
12
The Superoxide Dismutase Molecular Clock Revisited
Walter M. Fitch and Francisco J. Ayala
In 1985, Ayala, in his Wilhelmine E. Key lecture, spoke on the enormous difference between the way one might expect a molecular clock to work and the way Cu,Zn superoxide dismutase (SOD) seems to have evolved since the last common ancestor of the metazoans and fungi (Ayala, 1986). Even after an accepted point mutation (PAM) correction for multiple amino acid replacements at a single position in the sequence (Dayhoff, 1978), it was clear that the rates of change were discrepant; there were 10 replacements in the last 75 million years (Myr) but only 21 replacements in the first 600 Myr. This represents a nearly 4-fold difference in rates that could not be ascribed to an incorrect dating of the time of the common ancestor [75 million years ago (Mya) for cow-human; 1200 Mya for fungi-metazoans].
It seemed to one of us (W.M.F.) that the following could be true (or close to the truth): (i) the dates are correct, (ii) the observed differences are correct, and (iii) the clock is working. But these three assertions could all be true only if the correction for multiple replacements had been in error. Fitch and Markowitz introduced the concept of concomitantly variable codons (covarions), which asserts that at any one point in time and in any one lineage, there is a limited number of amino acid sequence positions that can tolerate an amino acid replacement and
Walter M. Fitch is professor and chairman of the Department of Ecology and Evolutionary Biology and Francisco J. Ayala is Donald Bren Professor of Biological Sciences at the University of California, Irvine.
perhaps some positions can never tolerate a replacement (Fitch and Markowitz, 1970). Thus multiple replacements at a site may occur more frequently than would be the estimate when the whole sequence is considered to be variable. However, as replacements are accepted, those very changes may affect and change the sites that are covariable. Thus the sites that have changed across very different taxa may be considerably greater than the number that are covariable in any given species. It seemed possible that the unrecognized presence of covarions could hide the extent of multiple replacements, thus causing the deeper (older) portions of the tree to appear as if evolving too slowly. As the other of us (F.J.A.) has embarked on a program of extensive SOD sequencing, it seems a propitious time to collaborate on a deeper evaluation of this problem.
From this collaboration, we have determined the number of covarions in SOD (28 in Drosophila) and shown that the molecular clock could be working fairly well. This is done by showing, by simulation, that the observed amino acid differences could have arisen if, among the fungi and metazoans, SOD had 44 of its codons permanently invariable and there was a high rate of exchange between codons that are covarions and those that are not variable at any particular time.
Methods
The data analyzed were 67 SOD sequences, aligned by eye. The sequences are from GenBank (see Figure 1 for sequence numbers), except for most of the flies, which came from the lab of F.J.A. The tree was obtained using the ANCESTOR program (Fitch, 1971; Fitch and Farris, 1974). The number of covarions was determined by the method of Fitch and Markowitz (1970).
Simulations were performed (Fitch and Ye, 1991) using a program for which the user prescribes five parameters: (i), the length of the sequence; (ii), the number of covarions; (iii), the persistence of the covarion set (the probability that no covarion will be exchanged for a presently invariable codon; if an exchange does occur, only one of the covarions is exchanged; the possibility of an exchange exists after each replacement); (iv), the number of alternative amino acids allowed at a site, and (v) the times at which the number of amino acid differences are to be determined. The times are in units of amino acid replacements and hence the clock is perfect in the simulations.
The divergence times can have any value but, to test the possibility that the SOD data can arise by the simulated clock, the replacements must be in the same proportion as the paleontological time estimates. The time estimates we used are given in Table 1.
The parameters used for the simulations were (i) length of 118 potentially variable amino acids (162 aligned positions minus 44 permanently invariable positions), (ii) number of covarions = 28 (determined as shown in Results), (iii) persistence = 0.01, (iv) half the variable sites had two alternative amino acids at each site; the other half had three alternatives, and (v) clock times are set by the paleontological dates in Table 1 with a rate of six replacements per 10 Myr.
Results
The SODs are abundant enzymes in aerobic organisms, with highly specific activity that protects the cell against the harmfulness of free oxygen radicals (Fridovich, 1986). The SODs have active centers that contain either iron, manganese, or both copper and zinc (Fridovich, 1986). The Cu,Zn SOD is a well-studied protein found in eukaryotes but also in some bacteria (Steinman, 1988). The amino acid sequence is known in many organisms, plants, animals, fungi, and bacteria. The three-dimensional structure for the bovine SOD has been determined at a 2-Å resolution (Tainer et al., 1982); it is conserved in humans (Getzoff et al., 1989) and presumably in Drosophila (Kwiatowski et al., 1992) and bacteria (Bannister and Parker, 1985). The amino acids essential for catalytic action (Tainer et al., 1983), as well as those for protein structures, are strongly conserved (Getzoff et al., 1989; Kwiatowski et al., 1992).
The Phylogeny. The tree used for this study is shown in Figure 1. It is not the most parsimonious. Rather, we required the tree to conform to what is believed to be correct on a priori grounds (i.e., based on knowledge that is independent of the SOD data). We only let parsimony dictate regions of the tree where other evidence does not seem to us to be determinative. The reason for this is that we wish to optimize the correctness of the tree, rather than its being most parsimonious, because estimates of divergence, against which clock measures are to be tested, will be more valid the closer the tree is to reality. The accuracy of the covarion estimate is similarly constrained. The most parsimonious tree we have found requires 1940 nucleotide substitutions; the tree in Figure 1 requires 1984. The sequences used were amino acid sequences, back translated into ambiguous codons so that the changes are in substitutions rather than replacements, although nearly all substitutions are replacement substitutions. The number of differences between pairs of sequences used for the clock test does not depend upon the topology of the tree. The average differences are shown in Table 1 for those contrasts for which there are reasonable, nonmolecular, paleontological dates.
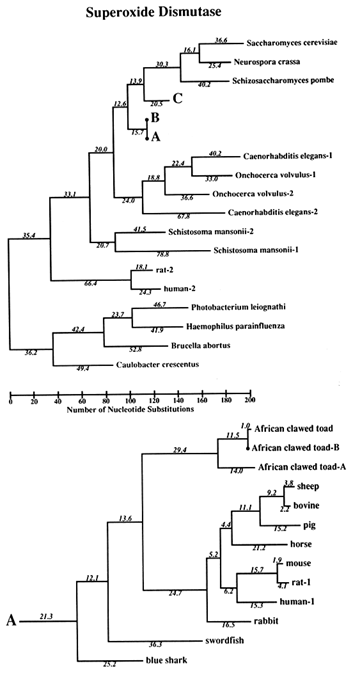
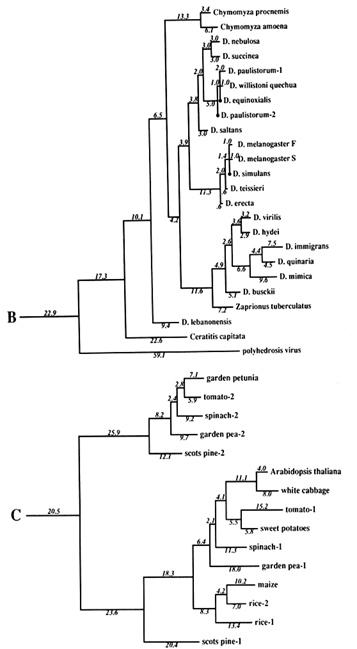
Figure 1 Superoxide dismutase tree. The amino acid sequences (back translated to ambiguous codons) were fit most parsimoniously to this tree, which is 1984 nucleotide substitutions long by the method of Fitch (1971) and Fitch and Farris (1974). It is not the most parsimonious tree, which was not used for reasons discussed in Results. Part C of the figure shows that the Cu,Zn SOD gene became duplicated early in plant evolution, at least before the divergence of gymnosperms and angiosperms. The GenBank numbers for these sequences are as follows: A00512, human-1 (Homo sapiens); S01134, rabbit (Oryctolagus cuniculus); P07632, rat-1 (Rattus norvegicus); JQ0915, (Continued) mouse (Mus musculus); P08294, human-2; X68041, rat-2; A00514, pig (Sus scrofa); A00513, bovine (Bos primigenius taurus); P09670, sheep (Ovis aries); A00515, horse (Equus caballus ); S05021, African clawed toad-A (Xenopus laevis); S05022, African clawed toad-B; S09568, African clawed toad-A P03946, swordfish (Xiphias gladius); S04623, blue shark (Prionace glauca); S84896, scots pine-1 (Pinus sylvestris); S00999, rice-1 (Oryza sativa); D01000, rice-2; A29077, maize (Zea mays); A25569, white cabbage (Brassica oleracea); X60935, Arabidopsis thaliana; X73139, sweet potatoes (Ipomoea batatas); S08350, tomato-1 (Lycopersicon esculentum); P22233, spinach-1 (Spinacia oleracea); M63003, garden pea-1 (Pisum sativum); S84902, scots pine-2; JS0011, spinach-2; S12313, garden pea-2; S08497, tomato-2; P10792, garden petunia (Petunia hybrida); M58687, Neurospora crassa; A36171, Saccharomyces cerevisiae (yeast); X66722, Schizosaccharomyces pombe (yeast); X57105, Onchocerca volvulus-1 (nematode); Z27080, Caenorhabditis elegans-1 (nematode); L20135, Caenorhabditis elegans-2; L13778, Onchocerca volvulus-2; M68862, polyhedrosis virus; A37019, Schistosoma mansonii -1 (liver fluke); M86867, Schistosoma mansonii-2; M84013 and M84012, Haemophilus parainfluenza; M55259, Caulobacter crescentus (bacterium); A00519, Photobacterium leiognathi (bacterium); A33893, Brucella abortus (bacterium). In part B, D. stands for Drosophila.
Covarions. Figure 2 shows a plot that estimates the fraction of the codons that are invariable, based on a two-Poisson fit. The plotted value for each clade is the average number (±SD) of substitutions from the root of the clade to the descendents at the tips. The average is obtained by weighing equally each bifurcating branch irrespective of the number of subsequent branchings. The γ intercept yields the estimate of the fraction of invariable codons in a single species and is 0.826. Thus, 0.174 is the fraction of codons that is covarying and, therefore, the number of covarions is 0.174 × 162 = 28. Strictly speaking, this number is for the Drosophila species. Other work (e.g., on cytochrome c where plants and mammals have the same number of covarions; see figure 5 of Fitch, 1976) suggests that the number of covarions is not highly variable across phyla, although this has not been widely tested.
Discussion
Interest in dating molecular evolutionary events extends back at least to Zuckerkandl and Pauling (1962) who observed 6, 36, and 78 differences between the human β subunit of hemoglobin and each of three other human globin subunits δ, γ, and α, respectively. Assuming a rate calculated from the divergence of the α subunit of human and horse hemoglobin, they transformed the above differences into the dates when the gene duplications occurred—namely, 44, 260, and 565 Mya. This calculation assumes that the number of replacements equals the
TABLE 1 Paleontological dates and numbers of replacements
|
|
Amino acid differences |
|||
|
Sister groups |
Mya |
Replacements |
Observed SOD |
Simulated |
|
D. nebulosa–D. melanogaster |
55 ± 5 |
33 |
18 ± 2 |
18 ± 2 |
|
D. hydei–D. melanogaster |
60 ± ? |
36 |
19 ± 3 |
20 ± 4 |
|
Chymomyza–D. melanogaster |
65 ± ? |
39 |
23 ± 2 |
20 ± 4 |
|
Homo sapiens–Bos taurus |
70 ± 10 |
42 |
27 ± 2 |
22 ± 4 |
|
Ceratitis–D. melanogaster |
100 ± ? |
60 |
31 ± 2 |
28 ± 3 |
|
Monocot–dicot |
125 ± ? |
74 |
28 ± 3 |
31 ± 5 |
|
Angiosperm–gymnosperm |
220 ± ? |
132 |
29 ± 7 |
42 ± 5 |
|
Frog–mammals |
350 ± 10 |
210 |
49 ± 2 |
53 ± 6 |
|
Fish–tetrapods |
400 ± 20 |
240 |
44 ± 4 |
56 ± 7 |
|
Yeast–Neurospora |
? |
|
46 ± 1 |
|
|
Insect–vertebrate |
580 ± 20 |
348 |
59 ± 3 |
60 ± 6 |
|
Fungi–metazoans |
1000 ± ? |
600 |
67 ± 4 |
66 ± 7 |
|
Replacements refer to the number of replacements used in the simulation, which is equivalent to six replacements every 10 Myr. Observed is the average number of amino acid differences observed between members of the two sister groups shown. The sister group names (e.g., Drosophila nebulosa–Drosophila melanogaster) should be understood as indicating the groups to which these species belong and not just two individual species. Simulated is the observed number of amino acid differences obtained after the number of replacements shown had been incorporated. The plus/minus values are crude estimates of error for Mya but are SDs for observed and simulated differences. The simulated values are based on 40 simulated instances for each entry. See Methods for details of the simulation. D. stands for Drosophila. |
||||
number of differences. It was soon recognized that changes could occur at positions where changes had already occurred. Margoliash and Smith (1965) introduced a correction as follows
where r is the number of replacements, n is the number of variable positions in the sequence, and d is the number of divergent positions (differences). It is interesting that Margoliash and Smith contemplated the possibility of invariable positions so early, even though the concern that this might be an important consideration was not present in the field for nearly 20 years, except for Fitch (Fitch and Markowitz, 1970; Shoemaker and Fitch, 1989).
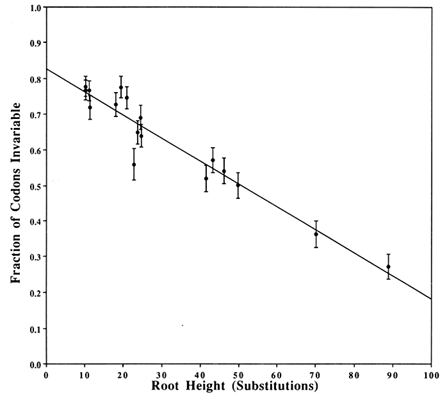
Figure 2 Estimation of the number of covarions. For each clade in the tree in Figure 1, we counted the number of codons suffering zero, one, two, … substitutions and fitted these data to two-Poissons by the method of Fitch and Markowitz (1970). One Poisson fits the varied codons and thus estimates a number of variable but unvaried codons. This number, subtracted from the number of codons with zero changes, is the number of invariable codons that, as a fraction of the total, is plotted on the y axis. On the x axis is plotted the weighted average of the number of substitutions from the root of the clade to the descendents of that root. Weighting is described in Results. The y intercept is the estimated fraction of codons that are not covarions. The vertical bars represent 1 SD of the estimate of the fraction of invariable codons.
In 1969, Jukes and Cantor (1969) introduced a correction that has become commonly used:
where n is the length of the sequence and d = d/n is the fraction of the variable sites that differ. This correction is for nucleotide substitutions,
not amino acid replacements, and, moreover, assumes that all sites are variable. For the first time, this equation recognizes that even two unrelated sequences will still have positions that match. The 3/4 and 4/3 terms reflect that there are four kinds of nucleotides and thus there are three ways in which a second nucleotide may not match the first. To make equation 2 suitable for replacements, the 3/4 and 4/3 should be 19/20 and 20/19, respectively. Note that equation 2, to be technically correct, requires n to be the number of variable sites, although generally n is taken as the length of the sequence, the implicit assumption being that all sites are variable.
Equation 2 assumes that every nucleotide (or every amino acid when modified as stated) is equally likely to be present at a position. The equation can be made more accurate if one knows the frequencies of the elements. If b = 1 - σp2i, where pi is the frequency of the ith element, and 1 ≤ i ≤ k, where k = 4 for nucleotides and k = 20 for amino acids, then
where b is the probability that two randomly drawn elements do not match.
The above modifications (equations 2 and 3) improve the estimate of r by recognizing and accounting for additional biological facts. However, there are other biological features that may be important. In particular, it may be important to know whether n is all sites or only a fraction thereof. Moreover, we may need to consider not only that n is less than the length of the sequence but also that the sites that make up n may not be the same (e.g., in fungi as in mammals). This is the concept incorporated in the notion of covarions, which is supported by reasonable evidence, derived not only from the observed fitting of Poisson distributions to data such as in Fitch and Markowitz (1970) but based also on a test that showed that the invariable positions of cytochrome c are, in fact, not the same in the fungi as in the metazoans (Fitch, 1971).
The first parameter determined was the number of covarions in SOD, estimated to be 28. The fixation of this number reduces the number of parameters that are free to change in order to get a good fit to the SOD observations. In a complementary vein, the 11 clades for which paleontological dates were estimated constitute a large extent of variant data, all of which must be fit to demonstrate that the SOD observations could arise via a perfect clock.
The second parameter fixed was the number of potentially variable codons. This was set rather arbitrarily at 118 by the following logic. Of the 162 codon positions, 44 were unvaried in our data set or were
positions present only in the liverfluke, nametode, and/or bacteria but not in the other sequences. If all 44 unvaried sites were permanently invariable sites within the plant-animal-fungal sequences, then 162 - 44 = 118 were potentially variable.
The third parameter set was the number of alternatives that, on average, are allowed in a variable site. A site that must have a negative charge (aspartate or glutamate) has only two alternatives. Other possible pairs include serine-threonine, phenylalanine-tyrosine, asparagine-glutamine, and lysine-arginine. At the other extreme, there may be sites at which any amino acid can be present, in which case there are 20 alternatives. If all possibly variable sites do vary at some point, then one would expect, that (α - 1)/α of them will differ in distant pairwise comparisons (α is the average number of alternatives at variable sites; it is like the 3/4 or 19/20, in Eq. 2 above). If α = 2.5, then one expects 1.5/2.5 = 0.6 of the potentially variable sites to differ when the number of replacements per site is large. This number may be estimated as 0.6 × 118 = 70.8 differences, a number slightly greater than the average number of differences observed between fungi and metazoans. Thus, we have set α at 2.5, although it may be somewhat low in view of the fact that the number is 3.1 in the Ayala (1986) data.
The other parameters are obtained by trying, in a hit-or-miss fashion, various combinations of the persistence and the number of replacements per million years. The results shown in Figure 3 were obtained by setting the persistence at 0.01 and the replacements per million years at 0.6. The persistence needs to be low for the vast majority of potentially variable sites to have been variable and experienced a replacement while variable—that is, to get an average of 67 differences in 118 codons after 600 replacements. A larger a plus a larger persistence could yield a comparable result but would make the short-duration times yield simulated differences that are too small (compared to observed differences) because too many replacements would occur in sites with prior replacements. A larger α and a smaller number of potentially variable codons would also improve the fit, but it does not seem correct to reduce the number of potentially variable codons below the number that had in fact varied at least once among the 67 sequences in the group. Data similar to that shown in Figure 3 were presented by Kwiatowski et al. (1991) who tried to fit them with both a double Poisson and a rectangular parabola. They found, as we do here, that it was difficult to fit well all the data at once.
We present the best fit that we have obtained, but there is no reason to believe that it is optimal. Therefore, we do not wish to assert that we have determined the real values of the parameters. What we do wish to assert, however, is that a reasonable model of the biological processes
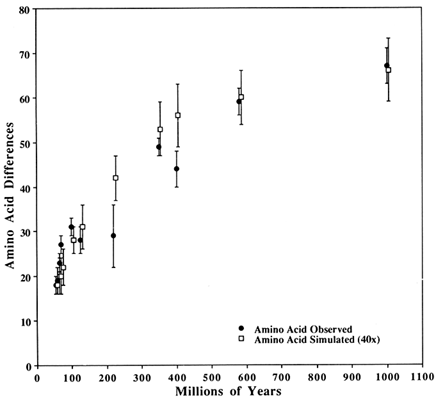
Figure 3 Comparison of observed SOD differences with those from a simulated perfect clock. The closed circles are observed differences in SOD; The open squares are simulated with a perfect clock (see text for clock parameters). The vertical bars show 1 SD about the mean. Observed and simulated values are for the same time despite their being horizontally offset slightly for clarity.
involved permits one to conclude that what at first appeared to be a very inaccurate clock (1) may have been inaccurate simply because the necessary corrections had not been made. In the same spirit as it was appropriate to correct Zuckerkandl-Pauling differences (1962) by accounting for multiple hits (Margoliash and Smith, 1965) and then correcting that for a finite number of alternative character states (Jukes and Cantor, 1969), it may then be necessary to take into account that the variable positions are not the same in different lineages.
Any estimate of divergence time, where the covarion process has not been taken into account, is in danger of significant error. One might naively believe that, if the differences between species are less than 10%,
no Poisson or Jukes-Cantor correction is needed. That is true only if all sites are variable. For example, if only 12 of 100 sites are variable, observing 10 differences implies 23.9 replacements [r = -12(19/20) ln[1 - (20/19)(10/12)], which is more than double the 10.6 replacements [r = -100(19/20)ln[1 - (20/19)(20/100)] calculated on the assumption that all 100 sites were variable. The example may well be somewhat extreme, but any minor problem gets amplified as the ancestor becomes increasingly distant.
We do not assert that the parameters used to get the fit in Figure 3 and Table 1 are necessarily correct, but we want to make five observations.
The first observation is that the implications of the parameter values are likely to be correct. These implications are (i) there are about 28 covarions that (ii) tend to turn over fairly often, although (iii ) the number of allowable alternatives at a variable position is, on average, limited (two to four alternatives), and (iv) that there is a sizeable number of positions (![]() 40) that cannot fix a replacement even though the covarions fix them (v) with a reasonable rate of approximately six replacements per 10 Myr.
40) that cannot fix a replacement even though the covarions fix them (v) with a reasonable rate of approximately six replacements per 10 Myr.
The second observation follows from the pattern seen in Figure 3 (and Table 1) that the simulated values are consistently lower than the observed ones during the first 100 Myr but consistently higher in the intermediate years (200–600 Mya). This pattern is expected if a sizeable fraction of the potentially variable codons has a significantly lower probability of becoming variable than the rest. This is biologically reasonable, although an alternative possibility is that the paleontological dates are either systematically underestimated for the early dates or overestimated for the intermediate dates. This would make it easier to obtain a better fit for both regions, simply by assuming a somewhat larger or smaller replacement rate respectively.
The third observation is that while we know of no good estimates of the divergence time of S. cerevisiae and N. crassa, our data, based simply on differences and not on covarions, place their divergence around 380 ± 40 Mya.
Fourth, we have tried a correction for superimposed replacements of the Jukes-Cantor type where b = 19/20 for the fungi–metazoan divergence. The value obtained is r = -162 × (0.95)ln[1 - 67/(0.95 × 162)] = 88 replacements, a 31% increase over the 67 observed differences, whereas the number of replacements required to obtain 67 differences in the simulations is 600, which is a 796% increase. Thus, corrections of the Jukes-Cantor type may still yield divergences that are gross underestimates if a covarion model is operating.
Fifth, the rate of amino acid replacement that fits our data is 6 × 10-7 replacements per year for the entire gene. This is the same as
(6 × 107)/[(3 nucleotides/covarion) × 28 covarions] = 7 × 10-9 replacement substitutions per variable nucleotide per year. This may be compared to the neutral rate, which, for cow and goat β hemoglobin pseudogenes (Li and Graur, 1991; p. 72) is 3.6 × 10-9 substitutions per nucleotide per year. Since only 3/4 of those substitutions lead to replacements, the neutral replacement substitution rate is 2.7 × 10-9. As the fitted replacement rate is 2.6 times the pseudogene replacement rate, it suggests that there may be positive selection occurring at the SOD locus.
In conclusion, the following observations may be derived from our analysis. First, a molecular clock (i.e., a particular gene or protein) may appear to be very unreliable, yet be fairly accurate. The apparent distortion may emerge because relevant components of the clock (such as covarions, persistence, and the number of alternatives allowed per site) have not been taken into account. As we have shown, the apparently erratic SOD becomes a fairly accurate clock when the appropriate components are taken into account.
The flip side of the observation just made is that inferences of divergence time between lineages derived from a particular clock cannot be assumed to be correct unless the relevant components of the clock have been ascertained. The apparent rate of SOD divergence observed among mammals or flies would yield grossly erroneous time estimates when simply extrapolated to the differences observed between the vertebrate classes (fish versus tetrapod or amphibian versus mammal) or between metazoans and fungi. The SOD ''clock" is a complex of several component parts subject to different constraints and that interact with each other.
Finally, divergence times inferred from a particular molecular clock are subject to the possibility of variations caused by natural selection or other extraneous factors. Thus, for example, some chloroplast genes evolve at rates that are similar in several grass lineages [such as maize and rice (Gaut et al., 1993)] but different from those observed in the tobacco lineage (Kwiatowski et al., 1991). Another example is the acceleration of the rate of lysozyme c evolution in ruminant lineages as lysozyme was recruited for a distinctive stomach function (Stewart and Wilson, 1987; Jolles et al., 1990). A conspicuous anomaly in the SOD data is that there are fewer amino acid differences between fish and tetrapods than between amphibians and mammals. Whether or not the differences are statistically significant, they would seem to support the wrong branching order among the corresponding lineages. The conclusion derived from this anomaly, as well as from the chloroplast, lysozyme, and other examples of uneven rates, is the obvious one that inferences based on a particular clock must be taken with caution, but they become
stronger as they become supported by several independent molecular clocks.
Summary
The Cu,Zn superoxide dismutase (SOD) was examined earlier by Ayala (1986) and found to behave in a very unclocklike manner despite (accepted point mutation, or PAM) corrections for multiple replacements per site. Depending upon the time span involved, rates could differ 5-fold. We have sought to determine whether the data might be clocklike if a covarion model were used. We first determined that the number of concomitantly variable codons (covarions) in SOD is 28. With that value fixed we found that the observations for SOD could fit reasonably well a molecular clock if, given 28 covarions, (i) there are approximately six replacements every 10 million years, (ii) the total number of codons is 162, (iii) the number of codons that are permanently invariable across the range of taxa from fungi to mammals is 44, and (iv) the persistence of variability is quite low (0.01). Thus, the inconsistent number of amino acid differences between various pairs of descendent sequences could well be the result of a fairly accurate molecular clock. The general conclusion has two sides: (i) the inference that a given gene is a bad clock may sometimes arise through a failure to take all the relevant biology into account and (ii) one should examine the possibility that different subsets of amino acids are evolving at different rates, because otherwise the assumption of a clock may yield erroneous estimates of divergence times on the basis of the observed number of amino acid differences.
We thank Ms. Helene Van for her valuable technical assistance. This work was supported by National Institutes of Health Grant GM42397 to F.J.A. (principal investigator) and W.M.F. (co-principal investigator).
References
Ayala, F. J. (1986) On the virtues and pitfalls of the molecular evolutionary clock. J. Hered. 77, 226–235.
Bannister, J. V. and Parker, M. W. (1985) The presence of a copper/zinc superoxide dismutase in the bacterium Photobacterium leiognathi: A likely case of gene transfer from eukaryotes to prokaryotes. Proc. Natl. Acad. Sci. USA 82, 149–152.
Dayhoff, M. O. (1978) Atlas of Protein Sequence and Structure (Natl. Biomed. Res. Found., Washington, DC).
Fitch, W. M. (1971) Toward defining the course of evolution: Minimum change for a specific tree topology. Syst. Zool. 20, 406–416.
Fitch, W. M. (1971) The non-identity of invariant positions in the Cytochrome c of different species. Biochem. Genet. 5, 231–241.
Fitch, W. M. (1976) The molecular evolution of cytochrome c in eukaryotes. J. Mol. Evol. 8, 13–40.
Fitch, W. M. and Farris, J. S. (1974) Evolutionary trees with minimum nucleotide replacements from amino acid sequences. J. Mol. Evol. 3, 263–278.
Fitch, W. M. and Markowitz, E. (1970) An improved method for determining codon variability in a gene and its application to the rate of fixations of mutations in evolution. Biochem. Genet. 4, 579–593.
Fitch, W. M. and Ye, J. (1991) Weighted parsimony: Does it work? In Phylogenetic Analysis of DNA Sequences, eds. Miyamoto, M. and Cracraft, J. (Oxford Univ. Press, New York), pp. 147–154.
Fridovich, I. (1986) Superoxide dismutase. Adv. Enzymol. 58, 61–97.
Gaut, B. S., Muse, S. V. and Clegg, M. T. (1993) Relative rates of nucleotide substitution in the chloroplast genome. Mol. Phylogenet. Evol. 2, 89–96.
Getzoff, E. D., Tainer, J. A., Stempien, M. M., Bell, G. I. and Hallewell, R. A. (1989) Evolution of CuZn superoxide dismutase and the Greek key beta-barrel structural motif. Proteins 5, 322–336.
Jolles, J., Prager, E. M., Alnemri, E. S., Jolles, P., Ibrahimi, I. M. and Wilson, A. C. (1990) Amino acid sequences of stomach and nonstomach lysozymes of ruminants. J. Mol. Evol. 30, 370–382.
Jukes, T. H. and Cantor, C. R. (1969) Evolution of protein molecules In Mammalian Protein Metabolism, ed. Munro, H. N. (Academic, New York), pp. 21–132.
Kwiatowski, J., Hudson, R. R. and Ayala, F. J. (1991) The rate of Cu,Zn Superoxide dismutase evolution. Free Radical Res. Commun. 13, 363–370.
Kwiatowski, J., Skarecky, D. and Ayala, F. J. (1992) Structure and sequence of the Cu,Zn Sod gene in the Mediterranean fruit fly, Ceratitis capitata: Intron insertion/deletion and evolution of the gene. Mol. Phylogenet. Evol. 1, 72–82.
Li, W. H. and Graur, D. (1991) Fundamentals of Molecular Evolution (Sinauer, Sunderland, MA).
Margoliash, E. and Smith, E. (1965) Structural and functional aspects of cytochrome c in relation to evolution. In Evolving Genes and Proteins , eds. Bryson, V. and Vogel, H. J. (Academic, New York), pp. 221–242.
Shoemaker, J. S. and Fitch, W. M. (1989) Evidence from nuclear sequences that invariable sites should be considered when calculating sequence divergence. Mol. Biol. Evol. 6, 270–289.
Steinman, H. M. (1988) Bacterial superoxide dismutases. Basic Life Sci. 49, 641–646.
Stewart, C. B. & Wilson, A. C. (1987) Sequence convergence and functional adaptation of stomach lysozymes from foregut fermenters. Cold Spring Harbor Symposium. Quant. Biol. 52, 891–899.
Tainer, J. A., Getzoff, E. D., Beem, K. M., Richardson, J. S. and Richardson, D. C. (1982) Determination and analysis of the 2Å structure of copper, zinc superoxide dismutase. J. Mol. Biol. 160, 181–217.
Tainer, J. A., Getzoff, E. D., Richardson, J. S. and Richardson, D. C. (1983) Structure and mechanism of copper, zinc superoxide dismutase. Nature (London) 306, 284–287.
Zuckerkandl, E. and Pauling, L. (1962) Molecular disease, evolution and genetic heterogeneity. In Horizons in Biochemistry (Academic, New York), pp. 189–225.
| This page in the original is blank. |
















