
Page 60
2
Genetic and Molecular Basis of DNA Typing
This chapter describes the two principal kinds of genetic systems used in forensic DNA typing. Both take advantage of the great molecular variability in the human population, which makes it very unlikely that two unrelated persons have the same DNA profile. The first kind involves highly variable chromosomal regions that differ in length; the length measures are imprecise, so statistical procedures are needed to address the uncertainty. In the second kind, genetic variability is less, but the gene determination is usually unambiguous. Before describing the systems, we set forth some principles of genetics and molecular biology necessary for understanding them.
Fundamentals of Genetics1
In higher organisms, the genetic material is organized into microscopic structures called chromosomes. A fertilized human egg has 46 chromosomes (23 pairs), which, with appropriate staining and microscopic techniques, are visible in the cell nucleus. The two members of a pair are homologous. One member of each pair comes from the sperm and the other from the egg. Through the process of chromosomal duplication and separation (mitosis) at the time of cell
1 Introductions to genetics and molecular biology are available in various textbooks. Mange and Mange (1994) have written an easy-to-read, yet quite complete elementary textbook of human genetics. The basics of forensic DNA technology are given by Kirby (1992). For more details, see Ballantyne et al. (1989), Lee and Gaensslen (1990), Pena et al. (1993), Saferstein (1993), and Weir ( 1995b). A clear summary of the general principles and techniques is given in the Summary and Chapters I and 2 of the 1992 report (NRC 1992).
Page 61
division, the two daughter cells and the parent all are identical in chromosomal content, and, with a few exceptions, all the cells in the body should have chromosomes identical with those of the fertilized egg. The process sometimes errs: some cells have too many or too few chromosomes, and some differentiated tissues (such as liver) might have some cells with a different chromosome number (Therman and Susman 1993). But for the most part, cells throughout the body are identical in chromosomal composition.2 The most important exception occurs in the development of the reproductive cells. During formation of sperms and eggs, the process of reduction division (meiosis)—a chromosomal duplication followed by two cell divisions—halves the number of chromosomes from 46 to 23. Thus, sperms and eggs have only one member of each chromosome pair. The double number, 46, is restored by fertilization. A cell (or organism) with two sets of chromosomes is diploid. A cell, such as an egg, with one set is haploid.
Chromosomes vary greatly in size, but the two members of a homologous pair (one maternal and one paternal) are identical in microscopic appearance, except for the sex-chromosome pair, X and Y, in which the male-determining Y is much smaller than the X. A set of 23 chromosomes with the genetic information they contain is termed the genome.
A chromosome is a very thin thread of DNA, surrounded by other materials, mainly protein. If straightened out, an average chromosome would be an inch or more long. But it is arranged as coils within coils and so can be packed into a cell only a thousandth of an inch in diameter. The DNA thread is not visible in an ordinary microscope, and a stained chromosome is more rod-like than thread-like during the mitotic stages when it is most visible.
The DNA thread is actually double—two strands coiled around each other like a twisted rope ladder with stiff wooden steps (Figure 2. 1). The basic chemical unit of DNA is the nucleotide, consisting of a base (a half-step in the ladder) and a sugar-phosphate complex (the adjacent section of the rope). There are four kinds of bases, designated A, G, T, and C; A stands for adenine, G for guanine, T for thymine, and C for cytosine. The nucleotides of one DNA strand pair up in a specific fashion with those of the other to form the ladder; because of their specific size and complementary shape, T always pairs with A, and G with C. A DNA strand has a chemical directionality that is defined by the antisymmetry of the chemical connections between the successive sugars and phosphates in the two strands. In double-stranded DNA, the two strands run in opposite directions.
Because of the pairing rule just described, if we know the sequence of nucleotides on one strand, we automatically know the sequence on the other strand. A short segment of double-stranded DNA is shown below; the arrows indicate opposite directionality of the two strands.
2 More important for our purpose, tissues with different numbers of chromosomes (except for some malignancies) have the same DNA content as diploid cells.
Page 62
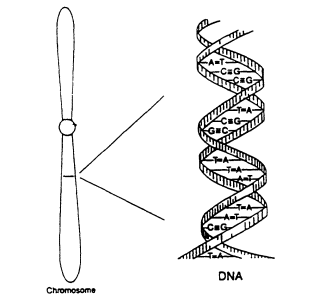
Figure 2.1
Diagram of the double-helical structure of DNA
in the chromosome. From NRC (1992).
![]() (1)
(1)
![]() (2)
(2)
Note that a T is always opposite an A and a G opposite a C. Because the chemical bonds holding the two bases (half-steps) together are weak, the two members of a base pair easily come apart; when that happens, the DNA ladder separates into two single strands. If a short single-strand segment, such as (1), is free in the cell, it will tend to pair with its complement, (2), even if the complement is part of a much longer piece of DNA. This process, termed hybridization, can occur in vitro and is one of the key properties that make DNA typing possible. In the laboratory, the two strands of DNA are easily separated by heat and rejoin at lower temperatures, so the process can be manipulated by such simple procedures as changing the temperature; chemical treatments can also be used.
The total DNA in a genome amounts to about 3 billion nucleotide pairs; because there are 23 chromosomes per genome, the average length of a chromosome is about 130 million nucleotide pairs. A gene is a segment of DNA, ranging from a few thousand to more than a hundred thousand nucleotide pairs, that contains the information for the structure of a functional product, usually a protein. The specific sequence of nucleotides in a gene acts as an encoded message that is translated into the specific amino acid sequence of a polypeptide or protein.
Page 63
The gene product might be detected only chemically or might lead to a visible trait, such as eye pigment. An alteration (mutation) of the gene might compromise the gene function and result in a disease, such as cystic fibrosis. The position on the chromosome where a particular gene resides is its locus.
Alternative forms of a gene, such as those producing normal and sickle-cell hemoglobin, are called alleles. If the same allele is present in both chromosomes of a pair, the person is homozygous; if the two alleles are different, the person is heterozygous. (The corresponding nouns are homozygote and heterozygote.) A person's genetic makeup is the genotype. Genotype can refer to a single gene locus with two alleles, A and a, in which case the three possible genotypes are AA, Aa, and aa; or it can be extended to several loci or even to the entire set of genes. In forensic analysis, the genotype for the group of analyzed loci is called the DNA profile. (The word fingerprint is sometimes used, but to avoid confusion with dermal fingerprints we shall use the word profile.)
The number of human genes is thought to be between 50,000 and 100,000; the number is quite uncertain. It is known, however, that genes make up only a small fraction of all the DNA in the genome. Even functional genes, especially larger ones, contain noncoding regions (introns). In fact, the great bulk of DNA has no known function. The chromosomal segments used most often in forensic analysis are usually in nonfunctional regions.
The sequence of nucleotides in the genome determines the genetic difference between one person and another. But the DNA of different persons is actually very similar. Corresponding sequences from the same genes in two people differ by an average of less than one nucleotide in 1,000 (Li and Sadler 1991). Yet the total number of nucleotides in a haploid genome is so large, about 3 billion, that any two people (unless they are identical twins) differ on the average in several million nucleotides. Most of the differences are outside the coding regions (genes), so the average number of nucleotide differences in the functional regions between two unrelated persons is much less. Nevertheless, the number of differences in the functional regions is large enough to account for the genetic diversity in the human population that is so apparent in such things as body shape, hair color, and facial appearance.
Before a cell divides, each chromosome is copied. In this process, the two strands of DNA in a short stretch separate, and each single strand copies its opposite, according to the A-T, G-C rule. The process proceeds, zipper-like, along the chromosome until there are two double strands where there was one before. (The entire chromosome is not actually copied sequentially from end to end—this would require more time than the interval between cell divisions; rather, there are multiple starting points along the chromosome.) When the cell divides, the two identical chromosomes, each half-old and half-new, go into separate daughter cells and ensure the genetic identity of the two cells.
Genes that are on the same chromosome are linked; that is, they tend to be inherited together. However, during the formation of a sperm or egg, the two
Page 64
members of a chromosomal pair line up side by side and randomly exchange parts, a process called crossing over or recombination. Therefore, genes that were once on the same chromosome might eventually be on a partner chromosome (Figure 2.2). Genes that are very close to one another on the same chromosome might remain associated for many generations before they are separated. Genes that are on nonhomologous chromosomes are inherited independently, as are genes far apart on the same chromosome. The allelic combinations eventually become randomized in the population, quickly if the loci are on nonhomologous chromosomes or far apart on the same chromosome, more slowly if the loci are closer together.
The process of DNA copying, although nearly exact, is not perfect, so a gene is sometimes changed to another form. This mistake, which can also happen in other ways (e.g., because of radiation and some chemicals), is called a mutation. Ordinarily this occurs very rarely; the probability of a typical gene's mutating is 1/100.000 or less per generation.3
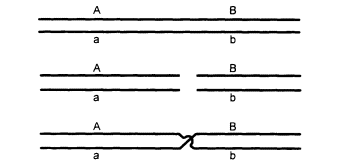
Figure 2.2
The exchange of parts between homologous chromosomes (crossing over).
The chromosomes pair (upper), break at corresponding points (middle), and exchange
parts (lower). The result is that genes that were formerly on the same strand are separated,
and vice versa. For example, alleles A and B, which were formerly on the same chromo-
some, are now on homologous chromosomes, and A and b, formerly separated, are now
together. (This shows only the two strands that participate in the event. Needless to say,
the actual molecular details of the process are more complicated.)
3 The human body has an enormous number of cells. Rarely, mutations occur in the body cells during development or later, after the organism has formed. Such so-called somatic mutations play an important role in the causation of cancer, but they are not a problem in forensic testing because the tiny fraction of mutant cells in a tissue sample are swamped by the much larger number of nonmutant cells. There is a remote possibility that a mutation might occur so early in embryonic development that DNA in eggs or sperm might differ from that in blood from the same person. We are not aware of any such instance in forensic work, although rare occurrences have been observed by researchers. Regardless, when any sample shows a three-allele pattern at one locus but not at others, additional testing should be done to resolve the uncertainty. If it should occur, it could lead to the conclusion that two samples of DNA from the same person came from two different persons.
Page 65
In view of the identical DNA composition in most cells, analysis of DNA from various tissues yields the same results. This is an important feature of DNA profiling because it means that cells from various parts of the body (such as blood, semen, skin, hair, and saliva) can be used.
VNTR Typing
The regions of DNA that have most often been used in forensic analysis have no product and no known function. They are known as minisatellites or variable-number tandem repeats (VNTRs). VNTR regions are not genes, and our interest in them is solely related to their use for identifying individuals. We therefore refer to them as markers.
In these regions, usually ranging from 500 to 10,000 nucleotide pairs, a core sequence of some 15-35 base pairs is repeated many times consecutively along the chromosome. In a VNTR, the number of repeats varies from person to person. At a given marker locus, sequences with different numbers of repeated units are called alleles, even though the word was originally applied to functional genes.
Because different alleles consist of different numbers of repeats, VNTR alleles can be identified by their lengths. If DNA fragments of different lengths are placed on a semisolid medium (gel) in an electric field, they migrate at different rates; different-sized fragments can therefore be identified by the distance they travel between electrodes in such a gel.
The VNTR loci chosen for forensic use are on different chromosomes, or sometimes very far apart on the same chromosome, so they are independently inherited. VNTR loci are particularly convenient for identification because they have a very large number of alleles, often a hundred or more.
One reason for the great variability of VNTRs is their high mutation rate, as much as 1% per generation (Jeffreys and Pena 1993). The repeated units predispose the chromosomes to mistakes in the process of replication and crossing over, thus increasing or decreasing the length (Armour and Jeffreys 1992; Olaisen et al. 1993). The large number of alleles means that the number of possible genotypes is enormous. For example, at a locus with 20 alleles, there are 20 homozygous genotypes, in addition to (20 x 19)/2 = 190 heterozygous ones, for a total of 210. With four such loci, the number of genotypes is 2104 or about 2 billion. With five loci, this number becomes more than 400 billion. The corresponding number of genotypes at a locus with 50 alleles is 1,275; the number for four such loci exceeds 2 trillion.
Another advantage of VNTRs for forensic work is that none of the alleles is very common. The different alleles are much more similar in frequency than multiple alleles of most genes. That is undoubtedly due to the high mutation rate and to the fact that most mutations increase or decrease the length of a VNTR by only one or a few units.
The essentials of the typing procedure are as follows (FBI 1990). The details
Page 66
vary somewhat from laboratory to laboratory; in a well-run operation, there are tests and checks at each stage to prevent errors. The technique is illustrated in Figure 2.3. First, the DNA is extracted from the source material and put into solution; the procedure differs according to whether the source is blood, 4 saliva, hair, semen, or other tissue. A portion of the DNA solution is tested to determine whether the amount and quality of DNA are sufficient for the analysis to be continued.
The next step involves cutting the DNA into small fragments. This is done with a restriction enzyme that recognizes a specific short DNA sequence and cuts the molecule at that point. For example, the enzyme HaeIII, widely used in forensic work, finds the sequence GGCC (CCGG on the other strand that is paired with it) wherever it exists and cuts both strands of the DNA between the G and the C. Thus, the DNA is cut into small pieces whose lengths are determined by the distances between successive GGCC sequences. This four-base sequence occurs millions of times in the genome, so the total DNA is chopped up into millions of fragments. Of course, the use of this enzyme generally requires that there be no GGCC sequences within any VNTR marker that will be analyzed; when such sequences are present, there are breaks within the VNTR leading to fragments of other sizes, and the analysis becomes more complicated.
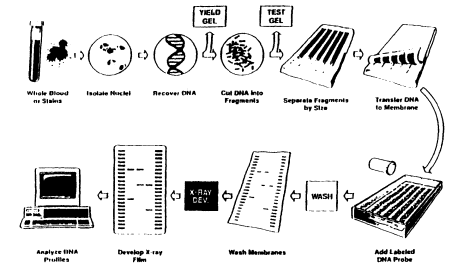
Figure 2.3
An outline of the DNA profiling process. The figure illustrates a procedure
for VNTR analysis of DNA extracted from whole blood. However,
nuclei are not usually isolated as a separate step in DNA typing.
4 Because red blood cells have no nuclei, they have no DNA. But white blood cells do have nuclei and are numerous enough for a small amount of blood usually to be sufficient for an analysis.
Page 67
The collection of fragments is then placed into a well on a flat gel, and the gel is placed in an electric field. After an appropriate length of time, the fragments migrate different distances in the electric field, depending mainly on their sizes, the smaller ones migrating more rapidly. This process is called electrophoresis. At this stage, the fragments are invisible. They are then chemically treated to separate the double strands into single ones.
Because the gels are difficult to work with, the single-stranded fragments are then transferred directly to a nylon membrane, to which they adhere. This process is called Southern blotting, named after its inventor. The fragments are then in the same positions on the membrane as they were on the gel. The next step is to flood the membrane with a single-stranded probe, a short segment of single-stranded DNA chosen to be complementary to a specific VNTR. The probe will hybridize with the DNA fragment that contains the target VNTR sequence and adhere to it. Any probe that does not bind to this specific DNA sequence is washed off. The probe also contains radioactive atoms. The nylon membrane is then placed on an x-ray film, and emissions from the probe expose the film at locations along the membrane where the probe has adhered to the VNTR. The film with its pictures of the radioactive spots is called an autoradiograph, or autorad. The process requires several days for sufficient radioactive decay to produce a visible band on the film.
Corresponding fragments from different persons differ in the number of repeat units; hence, the sizes of the fragments vary. That is reflected in their migrating at different rates in the electric field and showing up as bands in different positions on the autorad.
The number of different repeat units in VNTR markers can be very large. As a consequence, determining the exact number of repeats is beyond the resolving power of the usual laboratory technology, and analysis must allow for the resulting imprecision of the measurement. If two bands are visible on an autorad, the person is heterozygous. But if the bands occur in indistinguishable positions, so that only one is visible, the person is presumed to be homozygous. That causes no difficulty; treating a group of indistinguishable alleles as a single allele is a standard practice in traditional genetics.
Forensic VNTR DNA analysis involves testing at several loci, usually four or five, but often more. The analyst follows the procedure described above for one class of radioactive DNA (one probe). After an autoradiograph has been produced for one radioactive probe, this probe is washed off (stripped), another DNA probe targeting another VNTR locus on another chromosome is applied, and the procedure is repeated. The whole process is repeated for each of the multiple probes. Because it takes several days for sufficient radioactivity to be emitted to produce a visible band on the film, the entire process of four or five probes takes several weeks.
The position of a radioactively labeled band on the membrane is an indication of the size of the VNTR, usually expressed as the number of nucleotide pairs.
Page 68
Because of measurement uncertainty, the size of a band is not known exactly, and it is necessary to take this uncertainty into account in analyzing autorads (see Chapter 5).
Figure 2.4 shows an autorad for one locus (D1S7) in an actual case. (In this notation, the first number, I in this case, indicates that this locus is on chromosome

Figure 2.4
An autoradiograph from an actual case. This autorad illustrates restriction
fragment-length variation at the D1S7 locus. The lanes from left to right are: (1)
standard DNA fragment sizing ladder; (2) K562, a standard cell line
with two bands of known molecular weight; (3) within-laboratory blind quality-
control sample; (4) standard DNA-fragment sizing ladder; (5) DNA from the evidence
blood stain; (6) standard DNA-fragment sizing ladder; (7) DNA from the first victim; (8)
another sample from the first victim; (9) standard DNA-fragment sizing ladder; (10)
DNA from the second victim; (11)DNA from the first suspect; (12) DNA from the
second suspect; (13) standard DNA-fragment sizing ladder. Courtesy of the State of
California Department of Justice DNA Laboratory.
Page 69
number one.) Suspects S1 and S2 were charged with having beaten to death two victims, V1 and V2. Blood stains (E blood) were found on the clothing of S1. K562 is from a human cell line and is a widely used laboratory standard. Lanes 1, 4, 6, 9, and 13 show standard DNA fragments used as a molecular-weight sizing ladder. Using multiple lanes for the sizing ladder allows more accurate sizing of the DNA fragments. The quality-control lane (QC) is a blood stain given to the analyst at the beginning of the case, to be processed in parallel with the evidence sample; it is a blind test for the analyst and must meet laboratory specifications. In this particular case, full testing using 10 loci gave consistent matches between E blood and Victim 1.
Bands of similar size are often grouped into bins, sets of VNTR alleles of similar size. The usual width of a bin is about 10% of the mean size of the VNTR segment at the center of the bin. The alleles within a bin are treated as though they are a single allele. The words homozygous and heterozygous then apply to persons whose DNA falls into the same or different bins.
The presence of a single band in a lane might mean that the person is homozygous, but the person could also be heterozygous and the second band for some reason is not visible. Two bands might be so close together that they appear as one on the gel, a second band might be too faint to see (sometimes a problem with degraded material), or the second band might be from an allele so large or small as to fall outside the size range that can be distinguished by electrophoresis. There is a rule for dealing with this situation, and we discuss it in Chapter 4.
In an effort to avoid the use of radioactivity, some laboratories are beginning to use luminescent molecules as labels on their probes. An added benefit of this approach is that analysis of each probe can be completed within a single working day. As these methods are perfected and become more widespread, the time required for an analysis will be greatly reduced and the problems of disposal of radioactive waste circumvented.
PCR-Based Methods
The polymerase chain reaction (PCR) is a laboratory process for copying a chosen short segment of DNA millions of times. The process is similar to the mechanism by which DNA duplicates itself normally. The PCR process consists of three steps. First, each double-stranded segment is separated into two strands by heating. Second, these single-stranded segments are hybridized with primers, short DNA segments (20-30 nucleotides in length) that complement and define the target sequence to be amplified. Third, in the presence of the enzyme DNA polymerase, and the four nucleotide building blocks (A, C, G, and T), each primer serves as the starting point for the replication of the target sequence. A copy of the complement of each of the separated strands is made, so that there are two double-stranded DNA segments. This three-step cycle is repeated, usually 20-35 times. The two strands produce four copies; the four, eight copies; and so
Page 70
on until the number of copies of the original DNA is enormous. The main difference between this procedure and the normal cellular process is that the PCR process is limited to the amplification of a small DNA region. This region is usually not more than 1,000 nucleotides in length, so PCR methods cannot, at least at present, be used for large DNA regions, such as most VNTRs. There is a possibility that this limitation may soon be removed (Barnes 1994).
The PCR process is relatively simple and is easily carried out in the laboratory. Results can be obtained within a short time, often within 24 hours, in contrast with the several weeks required for a complete VNTR analysis. Because the amplification is almost unlimited, PCR-based methods make possible the analysis of very tiny amounts of DNA. This advantage makes the technique particularly useful for forensic analysis, in that the amount of DNA in some forensic samples, such as single shed hairs or saliva traces on cigarette butts, is minute. The technique extends DNA typing to evidence samples that at present cannot be typed with other approaches. Moreover, the small amount of DNA required for PCR analysis makes it easier to set aside portions of samples for repeat testing in the same or another laboratory. Amplification of samples that contain degraded DNA is also possible; this allows DNA typing of old and decayed samples, remains of fire and accident victims, decayed bodies, and so on.
There is another advantage of PCR-based methods. They usually permit an exact identification of each allele, in which case there are no measurement uncertainties. Thus, the calculations and statistical analysis associated with matching and binning of VNTRs are not needed. Nevertheless, ambiguity can sometimes arise if there are mutations that alter individual repeats, and binning or some other adjustment may be required.
Given those advantages, it is not surprising that PCR-based typing is widely and increasingly used in forensic DNA laboratories in this country and abroad. Many forensic laboratories carry out PCR-based typing along with VNTR typing. Some laboratories, particularly smaller ones, have gone exclusively to PCR techniques.
Once the amount of DNA is amplified by PCR methods, the analysis proceeds in essentially the same way as with VNTRs. There are minor procedural modifications, but the general procedures are the same—identification of fragments of different size by their migration in an electric field.
Another class of repeated units is STRs, short tandem repeats of a few nucleotide units. These are very common and are distributed widely throughout the genome (Edwards et al. 1992; Hammond et al. 1994). Because the total length is short, STRs can be amplified with PCR. Alleles differing in size can be resolved to the scale of single bases with both manual and automated sequencing technologies. Moreover, it has proved possible to co-amplify STRs at multiple loci, allowing significant increases in the speed of test processing (Klimpton et al. 1993; Hammond et al. 1994). They do not have as many alleles per locus as VNTRs, but that is compensated by the very large number of loci that are
Page 71
potentially usable. As more STRs are developed and validated, this system is coming into wide use.
Any procedure that uses PCR is susceptible to error caused by contamination leading to amplification of the wrong DNA. The amplification process is so efficient that a few stray molecules of contaminating DNA can be amplified along with the intended DNA. Most such mistakes are readily detected after the PCR analysis is completed because the contaminating DNA yields a weak pattern that differs from the predominant pattern. Most undetected contamination is likely to lead to a false-negative result; that is, a nonmatch might be declared when a match actually exists. Nevertheless, false-positive results are also possible, in which the profile from an evidence sample is falsely declared to match the genetic type of another person. That could happen, for example, if by mistake the same amplified sample were used twice in a given analysis, instead of two different samples. Procedures for minimizing the occurrence of errors are discussed in Chapter 3.
A second disadvantage of most markers used in PCR-based typing is that they have fewer alleles than VNTRs and the distribution of allele frequencies is not as flat. Hence, more loci are required to produce the same amount of information about the likelihood that two persons share a profile. Furthermore, some of these loci are functional (they are genes, not just markers). Those are more likely to be subject to natural selection and therefore might not conform strictly to some of the population-genetics assumptions used in evaluating the significance of a match (discussed in Chapter 4). In the future, loci that are brought on as markers should be chosen so as not to be linked to important disease-producing genes, so that the markers can more confidently be treated as neutral, and to provide greater assurance of genetic privacy. In fact, some three-base repeating units are the cause of severe human diseases (Wrogemann et al. 1993; Sutherland and Richards 1995), and even some VNTRs might have disease associations (Krontiris 1995). These are not used in forensics, however.
One application of PCR in forensic work has used the DQA locus (the gene is called DQA, its product, DQa) (Blake et al. 1992; Comey et al. 1993). In distinction to VNTRs, the alleles at this locus code for a protein. This locus is part of the histocompatibility complex, a group of highly variable genes responsible for recognizing foreign tissue. Eight alleles at the DQA locus have been identified, although only six are commonly used in forensic work. The different alleles can be distinguished by specific probes. With these six alleles there are 21 possible genotypes; six homozygous and 15 heterozygous.
Analysis of DQA uses the same DNA hybridization technique as VNTR analysis. In this case, probes specific for individual alleles are placed in designated locations on a membrane (because the probes, rather than the DNA to be typed, are fixed on the membrane, this is called a reverse blot). The amplified DNA is then added, and the DNA from whatever DQA alleles are present hybridizes with the appropriate probe. A stain reaction specific for double-stranded DNA
Page 72
shows up as a colored spot on the membrane wherever specific hybridization occurs. The positions of the colored spots on the membrane strip indicate which alleles are present.
The DQA system has several advantages. It is quick and reliable, so it is useful as a preliminary test. It can also be used, with other markers, as part of a more detailed DNA profile. In practice, a substantial fraction of suspects are cleared by DNA evidence, and prompt exclusion by the DQA test is obviously preferable to waiting months for results of a VNTR test. On the average, the DQA genotype of a given person is identical with that of about 7% of the population at large, so an innocent person can expect to be cleared in short order 93% of the time. This high probability might not be achieved if the sample includes DNA from more than one individual.
Another system that is beginning to be widely used is the Amplitype poly-marker (PM) DNA system. This system analyzes loci simultaneously: LDLR (low-density-lipoprotein receptor), GYPA (glycophorin A, the MN blood-groups), HBGG (hemoglobin gamma globin), D7S8 (an anonymous genetic marker on chromosome 7), and GC (group-specific component). There are two or three distinguishable alleles at each locus. The system has been validated with tests for robustness with respect to environmental insults (Herrin et al. 1994; Budowle, Lindsay, et al. 1995), and there is substantial information on population frequencies, which is discussed in Chapter 4.
Other PCR-based techniques have been or are being developed. For example, D1S80 is a VNTR in which the largest allele is less than 1,000 bp long. Its value for forensic analysis has been validated in a number of tests (Sajantila et al. 1992; Herrin et al. 1994; Budowle, Baechtel, et al. 1995; Cosso and Reynolds 1995). The locus consists of a 16-base unit that is repeated a variable number of times. There are more than 30 distinguishable alleles. The size classes are fully discrete, so usually each allele can be distinguished unambiguously. However, some ambiguous alleles are caused by insertion or deletion of a single base and these complicate the analysis.
Another class of genetic marker is mitochondrial DNA. Mitochondria are microscopic particles found in the cell, but outside the nucleus, so they are not associated with the chromosomes. The transmission of mitochondria is from mother to child; the sperm has very little material other than chromosomes. Ordinarily, all the mitochondrial particles in the cell are identical. There is no problem distinguishing heterozygotes from homozygotes, since only one kind of DNA is present. Since mitochondrial DNA is always transmitted through the female, all the children of one woman have identical mitochondrial DNA. Therefore, siblings, maternal half-siblings, and others related through female lines are as much alike in their mitochondrial DNA as identical twins. Mitochondrial DNA is particularly useful for associating persons related through their maternal lineage, for example, for associating skeletal remains to a family.
A highly variable region of mitochondrial DNA is used for forensic analysis.
Page 73
The techniques have been validated, and there is a growing body of frequency data. For a detailed account of the methodology and validation, see Wilson et al. (1993). A disadvantage for forensic use is that siblings cannot be distinguished, nor can other maternally related relatives, such as cousins related through sisters. Since mitochondria are inherited independently of the chromosomes, mitochondrial information can be combined with nuclear data to yield probabilities of a random match (see Chapter 4).
A promising technique is minisatellite repeat mapping, or digital typing, which, apart from length variation, detects sequence differences within the base sequences repeated in VNTRs (Jeffreys et al. 1991; Armour and Jeffreys 1992; Monckton et al. 1993). Although technical limitations still need to be overcome before this system can be used in forensic analysis, it could have a particular advantage, in that it uses the same loci that have already been extensively studied in various populations and subpopulations.
Table 2.1 summarizes the most widely used systems.
Conclusions
DNA analysis is one of the greatest technical achievements for criminal investigation since the discovery of fingerprints. Methods of DNA profiling are firmly grounded in molecular technology. When profiling is done with appropriate care, the results are highly reproducible. In particular, the methods are almost certain to exclude an innocent suspect.
One of the most widely used techniques today involves VNTRs. These loci are extremely variable, but individual alleles cannot be distinguished, because of intrinsic measurement variability, and the analysis requires statistical procedures. It involves radioactivity and requires a month or more for full analysis. PCR-based methods are prompt, require only a small amount of material, and can yield unambiguous identification of individual alleles. Various PCR methods, particularly STRs, are increasingly being used.
The state of the profiling technology and the methods for estimating frequencies and related statistics have progressed to the point where the admissibility of properly collected and analyzed DNA data should not be in doubt. We expect continued development of new and better methods and hope for prompt validation so that they can quickly be brought into use.
Page 74
| TABLE 2.1 Genetic Markers Used in Forensic Identification | ||||
| Nature of Variation at Locus | ||||
| Locus Example | Method of Detection | Number of Alleles | Diversitya | |
| Variable Number Tandem Repeat (VNTR)b | ||||
| D2S44 (core repeat 31 bp) | Intact DNA digested with restriction enzyme, producing fragments that are separated by gel electrophoresis; alleles detected by Southern blotting followed by probing with locus-specific radioactive or chemiluminescent probe | At least 75 (size range 700-8500 bp); allele size distribution continuous | ca. 95% in all populations studied | |
| DIS80 (core repeat 16 bp) | Amplification of allelic sequences by PCR; discrete allelic products separated by electrophoresis and visualized directly | ca. 30 (size range 350-1000 bp); alleles can be discretely distinguished | 80-90%, depending on population | |
| Short Tandem Repeat (STR)c | ||||
| HUMTHO 1 (tetranucleotide repeat) | Amplification of allelic sequences by PCR; discrete allelic products separated by electrophoresis on sequencing gels and visualized directly | 8 (size range 179203 bp); alleles can be discretely distinguished | 70-85%, depending on population | |
| Simple Sequence Variation d | ||||
| DQA (an expressed gene in the histocompatibility complex) | Amplification of allelic sequences by PCR; discrete alleles detected by sequence-specific probes | 8 (6 used in DQA kit) | 85-95%, depending on population | |
| Polymarker (a set of 5 loci) | Amplification of allelic sequences by PCR; discrete alleles detected by sequencespecific probes | Loci are bi- or triallelic; 972 genotypic combinations | 37-65%, depending on locus and population | |
| Mitochondrial DNA Control Region (Dloop) | Amplification of control-region sequence and sequence determination | Hundreds of sequence variants known | Greater than 95% | |
| aIn a randomly mating diploid population, diversity is the same as heterozygosity. In general, including haploid mitochondria, the value is 1 - Sipi2 (for explanation, see Chapter 4). | ||||
| bVNTR loci contain repeated core sequence elements, typically 15-35 bp in length. Alleles differ in number of repeats and are differentiated on the basis of size. | ||||
| c STR loci are like VNTR loci except that the repeated core sequence elements are 2-6 bp in length. Alleles differ in number of repeats and are differentiated on the basis of size. | ||||
| d Nucleotide substitution in a defined segment of a sequence. | ||||















