gene encoding photosystem I polypeptide I) and is 1694 bases long in the rice genome, making it one of the longest noncoding regions of the genome and the longest when introns are excluded (Figure 1). A pseudogene for the chloroplast gene rpl23 is also located within this noncoding segment (Hiratsuka et al., 1989). Hiratsuka et al. (1989) argued that a large inversion, unique to the grass family, arose through a recombinational interaction between nonhomologous tRNA genes, and the same process of recombination between short repeats has been invoked as the mechanism responsible for the origin of the rpl23 pseudogene Ψrpl23 (Ogihara et al., 1992).
The functional rpl23 gene of the rice genome is located in the inverted repeat about 27 kb away. Bowman et al. (1988) suggested that the pseudogene was being converted by the functional rpl23 gene because the genetic divergence among pseudogenes was lower than the divergence observed for the surrounding noncoding regions. Subsequent work, based on a phylogenetic analysis (Morton and Clegg, 1993), has provided additional support for a model of gene conversion between the rpl23 pseudogene and its functional counterpart in at least two lineages of the grass family.
Four independent deletion events of at least 850 bases in length have been observed spanning almost identical stretches of the noncoding region between rbcL and psaI (Morton and Clegg, 1993; Ogihara et al., 1988). Based on flanking sequence data, it has been suggested that recombination between short direct repeats was responsible for these
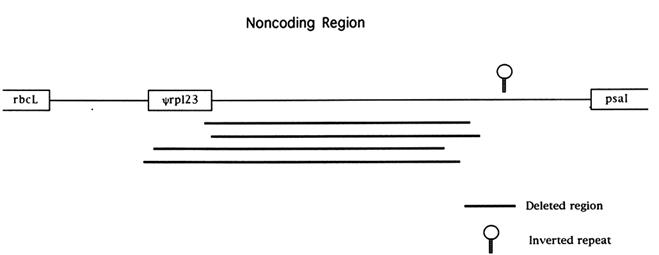
deletions (Ogihara et al., 1988). The four large deletions observed in the grasses all extend from within, or very close to, Ψrpl23 to an area about 400 bases upstream of psaI. The region spanned by the deletions has a low A+T content relative to other chloroplast noncoding sequences (including the surrounding noncoding sequences) and may have been a coding sequence inserted by recombination, as was Ψrpl23 , prior to the divergence of the grass family (Morton and Clegg, 1993). The rate of nucleotide substitution in this noncoding region is roughly equal to the rate of synonymous substitution (with the exception of Ψrpl23) for the neighboring rbcL gene (Morton and Clegg, 1993), but a large number of short insertion/deletions (indels) have occurred. In addition to both the high rate of indel mutation and the large deletions, an inverted repeat in the noncoding region about 300 bases upstream of the psal gene appears to be labile to complex rearrangement events in the grass family (Morton and Clegg, 1993).
A study of the noncoding region upstream of rbcL in the grass family revealed similar patterns of complex change (Golenberg et al., 1993). Indels were found to occur at a greater frequency than nucleotide substitutions, and more complex changes, including multiple tandem duplication events, were found to be confined to highly labile sites, resulting in similar although independent indels at identical sites. Taken together, both noncoding regions reveal a marked incidence of parallel mutations at labile sites that may be positively misleading for phylogenetic studies based on restriction fragment length polymorphism (RFLP) data.
Such complex patterns of mutation are not confined to noncoding sequences of the chloroplast. The chloroplast gene for the RNA polymerase subunit B", rpoC2, has an insertion within the coding sequence in rice relative to tobacco. This insertion has been shown to be confined to the grass family. A comparison of the insertion among members of the grass family revealed widespread diversity in terms of indel mutations as well as an increased rate of nucleotide substitution (Cummings et al., 1994).
The low noncoding content of the cpDNA generally and the high rate of large deletion events observed in the largest contiguous noncoding segment in the rice genome suggest that nonessential sequences are rapidly removed from the chloroplast genome as a result of both intra-and intermolecular recombination. This is supported by the rapid loss of all photosynthetic genes from the chloroplast genome of the nonphotosynthetic plant Epifagus (Wolfe et al., 1992). Given the low similarity of the noncoding sequence between the chloroplast genomes of rice and tobacco and the observed degree of variation in noncoding sequences
within the grasses, the conserved chloroplast genes appear to exist in a very fluid medium of surrounding noncoding sequence.
Evolution of Protein Coding Genes
Codon Bias of Chloroplast Genes. The codon utilization pattern of chloroplast genes of the green algae Chlamydomonas reinhardtii and Chlamydomonas moewusii appears to be closely adapted to the chloroplast tRNA population. In contrast, land plant chloroplast genes are dominated by a genomic bias towards a high A+T content, which is reflected in a high third-codon position A+T content. An interesting exception is the gene coding for the central protein of the photosystem II reaction center (denoted psbA) (Umesono et al., 1988). The psbA protein turns over at a very high rate and, consequently, is the most abundant translation product of the plant chloroplast (Mullet and Klein, 1987). The psbA gene of flowering plants has a codon use very similar to Chlamydomonas chloroplast genes (Morton, 1993). Given the tRNAs encoded by the sequenced chloroplast genomes, the pattern of codon bias of the Chlamydomonas genes appears to be the result of selection for an intermediate codon-anticodon interaction strength (Morton, unpublished data). Further, based on the fact that psbA is the sole flowering plant chloroplast gene following this codon utilization pattern, selection most likely acted on psbA codon use to increase translation efficiency (Morton, 1993).
Despite the difference in codon use by psbA relative to other plant chloroplast genes, this gene has a much lower bias in codon use than do Chlamydomonas chloroplast genes. In fact, there is good evidence that selection no longer acts on psbA codon use in flowering plant lineages; it is merely a remnant of the ancestral codon use. Further, the highly expressed gene rbcL displays a codon use more like psbA than does any other flowering plant chloroplast gene (Morton, 1994). These two factors, the apparently recent loss of selection on flowering plant psbA codon use and the similarity of rbcL to psbA, indicate that loss of selection for codon adaptation on plant chloroplast genes may have occurred at different times for each plant chloroplast gene, most likely as genome number increased over time (Morton, unpublished). Such a scenario may have important implications for studies of chloroplast origins because standard phylogenetic estimation methods are likely to be biased and, therefore, unreliable at very deep evolutionary levels. The codon bias results also suggest that nucleotide composition cannot be assumed to be at equilibrium, contrary to the assumptions incorporated in most distance estimators.
Relative Rates of Nucleotide Substitution. Early work on chloroplast sequences suggested that substitution rates do not follow a constant molecular clock. Rodermel and Bogorad (1987) first claimed that substitution rates in chloroplast loci can vary among evolutionary lineages. They found that the atpE gene had slower missense rates, and the atpH gene had slower synonymous rates, in grass lineages relative to dicot lineages. Similar comparisons of substitution rates in the rbcL locus indicated that both missense and synonymous rates were faster in grass species relative to palm species (Wilson et al., 1990).
These studies relied on fossil-based divergence times for substitution rate estimates. Fossil-based divergence times can have large errors that introduce large (and unmeasurable) uncertainty into rate estimates. Relative rate tests allow comparison of substitution rates between evolutionary lineages without dependence on knowledge of the time dimension. First utilized by Sarich and Wilson (1973), Wu and Li (1985) later extended relative rate tests to nucleotide sequences using a distance measure formulation. Subsequent refinements include a maximum likelihood relative test (Muse and Weir, 1992) and a simple counting method (Tajima, 1993). The power of these methods to detect deviations from a molecular clock depends on a number of factors (e.g., the number of substitution events in the lineages and the transition/transversion bias), but in many situations the three methods have comparable power (Muse and Weir, 1992; Tajima, 1993). Relative rates tests cannot detect changes in evolutionary rates if rates change proportionally among lineages (Fitch, 1976), although this precise condition seems unlikely to occur often. Relative rate tests may also fail to detect stochastic changes in rate within a lineage because the test compares average substitution rates. More importantly, relative rates contain less information on variation than do absolute rates, but absolute rates are difficult to estimate because of imperfect knowledge of the time dimension.
The large rbcL data base has facilitated the characterization of substitution rate variation in a number of evolutionary lineages. A few studies have applied relative rate tests to rbcL sequences from intrafamilial taxa (Soltis et al., 1990; Doebley et al., 1990; Bousquet et al., 1992a); on the whole, these studies have uncovered limited rate variation. Two studies have characterized rbcL rate variation over a wider sampling of plant taxa (Bousquet et al., 1992b; Gaut et al., 1992). Gaut et al. (1992) examined rbcL sequences from 35 monocotyledonous taxa and found substantial synonymous rate variation among evolutionary lineages, but little missense rate variation. The analyses revealed rate homogeneity for rbcL sequences within well-defined families, but substantial rate heterogeneity in interfamilial contrasts. The pattern of interfamilial rate variation
revealed the most rapid nucleotide substitution rate in the grass family, followed by families in the orders Orchidales, Liliales, Bromeliales, and Arecales (represented by the palms). Rates of synonymous nucleotide substitution in grass rbcL sequences exceed rates in other monocot sequences by as little as 130% and by as much as 800%.
Bousquet et al. (1992b) reported extensive rate variation among 15 rbcL sequences representing monocot, dicot, and gymnosperm taxa. They concluded that missense rate variation is more extensive than synonymous rate variation. This result differs with that reported by Gaut et al. (1992). The differences between these studies can probably be ascribed to the wide phylogenetic range of taxa surveyed. The relatively narrow monocot comparisons included few missense substitutions; the paucity of nonsynonymous substitutions may have made detection of missense rate heterogeneity difficult. On the other hand, the study of Bousquet et al. (1992b) may have underestimated synonymous rate variation because some of their sequence comparisons had probably been saturated for synonymous substitutions.
Clearly a simple stochastically constant molecular clock does not hold for the rbcL locus; however, there may be a generation-time effect (Li, 1993). While it is difficult to estimate generation times in most plant taxa, the monocot sequences show a clear negative correlation between the minimum generation time and substitution rates (Gaut et al., 1992). Given that rate variation in monocot sequences is largely synonymous (and therefore presumably close to neutral) and that rate variation is correlated with minimum generation time, rate variation at the rbcL locus of monocot taxa is consistent with neutral predictions. The conclusions of Bousquet et al. (1992b) are not as straightforward, but their results do indicate clear differences in substitution rates among annual and perennial taxa, a result not inconsistent with a generation-time effect. Bousquet et al. (1992b) also speculate that speciation rates influence substitution rates. Other factors hypothesized to contribute to rate variation among evolutionary lineages include polymerase fidelity (Wu and Li, 1985; Li et al., 1985), selection (Gillespie, 1986), and G + C content (Bulmer et al., 1991).
What is the pattern of rate variation at other chloroplast loci? If rate variation is predominantly a function of an evolutionary factor that affects the whole genome (like, presumably, the generation-time effect), then one would expect to find similar patterns of rate variation in most chloroplast loci. Conversely, widely divergent patterns of rate variation among chloroplast loci would argue for locus-specific factors (like selection) as the motive force behind substitution rate variation. Ideally, one should sample chloroplast loci from the taxa used in the rbcL studies to directly compare patterns of rate variation among loci. Unfortunately,
data for these kinds of studies are not yet available. As a first step in the analysis of rate variation among chloroplast loci, Gaut et al. (1993) examined rate heterogeneity among a number of chloroplast loci from three taxa (maize, rice, and tobacco, using Marchantia as an outgroup). Comparison of sequence data from the maize and rice chloroplast genomes revealed little rate heterogeneity (using tobacco as an outgroup). However, comparisons of sequence data from rice and tobacco (using Marchantia as the outgroup) revealed much heterogeneity: significant deviation from a molecular clock was detected at 14 of 40 loci. All 14 loci have accelerated substitution rates in rice relative to tobacco, suggesting concerted rate increases in the rice lineage. In addition, 17 loci had nonsignificantly accelerated rates in rice, while the remaining 9 loci had nonsignificantly slower rates in rice, relative to tobacco.
Interestingly, many of the 14 loci that exhibit significant rate heterogeneity between rice and tobacco lineages encode protein products of related function. For example, three of the four loci that encode RNA polymerase subunits demonstrate rate heterogeneity between rice and tobacco lineages. Further analysis of RNA polymerase genes suggests rate acceleration with subsequent rate deceleration (B. S. Gaut, unpublished data), perhaps indicating the episodic rate pattern thought to result from selective pressures (Gillespie, 1986).
Patterns of Amino Acid Replacement in the RuBisCo Protein
The question of site-dependent probabilities of amino acid replacement can be addressed in considerable detail by using the very large rbcL data base. This large data base may be used to ask whether models of nucleotide substitution provide an acceptable fit to the data, and it is even more important to ask whether the pattern of accepted amino acid change provides useful information on protein adaptation. The three-dimensional structure of the RuBisCo protein has been determined for a wide phylogenetic range of species (Chapman et al., 1988; Knight et al., 1989; Schneider et al., 1990). The pattern of amino acid replacement can be mapped onto the physical structure to identify major constraints in molecular change. Such an analysis, when placed in a phylogenetic context, may also help to identify amino acid replacement of functional importance.
The large subunit of RuBisCo contains two domains: (i) a carboxyl-terminal C domain that includes (a) an α/β-barrel consisting of alternating α-helices and β-strands, with the parallel β-strands forming an interior barrel; (b) an interior extension with an α-helix followed by a pair of antiparallel β-strands; and (c) a terminal extension of two to four
α-helices; and (ii) an amino-terminal N domain consisting of a five-stranded antiparallel β-sheet and four or five α-helices that form a lid-like structure covering the barrel of an adjacent large subunit. Furthermore, the active site is well known. It consists of charged and polar residues at the carboxyl-terminal ends of the β-strands in the α/β-barrel of the C domain of one subunit and asparagine and glutamic acid residues from the lid of the adjacent N domain.
One approach to examining patterns of amino acid replacements in a structural context is to map amino acid replacements on a fully resolved, unambiguous tree. Since the evolutionary relationships among all of the >750 taxa are not known to any degree of certainty, a subset of the taxa were chosen for this analysis. We used 105 taxa, including three prokaryotes, four algae (including Charophytes), five bryophytes and ferns, eight gymnosperms, and 85 angiosperms (for details, contact the authors). In general, conventional phylogenetic relationships that were supported by the analysis of the rbcL nucleotide sequence data (Chase et al., 1993; Duvall et al., 1993) were used to construct this tree. The amino acid sequences for the 105 taxa were translated from the nucleotide sequences and aligned (along with the >700 amino acid sequences in the large data set) by using the following method. A preliminary alignment obtained using CLUSTAL V (Higgins et al., 1992) was refined by aligning similar, presumably homologous features of the solved three-dimensional crystal structures for the RuBisCo large subunit. Nine gaps were required to align the sequences. There were 494 sites in the aligned data set. The computer program MACCLADE version 3.04 (Maddison and Maddison, 1992) was then used to locate amino acid replacement on branches of the tree and to count the total number of amino acid changes required through the phylogeny.
Of the 1350 amino acid replacements; 762 could be unambiguously inferred; 568 and 182 were in α-helices and β-strands, respectively. Only 488 (36%) of the replacements were in the α/β-barrel structure, which constitutes 46% of the sites. For the complete sequence, the most common unambiguous replacements were Glu -> Asp, and Ala -> Ser (40 and 35 changes, respectively). When the number and types of replacements were examined for the various structures, interesting patterns emerged. Figure 2 shows the fractions of sites with replacements and the number of changes for the complete sequences, α-helices, β-strands, and other structures. The distributions are highly skewed, indicating that some sites may accept as many as 26 replacement events while other sites do not accept amino acid replacements. [Character state distributions (amino acids at a given site) for the 105 taxon dendrogram does not allow unequivocal reconstruction of the particular residues at all nodes in the dendrogram. For these residues the number
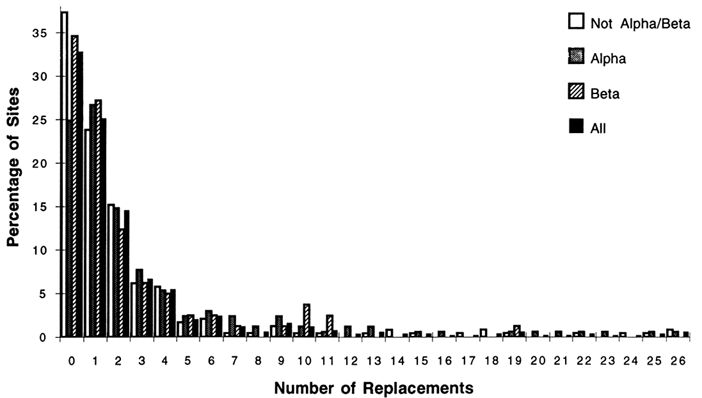
Figure 2 The percentage of sites with replacements versus the number of changes at a given site for the complete sequences, α-helix, β-strand, and other structures. The replacements were inferred for a dendrogram of 105 taxa (see text) by using the computer program MACCLADE version 3.04. Of the 494 aligned positions, 169 lie in α-helices, and 81 lie in β-strands.
of unambiguous replacements underestimates the total number of replacements.]
For the complete sequence, 33% of all sites are unvaried, showing no changes. For α-helices, β-strands, and non-α/β structures, the percentages of unvaried sites are 25%, 35%, and 37%, respectively. The length of the tails of the distributions also differ among structural classes; there are as many as 26 unambiguous changes for the α and non-α/β structural classes, while the most changes allowed for sites in β-strands is 19. The unambiguous amino acid replacements that were most common among all sites in the α-helical regions were Ala ⇒ Ser (31 changes), Glu ⇒ Asp (15), and Ser ⇒ Thr (15). For the β regions among all sites, the most common replacements were Ile ⇒ Met (9) and Ile ⇒ Val (8). The most common replacements for the non-α/β regions were Glu ⇒ Asp (21) and Tyr ⇒ Phe (14). These are all fairly conservative replacements. More noteworthy is the fact that the predominant changes for the b-strands involve replacements among nonpolar residues.
Tables 1A and 1B show the sites that are most variable for α and non-α/β (>19 changes) and β (>8 changes) structures. Many of the highly variable sites for all of the structural classes involve replacements among nonpolar residues. Also interesting is the replacement of Ala-103 by Cys. This change apparently occurs in six different lineages. In none of the lineages is the change reversed.
What conclusions may we draw from these analyses of the patterns of amino acid variation? The analyses suggest that in angiosperms, amino acid replacements among hydrophobic (nonpolar) residues occur more frequently at some sites in the large subunit of RuBisCo. While the frequency of the highly variable sites and the degree of variability differ for β-strand sites as compared to other structural regions, these variable sites may be found throughout the large subunit sequence. This sort of variability may have an impact on the use of nucleotide sequence data for phylogeny reconstruction because methods of phylogenetic inference assume the probability of replacement is independent of site. The fact that third-nucleotide positions in a codon differ in substitution rate compared to first and second position sites is widely appreciated, but this is a simple and easily corrected source of variation. It is much more difficult to correct for complex site-dependent probabilities that may also change between major evolutionary lineages.
Some of the more variable sites show high levels of replacement for amino acids that are coded for by codons with third-site differences [Asp (GAY) ![]() Glu (GAR), Ile (ATH)
Glu (GAR), Ile (ATH) ![]() Met (ATG)], where Y = T or C, R = G or A, and H = A, C, or T. The fact that these replacements are fairly frequent and readily tolerated is concordant with the higher rate of
Met (ATG)], where Y = T or C, R = G or A, and H = A, C, or T. The fact that these replacements are fairly frequent and readily tolerated is concordant with the higher rate of
TABLE 1 Highly variable sites in RuBisCo large subunits in the phylogenetic tree for 105 taxa for α and non-α/β sites (>20 replacements per site) and for β-strand sites (>9 replacements per site)
|
a. α and non-α/β sites |
||||
|
Sitea |
Replacements |
Statesb |
Consistency index |
Predominant replacementc |
|
32 |
22 |
2 |
0.05 |
Asp ⇒ Glu (6) |
|
95 |
26 |
5 |
0.15 |
|
|
99 |
25 |
7 |
0.24 |
|
|
146 α |
23 |
6 |
0.22 |
|
|
149 α |
20 |
7 |
0.30 |
|
|
229 α |
21 |
4 |
0.14 |
Ile ⇒ Leu (13) |
|
255 α |
26 |
5 |
0.15 |
Ile⇒Met (9) |
|
259 α |
22 |
7 |
0.27 |
|
|
332 |
24 |
5 |
0.17 |
|
|
453 |
26 |
6 |
0.19 |
|
|
b. β stand sites |
||||
|
Sitea |
Replacements |
Statesb |
Consistency index |
Predominant replacementc |
|
90 |
9 |
7 |
0.67 |
|
|
103 |
10 |
3 |
0.20 |
Ala ⇒ Cys (6) |
|
313 |
19 |
3 |
0.11 |
Ile ⇒ Met (8) |
|
330 |
10 |
4 |
0.30 |
Ile ⇒ Val (3) |
|
357 |
10 |
4 |
0.30 |
Phe ⇒ Tyr (6) |
|
358 |
11 |
5 |
0.36 |
Ile ⇒ Val (3) |
|
a Site is the position number in the 494 aligned positions. An a is appended to the number of the residue for those in α-helices to distinguish them from non-α/β sites. b States is the number of different kinds of amino acids found at the indicated site. c Predominant replacement indicates the most frequent unambiguously inferred amino acid replacement in the character state reconstruction for the dendrogram at the indicated site; the number of unambiguously inferred independent occurrences is in parentheses. The number of unambiguous replacements is an underestimate of the true number of replacements for some sites (see text). |
||||
substitution for third-position sites. Other third-position substitutions [e.g., Gln (CAR) ![]() His (CAY), Lys (AAR)
His (CAY), Lys (AAR) ![]() Asn (AAY), Arg (AGR)
Asn (AAY), Arg (AGR) ![]() Ser (AGY)] are not among the replacements seen for highly variable sites; furthermore, replacements such as Ser (TCN) ⇒ Ala (GCN), Ile (ATH) ⇒ Val (GTN), and Phe (TTY) ⇒ Tyr (TAY) are similarly favorable even though they require first- or second-position substitutions. The rate heterogeneities that follow from such patterns make methods of phylogenetic inference using a simple weighting scheme for different codon positions problematical.
Ser (AGY)] are not among the replacements seen for highly variable sites; furthermore, replacements such as Ser (TCN) ⇒ Ala (GCN), Ile (ATH) ⇒ Val (GTN), and Phe (TTY) ⇒ Tyr (TAY) are similarly favorable even though they require first- or second-position substitutions. The rate heterogeneities that follow from such patterns make methods of phylogenetic inference using a simple weighting scheme for different codon positions problematical.
Patterns of Intron Evolution
Data from the plastid genomes that have been completely sequenced reveal that introns are a general feature of these genomes. Each of the three classes of introns (groups I, II, and III) have a characteristic secondary structure that is related to the mechanisms by which the intervening sequence is excised from RNA precursors. The secondary structure requirements, along with other constraints, appear to limit the acceptance of mutational changes in intron sequences through evolutionary time. Group I introns, which were the first self-splicing introns identified (Zaug and Cech, 1980), are found in a single tRNA gene, trnL(UAA), from a number of plastid genomes. Group III introns have thus far only been identified in the euglenophytes Euglena gracilis and Astasia longa (Christopher and Hallick, 1989) and appear to be truncated (or streamlined) group II introns. It is noteworthy that group I introns are absent from the newly sequenced plastid genomes of Euglena gracilis (Hallick et al., 1993) and beechdrops, Epifagus virginiana (Wolfe et al., 1992). Both genomes are atypical, however: the Euglena genome is markedly different in structural organization and may have had a separate origin from the plastid genomes of land plants; beechdrops is a nonphotosynthetic parasitic plant, and its genome is highly reduced, lacking trnL (UAA) among other genes. Although comparative sequence analyses of both group I and group III introns could provide information about rates and mechanisms of chloroplast DNA evolution, there are few comparative data, and consequently our discussion will be limited to group II introns.
Group II introns form the most numerous and best characterized class of plastid intervening sequences. They are found in both protein-coding genes and tRNA genes. The secondary structure of group II introns is characterized by six domains (I–VI) (reviewed in Michel et al., 1989). Domain I has a complicated structure and contains sites that probably form base pairs with the 5' exon and are important for intron processing (Jacquier and Michel, 1987). Domains II–VI are typically simple stem–loop structures. Domains V and VI have also been shown to be required for proper processing of the transcript (Schmelzer and Müller, 1987; Jarrell et al., 1988). Learn et al. (1992) examined the evolutionary constraints on the various domains of group II introns in a comparative study of the intron found in a tRNA gene, trnV(UAC), from seven land plants. They found that domain II evolves most rapidly, comparable to the synonymous substitution rate of protein-coding genes, consistent with the finding that domain II may be dispensable in self-splicing introns (Kwakman et al., 1989). Portions of domain I (that are important in binding to the 5' exon) and domain V evolve at the slowest rates,
comparable to missense rates for a number of protein-coding genes. These results illustrate that evolutionary rates within group II introns vary and appear to be related to the functional importance of intron structural features.
Conclusions
The uniformitarian assumption plays a fundamental role in the science of molecular evolution, just as it did in the evolutionary paleontology of G. G. Simpson. We must assume that the kinds of mutational changes that we can demonstrate in ''microscopic detail" from comparative sequence analyses are the substance of molecular evolutionary change. When viewed in detail, the patterns of mutational change in the chloroplast genome are complex, and they belie the notion that the cpDNA is a staid and conservative molecule.
The use of cpDNA RELPs and cpDNA gene sequence data for phylogenetic inference has had an enormous impact on studies of plant phylogenetics and systematics. This has been facilitated by the belief that cpDNA-based mutational change is regular. By and large, the assumption of statistical regularity is adequate, and cpDNA data are an important addition to the previous evidence available to students of plant evolution. Nevertheless, it is important to investigate the ways that mutational change in this genome departs from the kinds of regularity assumed by most methods of phylogenetic inference. When this question is asked, we find that noncoding regions exhibit a number of mutational mechanisms. Some sites are clearly labile to small indel mutations, and in at least one case, large deletions also appear to be site dependent. Complex recombinational processes are also found to influence the evolution of at least some noncoding regions of cpDNA. Group II introns show a strong relationship between structure and probability of mutational change. Analyses of protein-coding genes also reveal complex patterns of mutational change. Finally, rates of nucleotide substitution are quite variable at the level of order and above. Current data suggest that much of the variation in evolutionary rate can be accounted for by the generation-time effect hypothesis, although this requires further investigation. There are also interesting patterns of codon bias. When land plant and algal genes are compared, it is evident that patterns of selection for codon utilization have changed over evolutionary time and that models that assume equilibrium nucleotide frequencies are likely to be violated. Patterns of amino acid replacement in the rbcL gene also reveal substantial variation in site-dependent probabilities of substitution. Taken in toto, these complexities in mutational change should motivate the development of more realistic algo-
rithms for phylogenetic inference when based on molecular data. In the interim, one must view molecular phylogenetic reconstructions as approximate, especially at very deep levels of evolution.
Comparative sequence analyses have utility beyond the study of phylogeny. The pattern of amino acid replacements observed over evolutionary time reflects the pattern of functional constraints that are evolutionary time reflects the pattern of functional constraints that are imposed by natural selection on the protein molecule. Natural selection is a sensitive filter because it is capable of detecting subtle changes associated with small fitness effects. Regions that do not accept change are clearly strongly constrained by functional requirements, but those regions that do accept change may be simply unconstrained, or in a few cases they may reflect responses to adaptive change. It is difficult to distinguish these latter two possibilities, but unusual patterns, such as the repeated replacement of Ala-103 by Cys, may be suggestive of adaptive change.
Comparative molecular sequence analyses also reveal aspects of the evolutionary process that would otherwise be opaque. For example, the recombinational processes that acted to convert Ψrpl23 to the functional gene are only resolvable at the sequence level. Similarly, the identification of labile sites for indel mutation, and more importantly, the identification of local sequence features that may promote indel mutation, depend on a substantial comparative sequence base. When viewed broadly, comparative sequence analyses reveal the rich variety of mutational mechanisms that subsume the processes of molecular evolution.
Summary
The chloroplast genome (cpDNA) of plants has been a focus of research in plant molecular evolution and systematics. Several features of this genome have facilitated molecular evolutionary analyses. First, the genome is small and constitutes an abundant component of cellular DNA. Second, the chloroplast genome has been extensively characterized at the molecular level providing the basic information to support comparative evolutionary research. And third, rates of nucleotide substitution are relatively slow and therefore provide the appropriate window of resolution to study plant phylogeny at deep levels of evolution. Despite a conservative rate of evolution and a relatively stable gene content, comparative molecular analyses reveal complex patterns of mutational change. Noncoding regions of cpDNA diverge through insertion/deletion changes that are sometimes site dependent. Coding genes exhibit different patterns of codon bias that appear to violate the equilibrium assumptions of some evolutionary models. Rates of molec-
ular change often vary among plant families and orders in a manner that violates the assumption of a simple molecular clock. Finally, protein-coding genes exhibit patterns of amino acid change that appear to depend on protein structure, and these patterns may reveal subtle aspects of structure/function relationships. Only comparative studies of molecular sequences have the resolution to reveal this underlying complexity. A complete description of the complexity of molecular change is essential to a full understanding of the mechanisms of evolutionary change and in the formulation of realistic models of mutational processes.
Support from National Institutes of Health Grants GM 45144 (to M.T.C.), GM 15528 (to B.S.G.) and GM 45344 (to North Carolina State University) is gratefully acknowledged. The order of authorship is listed alphabetically. All authors made an equal contribution to this article.
References
Bousquet, J., Strauss, S. H. & Li, P. (1992a) Complete congruence between morphological and rbcL-based molecular phylogenies in birches and related species (Betulaceae). Mol. Biol. Evol. 9, 1076–1088.
Bousquet, J., Strauss, S. H., Doerksen, A. H. & Price, R. A. (1992b) Extensive variation in evolutionary rate of rbcL gene sequences among seed plants. Proc. Natl. Acad. Sci. USA 89, 7844–7848.
Bowman, C. M., Barker, R. F. & Dyer, T. (1988) In wheat ctDNA, segments of ribosomal protein genes are dispersed repeats, probably maintained by nonreciprocal recombination. Curr. Genet. 14, 127–136.
Bulmer, R., Wolfe, K. H. & Sharp, P. M. (1991) Synonymous nucleotide substitution rates in mammalian genes: implication for the molecular clock and the relationship of mammalian orders. Proc. Natl. Acad. Sci. USA 88, 5974–5978.
Chapman, M. S., Suh, S. W., Curmi, P. M. G., Cascio, D., Smith, W. W. & Eisenberg, D. S. (1988) Tertiary structure of plant RuBisCO: Domains and their contacts. Science 241, 71–74.
Chase, M. W., Soltis, D. E., Olmstead, R. G., Morgan, D., Les, D. H., Duvall, M. R., Price, R. A., Hills, H. G., Qiu, Y.-L., Kron, K. A., Rettig, J. H., Conti, E., Palmer, J. D., Manhart, J. R., Sytsma, K. J., Michaels, H. J., Kress, W. J., Karol, K. G., Clark, W. D., Hedren, M., Gaut, B. S., Jansen, R. K., Kim, K.-J., Wimpee, C. F., Smith, J. F., Furnier, G. R., Straus, S. H., Xiang, Q.-Y., Plunkett, G. M., Soltis, P. S., Swensen, S. M., Williams, S. E., Gadek, P. A., Quinn, C. J., Equiarte, L. E., Golenberg, E., Learn, G. H., Jr., Graham, S. W., Barrett, S. C. H., Dayanandan, S. & Albert, V. A. (1993) Phylogenetics of seed Plants: an analysis of nucleotide sequences from the plastid gene rbcL. Ann. Mo. Bot. Gard. 80, 528–580.
Christopher, D. A. & Hallick, R. B. (1989) Euglena gracilis chloroplast ribosomal protein operon: a new chloroplast gene for ribosomal protein L5 and a description of a novel organelle intron category designated group III. Nucleic Acids Res. 17, 7591–7608.
Clegg, M. T. (1993) Chloroplast gene sequences and the study of plant evolution. Proc. Natl. Acad. Sci. USA 90, 363–367.
Clegg, M. T., Learn, G. H. & Golenberg, E. M. (1991) Molecular evolution of Chloroplast DNA. Chapter 7 In Evolution at the Molecular Level, eds. Selander, R. K., Clark, A. G. & Whittam, T. S. (Sinauer, Sunderland, MA), pp. 135–149.
Cummings, M. P., Mertens King, L. & Kellogg, E. A. (1994) Slipped-strand mispairing in a plastid gene: rpoC2 in grasses (Poaceae) Mol. Biol. Evol. 11, 1–8.
Doebley, J., Durbin, M., Golenberg, E. M., Clegg, M. T. & Ma, D. P. (1990) Evolutionary analysis of the large subunit of carboxylase (rbcL) nucleotide sequence among the grasses (Gramineae). Evolution 44, 1097–1108.
Downie, S. R. & Palmer, J. D. (1991) Use of chloroplast DNA rearrangements in reconstructing plant phylogeny. In Molecular Systematics of Plants , eds. Soltis, P. S., Soltis, D. E. & Doyle, J. J. (Chapman and Hall, New York), pp. 14–35.
Duvall, M. R., Clegg, M. T., Chase, M. W., Clark, W. D., Kress, W. J., Hills, H. G., Eguiarte, L. E., Smith, J. F., Gaut, B. S., Zimmer, E. A. & Learn, G. H., Jr. (1993) Phylogenetic hypotheses for the monocotyledons constructed from rbcL sequence data. Ann. Mo. Bot. Gard. 80, 607–619.
Fitch, W. M. (1976) Molecular evolutionary clocks. In Molecular Evolution , ed., Ayala, F. J. (Sinauer, Sunderland, MA), pp. 160–178.
Gantt, J. S., Baldauf, S. L., Calie, P. J., Weeden, N. F. & Palmer, J. D. (1991) Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO J. 10, 3073–3078.
Gaut, B. S., Muse, S. V. & Clegg, M. T. (1993) Relative rates of nucleotide substitution in the chloroplast genome. Mol. Phylogenet. Evol. 2, 89–96.
Gaut, B. S., Muse, S. V., Clark, W. D. & Clegg, M. T. (1992) Relative rates of nucleotide substitution at the rbcL locus of monocotyledonous plants. J. Mol. Evol. 35, 292–303.
Gillespie, J. H. (1986) Natural selection and the molecular clock. Mol. Biol. Evol. 3, 138–155.
Golenberg, E. M., Clegg, M. T., Durbin, M. L., Doebley, J. & Ma, D. P. (1993) Evolution of a noncoding region of the chloroplast genome. Mol. Phylogenet. Evol. 2, 52–64.
Gray, M. W. (1993) Origin and evolution of organelle genomes. Curr. Opin. Genet. Dev. 3, 884–890.
Hallick, R. B., Hong, L., Drager, R. G., Favreau, M. R., Monfort, A., Orsat, B., Spielmann, A. & Stutz, E. (1993) Complete sequence of Euglena gracilis chloroplast DNA. Nucleic Acids Res. 21, 3537–3544.
Higgins, D. G., Bleasby, A. J. & Fuchs, R. (1992) Phylogenetic hypotheses for the monocotyledons constructed from rbcL sequence data. Comp. Appl. Biosci. 8, 189–191.
Hiratsuka, J., Shamida, H., Whittier, R., Ishibashi, T., Sakamoto, M., Mori, M., Knodo, C., Honji, Y., Sun, C.-R., Meng, B.-Y., Li, Y.-Q., Kano, A., Nishizawa, Y., Hirai, A., Shinozaki, K. & Sugiura, M. (1989) The complete sequence of the rice (Oryza sativa) chloroplast genome: Intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. 217, 185–194.
Jacquier, A. & Michel, F. (1987) Multiple exon-binding sites in class II self-splicing introns. Cell 50, 17–29.
Jarrell, K. A., Dietrich, R. C. & Perlman, P. (1988) Group II intron domain 5 facilitates a trans-splicing reaction. Mol. Cell. Biol. 8, 2361–2366.
Knight, S., Andersson, I. & Branden, C.-I. (1989) Reexamination of the three-dimensional structure of RuBisCo from higher plants. Science 244, 702–705.
Kwakman, J. H., Konings, D., Pel. H. J. & Grivell, L. A. (1989) Structure-function relationships in a self-splicing group II intron: a large part of domain II of the mitochondrial intron aI5 is not essential for self-splicing. Nucleic Acids Res. 17, 4205–4216.
Learn, G. H., Jr., Shore, J. S., Furnier, G. R., Zurawski, G. & Clegg, M. T. (1992) Constraints on the evolution of chloroplast introns: the intron in the gene encoding tRNA-Val(UAC). Mol. Biol. Evol. 9, 856–871.
Li, W.-H. (1993) So, what about the molecular clock hypothesis? Curr. Opin. Genet. Dev. 3, 896–901.
Li, W.-H., Luo, C.-C., & Wu, C.-I. (1985) Evolution of DNA sequences. In Molecular Evolutionary Genetics, ed. MacIntyre, R. J. (Plenum, New York), pp. 1–94.
Maddison, W. P. and D. R. Maddison (1992) MacClade: Analysis of phylogeny and character evolution. Version 3.0. Sinauer Associates, Sunderland, Massachusetts.
Michel, F., Umesono, K. & Ozeki, H. (1989) Comparative and functional anatomy of group II catalytic introns—a review. Gene 82, 5–30.
Morton, B. R. (1993) Chloroplast DNA codon use: evidence for selection at the psbA locus based on tRNA availability. J. Mol. Evol. 37, 273–280.
Morton, B. R. (1994) Codon use and the rate of divergence of land plant chloroplast genes. Mol. Biol. Evol. 11, 231–238.
Morton, B. R. & Clegg, M. T. (1993) A chloroplast DNa mutational hotspot and gene conversion in a noncoding region near rbcL in the grass family (Poaceae). Curr. Genet. 24, 357–365.
Mullet, J. E. & Klein, R. R. (1987) Transcription and RNA stability are important determinants of higher plant chloroplast RNA levels. EMBO J. 6, 1571–1579.
Muse, S. V. & Weir, B. S. (1992) Testing for equality of evolutionary rates. Genetics 132, 269–276.
Ogihara, Y., Terachi, T. & Sasakuma, T. (1988) Intramolecular recombination of chloroplast genome mediated by short direct-repeat sequences in wheat species. Proc. Natl. Acad. Sci. USA 85, 8573–8577.
Ogihara, Y., Terachi, T. & Sasakuma, T. (1992) Structural analysis of length mutations in a hot-spot region of wheat chloroplast DNAs. Curr. Genet. 22, 251–258.
Ohyama, K., Fukuzawa, H., Kohchi, T., Shirai, H., Sano, T., Sano, S., Umesono, K., Shiki, Y., Takeuchi, M., Chang, Z., Aota, S.-I., Inokuchi, H. & Ozeki, H. (1986) Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature (London) 322, 572–574.
Palmer, J. D. (1985) Comparative organization of chloroplast genomes. Annu. Rev. Genet. 19, 325–354.
Palmer, J. D. (1991) Plastid chromosomes: structure and evolution. In Cell Culture and Somatic Cell Genetics of Plants, eds. Bogorad, L. & Vasil, K. (Academic, New York), Vol. 7A, pp. 5–53.
Rodermel, S. R. & Bogorad, L. (1987) Molecular evolution and nucleotide sequences of the maize plasmid genes for the subunit of CF1 (atpA) and the proteolipid subunit of CF0 (atpH). Genetics 116, 127–139.
Sarich, V. M. & Wilson, A. C. (1973) Generation time and genomic evolution in primates. Science 179, 1144–1147.
Schmelzer, C. & Mueller, M. W. (1987) Self-splicing of group II introns in vitro: lariat formation and 3' splice site selection in mutant RNAs. Cell 51, 753–767.
Schneider, G., Lindqvist, Y. & Lundqvist, T. (1990) Crystallographic refinement and structure of ribulose-1, 5-biphosphate carboxylase from Rhodospirillum rubrum at 1.7 A resolution. J. Mol. Biol. 211, 989–1008.
Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi, T., Hayashida, N., Matsubayashi, T., Zaita, N., Chunwongse, J., Obokata, J., Yamaguchi-Shinozaki, K., Ohto, C., Torazawa, K., Meng, B. Y., Sugita, M., Deno, H., Kamogashira, T., Yamada, K., Kusuda, J., Takaiwa, F., Kato, A., Tohdoh, N., Shimada, H. & Sugiura, M. (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 5, 2043–2049.
Soltis, D. E., Soltis, P. S., Clegg, M. T. & Durbin, M. (1990) rbc L sequence divergence and phylogenetic relationships in Saxifragaceae sensu lato. Proc. Natl. Acad. Sci. USA 87, 4640–4644.
Tajima, F. (1993) Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 135, 599–607.
Umesono, K., Inokuchi, H., Shiki, Y., Takeuchi, M., Chang, Z., Fukuzawa, H., Kohchi, T., Shirai, H., Ohyama, K. & Ozeki, H. (1988) Structure and organization of Marchantia polymorpha chloroplast genome II. Gene organization of the large single copy region from rps'12 to atpB. J. Mol. Biol. 203, 299–331.
Wakasugi, T., Tsudzuki, J., Itoh, S., Nakashima, K., Tsudzuki, T., Shibata, M. and M. Sugiura (1993) The entire structure of the chloroplast genome from black pine (Pinus Thubergii). XV International Botanical Congress Abstract 7.2.1-6. p. 161.
Wilson, M., Gaut, B. & Clegg, M. T. (1990) Chloroplast DNA evolves slowly in the palm family (Arecaceae). Mol. Biol. Evol. 7, 303–314.
Wolfe, K. H., Morden, C. W. & Palmer, J. D. (1992) Function and evolution of a minimal plastid genome from a nonphotosynthetic parasitic plant. Proc. Natl. Acad. Sci. USA 89, 10648–10652.
Wu, C.-I. & Li, W.-H. (1985) Evidence for higher rates of nucleotide substitution in rodents than in man. Proc. Natl. Acad. Sci. USA 82, 1741–1745.
Zaug, A. J. & Cech, T. R. (1980) In vitro splicing of the ribosomal RNA precursor in nuclei of Tetrahymena. Cell 19, 331–338.




















