
Page 9
Overview
This overview describes the essentials of the subject with a minimum of jargon, statistics, and technical details. The aim is to present technical information in nontechnical language, but without distorting the meaning by oversimplifying. Although this overview is intended to be self-contained, we shall refer to relevant sections in the main report for fuller explanations, corroborative details, and justification of recommended procedures. We have included an illustrative example at the end of the overview. The glossary and the list of abbreviations at the end of the report may be useful.
Introduction
DNA typing, with its extremely high power to differentiate one human being from another, is based on a large body of scientific principles and techniques that are universally accepted. These newer molecular techniques permit the study of human variability at the most basic level, that of the genetic material itself, DNA. Standard techniques of population genetics and statistics can be used to interpret the results of forensic DNA typing. Because of the newness of the techniques and their exquisite discriminating power, the courts have subjected DNA evidence to extensive scrutiny. What at first seemed like daunting complexity in the interpretation of DNA tests has sometimes inhibited the full use of such evidence. An objective of this report is to clarify and explain how DNA evidence can be used in the courtroom.
If the array of DNA markers used for comparison is large enough, the chance that two different persons will share all of them becomes vanishingly small. With
Page 10
appropriate DNA test systems, the uniqueness of any individual on the planet (except an identical twin) is likely to be demonstrable in the near future. In the meantime, the justification for an inference that two identical DNA profiles come from the same person rests on probability calculations that employ principles of population genetics. Such calculations are, of course, subject to uncertainty. When in doubt, we err on the side of conservatism (that is, in favor of the defendant). We also discuss ways of keeping laboratory and other errors to a minimum. We emphasize that DNA analysis, when properly carried out and interpreted, is a very powerful forensic tool.
Our Assignment
This committee was asked to update an earlier report, prepared for the National Research Council (NRC) in 1992. There are two principal reasons why such an update is needed. First, forensic science and techniques have progressed rapidly in recent years. Laboratory standards are higher, and new DNA markers are rapidly being introduced. An abundance of new data on DNA markers in different population groups is now available, allowing estimates of the frequencies of those markers in various populations to be made with greater confidence. Second, some of the statements in the first report have been misinterpreted or misapplied in the courts.
This report deals mainly with two subjects:
The first involves the laboratory determination of DNA profiles. DNA can be obtained in substantial amounts and in good condition, as when blood or tissue is obtained from a person, or it can be in limited amounts, degraded, or contaminated, as in some samples from crime scenes. Even with the best laboratory technique, there is intrinsic, unavoidable variability in the measurements; that introduces uncertainty that can be compounded by poor laboratory technique, faulty equipment, or human error. We consider how such uncertainty can be reduced and the risk of error minimized.
The second subject is the interpretation of a finding that the DNA profile of a suspect (or sometimes a victim) matches that of the evidence DNA, usually taken from the crime scene. The match might happen because the two samples are from the same person. Alternatively it might be that the samples are from different persons and that an error has occurred in the gathering of the evidence or in the laboratory. Finally, it might be that the samples are from different people who happen to have the same DNA profile; the probability of that event can be calculated. If the probability is very low, then either the DNA samples are from the same person or a very unlikely coincidence has occurred.
The interpretation of a matching profile involves at least two types of uncertainty. The first arises because the US population is not homogeneous. Rather it consists of different major races (such as black and white), within which there
Page 11
are various subgroups (e.g., persons of Italian and Finnish ancestry) that are not completely mixed in the "melting pot." The extent of such population structure and how it can be taken into account are in the province of population genetics.
The second uncertainty is statistical. Any calculation depends on the numbers in available databases. How reliable are those numbers and how accurate are the calculations based on them and on population genetic theory? We discuss these questions and give answers based on statistical theory and empirical observations.
Finally, some legal issues are discussed. We consider how the courts have reacted to this new technology, especially since the 1992 NRC report.
That earlier report considered a number of issues that are outside our province. Issues such as confidentiality and security, storage of samples for possible future use, legal aspects of data banks on convicted felons, non-DNA information in data banks, availability and costs of experts, economic and ethical aspects of new DNA information, accountability and public scrutiny, and international exchange of information are not in our charge.
As this report will reveal, we agree with many recommendations of the earlier one but disagree with others. Since we make no attempt to review all the statements and recommendations in the 1992 report, the lack of discussion of such an item should not be interpreted as either endorsing or rejecting it.
DNA Typing
DNA typing for forensic purposes is based on the same fundamental principles and uses the same techniques that are routinely employed in a wide variety of medical and genetic situations, such as diagnosis and gene mapping. Those methods analyze the DNA itself. That means that a person's genetic makeup can be determined directly, not indirectly through gene products, as was required by earlier methods. DNA is also resistant to many conditions that destroy most other biological compounds, such as proteins. Furthermore, only small amounts of DNA are required; that is especially true if PCR (polymerase chain reaction) methods, to be described later, are employed. For those reasons, direct DNA determinations often give useful results when older methods, such as those employing blood groups and enzymes, do not.
We emphasize that one of the most important benefits of DNA technology is the clearing of falsely-accused innocent suspects. According to the FBI, about a third of those named as the primary suspect in rape cases are excluded by DNA evidence. Cases in which DNA analysis provides evidence of innocence ordinarily do not reach the courts and are therefore less widely known. Prompt exclusions can eliminate a great deal of wasted effort and human anguish.
Before describing the techniques of DNA identification, we first provide some necessary genetic background and a minimum vocabulary.
Page 12
Basic Genetic Principles
Each human body contains an enormous number of cells, all descended by successive divisions from a single fertilized egg. The genetic material, DNA, is in the form of microscopic chromosomes, located in the inner part of the cell, the nucleus. A fertilized egg has 23 pairs of chromosomes, one member of each pair having come from the mother and the other from the father. The two members of a pair are said to be homologous. Before cell division, each chromosome splits into two. Because of the precision of chromosome distribution in the cell-division process, each daughter cell receives identical chromosomes, duplicates of the 46 in the parent cell. Thus, each cell in the body should have the same chromosome makeup. This means that cells from various tissues, such as blood, hair, skin, and semen, have the same DNA content and therefore provide the same forensic information. There are some exceptions to the rule of identical chromosomes in every cell, but they do not affect the conclusion that diverse tissues provide the same information.
The most important exception occurs when sperm and eggs are formed. In this process, each reproductive cell receives at random one representative of each pair, or 23 in all. The double number, 46, is restored by fertilization. With the exception of the sex chromosomes, X and Y (the male-determining Y is smaller than the X), the two members of a pair are identical in size and shape. (It might seem puzzling that sperm cells, with only half of the chromosomes, can provide the same information as blood or saliva. The reason is that DNA from many sperm cells is analyzed at once, and collectively all the chromosomes are represented.)
A chromosome is a very thin thread of DNA, surrounded by other materials, mainly protein. (DNA stands for deoxyribonucleic acid.) The DNA in a single chromosome, if stretched out, would be an inch or more in length. Remarkably, all that length is packed into a cell nucleus some 1/1,000 inch in diameter. The DNA is compacted by coils within coils.
The DNA thread is actually double, consisting of two strands twisted to form a helix (Figure 0.1). Each strand consists of a string of bases held together by a sugar-phosphate backbone. The four bases are abbreviated A, T, G, and C (these stand for adenine, thymine, guanine, and cytosine, but we shall employ only the abbreviations). In double-stranded DNA, the bases line up in pairs, an A opposite a T and a G opposite a C:
| C A T T A G A C T G A T |
Thus, if the sequence of bases on one strand is known, the other is determined.
Prior to cell division, the double strand splits into two single strands, each containing a single base at each position. There are free-floating bases in the cell nucleus, and these attach to each single strand according to the A-T, G-C pairing rule. Then they are tied together and zipped up by enzymes. In this way, each
Page 13
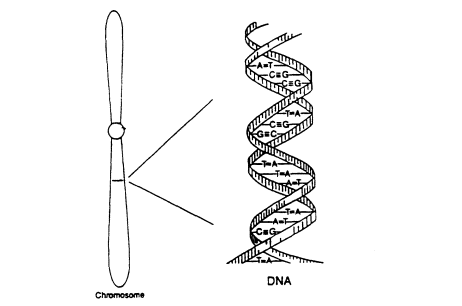
Figure 0.1
Diagram of a chromosome, with a small region
expanded to show the double-helical structure of DNA. The ''steps
" of the twisted ladder are four kinds of base pairs, AT, TA, GC,
or CG. From NRC (1992).
DNA double helix makes a copy of itself. There are then two identical double strands, each half old and half new, and one goes to each daughter cell. That accounts for the uniformity of DNA makeup throughout the body. The total number of base pairs in a set of 23 chromosomes is about 3 billion.
A gene is a stretch of DNA, ranging from a few thousand to tens of thousands of base pairs, that produces a specific product, usually a protein. The order of the four kinds of bases within the gene determines its function. The specific base sequence acts as an encoded message written in three-letter words, each specifying an amino acid (a protein building block). In the diagram above, CAT specifies one amino acid, TAG another, ACT a third, and so on. These amino acids are joined together to make a chain, which folds in various ways to make a three dimensional protein. The gene product may be detected by laboratory methods, as with blood groups, or by some visible manifestation, such as eye color.
The position that a gene occupies along the DNA thread is its locus. In chemical composition, a gene is no different from the rest of the DNA in the chromosome. Only its having a specific sequence of bases, enabling it to encode a specific protein, makes each gene unique. Genes are interspersed among the rest of the DNA and actually compose only a small fraction of the total. Most of the rest has no known function.
Page 14
Alternative forms of a gene, for example those producing normal and sicklecell hemoglobin, are called alleles. The word genotype refers to the gene makeup. A person has two genes at each locus, one maternal, one paternal. If there are two alleles, A and a, at a locus, there are three genotypes, AA, Aa, and aa. The word genotype can be extended to any number of loci. In forensic work, the genotype for the group of analyzed loci is called the DNA profile. (We avoid the word fingerprint to prevent confusion with dermal fingerprints.) If the same allele is present in both chromosomes of a pair, the person with that pair is homozygous. If the two are different, the person is heterozygous. (The corresponding nouns are homozygote and heterozygote.) Thus, genotypes AA and aa are homozygous and Aa is heterozygous.
Genes on the same chromosome are said to be linked, and they tend to be inherited together. They can become unlinked, however, by the process of crossing over, which involves breakage of two homologous chromosomes at corresponding sites and exchange of partners (Figure 0.2). Genes that are on nonhomologous chromosomes are inherited independently, as are genes far apart on the same chromosome.
Occasionally, an allele may mutate; that is, it may suddenly change to another allele, with a changed or lost function. When the gene mutates, the new form is copied as faithfully as the original gene, so a mutant gene is as stable as the gene before it mutated. Most genes mutate very rarely, typically only once in some 100,000 generations, but the rates for different genes differ greatly. Mutations can occur in any part of the body, but our concern is those that occur in the reproductive system and therefore can be transmitted to future generations.
Forensic DNA Identification
VNTRs
One group of DNA loci that are used extensively in forensic analysis are those containing Variable Numbers of Tandem Repeats (VNTRs). These are not genes, since they produce no product, and those that are used for forensic determinations have no known effect on the person. That is an advantage, for it means that VNTRs are less likely to be influenced by natural selection, which could lead to different frequencies in different populations. For example, several genes that cause malaria resistance are more common in people of Mediterranean or African ancestry, where malaria has been common.
A typical VNTR region consists of 500 to 10,000 base pairs, comprising many tandemly repeated units, each some 15 to 35 base pairs in length. The exact number of repeats, and hence the length of the VNTR region, varies from one allele to another, and different alleles can be identified by their lengths. VNTR loci are particularly convenient as markers for human identification because they have a very large number of different alleles, often a hundred or more, although
Page 15
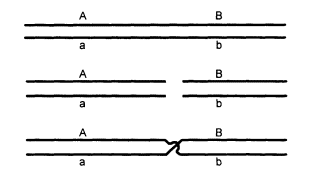
Figure 0.2
Diagram of crossing over. The chromosomes pair
(upper diagram), break at corresponding points (middle),
and exchange parts. The result is that alleles A and B,
which were formerly on the same chromosome,
are now on different chromosomes.
only 15 to 25 can be distinguished practically, as we explain later. (The word allele is traditionally applied to alternative forms of a gene; here we extend the word to include nongenic regions of DNA, such as VNTRs.)
VNTRs also have a very high mutation rate, leading to changes in length. An individual mutation usually changes the length by only one or a few repeating units. The result is a very large number of alleles, no one of which is common. The number of possible genotypes (pairs of alleles) at a locus is much larger than the number of alleles, and when several different loci are combined, the total number of genotypes becomes enormous.
To get an idea of the amount of genetic variability with multiple alleles and multiple loci, consider first a locus with three alleles, A1, A2, and A3. There are three homozygous genotypes, A1A1, A2A2, and A3A3, and three heterozygous ones, A1A2, A1A3, and A2A3. In general, if there are n alleles, there are n homozygous genotypes and n(n- 1)/2 heterozygous ones. For example, if there are 20 alleles, there are 20 + (20 X 19)/2 = 210 genotypes. Four loci with 20 alleles each would have 210 X 210 X 210 X 210, or about 2 billion possible genotypes.
For a genetic system to be useful for identification, it is not enough that it yield a large number of genotypes. The relative frequencies of the genotypes are also important. The more nearly equal the different frequencies are, the greater the discriminatory power. VNTRs exhibit both characteristics.
DNA Profiling
Genetic types at VNTR loci are determined by a technique called VNTR profiling. Briefly, the technique is as follows (Figure 0.3). First, the DNA is extracted from whatever material is to be examined. The DNA is then cut by a specific enzyme into many small fragments, millions in each cell. A tiny fraction of those fragments includes the particular VNTR to be analyzed. The fragmented
Page 16
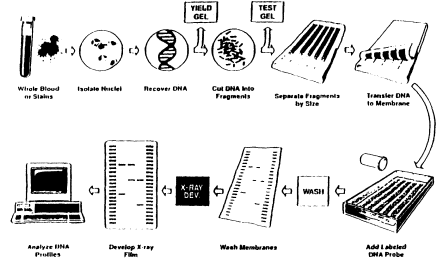
Figure 0.3
An outline of the DNA profiling process.
DNA is then placed in a small well at one edge of a semisolid gel. Each of the different DNA samples to be analyzed is placed in a different well. Additional wells receive various known DNA samples to serve as controls and fragment-size indicators. Then the gel is placed in an electric field and the DNA migrates away from the wells. The smaller the fragment, the more rapidly it moves. After a suitable time, the electric current is stopped, and the different fragments will have migrated different distances, the shorter ones for greater distances.
In the process, the DNA fragments are denatured, meaning that the double strands in each fragment are separated into single strands. The fragments are then transferred by simple blotting to a nylon membrane, which is tougher and easier to handle than the gel and to which the single-stranded fragments adhere. Then a radioactive probe is added. A probe is a short section of single-stranded DNA complementary to the specific VNTR of interest, meaning that it has a C where the VNTR has a G, an A where the VNTR has a T, and so on, so that the probe is specifically attracted to this particular VNTR. When the membrane is placed on a photographic film, the radioactive probes take a picture of themselves, producing dark spots on the film at positions corresponding to the particular DNA fragments to which the probe has attached. This photo is called an autoradiograph, or autorad for short.
The two DNA samples to be compared (usually from the evidence, E, and from a suspect, S) are placed in separate lanes in the gel, with DNA in several other lanes serving as different kinds of controls. Because of the large number of VNTR alleles, most loci are heterozygous, and there will usually be two bands
Page 17
in each lane. If the two DNA samples, E and S, came from the same individual, the two bands in each lane will be in the same, or nearly the same, positions; if the DNA came from different persons, they will usually be in quite different positions. The sizes of the fragments are estimated by comparison with a "ladder" in which the spots are of known size.
Figure 0.4 shows an example. In this case, the question is whether either of two victims, V1 and V2, match a blood stain, called E blood in the figure,

Figure 0.4
An autorad from an actual case, illustrating fragment-length variation at the D1S7 locus.
The lanes from left to right are: (1) standard DNA ladder, used to estimate sizes;
(2) K562, a standard cell line with two bands of known size, used as a control: (3)
within-laboratory quality control sample; (4) standard ladder; (5) DNA from blood at the
crime scene; (6) standard ladder; (7) DNA from the first victim: (8) another sample from
the first victim; (9) standard ladder; (10) DNA from the second victim; (11) DNA from
the first suspect; (12) DNA from the second suspect; (13) standard ladder. Courtesy of
the State of California Department of Justice DNA Laboratory.
Page 18
that was found on the clothing of suspect S 1. S2 is a second suspect in the case. The sizing ladders are in lanes 1, 4, 6, 9, and 13; these are repeated in several lanes to detect possible differences in the rate of migration in different lanes. K562 and QC are other controls. On looking at the figure, one sees that the evidence blood (E blood) is not from V2 (or from S1 or S2), since the bands are in quite different positions. However, it might well be from V , since the bands in E and V1 are at the same position.
After such an analysis, the radioactive probe is washed off the membrane. Then a new probe, specific for another VNTR locus, is added and the whole process repeated. This is continued for several loci, usually four or more. There is a practical limit, however, since the washing operation may eventually remove some of the DNA fragments, making the bands on the autorad weak or invisible. In the example in Figure 0.4, testing at 9 additional loci gave consistent matches between E blood and Victim 1, leaving little doubt as to the source of the blood.
In most laboratories, the sizes of the fragments are measured by a computer, which also does the calculations that are described below.
A DNA fragment from the evidence is declared to match the one from a suspect (or, in the case of Figure 0.4, from a victim) if they are within a predetermined relative distance. If the bands do not match, that is the end of the story: the DNA samples did not come from the same individual. If the DNA patterns do match, or appear to match, the analysis is carried farther, as described in the next section.
A difficulty with VNTRs using radioactive probes is the long time required to complete the analysis. One or two weeks are needed for sufficient radiation to make a clear autorad, and, as just described, the different loci are done in succession. As a result, the process takes several weeks. Some newer techniques use luminescent chemicals instead of radioactive ones. As such techniques are perfected and come into wider use, the process will speed up considerably.
Matching and Binning of VNTRs
Because of measurement uncertainty, the estimates of fragment sizes are essentially continuous. The matching process consists of determining whether two bands are close enough to be within the limits of the measurement uncertainty. After the two bands have been determined to match, they are binned. In this process, the band is assigned to a size class, known as a bin. Two analytical procedures are the-fixed-bin and the floating-bin methods. The floating-bin method is statistically preferable, but it requires access to a computerized data base. The fixed-bin method is simpler in some ways and easier for the average laboratory to use; hence, it is more widely employed. Only the fixed-bin method is described here, but the reader may refer to Chapter 5 (p 142) for a description of floating-bin procedures.
Page 19
A match between two different DNA sources (e.g., evidence and suspect DNA) is typically determined in two stages. First is a visual examination. Usually the bands in the two lanes to be compared will be in very similar positions or in clearly different positions. In the latter case, there is no match, and the DNA samples are assumed to have come from different persons. In Figure 0.4, only the bands of V1 match the evidence blood. The role of a visual test is that of a preliminary screen, to eliminate obvious mismatches from further study and thereby save time and effort.
The second, measurement-confirmation step is based on the size of the fragment producing the band, as determined by size standards (the standard ladders) on the same autorad (Figure 0.4). The recorded size is subject to measurement uncertainty, which is roughly proportional to the fragment size. Based on duplicate measurements of the same sample in different laboratories, roughly 2/3 of the measurements are within 1% of the correct value. In practice, a value larger than 1%, usually 2.5%—although this varies in different laboratories—is used to prevent the possible error of classifying samples from the same person as being different. The measurement with 2.5% of its value added and subtracted yields an uncertainty window.
Two bands, say from suspect and evidence, are declared to match if their uncertainty windows overlap; otherwise a nonmatch is declared. Compare the top two diagrams in Figure 0.5.
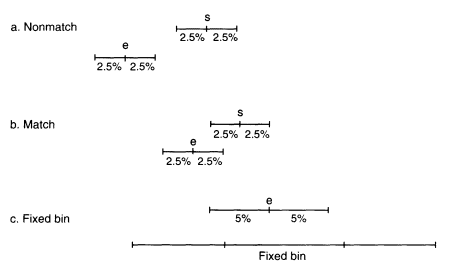
Figure 0.5
Diagrams showing the extent of the uncertainty windows (a,b) and the match window
(c). In the top group, the uncertainty windows do not overlap; in the second they do.
The bottom diagram shows the match window of a fragment along with the fixed
bin. The match window overlaps bins 10 and 11.
Page 20
The match window is the evidence measurement with 5% of its value added and subtracted. This is compared with the bins in the database (such as those in Table O.1). If the upper and lower values lie within a bin, then the frequency of that bin is used to calculate the probability of a random match. Often, two or more bins will be overlapped by the match window. In Figure 0.5, the match window overlaps bins 10 and 11. When that happens, we recommend that the bin with the highest frequency be used. (The 1992 NRC report recommends taking the sum of the frequencies of all overlapped bins, but empirical studies have shown that taking the largest value more closely approximates the more accurate floating-bin method.)
Frequency estimates for very rare alleles have a larger relative uncertainty than do those for more common alleles, because the relative uncertainty is largely
| TABLE 0.1 Bin (Allele) Frequencies at Two VNTR Loci (D2S44 and D17S79) in the US White Populationa | |||||||
| D2S44 | D17S79 | ||||||
| Bin | Size Range | N | Prop. | Bin | Size Range | N | Prop. |
| 3 | 0- 871 | 8 | 0.005 | 1 | 0- 639 | 16 | 0.010 |
| 4 | 872- 963 | 5 | 0.003 | 2 | 640- 772 | 5 | 0.003 |
| 5 | 964-1,077 | 24 | 0.015 | 3 | 773- 871 | 11 | 0.007 |
| 6 | 1,078-1,196 | 38 | 0.024 | 4 | 872-1,077 | 6 | 0.004 |
| 7 | 1,197-1,352 | 73 | 0.046 | 6 | 1,078-1,196 | 23 | 0.015 |
| 8 | 1,353-1,507 | 55 | 0.035 | 7 | 1,197-1,352 | 348 | 0.224 |
| 9 | 1,508-1,637 | 197 | 0.124 | 8 | 1,353-1,507 | 307 | 0.198 |
| 10 | 1,638-1,788 | 170 | 0.107 | 9 | 1,508-1,637 | 408 | 0.263 |
| 11 | 1,789-1,924 | 131 | 0.083 | 10 | 1,638-1,788 | 309 | 0.199 |
| 12 | 1,925-2,088 | 79 | 0.050 | 11 | 1,789-1,924 | 44 | 0.028 |
| 13 | 2,089-2,351 | 131 | 0.083 | 12 | 1,925-2,088 | 50 | 0.032 |
| 14 | 2,352-2,522 | 60 | 0.038 | 13 | 2,089-2,351 | 16 | 0.010 |
| 15 | 2,523-2,692 | 65 | 0.041 | 14 | 2,352- | 9 | 0.006 |
| 16 | 2,693-2,862 | 63 | 0.040 | ||||
| 17 | 2,863-3,033 | 136 | 0.086 | 1,552 | 0.999 | ||
| 18 | 3,034-3,329 | 141 | 0.089 | ||||
| 19 | 3,330-3,674 | 119 | 0.075 | ||||
| 20 | 3,675-3,979 | 36 | 0.023 | ||||
| 21 | 3,980-4,323 | 27 | 0.017 | ||||
| 22 | 4,324-5,685 | 13 | 0.008 | ||||
| 25 | 5,686- | 13 | 0.008 | ||||
| 1,584 | 1.000 | ||||||
| aD2 and D17 indicate that these are on chromosomes 2 and 17. N is the number of genes (twice the number of persons). Each bin includes a range of sizes (in base pairs), grouped so that no bin has fewer than five genes in the data set; this accounts for nonconsecutive bin numbers. Data from FBI (1993), p 439, 530. FBI (1993), p 439, 530. | |||||||
Page 21
determined by the absolute number of alleles in the database. To reduce such uncertainty, it is customary for the data to be rebinned. This involves merging all bins with an absolute number fewer than five genes into adjacent bins, so that no bin has fewer than five members. We endorse this practice, not only for fixed bins but also for floating bins, and not only for VNTRs but also for rare alleles in other systems.
Allele (Bin) Frequencies
Databases come from a variety of sources, which we shall discuss later. Each bin is assigned a number, 1 designating the smallest fragments. Table O.1 shows the size range and frequencies of the bins at two loci, D2S44 and D17S79, for the US white population. The first number in the locus designation tells us on which chromosome this locus lies. The second is an arbitrary number that designates the site of the locus on the chromosome. D2S44 is site 44 on chromosome number 2; D17S79 is site 79 on chromosome 17. The data in the table have been rebinned so that no bin has fewer than 5 representatives in the database. D2S44 is more useful for forensic purposes than D17S79 because it has a larger range of sizes, from less than 871 to more than 5,686 base pairs, and because the different bins have more nearly equal frequencies.
Figure 0.6 shows a graph of the frequencies of each bin in three populations for D2S44. The top two graphs are from white populations in Georgia and Illinois. Note that the distributions are quite similar. In both states, bin 8 is the commonest; bins 14, 15, and 16 are relatively rare; and the extremes at both ends of the distribution have very low frequencies. Using the Georgia database for an Illinois crime would not introduce much error. In contrast, the distribution for blacks, shown in the bottom graph, is clearly different. That argues for using separate databases for different racial groups. Nevertheless, the most striking feature of the graphs is that the variability among individuals within a population is greater than that between populations.
We have now described procedures for matching and binning and for determining the bin (allele) frequency. We next wish to combine these frequencies to determine the frequency of a multilocus profile. That will be taken up later, in the section on population genetics.
PCR-Based Systems
The polymerase chain reaction (PCR) is a method for greatly amplifying a short segment of DNA, copying the sequence in a way somewhat like that which occurs naturally in the cell (the procedure is described in Chapter 2, p 69). Most PCR-based typing systems allow alleles to be identified as discrete entities, thus avoiding most of the statistical issues that arise in matching and binning of VNTR alleles.
Page 22
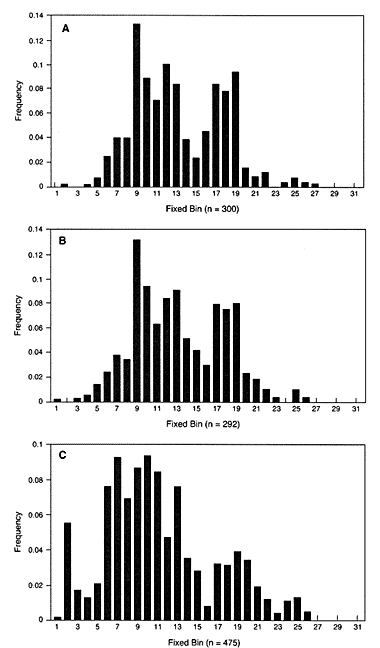
Figure 0.6
The distribution of bin sizes for locus D2S44. The horizontal axis gives
the bin number and the vertical column gives the relative frequency of
this bin in the database: (A) Illinois white population, (B) Georgia white
population. (C) US black population.
Page 23
The PCR process has several additional advantages over the procedures used with VNTRs. It is relatively simple and easily carried out in the laboratory. Results are obtained in a short time, often within 24 hours. Because of their almost unlimited capacity for amplification, PCR-based methods permit the analysis of extremely tiny amounts of DNA, thus extending the typing technique to samples too small to be used with other approaches (e.g., DNA from a cigarette butt). Moreover, the small sample size required for PCR analysis makes it easier to set aside portions of samples for duplicate testing to verify results or detect possible errors.
There are also disadvantages. One is that any procedure that uses PCR methodology is susceptible to error by contamination. If the contaminating DNA is present at a level comparable to the target DNA, its amplification can confound the interpretation of typing results, possibly leading to an erroneous conclusion. A second disadvantage is that most PCR loci have fewer alleles than VNTRs. That means that more loci are required to produce the same degree of discrimination of DNA from different persons. Third, some PCR loci are associated with functional genes, which means that they may have been subject to natural selection, possibly leading to greater differences among population subgroups than is found with VNTRs. In developing new systems, it is desirable to choose loci that are not associated with disease-causing genes. These are all problems that can be minimized by proper choice of markers and by care and good technique.
One PCR-based genetic marker, DQA, is widely used. It is quick and reliable, and that makes it particularly useful as a preliminary test. On the average, about 7% of the population have the same DQA type, so that different individuals will be distinguished about 93% of the time. Thus, a wrongly accused person has a good chance of being quickly cleared. Other systems are already in use or are being developed. Eventually, we expect such exact determinations to replace current VNTR methods, with a resulting simplification and speed of analysis and reduction of statistical uncertainties.
One of the most promising of the newer techniques involves amplification of loci containing Short Tandem Repeats (STRs). STRs are scattered throughout the chromosomes in enormous numbers, so that there is an almost unlimited potential for more loci to be discovered and validated for forensic use. Individual STR alleles can usually be individually identified, circumventing the need for matching and binning.
We affirm the statement of the 1992 report that the molecular technology is thoroughly sound and that the results are highly reproducible when appropriate quality-control methods are followed. The uncertainties that we address in this report relate to the effects of possible technical and human errors and the statistical interpretation of population frequencies, not to defects in the methodology itself.
Assuring Laboratory Accuracy
The best assurance of accuracy is careful design and statistical analysis, coupled with scrupulous attention to details. The maintenance of high laboratory
Page 24
standards rests on a foundation of sound quality control and quality assurance. Quality control (QC) refers to measures taken to ensure that the DNA typing and interpretation meet a specified standard. Quality assurance (QA) refers to steps taken by the laboratory to monitor, verify, and document its performance. Regular proficiency testing and regular auditing of laboratory operations are both essential components of a QA program.
Specific and detailed guidelines on QC and QA have been developed by the Technical Working Group on DNA Analysis Methods (TWGDAM), a group of forensic DNA analysts from government and private laboratories. These guidelines define currently accepted practice. They have been endorsed by the American Society of Crime Laboratory Directors/Laboratory Accreditation Board. These and other organizations provide standards for accreditation. Requirements for accreditation include extensive documentation of all aspects of laboratory operations, proficiency testing, internal and external audits, and a plan to address and correct deficiencies.
The DNA Identification Act of 1994 established a federal framework for setting national standards on quality assurance and proficiency testing. These standards are to be developed by a DNA Advisory Board, appointed by the FBI from a list of nominations made by the National Academy of Sciences and professional societies representing the forensic community. This Advisory Board is now in place and is formulating mechanisms for accreditation and quality control.
We believe that proficiency testing is of great value. These tests can be either open or blind. TWGDAM recommends one fully blind proficiency test per laboratory per year, if such a program can be implemented.
In open proficiency tests, the analyst knows that a test is being conducted. In blind proficiency tests, the analyst does not know that a test is being conducted. A blind test is therefore more likely to detect such errors as might occur in routine operations. However, the logistics of constructing fully blind proficiency tests are formidable. The ''evidence" samples have to be submitted through an investigative agency so as to mimic a real case, and unless that is done very convincingly, a laboratory might well suspect that it is being tested.
Whichever kind of test is used, the results are reported and, if errors are made, needed corrective action is taken. Several tests per year are mandated by the various accrediting organizations.
Some commentators have argued that the probability of a laboratory error leading to a reported match for samples from different individuals should be estimated and combined with the probability of randomly drawing a matching profile from the population. We believe this approach to be ill advised. It is difficult to arrive at a meaningful and accurate estimate of the risk of such laboratory errors. For one thing, in this rapidly evolving technology, it is the current practice and not the past record of a laboratory that is relevant, and that necessarily means smaller numbers and consequent statistical uncertainty. For
Page 25
another, the number of proficiency tests required to give an accurate estimate of a low error rate (and it must be low to be acceptable) is enormous and would be outlandishly expensive and disruptive. We believe that such efforts would be badly misplaced and would use resources that could much better be used in other ways, such as improving laboratory standards.
No amount of attention to detail, auditing, and proficiency testing can completely eliminate the risk of error. There is a better approach, one that is in general agreement with the 1992 NRC report: wherever feasible, evidence material should be separated into two or more portions, with one or more portions reserved for possible duplicate tests. Only an independent retest can satisfactorily resolve doubts as to the possibility that the first test was in error. It is usually possible to preserve enough material for possible repeat tests. Even if VNTR tests consume most of the material, it should almost always be possible to reserve enough for independent PCR-based confirmatory tests. The best protection an innocent suspect has from a false match is an independent test, and that opportunity should be made available if at all possible.
Even the strongest evidence will be worthless—or worse, might possibly lead to a false conviction—if the evidence sample did not originate in connection with the crime. Given the great individuating potential of DNA evidence and the relative ease with which it can be mishandled or manipulated by the careless or the unscrupulous, the integrity of the chain of custody is of paramount importance. This means meticulous care, attention to detail, and thorough documentation of every step of the process, from collecting the evidence material to the final laboratory report.
Population Genetics
If the DNA profile from the evidence sample and that of the suspect match, they may have come from the same person. Alternatively, they might represent a coincidental match between two persons who happen to share the profile. To assess the probability of such a coincidental match, we need to know the frequency of the profile in the population.
Ideally, we would know the frequency of each profile, but short of testing the whole population we cannot know that. We must therefore rely on samples from the population, summarized in a database. Furthermore, the probability of a specific profile is very small, much smaller than the reciprocal of the number of people represented in the database. That means that the great majority of profiles are not found in any database. The analyst must therefore estimate the frequency of a profile from information about the component allele frequencies. That requires some assumptions about the relation between allele frequencies and profile frequencies; it also requires modeling.
Page 26
Randomly Mating Populations
The simplest assumption relating allele and genotype frequencies is that mates are chosen at random. Perhaps surprisingly, such an assumption provides a good approximation to reality for forensic markers. Of course, matings in the United States are not literally at random; two persons from Oregon are much more likely to be mates than are a person from Oregon and one from Florida. But random-mating genotype proportions occur when mating frequencies are determined by the frequencies of the markers. And if the marker frequencies are the same in Oregon, Florida, and other states, that could lead to random-mating proportions throughout the nation, even though the United States is far from a random-mating unit.
Of course, for some traits the population is not in random-mating proportions. Mates are often chosen for physical and behavioral characteristics. But obviously, VNTRs and other forensic markers are not the basis for choice. For example, people often choose mates with similar height, but unless a forensic marker is closely linked to a possible major gene for height, the forensic genotypes will still be in random-mating proportions.
The simplest way to deal with random mating is to take advantage of the convenient fact that random mating of persons has the same genetic consequences as random combination of eggs and sperm. Suppose that at the A locus, 1/10 of the alleles are A1 and 1/25 are A2 Then 1/10 of the eggs carry allele A1 and of these 1/10 will be fertilized by A1 sperm, so 1/10 of 1/10, that is 1/10 X 1/10 = (1/10)2 = 1/100 of the fertilized eggs will be of genotype A1A1. Similarly 1/25 of the A1 eggs will be fertilized by A2 sperm, leading to (1/10) X (1/25) = 1/250 A1A2 individuals. However, the A1A2 genotype can also be produced,with equal frequency, by A2 eggs fertilized by Al sperm, so the total frequency of A1A2 genotypes is twice the product of the allele frequencies, or 1/125. Therefore, the frequencies of the genotypes are:
![]()
It is conventional in general formulations to use letters instead of numerical fractions. If we designate the frequency of allele A1 by p1 and of allele A2 by P2 (in this example, p1 = 1/10 and p2 = 1/25), the genotype frequencies are
![]() (O.1a)
(O.1a)![]() (O.1b)
(O.1b)
Populations in which the genotypes are in random-mating proportions are said to be in Hardy-Weinberg (HW) ratios, named after G. H. Hardy and W. Weinberg, the discoverers of this simple principle.
How well do actual populations agree with HW ratios? One example is given in Table 4.3 (p 94). M and N are two alleles at a blood-group locus. Six
Page 27
studies were done in the white population of New York City, a population that is genetically quite heterogeneous. The data came from blood donors, persons involved in paternity cases, patients, and hospital staff. They involve six studies over a period of almost 40 years. The total number was 6,001 persons, or 12,002 genes. Yet, as the table shows, the overall frequency of heterozygotes is within 1% of its HW expectation. For traits that are not involved in mate selection, the genotypes in actual populations are very close to HW proportions.
With continued random mating, alleles at different loci, even if initially linked on the same chromosome, become separated by crossing over and eventually reach linkage equilibrium (LE). At LE, the frequency of a composite genetic profile is the product of the genotype frequencies at each constituent locus. The rate of approach to LE depends on how close together the loci are on the chromosome. Loci on nonhomologous chromosomes, as almost all forensic loci are, approach LE quickly: the departure from LE is halved each generation. After half a dozen generations, LE can be assumed with sufficient accuracy for forensic purposes. Confirming this, in the large TWGDAM data set, the departure of two-locus pairs from LE in the white population was less than half a percent, and only slightly larger in blacks and Hispanics (Table 4.7, p 110). The deviations from expectations in individual cases were small and in both directions, as expected. Under HW and LE assumptions, the expected proportion of a specific genetic profile can be readily computed by calculating the genotype frequencies at each locus and multiplying them. In the forensic literature, that calculating procedure is called the product rule.
For illustration, suppose that at a second locus the two relevant alleles are B1 and B2, with frequencies 1/15 and 1/40. Then the frequency of genotype B1B2 is 2(1/15)(1/40) = 1/300. Now, putting this together with the A locus considered above, we find that the frequency of the composite genotype A1A1 B1B2 is 1/100 X 1/300 = 1/30,000. And likewise for more than two loci; genotype frequencies at each locus are multiplied.
Such estimates of the frequency of a particular profile in a population are, of course, subject to uncertainty. Even moderate-sized DNA databases (drawn from samples of several hundred persons) are subject to statistical uncertainty, and in smaller ones, the uncertainty is greater. In addition, the database might not properly represent the population that is relevant to a particular case. Finally, the assumptions of HW and LE, although reasonable approximations for most populations, are not exact. We shall elaborate on this point later, but to anticipate, we believe that it is safe to assume that the uncertainty of a profile frequency calculated by our procedures from adequate databases (at least several hundred persons) is less than a factor of about 10 in either direction. To illustrate, if a profile frequency is calculated to be one in 100 million, it is safe to say that the true value is almost always between one in 10 million and one in a billion.
We now consider modifications of the product-rule calculations to make them more realistic in the face of uncertainties.
Page 28
Population Structure
The population of the United States is made up of subpopulations descended from different parts of the globe and not fully homogenized. The authors of the 1992 NRC report were concerned that profile frequencies calculated from population averages might be seriously misleading for particular subpopulations. Extensive studies from a wide variety of databases show that there are indeed substantial frequency differences among the major racial and linguistic groups (black, Hispanic, American Indian, east Asian, and white). And within these groups, there is often a statistically significant departure from random proportions. As we said earlier, those departures are usually small, and formulae based on random mating assumptions are usually quite accurate. So, the product rule, although certainly not exact for real populations, is often a very good approximation.
The main reason for departures from random-mating proportions in forensic DNA markers is population structure due to incomplete mixing of ancestral stocks. Suppose that we estimate genotype frequencies in a subgroup by applying the product rule to allele frequencies based on overall population averages. To the extent that the subgroups have different allele frequencies, such an estimate will be too high for heterozygotes and too low for homozygotes. The reason for this is that matings within a subgroup will tend to be between (perhaps distant) relatives, and relatives share alleles. Thus, matings within a subgroup will produce more homozygotes and fewer heterozygotes than if the mates were chosen at random from the whole population.
In contrast to this systematic effect on homozygote and heterozygote frequencies, departures from LE because of population substructure are largely random and are not predictable in direction. Consequently, when several loci are involved, deviations in opposite directions tend to cancel.
Dealing with Subpopulations
The writers of the 1992 NRC report were concerned that there might be important population substructure and recommended an interim ceiling principle (discussed later in this overview) to address that concern. We take a different tack. We assume that there is undetected substructure in the population and adjust the product rule accordingly. There is a simple procedure for doing this. Since using the HW rule for heterozygote frequencies provides an overestimate if there is substructure, we employ the product rule as a conservative estimate for heterozygotes. But we need a modification to correct the opposite bias in the homozygote estimates.
For VNTRs, a single band in a lane does not necessarily imply a homozygote. It might be a heterozygote with two alleles too close together to distinguish, or one of the alleles, for any of several reasons, might not be detected. It has become
Page 29
standard practice in such cases to replace p2, the homozygote frequency as estimated by Equation O.1a (p 26), by 2p, where p is the bin frequency. It is easily shown (Chapter 4, p 105) that this substitution provides a conservative correction for homozygotes. So we follow earlier recommendations (e.g., the 1992 report) to use the product rule for VNTRs and to replace p2 by 2p for all single bands. This is called the 2p rule. It is illustrated in the example at the end of this overview.
The 2p rule has been criticized as being more conservative than necessary. However, with VNTRs, double bands greatly outnumber single bands, so the bias is usually not great. We retain the rule for two reasons: It is conservative, and it is thoroughly ingrained in standard forensic practice. We caution, however, that it was intended for criminal cases and might not be appropriate for other applications, such as determining paternity. It should not be used except as a conservative modification for rare alleles when heterozygotes may appear to be homozygotes.
Another rule is applicable when there is no problem in distinguishing homozygotes from heterozygotes, as with most PCR-based systems. The procedure is to replace p2 with the expression ![]() , where
, where ![]() is an empirically determined measure of population subdivision. The measured value of
is an empirically determined measure of population subdivision. The measured value of ![]() is usually considerably less than 0.01 for forensic markers in the United States, so we recommend 0.01 as a conservative value, except for very small, isolated populations of interrelated people, where 0.03 may be more appropriate.
is usually considerably less than 0.01 for forensic markers in the United States, so we recommend 0.01 as a conservative value, except for very small, isolated populations of interrelated people, where 0.03 may be more appropriate.
Persons from the Same Subpopulation
Usually, the subgroup to which the suspect belongs is irrelevant, since we want to calculate the probability of a match on the assumption that the suspect is innocent and the evidence DNA was left by someone else. The proper question is: What is the probability that a randomly chosen person, other than the suspect, has the genetic profile of the evidence DNA? That is the question we have dealt with so far. In some cases, however, it may be known that the suspect(s) is(are) from the same subpopulation as the source of the evidence DNA. An instance would be a crime committed in a small, isolated village, with all potential suspects from the same village. Ideally, the calculation should be based on the allele frequencies in that particular village, but usually such frequencies will not be known.
An alternative is to measure the degree of population subdivision and, using that, to write expressions for the conditional probability that, given the genotype of the first person, a second person from the same subgroup will have that genotype. The appropriate expressions for the match probability are
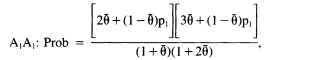 (0.2a)
(0.2a)
Page 30
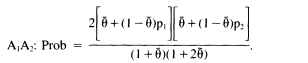 (0.2b)
(0.2b)
Although these expressions might appear complex, they are actually a straightforward adjustment of the standard HW formulae. Notice that if ![]() , the formulae are p12 and 2p1p2, the HW formulae. As before, p1 and p2 are obtained from the frequencies in the database. We suggest 0.01 as a suitable value of
, the formulae are p12 and 2p1p2, the HW formulae. As before, p1 and p2 are obtained from the frequencies in the database. We suggest 0.01 as a suitable value of ![]() . If the population is very small and isolated, or if a still more conservative estimate is desired, 0.03 can be used.
. If the population is very small and isolated, or if a still more conservative estimate is desired, 0.03 can be used.
As an example, consider the A locus already used (p 26), in which p1 = 1/10 and p2 = 1/25. Then the match probability for the heterozygote, A1A2, is 2(1/10)(1/25) = 1/125 or 0.008 when ![]() , 0.0105 when
, 0.0105 when ![]() , and 0.0160 when
, and 0.0160 when ![]() . Clearly, this calculation is more conservative than the simple product rule.
. Clearly, this calculation is more conservative than the simple product rule.
Some Statistical Considerations
The Reference Database
Ideally, the reference data set, from which profile frequencies are calculated, would be a simple random sample or a scientifically structured random sample from the relevant population. But this can be an impracticable ideal. For one thing, it is not always clear which population is most relevant. Should the sample be local or national? Should it include both sexes? If only males, should it include only those in the ages that commit most crimes? For another thing, random sampling is usually difficult, expensive, and impractical, so we are often forced to rely on convenience samples. Databases come from such diverse sources as blood banks, paternity-testing laboratories, laboratory personnel, clients in genetic-counseling centers, law-enforcement officers, and people charged with crimes. The saving point is that the DNA markers in which we are interested are believed theoretically and observed empirically to be essentially uncorrelated with the rules by which the samples are chosen.
We are confident that these convenience samples are appropriate for forensic uses, mainly for two reasons. First, the loci generally used for identification are usually not parts of functional genes and therefore are unlikely to be correlated with any behavioral or physical traits that might be associated with different subsets of the population. Second, empirical tests have shown only very minor differences among the frequencies of DNA markers from different subpopulations or geographical areas.
Indeed, samples from different subgroups often show statistically significant differences. This is especially true if the sample sizes are large, since in large samples, small differences can be statistically significant. But we are more con-
Page 31
cerned with the magnitude of the difference and the uncertainty in our calculations than with formal statistical significance. We shall deal with this further on.
Match Probability, Likelihood Ratio, and Two Fallacies
Forensic calculations are conventionally presented in one of two ways: the probability of a random match (called the match probability), calculated from the frequencies of DNA markers in the database; and the likelihood ratio (LR). The LR is the ratio of the probability of a match if the DNA in the evidence sample and that from the suspect came from the same person to the probability of a match if they came from different persons. Since the probability of a match when the samples came from the same person is one (unless there has been a mistake), the likelihood ratio is simply the reciprocal of the match probability.
A likelihood ratio of 1,000 says that the profile match is 1,000 times as likely if the DNA samples came from the same person as it would be if they came from two randomly chosen members of the population. It does not say that if the DNA samples match then they are 1,000 times as likely to have come from the same person as from different persons. It is important to keep this distinction straight. The misstatement, a logical reversal of the first, is an example of "the prosecutor's fallacy."
Although in the simplest cases the match probability and the likelihood ratio provide the same information (because one is the reciprocal of the other), there are cases in which the likelihood ratio is conceptually simpler. One such case happens with a mixed sample. This is illustrated in Chapter 5 (p 129) with an example in which the evidence sample has four bands, two of which are shared with the suspect. The match-probability approach, used in the 1992 NRC report, ignores some of the data, whereas a complete analysis is easily obtained by using the LR.
The second fallacy is "the defendant's fallacy." That is to assume that in a given population, anyone with the same profile as the evidence sample is as likely to have left the sample as is the suspect. If 100 persons in a metropolitan area are expected to have the same DNA profile as the evidence sample, it is a fallacy to conclude that the probability that the suspect contributed the sample is only 1/100. The suspect was originally identified by other evidence; such evidence does not exist for the 99 other persons expected to have the same profile. However, if the suspect was found through a search of a large DNA database, that changes the situation, as we shall soon discuss.
Bayes's Theorem
The reason that the prosecutor's fallacy is inviting is that, even though it gives a wrong answer, it purports to answer the question in which the court is really interested—namely, what is the probability that the evidence sample and
Page 32
the suspect sample came from the same person? Neither the match probability nor the likelihood ratio gives this. Yet, the latter can be used to obtain this probability, provided we are willing to assume a value for the prior probability that the two samples have a common source. The prior probability that the two samples came from the same person is the probability of that event based on evidence other than the DNA.
The principle is more easily expressed if stated as odds rather than probability. (Odds are the ratio of the probability that an event will occur to the probability that it will not: Odds = Prob/(l - Prob); if the probability is 2/3, the odds in favor are 2/1, or as conventionally written, 2:1.) Specifically, the final (posterior) odds that the suspect and evidence DNA came from the same person are the prior odds multiplied by the likelihood ratio (LR):
![]()
In other words, whatever you believe the odds to be without the DNA evidence, they are multiplied by LR when the DNA evidence is included. Although this rule (Bayes's Theorem) is routinely used in paternity cases, it has hardly ever been used in criminal cases not involving proof of paternity.
Since the prior odds are hardly ever known even approximately and are usually subjective, a practice that has been advocated is to give posterior odds (or probabilities) for a range of prior odds (or probabilities). If the likelihood ratio is very high, uncertainty about the value of the prior probability may make little difference in the court's decision.
Suppose that the LR is one million. If the prior odds are 1:10, the posterior odds are 100,000:1; if the prior odds are 1:100, the posterior odds are still 10,000:1.
Suspect Identified by Database Search
A special circumstance arises when the suspect is identified not by an eyewitness or by circumstantial evidence but rather by a search through a large DNA database. If the only reason that the person becomes a suspect is that his DNA profile turned up in a database, the calculations must be modified. There are several approaches, of which we discuss two. The first, advocated by the 1992 NRC report, is to base probability calculations solely on loci not used in the search. That is a sound procedure, but it wastes information, and if too many loci are used for identification of the suspect, not enough might be left for an adequate subsequent analysis. That will become less of a problem as STRs and other systems with many loci become more widely used.
A second procedure is to apply a simple correction: Multiply the match probability by the size of the database searched. This is the procedure we recommend.
The analysis assumes that the database, although perhaps large, is nevertheless a small fraction of the whole population. At present, that is the usual situation. However, as the databases grow large enough to be a substantial fraction of the
Page 33
population, a more complicated calculation is required. Although such a calculation can be straightforward, it is best handled on a case-by-case basis.
Uniqueness
Another issue—one that has not been resolved by the courts—is uniqueness. The 1992 NRC report said: "Regardless of the calculated frequency, an expert should—given ... the relatively small number of loci used and the available population data—avoid assertions in court that a particular genotype is unique in the population." Some courts have held that statements that a profile is unique are improper. Yet, with existing databases and afortiori with larger numbers of loci, likelihood ratios much higher than the population of the world are often found. An LR of 60 billion is more than 10 times the world population. Should a profile that rare be regarded as unique?
The definition of uniqueness is outside our province. It is for the courts to decide, but in case such a decision is to be made, we show how to do the relevant calculations. Before a suspect has been profiled, the probability that at least one other person in a population of N unrelated persons has the profile of the evidence DNA is at most NP, where P is the probability of the profile. Then the probability that the profile is unique is at least 1 - NP.
Suppose the calculated profile probability P = 1/(60 billion) and the world population N is taken as 6 billion. Then NP = 1/10. The probability that the profile is unique, except possibly for relatives, is at least about 9/10.
Uncertainty About Estimated Frequencies
Match probabilities are estimated from a database, and such calculations are subject to uncertainties. The accuracy of the estimate will depend on the genetic model, the actual allele frequencies, and the size of the database. In Chapter 5 (p 146) we explain how to compute confidence limits on the probabilities, if the databases are regarded as random samples from the populations they represent. That, however, includes only part of the uncertainty. Remaining is the uncertainty due, not to the small sample size, but to the possibilities that the database is not representative of the population of interest or that the mathematical model might not be fully appropriate. We therefore take a more realistic, empirical approach. As mentioned earlier, the uncertainty of a profile-frequency calculation that uses our methods and an adequate database (at least several hundred persons) is less than about 10-fold in either direction. We now explain where this conclusion comes from.
We used the published data and graphs assembled from around the world by the FBI to determine the extent of error if an incorrect database is used. That should provide an upper limit for the uncertainty with the correct database. For example, suppose a crime is committed in Colorado by a man known to be white.
Page 34
In the absence of a local database, a national white database is used. Graphs (examples are Figures 5.3 and 5.4, p 150 and 152) show that the individual values that are possibly incorrectly estimated lie within 10-fold above and below the ''correct" value. We conclude that it is reasonable to regard calculated multilocus match probabilities as accurate within a factor of 10 either way. This is true for various subsets within the white, black, Hispanic, and east Asian populations. However, if the database from the wrong racial group is used, the error may be larger (Figure 5.5, p 153). That argues for the use of the correct racial database if that can be ascertained; otherwise, calculations should be made for all relevant racial groups, i.e., those to which possible suspects belong. The databases should be large enough to have some statistical accuracy (at least a few hundred persons), and alleles represented fewer than five times should be rebinned (grouped so that no bin has fewer than five).
Additional information comes from comparison of profiles within the databases. An early study used FBI and Lifecodes data for blacks, whites, Southeast Hispanics, and Southwest Hispanics. Among 7,628,360 pairs of profiles from within those databases, no four- or five-locus matching profiles were found, and only one three-locus match was seen. A newer and more extensive analysis, compiling data from numerous TWGDAM sources, summarized a large number of profiles from white, black, and Hispanic databases. Of 58 million pairwise comparisons within racial groups, only two possible four-locus matches were found, and none were found for five or six loci.
We conclude that, when several loci are used, the probability of a coincidental match is very small and that properly calculated match probabilities are correct within a factor of about 10 either way. If the calculated probability of a random match between the suspect and evidence DNA is 1/(100 million), we can say with confidence that the correct value is very likely between l/(10 million) and 1/(l billion).
PCR-Based Tests
As already mentioned, PCR-based tests have a number of advantages. They include the ability to identify individual alleles, as well as simplicity and quick turn-around. But there are disadvantages. Most of the loci used have a small number of alleles, so that many more loci are required for the same statistical power as provided by a few VNTRs. STRs are also based on repeating units, have a high mutation rate (although not as high as some VNTRs), have a fairly large number of alleles, and are usually capable of unique allelic identification. With 12 STR loci, there is discriminatory power comparable to that of four or five VNTRs, and comparisons between geographical and racial groups show similarities and differences comparable to those of VNTRs.
The quantity ![]() , which we use as a measure of population substructure, is determined largely by the population history rather than by the frequency of the
, which we use as a measure of population substructure, is determined largely by the population history rather than by the frequency of the
Page 35
alleles involved. It is also very small, less than about 0.01 in the United States. There has not been the extensive sampling of subpopulations and geographical areas for PCR-based systems that has been done with VNTRs. New data show low values of ![]() and good agreement with HW and LE. The uncertainty range appears to be about the same as that for VNTRs. We therefore believe that STRs can take their place along with VNTRs as forensic tools. They circumvent most of the matching and binning problems that VNTRs entail.
and good agreement with HW and LE. The uncertainty range appears to be about the same as that for VNTRs. We therefore believe that STRs can take their place along with VNTRs as forensic tools. They circumvent most of the matching and binning problems that VNTRs entail.
The Ceiling Principles
The most controversial recommendations of the 1992 NRC report are the ceiling principle and the interim ceiling principle. They were intended to place a lower limit on the size of the profile frequency by setting threshold values for allele frequencies used in calculations. The ceiling principle calls for sampling 100 persons from each of 15-20 genetically homogeneous populations spanning the racial and ethnic diversity of groups represented in the United States. For each allele, the highest frequency among the groups sampled, or 5%, whichever is larger, would be used. Then the product rule would be applied to those values to determine the profile frequency. But the data needed for applying this principle have not been gathered. We share the view of those who criticize it on practical and statistical grounds and who see no scientific justification for its use.
The 1992 report recommended further that until the ceiling principle could be put into effect, an interim ceiling principle be applied. In contrast to the ceiling principle, the interim ceiling principle has been widely used, and sometimes misused. The rule says: "In applying the multiplication [product] rule, the 95% upper confidence limit of the frequency of each allele should be calculated for separate US 'racial' groups and the highest of these values or 10% (whichever is larger) should be used. Data on at least three major 'races' (e.g., Caucasians, blacks, Hispanics, east Asians, and American Indians) should be analyzed."
The interim ceiling principle has the advantage that in any particular case it gives the same answer irrespective of the racial group. That is also a disadvantage, for it does not permit the use of well-established differences in frequencies among different races; the method is inflexible and cannot be adjusted to the circumstances of a particular case. The interim ceiling principle has been widely criticized for other reasons as well, and we summarize the criticisms in Chapter 5 (p 157). We agree with those criticisms.
Our view is that sufficient data have been gathered to establish that neither ceiling principle is needed. We have given alternative procedures, all of which are conservative but less arbitrary.
Although we recommend other procedures and believe that the interim ceiling principle is not needed, we recognize that it has been used and some will probably continue to use it. To anticipate this possibility, we offer several suggestions in
Page 36
Chapter 5 that will make the principle more workable and less susceptible to creative misapplications.
DNA in the Courts
Prior to 1992, there was controversy over our two main issues, laboratory error and population substructure. The 1992 NRC report was intended to resolve the controversy, but the arguments went on. One reason is that the scientific community has not spoken with one voice; defense and prosecution witnesses have given highly divergent statistical estimates or have disagreed as to the validity of all estimates. For this reason, some courts have held that the analyses are not admissible in court. The courts, however, have accepted the soundness of the typing procedures, especially for VNTRs. The major disagreement in the courts has been over population substructure and possible technical or human errors. The interim ceiling principle, in particular, has also been the subject of considerable disagreement. We hope that our report will ease the acceptance of DNA analysis in the courts and reduce the controversy.
We shall not summarize the various court findings and opinions here. The interested reader can find this information in Chapter 6, which also discusses the implications that our recommendations could have on the production and introduction of DNA evidence in court proceedings.
Conclusions and Recommendations
Conclusions and recommendations are given at the ends of the chapters in which the relevant subject is discussed. For convenience, they are repeated here.
Admissibility of DNA Evidence (Chapter 2)
DNA analysis is one of the greatest technical achievements for criminal investigation since the discovery of fingerprints. Methods of DNA profiling are firmly grounded in molecular technology. When profiling is done with appropriate care, the results are highly reproducible. In particular, the methods are almost certain to exclude an innocent suspect.
One of the most widely used techniques involves VNTRs. These loci are extremely variable, but individual alleles cannot be distinguished, because of intrinsic measurement variability, and the analysis requires statistical procedures. The laboratory procedure involves radioactivity and requires a month or more for full analysis. PCR-based methods are prompt, require only a small amount of material, and can yield unambiguous identification of individual alleles.
The state of the profiling technology and the methods for estimating frequencies and related statistics have progressed to the point where the admissibility of properly collected and analyzed DNA data should not be in doubt. We expect
Page 37
continued development of new and better methods and hope for their prompt validation, so that they can quickly be brought into use.
Laboratory Errors (Chapter 3)
The occurrence of errors can be minimized by scrupulous care in evidence-collecting, sample-handling, laboratory procedures, and case review. Detailed guidelines for QC and QA (quality control and quality assurance), which are updated regularly, are produced by several organizations, including TWGDAM. ASCLD-LAB is established as an accrediting agency. The 1992 NRC report recommended that a National Committee on Forensic DNA Typing (NCFDT) be formed to oversee the setting of DNA-analysis standards. The DNA Identification Act of 1994 gives this responsibility to a DNA Advisory Board appointed by the FBI. We recognize the need for guidelines and standards, and for accreditation by appropriate organizations.
Recommendation 3.1. Laboratories should adhere to high quality standards (such as those defined by TWGDAM and the DNA Advisory Board) and make every effort to be accredited for DNA work (by such organizations as ASCLD-LAB).
Proficiency Tests
Regular proficiency tests, both within the laboratory and by external examiners, are one of the best ways of assuring high standards. To the extent that it is feasible, some of the tests should be blind.
Recommendation 3.2: Laboratories should participate regularly in proficiency tests, and the results should be available for court proceedings.
Duplicate Tests
We recognize that no amount of care and proficiency testing can eliminate the possibility of error. However, duplicate tests, performed as independently as possible, can reduce the risk of error enormously. The best protection that an innocent suspect has against an error that could lead to a false conviction is the opportunity for an independent retest.
Recommendation 3.3: Whenever feasible, forensic samples should be divided into two or more parts at the earliest practicable stage and the unused parts retained to permit additional tests. The used and saved portions should be stored and handled separately. Any additional tests should be performed independently of the first by personnel not involved in the first test and preferably in a different laboratory.
Page 38
Population Genetics (Chapter 4)
Sufficient data now exist for various groups and subgroups within the United States that analysts should present the best estimates for profile frequencies. For VNTRs, using the 2p rule for single bands and HW for double bands is generally conservative for an individual locus. For multiple loci, departures from linkage equilibrium are not great enough to cause errors comparable to those from uncertainty of allele frequencies estimated from databases.
With appropriate consideration of the data, the principles in this report can be applied to PCR-based tests. For those in which exact genotypes can be determined, the 2p rule should not be used. A conservative estimate is given by using the HW relation for heterozygotes and a conservative value of ![]() in Equation 4.4a for homozygotes.
in Equation 4.4a for homozygotes.
Recommendation 4.1: In general, the calculation of a profile frequency should be made with the product rule. If the race of the person who left the evidence-sample DNA is known, the database for the person's race should be used; if the race is not known, calculations for all racial groups to which possible suspects belong should be made. For systems such as VNTRs, in which a heterozygous locus can be mistaken for a homozygous one, if an upper bound on the genotypic frequency at an apparently homozygous locus (single band) is desired, then twice the allele (bin) frequency, 2p, should be used instead of p2. For systems in which exact genotypes can be determined, ![]() should be used for the frequency at such a locus instead of p2. A conservative value of
should be used for the frequency at such a locus instead of p2. A conservative value of ![]() for the US population is 0.01; for some small, isolated populations, a value of 0.03 may be more appropriate. For both kinds of systems, 2pipj should be used for heterozygotes.
for the US population is 0.01; for some small, isolated populations, a value of 0.03 may be more appropriate. For both kinds of systems, 2pipj should be used for heterozygotes.
A more conservative value of ![]() = 0.03 might be chosen for PCR-based systems in view of the greater uncertainty of calculations for such systems because of less extensive and less varied population data than for VNTRs.
= 0.03 might be chosen for PCR-based systems in view of the greater uncertainty of calculations for such systems because of less extensive and less varied population data than for VNTRs.
Evidence DNA and Suspect from the Same Subgroup
Sometimes there is evidence that the suspect and other possible sources of the sample belong to the same subgroup. That can happen, e.g., if they are all members of an isolated village. In this case, a modification of the procedure is desirable.
Recommendation 4.2: If the particular subpopulation from which the evidence sample came is known, the allele frequencies for the specific subgroup should be used as described in Recommendation 4.1. If allele frequencies for the subgroup are not available, although data for the full population are, then the calculations should use the population-structure Equations 4.10 for each locus, and the resulting values should then be multiplied.
Page 39
Insufficient Data
For some groups—and several American Indian and Inuit tribes are in this category—there are insufficient data to estimate frequencies reliably, and even the overall average might be unreliable. In this case, data from other, related groups provide the best information. The groups chosen should be the most closely related for which adequate databases exist. These might be chosen because of geographical proximity, or a physical anthropologist might be consulted. There should be a limit on the number of such subgroups analyzed to prevent inclusion of more remote groups less relevant to the case.
Recommendation 4.3: If the person who contributed the evidence sample is from a group or tribe for which no adequate database exists, data from several other groups or tribes thought to be closely related to it should be used. The profile frequency should be calculated as described in Recommendation 4.1 for each group or tribe.
Dealing with Relatives
In some instances, there is evidence that one or more relatives of the suspect are possible perpetrators.
Recommendation 4.4: If the possible contributors of the evidence sample include relatives of the suspect, DNA profiles of those relatives should be obtained. If these profiles cannot be obtained, the probability of finding the evidence profile in those relatives should be calculated with Formulae 4.8 or 4.9.
Statistical Issues (Chapter 5)
Confidence limits for profile probabilities, based on allele frequencies and the size of the database, can be calculated by methods explained in this report. We recognize, however, that confidence limits address only part of the uncertainty. For a more realistic estimate, we examined empirical data from the comparison of different subpopulations and of subpopulations within the whole. The empirical studies show that the differences between the frequencies of the individual profiles estimated by the product rule from different adequate subpopulation databases (at least several hundred persons) are within a factor of about 10 of each other, and that provides a guide to the uncertainty of the determination for a single profile. For very small estimated profile frequencies, the uncertainty can be greater, both because of the greater relative uncertainty of individually small probabilities and because more loci are likely to be multiplied. But with very small probabilities, even a larger relative error is not likely to change the conclusion.
Database Searches
If the suspect is identified through a DNA database search, the interpretation of the match probability and likelihood ratio given in Chapter 4 should be modified.
Page 40
Recommendation 5.1: When the suspect is found by a search of DNA databases, the random-match probability should be multiplied by N, the number of persons in the database.
If one wishes to describe the impact of the DNA evidence under the hypothesis that the source of the evidence sample is someone in the database, then the likelihood ratio should be divided by N. As databases become more extensive, another problem may arise. If the database searched includes a large proportion of the population, the analysis must take that into account. In the extreme case, a search of the whole population should, of course, provide a definitive answer.
Uniqueness
With an increasing number of loci available for forensic analysis, we are approaching the time when each person's profile is unique (except for identical twins and possibly other close relatives). Suppose that, in a population of N unrelated persons, a given DNA profile has probability P. The probability (before a suspect has been profiled) that the particular profile observed in the evidence sample is not unique is at most NP.
A lower bound on the probability that every person is unique depends on the population size, the number of loci, and the heterozygosity of the individual loci. Neglecting population structure and close relatives, 10 loci with a geometric mean heterozygosity of 95% give a probability greater than about 0.999 that no two unrelated persons in the world have the same profile. Once it is decided what level of probability constitutes uniqueness, appropriate calculations can readily be made.
We expect that the calculation in the first paragraph will be the one more often employed.
Matching and Binning
VNTR data are essentially continuous, and, in principle, a continuous model should be used to analyze them. The methods generally used, however, involve taking measurement uncertainty into account by determining a match window. Two procedures for determining match probabilities are the floating-bin and the fixed-bin methods. The floating-bin method is statistically preferable but requires access to a computerized database. The fixed-bin method is more widely used and understood, and the necessary data tables are widely and readily available. When our fixed-bin recommendation is followed, the two methods lead to very similar results. Both methods are acceptable.
Recommendation 5.2: If floating bins are used to calculate the random-match probabilities, each bin should coincide with the corresponding match window. If fixed bins are employed, then the fixed bin that has the largest frequency among those overlapped by the match window should be used.
Page 41
Ceiling Principles
The abundance of data in different ethnic groups within the major races and the genetically and statistically sound methods recommended in this report imply that both the ceiling principle and the interim ceiling principle are unnecessary.
Further Research
The rapid rate of discovery of new markers in connection with human gene-mapping should lead to many new markers that are highly polymorphic, mutable, and selectively neutral, but which, unlike VNTRs, can be amplified by PCR and for which individual alleles can usually be distinguished unambiguously with none of the statistical problems associated with matching and binning. Furthermore, radioactive probes need not be used with many other markers, so identification can be prompt and problems associated with using radioactive materials can be avoided. It should soon be possible to have systems so powerful that no statistical and population analyses will be needed, and (except possibly for close relatives) each person in a population can be uniquely identified.
Recommendation 5.3: Research into the identification and validation of more and better marker systems for forensic analysis should continue with a view to making each profile unique.
Legal Issues (Chapter 6)
In assimilating scientific developments, the legal system necessarily lags behind the scientific world. Before making use of evidence derived from scientific advances, courts must scrutinize the proposed testimony to determine its suitability for use at trial, and controversy within the scientific community often is regarded as grounds for the exclusion of the scientific evidence. Although some controversies that have come to closure in the scientific literature continue to limit the presentation of DNA evidence in some jurisdictions, courts are making more use of the ongoing research into the population genetics of DNA profiles. We hope that our review of the research will contribute to this process.
Our conclusions and recommendations for reducing the risk of laboratory error, for applying human population genetics to DNA profiles, and for handling uncertainties in estimates of profile frequencies and match probabilities might affect the application of the rules for the discovery and admission of evidence in court. Many suggestions can be offered to make our recommendations most effective: for example, that every jurisdiction should make it possible for all defendants to have broad discovery and independent experts; that accreditation, proficiency testing, and the opportunity for independent testing (whenever feasible) should be prerequisites to the admission of laboratory findings; that in resolving disputes over the adequacy or interpretation of DNA tests, the power
Page 42
of the court to appoint its own experts should be exercised more frequently; and that experts should not be barred from presenting any scientifically acceptable estimate of a random-match probability. We have chosen, however, to make no formal recommendations on such matters of legal policy; we do, however, make a recommendation concerning scientific evidence—namely, the need for behavioral research that will assist legal decision makers in developing standards for communicating about DNA in the courtroom.
Recommendation 6.1: Behavioral research should be carried out to identify any conditions that might cause a trier of fact to misinterpret evidence on DNA profiling and to assess how well various ways of presenting expert testimony on DNA can reduce such misunderstandings.
We trust that our efforts to explain the state of the forensic science and some of the social-science findings that are pertinent to resolving these issues will contribute to better-informed judgments by courts and legislatures.
Illustrative Example
A Typical Case
As an illustration, we have chosen an example that involves VNTR loci. The methods used for the other systems are very similar, except that they usually do not involve the complications of matching and binning, so the more complicated situation is better for illustration. We shall analyze the same data in several ways.
Suppose that samples of blood are obtained from a crime scene and DNA from two suspects, 1 and 2. We should like to know whether the profile of either suspect matches the profile of the evidence DNA.
First we isolate the DNA from the three samples, making sure that all three have been handled separately and that each step in the chain of custody has been checked and documented. The DNA is first cut into small segments by an enzyme, Hae III. The fragments from the evidence sample (E) and from the two suspects (S1 and S2) are placed in small wells in the gel, each sample in a separate lane. Along with these three are a number of controls, as illustrated in Figure 0.4, each with its own lane. The laboratory has been careful not to put any of the three DNA samples into adjacent lanes to prevent possible leakage of DNA into the wrong lane.
After being placed in an electric field for a carefully determined time, the DNA in all the lanes is transferred by blotting to a nylon membrane (stronger and easier to handle than the gel). Then a radioactive probe that is specific for locus D2S44 is flooded onto the membrane. The probe adheres to the corresponding region in the DNA sample, and any nonadhering probes are washed off. The membrane is then placed in contact with a photographic film to prepare an autorad. Figure 0.7 illustrates the result in this case.
Page 43
The rough size of the fragment can be determined from the scale in the figure. In practice, the scale is a ladder, a group of DNA fragments that differ from each other in increments of approximately 1,000 base pairs (the ladder can be seen in Figure 0.4) It is immediately apparent (Figure 0.7) that E and S1 match as far as the eye can tell, but that S2 is clearly different. That alone is sufficient to exclude S2 as a suspect. The sizes of the six bands are determined by comparison with the ladder. This operation is ordinarily done by a computer programmed to scan the autorad and measure the sizes of the bands.
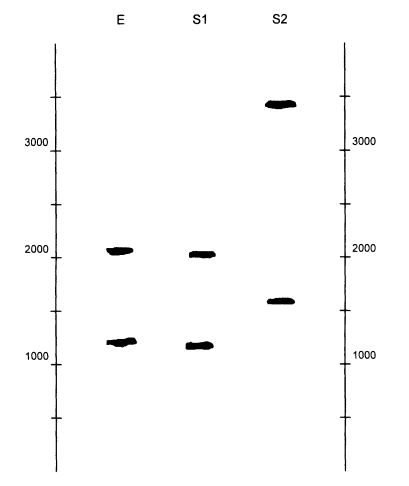
Figure 0.7
Diagram of a hypothetical autorad for evidence DNA (E) and two suspects (S1 and S2). Note that E and
S1 appear to match, whereas S2 is clearly not the source of the evidence DNA. The numbers at the
two sides are numbers of base pairs.
Page 44
| TABLE 0.2 The Uncertainty Windows for a VNTR Marker (Probe D2S44) in an Illustrative Example | ||||
| Source | Band | Size | 2.5% | Uncertainty |
| E | Larger | 1,901 | 48 | 1,853-1,949 |
| Smaller | 1.137 | 28 | 1,109-1,165 | |
| S1 | Larger | 1,876 | 47 | 1,829-1,923 |
| Smaller | 1,125 | 28 | 1,097-1,153 | |
| S2 | Larger | 3,455 | 86 | 3,369-3,541 |
| Smaller | 1,505 | 38 | 1,467-1.543 | |
The calculations (or computer output) are shown in Table 0.2. The measured value of each band is given, along with upper and lower limits of the uncertainty window, which spans the range from 2.5% below to 2.5% above the measured value. Comparing the uncertainty window of S1 and E for the smaller band, we see that the windows overlap; the upper limit of Sl, 1,153, is within the range, 1,109 to 1,165, of E. Likewise, the uncertainty windows of the larger bands also overlap. In contrast, the uncertainty windows for the two bands from S2 do not overlap any of the evidence bands. So our visual impression is confirmed by the measurements. S2 is cleared, whereas S1 remains as a possible source of the evidence DNA.
The next step is to compute the size of the match window (Table 0.3), which will be used to find the frequency of this marker in a relevant database of DNA marker frequencies. This is the measurement E plus and minus 5% of
| TABLE 0.3 Match Windows and Frequencies for Several VNTR Markers in an Illustrative Example | ||||||
| Locus | Band | Size | 5% | Match Window | Bin(s) | Freq. |
| D2S44 | Larger | 1,901 | 95 | 1,806-1,996 | 11, 12 | 0.083 |
| Smaller | 1,137 | 57 | 1,080-1,194 | 6 | 0.024 | |
| D17S79 | Larger | 1,685 | 84 | 1,601-1,769 | 9, 10 | 0.263 |
| Smaller | 1,120 | 56 | 1,064-1,176 | 4, 6 | 0.015 | |
| D1S7 | Single | 14 | 0.068 | |||
| D4S139 | Larger | 10 | 0.072 | |||
| Smaller | 13 | 0.131 | ||||
| D10S28 | Larger | 9 | 0.047 | |||
| Smaller | 16 | 0.065 | ||||
| Probability of a random match, 5 loci: P = 2(0.083)(0.024) x 2(0.263)(0.015) x 2(0.068) x 2(0.072)(0.131) x 2(0.047)(0.065) Uncertainty range: 1/(200 million) to 1/(20 billion) | ||||||
Page 45
its value. So for the larger band the limits are 1,901-95 and 1,901 + 95, or 1,806 to 1,996. We then look at a bin-frequency table, shown in Table 0.1 (p 20). The table shows that the lower limit, 1,806, lies in bin 11, and the upper limit, 1,996, is in bin 12. Notice that the frequency of the alleles in bin 11 is 0.083 and that in bin 12 is 0.050, so we take the larger value, 0.083. This is shown as the frequency in the rightmost column of Table 0.3.
Continuing, we find the size of the smaller band of E is 1,137, and its lower and upper limits are 1,080 and 1,194. Both of these values are within bin 6 in Table 0.1. Its frequency is 0.024, shown in the right column of Table 0.3.
Now the membrane is ''stripped," meaning that the probes are washed off. Then the membrane is flooded with a new set of probes, this time specific for locus D17S79. Assume that the measurements of E are 1,685 and 1,120, and that the uncertainty windows of E and S1 again overlap. The ± 5% match window for the larger band is 1,601 to 1,769, and comparing this with Table 0.1 shows that the match window overlaps bins 9 and 10, of which 9 has the higher frequency, 0.263. In the same way, the match window for the smaller band overlaps bands 4 and 6, and the larger frequency is 0.015.
Again, the membrane is stripped and a new probe specific for DI S7 is added. This time, there is only one band. The individual is either homozygous, or heterozygous and the second band did not appear on the gel. So we apply the 2p rule, doubling the frequency from 0.068 to 0.136. Now the process is continued through two more probes, D4S139 and D10S28, with the frequencies shown in the Table 0.3. (If you wish, you may verify these numbers from Table 4.5, p 101, which also shows frequencies for black and Southeastern Hispanic databases.)
The next step is to compute the probability that a randomly chosen person has the same profile as the evidence sample, E. For this, we use the product rule with the 2p rule for the single band. For each double band, we compute twice the product of the two frequencies. For the single band, we use twice the allele frequency. Thus, going down through the table, the probability is
![]()
The maximum uncertainty of this estimate is about 10-fold in either direction, so the true value is estimated to lie between 1 in 200 million and 1 in 20 billion.
Suspect Found by Searching a Database
In the example above, we assumed that the suspect was found through an eyewitness, circumstantial evidence, or from some other information linking him to the crime. Now assume that the suspect was found by searching a database. If the database consists of 10,000 profiles, we follow the rule of multiplying the calculated probability by that number. Thus, the match probability, instead of one in 2 billion, is 10,000 times greater, or one in 200,000.
Page 46
Suspect and Evidence from the Same Subpopulation
It might be that the crime took place in a very small, isolated village, and the source of the evidence and suspect are both known to be from that village. In that case, we use the modified Equation 0.2b.
Consider first D2S44, in which p1 = 0.083 and p2 = 0.024. Suppose that the village is very small and that we wish to be very conservative, so we take ![]() . The probability from Equation 0.2b is
. The probability from Equation 0.2b is
![]()
Continuing in the same way through the other four loci, using Equation 0.2a for D1S7, and multiplying the results gives about 1/(600 million).
A PCR-Based System
We shall not give a specific example for a PCR-based system. The reason is that the situation is simpler, since there is usually no matching and binning. The detailed procedures are specific for each system and will not be repeated here. The techniques in general (e.g., for STRs) are the same as for VNTRs. They involve positions of bands in gels and photographs of the bands. The methods often use chemical stains rather than radioactive probes; that saves time. The allele frequency is determined directly from the database, and the calculations of match probabilities and likelihood ratios are exactly the same as those just illustrated.






































