
4
Ethics and Public Policy
This chapter was drawn largely from the workshop Session III: Ethics and Public Policy. Thus, the majority of the chapter summarizes workshop presentations. Where useful for background, some sections have been supplemented with additional information. The chapter, however, is not intended as an in-depth analysis and summary of the complex issues of ethics and public policy related to xenotransplantation. Xenotransplantation, no matter how scientifically promising or potentially lifesaving, poses critical questions demanding a broad societal examination that considers public health; the perspectives of transplant physicians, nurses, and staff; and the needs and views of patients and their families. This chapter explores the potential impact of xenotransplantation on physicians and health care providers, individual patients, and society. Consideration is given to the value and quality of human life, special problems of informed consent, and the use of animals. The chapter also addresses issues regarding the economic and regulatory impact of xenotransplantation.
Patients, Ethics, And Society
Patients' Perspectives
One of the most compelling parts of the workshop was a panel that included patients, family members, and advocates. The individuals on this panel discussed their experiences waiting for organs, undergoing allotransplants, or working to obtain experimental xenotransplants for others. The
nature and style of the arguments in other parts of the workshop, based on scientific and philosophical reasoning, were in stark contrast to the personal stories and perceptions of those whose lives had been directly affected by diseases for which transplants or novel medical therapies offered the only hope of survival.
One of the clearest messages conveyed how patients often feel excluded from decisions made by the medical, scientific, ethical, and public policy communities. The debate about transplanting baboon bone marrow into an AIDS patient in San Francisco prominently displayed this message for two reasons. First, the experiment in question was in the process of being considered for approval by the Food and Drug Administration (FDA). Second, AIDS patient activists in favor of the experiment represent a well-organized and sophisticated patient advocacy group that has been successful in bringing patient perspectives to debates about the accessibility and applications of new AIDS therapies. A leader from this community pointedly reminded the committee that it should have included a patient representative.
Brenda L. from Project Inform in San Francisco presented the position of AIDS activists by directly addressing some of the most common conflicts among stakeholder groups. For example, regarding research into new therapies, she asked:
What is the ultimate goal? Is it to answer questions that pertain to the public health or is it to answer interesting academic questions? Compounded with this is the fact that it is difficult to know if those interesting questions are going to impact on the public health or if they will just raise more interesting academic questions. One has to wonder if some of the delays aren't just due to pure academic rhetoric flying back and forth. One knows that a lot of delays are happening because the scientists have proprietary interests over their own science. Sometimes industry and their proprietary interests get involved. That invariably ends up hurting the patient population.
On issues of informed consent, the AIDS bone marrow experiment is unique in that the patient actively participated in much of the planning and development of the experimental procedure. Nevertheless, Ms. L. spoke about the subjects in early transplant experiments in terms with which many physicians and ethicists would likely agree.
As with any new procedure, there will be those who will sacrifice their lives in the name of science. The history of transplantation clearly has been such that most people who participate in earlier studies don't fare as well. These first patients carried a heavier burden of risk. However, if it weren't for their participation, such as in the early lung transplants, and some of them actually sacrificing
their lives to participate in research, we would not be where we are today with organ transplantation.
Although much in the public eye, the application of xenotransplants for AIDS is but one potential benefit from a patient perspective. There are thousands of people with kidney, liver, and heart disease who also face the real possibility that lifesaving therapy will not be available to them. This view was voiced by another member of the panel, Len K., a 38-year-old man who had been waiting for a kidney transplant for a year and a half and had undergone peritoneal dialysis. 1 His kidney disease, caused by severe diabetes and hypertension, forced him to be on Social Security Disability and created financial and health-related insecurities that seriously disrupted the lives of Len and his wife Maryanne.
I am comfortable on disability … on the one hand, the idea of disability, in terms of my mental state, is pretty good, and I am adjusting to it well. On the other hand, when I go to a conference, or hear about people my age or people I grew up with or worked with, and I see them get promotions, advance in their careers, or have families, that gets me down … there has been an impact, I know, on Maryanne. With my constant illness and with a variety of major events such as triple-bypass surgery, kidney failure, arthritis, and a variety of skin diseases—mostly related to the dialysis—it takes an effect on Maryanne. I think that a lesser woman would have left me … given what Maryanne has put up with, I think it is amazing.
Three other persons shared their experiences with workshop participants. Gloria B. and Calvin W. had received organ transplants. The third person, Evelyn W., was Calvin's mother, who spoke about the issues confronted by families. Gloria B., a mother of two, had experienced acute liver failure due to an extremely severe case of hepatitis A. By the time she reached the first hospital, Gloria was near death and only occasionally conscious. Her family was forced to make the decision to put her on artificial life support or to let her die. However, another option became available when she was transferred to a hospital capable of doing liver transplants and, fortunately, a liver became available. Gloria's family agreed to the liver transplant immediately. Despite their questions, with time running out, they felt they had to proceed.
Calvin W. also contracted hepatitis, but as an infant in 1972. For years, Calvin's mother and doctors did what they could to ''patch him up" from numerous complications of the disease. In 1986, when liver transplants became available for pediatric patients, only a very few had been done. Calvin, however, was deteriorating rapidly and was given a transplant, after extensive discussions involving Calvin, his family, and his physicians. Even Calvin's doctor, however, expected that a second transplant would eventually be necessary. Getting Calvin to the second transplant was a particular challenge because a suitable donor was not available quickly. To "bridge" Calvin and buy him time, doctors offered to perform a procedure in which Calvin's blood would be perfused through a pig's liver—essentially a temporary xenotransplant. Calvin's mother, Evelyn, recalled the decision this way:
When they offered me the pig, I didn't think about the fact that it was a pig. I thought, "We are losing this battle, and desperate men take desperate measures." We were desperate … At that point Calvin could not speak for himself, and I was his proxy. As such I was going to do everything I possibly could to see him through this. If he did not want it done, then he was going to have to tell me that when this was all over.
Both Calvin's second liver transplant and Gloria's first liver transplant have been, from a medical point of view, successful, but what about their lives afterwards? Their stories reflect those of many transplant patients in that the time following even successful transplants can be difficult for a variety of reasons. For example, Calvin's family had adequate medical insurance to cover nearly the entire cost of the transplants. Indeed, his family had been more adversely affected financially by the frequency and expense of medical care before the transplants. In contrast, Gloria B.'s family struggled for more than a year to get its health maintenance organization to pay $300,000 toward the $500,000 expense of the transplant. A large portion (the average is approximately 82 percent) of Len K.'s kidney transplant will be paid entirely by Medicare's End Stage Renal Disease Entitlement Program. For many others, no insurance or Medicare program is available, and because of lack of money, many of these patients will die.
Although economic issues loom large for transplant patients, they face other serious concerns related to quality of life and their complex reactions to the fact that another person's organ resides within their bodies. Also, lifelong immunosuppressive therapy has significant side effects, which often make it difficult for patients to take these medications as directed. Feelings expressed by patients are by no means homogeneous in this regard. According to Calvin,
I really didn't realize the value of the gift with the first transplant. Although I was worried and concerned, I think I took the donation for granted a little. Now, after the second transplant, I appreciate the donor more, including taking the medication and everything.
Gloria, however, said,
My immediate reaction was—I wouldn't say anger—probably a bit of disgust with myself mixed with confusion because the whole process was so foreign to me and I felt almost like a guinea pig, they were just doing anything to keep me alive. I wondered how important is that, because now I am going to deal with having this organ inside me that does not belong to me. Left up to me and as I didn't have any prior knowledge [about transplantation and donors], and I had some say-so in the matter, I'm not so sure I would have made the same decision for myself.
Reactions such as these are by no means unique, and they illustrate the deep complexity of patients' views about their health, their lives, and the means by which their lives are preserved. When asked, some patients embrace xenotransplantation as a wonderful answer to their prayers for a reasonable medical treatment. Others, for equally important personal reasons, reject such treatment. Despite the complexity of their reactions, one response seemed almost universal among transplant patients: they want to be heard and to participate in decisionmaking. Such decisionmaking occurs at many levels, and achieving maximal patient participation is challenging. Although AIDS activists have had a large voice in regulatory matters concerning AIDS therapies, few other patient groups have had such a voice. Many patients, who are healthy enough, are actively involved in decisionmaking with their physicians, but this partnership varies greatly from physician to physician and from patient to patient. Rarely do patients and scientists participate together in decisionmaking; indeed, each is likely to bring different vocabularies and perceptions to issues, which would require extensive translation for effective communication. Fortunately, protections that encourage, and at points require, patient–physician and patient–researcher communication exist in the form of guidelines for research with human subjects.
Informed Consent
Informed consent of research subjects is required in all clinical research conducted in the United States.2 The overall purpose of informed consent is to give potential subjects information that will enable them to make freely chosen, knowledgeable, and careful decisions about whether they wish to participate in research. Federal regulations require that the consent form contain a description of the nature and purpose of the research and its risks, benefits, and alternatives, among other requirements (45 CFR 46). The form must be approved by the local institutional review board (IRB) before it is given by the physician/researcher to a prospective participant for his or her consideration.
At this early stage, xenotransplant research carries high risks and high uncertainties in the setting of possibly desperate patient need, a situation that places added weight on informed consent. For example, more research needs to be done on the psychological, religious, and social interpretations of xenotransplants for patients and their families. In addition, for the patient, the risks are especially great relative to the individual benefits, and for the community, there is a possible public health risk from animal pathogens or new infectious agents.
The history of organ transplantation is replete with instances of medical community enthusiasm tending to underestimate risks and exaggerate individual benefits of new medical and surgical interventions (Arnold, 1995; Fox and Swazey, 1992). Overly optimistic judgments have appeared repeatedly in public statements by treating physicians and in consent forms. In one example, the permission form signed by the parents of Baby Fae—the neonate who received a baboon heart in 1985 for treatment of a congenital defect—clearly overstated the benefits in light of what was known at the time: "Long-term survival with appropriate growth and development may be possible following heart transplantation … this research is an effort to provide your baby with some hope of immediate and long-term survival." Baby Fae survived four weeks, after which no whole organ xenotransplants were attempted for eight years. Given that none of the more than 20 xenotransplant patients has survived longer than 9 months, and most for considerably shorter periods, xenotransplants must be considered extremely risky. The benefits, if
any, in the short term are likely to be highly limited. The patient may die immediately, may experience increased pain and suffering before dying, and/or may survive somewhat longer than otherwise. In contrast, the benefits to "society" may be significant because the new knowledge gained may benefit future patients.3 In this kind of circumstance, when the personal benefits are likely to be disproportionately low relative to the high degree of risk, informed consent assumes even greater prominence.
The former director of the National Institutes of Health (NIH) Office of Protection from Research Risks, Charles McCarthy, has offered suggestions for the content of informed consent for xenotransplant recipients. These suggestions, which are based on criteria listed in federal regulations, include a clear statement of the early stage of the research; mortality and morbidity data from previous human recipients (including quality of life); the option of no treatment; a fair estimate of the risks and of the time that will elapse between the xenotransplant procedure and the availability of a human organ (if the xenotransplant is to serve as a "bridge"); and disclosure of the degree of media attention on patients and their families and of the likelihood of offers to "sell" their story (McCarthy, 1995).
Apart from obligations to patients, what are investigators' obligations to provide information to, or seek some form of consent from, health care workers, family members, and the public? These groups may bear a risk, however difficult to quantify, of unwitting exposure to emerging pathogens.
There is no legal requirement for informed consent of "third parties" (i.e., people who are in contact with the research subject). Third-party risks are not normally evaluated in the course of the IRB approval process, and if the IRB does approve a research project involving a human subject, neither disclosure nor the consent of third parties is required. In the absence of legal mandates, is there an ethical obligation? What follows is first a discussion of the capability of the IRB to assess third-party risks and then a discussion of disclosure and/or approval of third parties.
Some have suggested that IRBs lack formal guidance in defining and assessing third-party risks (Dresser, 1995). The only explicit guidance on third-party risks comes from the field of gene therapy. When investigators develop gene therapy protocols for review by the NIH Recombinant DNA Advisory Committee and by the FDA, they are guided by the "NIH Guidelines for Research Involving Recombinant DNA Molecules." One of the appendixes to
these guidelines lists the following key public health questions that investigators should address:
- On what basis are potential public health benefits or hazards postulated?
- Is there a significant possibility that the added DNA will spread from the patient to other persons or to the environment?
- What precautions will be taken against such spread (e.g., patients sharing a room, health care workers, or family members)?
- What measures will be undertaken to mitigate the risks, if any, to public health?
- In light of possible risks to offspring, including vertical transmission, will birth control measures be recommended to patients? Are such concerns applicable to health care personnel?
These are complex questions, the answers to which are fraught with unknowns. If they are addressed, another set of questions will emerge related to the content of and process for public disclosure and/or approval.
As a practical matter, obtaining informed consent of health care workers and families is much easier than obtaining informed consent from the community. Informed consent of communities is traditionally undertaken, not individually, but through public hearings, advisory bodies, and a variety of legislative and executive branch processes. Many workshop participants believed that addressing such issues for xenotransplants will require extensive discussion involving government agencies, patients and their families, and the public.
Justice and Fairness Issues: Organ Allocation and Research
There are two major questions of justice and fairness that must be addressed in the consideration of xenotransplants: organ allocation and access to research. The United States has already developed a system for allocating human organs. The system was designed with patient and public input and with the input of physicians, ethicists, economists, and many other professionals. Despite this, support may be fragile because the scarcity is so acute that it fosters perceptions—however justified—of inequality, of organs going to the wealthiest and most powerful (Bowman, 1995).
A system for organ allocation was established by the National Organ Transplant Act (NOTA) of 1984. The system was designed to eliminate inequities in the distribution of whole organs, and the legislation created a voluntary national system of organ allocation that has operated with the
support and compliance of the transplant community. This national system, the Organ Procurement and Transplantation Network, was established by the U.S. Department of Health and Human Services and is operated under contract by the Health Resources and Services Administration with the United Network for Organ Sharing (UNOS). The role of UNOS is to link all organ procurement organizations with transplant centers. It operates a national computerized waiting list of patients in need of organs, whom it matches with donors when organs become available.
The board of UNOS, in consultation with the public, set down principles for the allocation of scarce organs and created a formula for their allocation to those on the waiting list. The formula varies according to the organ, but in general it is based on medical, scientific, and ethical criteria, such as histocompatibility, blood type, waiting time, logistics, and medical urgency. The ability to pay for the transplant is not a factor in the allocation of organs to patients on the waiting list (although it does factor in elsewhere, as discussed below). When an organ becomes available, UNOS generates a list of eligible patients in priority order. If the local organ procurement organization violates the ranking or other UNOS policies, its membership may be threatened. The development of more stringent sanctions—such as the loss of Medicare and Medicaid accreditation—is in progress, based on the passage of recent federal legislation.
Inequities can occur before placement on the waiting list. Two critical stages precede placement on a waiting list: patients need to be referred to a transplant center, and they need to be accepted by the center (Moskop, 1991). Inability to pay factors in at both stages. Because most private and public insurance covers transplants (see section on economics below), patients who are unable to pay are either uninsured or underinsured and account for 26 percent of the U.S. population (Evans, 1989). These patients may receive medical care sporadically, if at all. If they receive care and are diagnosed adequately, they still may be less likely to be referred to a transplant center because of their inability to pay. If they are referred, the transplant center generally requires assurance of payment before placing a patient on the waiting list (Evans, 1989). Transplant centers justify this practice on the grounds that, in order to recover losses, they must charge higher rates to all patients. This barrier to transplantation, created by inability to pay, is often referred to as the "green screen." Because of this barrier, many believe that the need for organs exceeds the actual number on the waiting list, but the extent of the real demand is difficult to document.
The green screen is less likely to operate for patients needing kidneys rather than other organs.4 Inability to pay was virtually eliminated as a factor in access to kidney dialysis and transplantation with the passage in 1972 of legislation extending Medicare benefits to patients with end stage renal disease (ESRD). These benefits are available to more than 90 percent of the U.S. population (Evans, 1989). The significance of this legislation is best demonstrated by a study that compared the composition of dialysis patients before and after the legislation's passage. It revealed that before 1973, many patients were selected on the basis of social status rather than medical suitability. Afterwards, access was greatly expanded, irrespective of income, race, gender, and education (Evans et al., 1981). For example, the percentage of African Americans in the hemodialysis population climbed from 7 to 34.9 percent, the latter figure reflecting the disproportionately high prevalence of ESRD among African Americans in the U.S. population. Likewise, African Americans represent 33.7 percent of patients waiting for kidney transplants (UNOS, 1994). Ironically, African Americans experience the longest waiting times for kidneys, once they have been placed on the waiting list. Their median number of days on the list is 74 percent greater than that of whites. This disparity is due to the greater difficulty in finding suitable tissue matches for African American patients. Although the organ donation rate among African Americans is proportional to their prevalence in the general population (12 percent), it is much lower than their prevalence in the ESRD population (UNOS, 1994). Further, although suitable tissue matches are possible between African Americans and caucasians or other ethnic groups, there is a higher probability of a match between two African Americans. Thus, the total probability of finding matches for African Americans among the general organ donor population is reduced, decreasing the number of suitable organs available.
The implications of the present system of organ allocation for xenotransplants first involve the pressure of the organ shortage and its effects on the search for alternatives. From the patient's perspective, there is much confusion and uncertainty about the true fairness or equity of the present system. Whether or not disparities will result in higher rates of xenotransplants among specific groups is not clear. If such a disparity occurs, further questions will have to be addressed. For example, will the disparity differentially affect specific ethnic or age groups? In part, the importance of these questions will depend on whether or not xenotransplants are (1) as successful as allotransplants and (2) equal to allotransplants in terms of expense. If the answers to
both of these questions show xenotransplants to be equal to allotransplants, possible disparities across regions or ethnic groups may not constitute a lack of fairness or equity. Such answers, however, await a long period of research and technological development.
Equitable access to research, then, is the other major concern about xenotransplants in particular. The significance of "equitable access" has changed dramatically over time. Historically, concerns centered on unequal burdens or levels of exploitation of religious groups, minorities, and the disabled (Bowman, 1995). The Nazi medical experiments and the Tuskegee syphilis study represent egregious examples of inequality and exploitation. The legal and ethical commitment to informed consent and equitable selection of research subjects evolved out of these shameful legacies. The U.S. Department of Health and Human Services Regulations for the Protection of Human Subjects attest to the preeminence of informed consent and also mandate the IRB to ensure that the "selection of subjects is equitable" (45 CFR 46).
Although specific groups of people were disproportionally harmed in research in the past, the current emphasis is that historically oppressed groups not be deprived of research participation. Research participation is seen now by many as a benefit, a possible lifesaving alternative to conventional treatments. The women's movement claims as one of its successes, for example, the passage of legislation5 requiring the participation of women and minorities in clinical research on conditions that affect them. However, given the historical shifts back and forth, it is difficult to predict whether a positive perception of research by the public will persist in the future.
In light of the scarcity of organs, access to xenotransplant research is likely to be highly sought by many individuals. Researchers may be pressured by patients, families, and their advocates. The situation may become analogous to that immediately before the passage of NOTA, when some families took to the airwaves to dramatize their plight, a practice that has continued over time. More recently, when approval for one of the first xenogeneic bone marrow experiments for AIDS patients was delayed for almost two years, one of the investigators, Suzanne Ildstad, publicly reported being approached by several foreign governments to conduct the trial in their countries. Sensitivity about research is, nevertheless, still a strong motivating factor in minority communities. This distrust could easily lead to the perception that animal organs as experimental therapies will be offered to desperately sick people who lack the financial resources for allotransplants or are members of racial or ethnic minorities.
Social Acceptance of Xenotransplants
It is difficult to assess how a society as pluralistic as our own will view an unusual or innovative medical technology. As the workshop made clear, there are many stakeholder groups involved in the development of xenotransplantation. These groups—including patients, physicians, scientists, public health officials, biotechnology researchers and investors, ethicists, and others—often have conflicting priorities and concerns. Will xenotransplantation rouse community fears and anxieties? Will the public demand that xenotransplantation not be undertaken because of the risk of infections or will the public by and large embrace xenotransplantation as a potentially lifesaving procedure? Will such approval be dependent on the species of animal used, (i.e., pigs instead use of baboons)? Human organ donation serves as an initial backdrop for understanding and anticipating broader societal reactions to xenotransplantation.
The public endorses human organ transplantation—at least on the surface. There is widespread public acceptance of organ donation and transplantation, as evidenced by a 1993 Gallup survey sponsored by the Partnership for Organ Donation (Spital, 1995).6 In the survey of more than 400 adults, 85 percent expressed support for organ donation, although the level was somewhat lower for minority respondents. The survey found that people generally believe that organ donation allows something positive to emerge from a person's death and helps families to cope with their grief. Nevertheless, only 52 percent of these respondents had actually discussed their wishes to donate organs with a family member, and only 28 percent had ever formally granted permission for organ donation.
The disparity between attitudes and deeds may reflect many factors, including the relatively recent emergence of organ transplantation and the perceived conflicts with certain religious traditions regarding the body, as well as deep-rooted fears and anxieties about death, dismemberment, having living parts of another's body within oneself, and dehumanization. In addition, some persons are suspicious about the degree to which organ procurement fosters exploitation and premature harvesting of organs, although such practices clearly violate medical ethics and legal mandates. Such public unease may indicate a lack of trust in the medical profession.
The complex emotional underpinnings of organ donation may be difficult to capture or, indeed, may be concealed in public surveys. For example, each person responding to a survey could do so from a variety of viewpoints,
including that of a patient needing a transplant, a family member of a dying potential organ donor, or a potential organ donor. For each viewpoint, multiple psychological, cultural, religious, and other factors come into play that reflect a respondent's deepest beliefs about, and concepts of, self and those concerning relations to known and unknown others. Yet, social altruism is a strong value in this society, and some argue that surveys regarding organ transplantation are overly positive due to the eagerness of respondents to affirm what is widely expressed as a public good—the saving of another life (Siminoff et al., 1995).
Recent public policies and proposals designed to foster donation may have had the unintended consequence of heightening apprehensions (Fox, 1995). For example, although at least 26 states have passed laws requiring that relatives of a dying patient be asked about donating the person's organs for transplant, the number of organs donated has not increased appreciably. Furthermore, proposals of presumed consent (the assumption that, in the absence of specific instructions to the contrary, a dying patient would consent to donation) and financial incentives for donation have not been implemented in the United States, but have been tried in Europe. The fact that the overall rate of organ donation remains about the same has led to even more aggressive steps to increase donation, such as the American Medical Association (AMA) endorsement of the use of tissue and organs from anencephalic babies in whom the brain, particularly the cerebral cortex, fails to develop normally. Within months, the AMA rescinded this endorsement (AMA, 1995; Gianelli, 1995). Some social observers express concern that such steps conflict with established criteria for organ donations and, more importantly, will diminish the dignity and value of human life. Some argue that as our society moves away from an inviolate and unique regard for human life, dignity, and death, the introduction of xenotransplantation may exacerbate the problem by blurring the boundaries between humans and animals.
Transplant surgeons and ethicists also voice concerns that exaggeration of the scientific potential of xenotransplants will actually reduce the number of human organs now being donated. They view public support for voluntary cadaveric organ donation as tenuous enough to be undermined easily. Although they share the hope that xenotransplantation can ameliorate the organ shortage, they are concerned that the "unnaturalness" of, or aversion to, xenotransplantation may erode public support for allotransplant donation. Indeed, the prospect of reliance on xenotransplants may undermine the symbolism of organ donation as an ultimate human gift. Sociologist of medicine Renee Fox stated at the workshop, "We would progressively lose what is perhaps the deepest and highest symbolic moral and existential significance of organ transplantation, its gift exchange dimension … that the living parts of persons are offered in life or in death to known or unknown others, to our strangers and our enemies as well as to our kin, in the form of a gift beyond duty and claim, beyond reckoning and rules" (Fox, 1995).
In contrast to these views, others argue that reliance on xenotransplants would bolster human dignity (Rothman, 1995). Alleviating the organ shortage may strengthen our respect for human life by curtailing exploitative practices of human organ procurement in some Third World countries. The prospect of a plentiful supply of organs might undercut the practice in India and some other countries of selling organs from live donors. Other practices that might be eliminated occur in China, where organs are harvested after death from prisoners without their consent. Such hopes, however, may not be realized if cultural values against the use of animal organs in these countries outweigh values about human life. Alternatively, if xenotransplantation requires special animal facilities and increased expenditures, economically depressed countries may continue exploitative practices to procure human organs.
A final concern is that the use of xenotransplantation may complicate, rather than solve, the organ shortage. There are limits to the availability of nonhuman primates, and there may well be limits on the availability of transgenic swine (if they prove to be a successful organ source). Regulations concerning animal testing for infectious diseases are among the factors that will dictate the feasibility and expense of producing organs from these animals. Moreover, should xenotransplants prove successful as ''bridges" to human organ transplants, the shortage of human organs will continue to be a problem. In fact, the shortage may get worse as the demand likely increases. This is precisely what happened after the first successful kidney transplants in the 1960s—the organ shortage grew along with increasing medical achievements.
Social Acceptance of Infectious Disease Risk
The possibility of disease transmission from xenotransplantation and its effect on public perception of the procedure must be addressed. The literature on risk perception discussed in an earlier report by the National Research Council (1989) makes clear that public perception of risk almost always differs from expert judgment of risk. Public perceptions are influenced not only by scientific probabilities, but also by a host of psychological, social, economic, ethical, political, and other concerns.
Many of the conditions surrounding infectious disease risk from xenotransplants contribute to heightened public concern. The mere fact that the public derives no direct benefit from the procedure may tend to alter the risk-benefit equation by enhancing perceived risks. Lifesaving xenotransplants benefit the individual recipient and family, rather than society. Other features of xenotransplants likely to heighten public concern include scientific uncertainty about the likelihood of disease transmission; intense media attention to the first xenotransplants; a cultural distrust of science, particularly
if industry investment is at stake (i.e., transgenic pigs, encapsulants); the esoteric nature of xenotransplantation science, which combines such specialized fields as immunology, molecular biology, and virology; the involuntary nature of exposure to disease (if the risk is airborne); and the possibility of a latent, occult illness that might not become manifest for years. More than a decade's worth of experience with the HIV (human immunodeficiency virus) pandemic is sufficient to alert the public to the sometimes delayed expression of zoonotic infection. In addition, current media interest in deadly viruses, as evident in books and movies about the monkey colony infected by Ebola virus in Virginia, has certainly captured, and possibly sensitized, public attention and concern.
There have also been periods of intense public outcry surrounding the introduction of recombinant DNA technology since the 1970s. The concerns have often centered on the possibility that a hybrid virus would escape the laboratory and infect humans. Even though the scientific community had acted earlier to impose a moratorium until safety guidelines were in place, communities remained distrustful. Citizens of Cambridge, Massachusetts, opposed to recombinant DNA research at Harvard and the Massachusetts Institute of Technology, were successful in convincing their city council to impose a moratorium on the research until a citizen review board could evaluate the problem and recommend action. The board, composed entirely of nonscientists, agreed to permit the research to proceed, provided additional public safety measures were imposed. Whether similar community response will occur with xenotransplantation remains to be seen.
Although the various measures outlined in Chapter 3 and recommended in Chapter 5 will reduce the likelihood of any harm to the public health from xenotransplantation, these measures cannot eliminate all risks. Refusal to proceed, however, to avoid any possibility of harm to public health would likely forestall benefits for those whose health or continued life depends on successful xenotransplantation research. During the workshop the point was made repeatedly that, in the simplest terms, we as a society are obliged to choose between two risks of harm: to those who will suffer from illnesses potentially treatable by xenografts versus those who might suffer from infectious diseases potentially let loose in the general population by xenotransplantation. Ensuing committee discussion and correspondence was considerable on this topic and in addressing this choice, some committee members were guided by what they regarded as the moral imperative that our own humanity is diminished if, in order to protect ourselves, we turn away from others whose suffering is both clearly visible to us and more clearly devastating in its impact on them. This viewpoint, then, further holds that we are morally obliged not only as individuals, but as a community, to accept some risk to ourselves to save our fellow human beings from more certain harm. Accordingly, and given this choice, we are also obliged to do all within our power as a society to
protect the community from the possibility of infectious disease as we proceed with xenotransplantation.
Value And Use Of Animals
The use of animals in scientific research has been decried by some as unethical, a viewpoint that arose in the mid-eighteenth century and is held today by a number of spokespersons such as Tom Reagan and Peter Singer, philosophers, and Ingrid Newkirk, director of People for the Ethical Treatment of Animals. In fact, this viewpoint has engendered a debate that has become increasingly acrimonious in the past two decades. The following discussion of this issue begins by tracing briefly the history of philosophical and ethical thought on the use of animals in research. It then focuses specifically on the use of animals as a source of organs. The history of attitudes regarding xenotransplantation will be reviewed briefly, followed by modern-day reflections on the use of nonhuman primates and swine as a source of cells, tissues, and organs for transplantation.
Ethical Theory
René Descartes (1596–1650), the seventeenth century mathematician and philosopher, concluded that animals were automatons with the semblance of life, but without feelings or emotions. Because animals reacted only reflexively to external stimuli and did not have the capacity to suffer, humans could treat animals as they wished. Few people agree with this formulation today, primarily because it does not accord with their experience with pets and other animals. Instead, notions of stewardship toward animals, which existed before Descartes, have been more fully developed. Ethical theories have been proposed by philosophers and ethicists to formalize ideas of stewardship toward animals and to critically assess how these feelings may be applied to the use of animals by humans.
Ethicists address the general issue of what people ought to do and how they ought to behave. Moral arguments are developed and examined to determine if they can rationally support the actions and behavior of people. That is, ethicists study the question of what actions are considered good or bad and develop criteria for classifying decisions as good or bad. It is a rational approach to problem solving that relies on facts and logical inference, thereby enabling sound judgments. However, there is not yet a single, widely accepted and defensible theory that outlines rules of conduct toward all animals, whether they are used for scientific research, entertainment, food, or clothing.
Thus, at the current stage of development, ethical theories inform the present debate but do not settle it.
Two theories concerning the use of animals have been championed in the recent past: a utilitarian approach and a rights approach. For the purpose of efficient analysis, human activity can be divided into three components: the person performing the action, the action itself, and the consequences of that action. Utilitarian theory focuses on the consequences of an action and, hence, is a form of consequentalism, whereas rights theory considers the action itself. The utilitarian approach is discussed first, followed by the rights approach.
Utilitarianism was first enunciated by Jeremy Bentham (1748–1832), an English jurist and philosopher, and expanded by John Stuart Mill (1806–1873), an English philosopher and economist. A type of consequentialist or teleological theory, utilitarianism is best understood as a risk-benefit analysis in which the sum of the goods brought about by a certain action is weighed against the sum of the harms caused by that action. Originally, good was defined as pleasure and harm as pain, suffering, or decreased happiness.
This approach seems to accord with the way people often think about moral decisions, including the use of animals in research. Finding a new vaccine, such as the polio vaccine, or finding a cure for a disease far outweighs any harm that may befall the animal. This analysis, however, does not take into account the capacity of the animal to feel pleasure or happiness or to be free of pain and suffering. Peter Singer, a contemporary Australian philosopher, adds the loss of this good for the animal to the utilitarian analysis of the use of animals in research. Singer notes that much research involving animals appears to produce no useful results, and there may be an alternative that maximizes good without using animals in research. He concludes that the vast quantity of such research is not morally justified. Because utilitarian theory requires an estimation of the probability that a certain action will produce a good consequence, if most research produces no useful result, it is morally groundless or problematic. Alternatives to the use of animals in research may include conducting public health research or epidemiological studies instead of using animals. For example, if an epidemiological study shows that the incidence of heart attack drops when fat and cholesterol intake are decreased and when the individual exercises, then public education would be a way to maximize the good far more than animal research. This analysis ignores scientific questions concerning the links between myocardial infarction and cholesterol or exercise.
Two sets of criticisms of utilitarian theory include its not taking into account the worth of the individual and its defining ultimate good as hedonistic pleasure, rather than any number of other possible goods. Because some people are willing to grant sentient animals some moral status, the individual animal's interests must be considered when assessing the probable consequences of an action. This does not mean that an animal is to be accorded an equal moral
status to humans or that the life of a human is equal to the life of an animal. It is clear that the quality of human life in its totality (including cognitive development and the ability to feel and discuss emotions) is far richer and more complex than the life of any animal. Thus, one should give more weight to human than to animal life. The notion of complexity of life can be extended to our intuitive ideas that the life of a nonhuman primate is worth more than that of a mouse, which in turn is more complex than a paramecium. Thus, according to Singer, a more complete utilitarian analysis would include the rights of individuals as autonomous agents and a broad definition of "the good" to encompass the complexity of life.
The second approach to ethics, the rights approach, is indebted to the philosophy of Immanuel Kant (1724–1804), a German philosopher, who stated that humans are ends in themselves and thus have certain rights. The rest of nature is viewed as existing to serve human interests; in particular, animals exist "merely as a means to an end. That end is the good of humankind." According to this formulation, humans are morally free to use animals as they wish, provided that the animals are not treated cruelly (because to do so might make humans cruel to each other). This approach views acts as right or wrong regardless of their consequences. Thus, rights or deontological theorists reject the utilitarian approach. Instead, their view is that what is right does not depend on the value of the consequence, but on appropriate treatment of the individual. Furthermore, rights theory focuses on the inherent rights of individuals, not on the sum of the interests of a group of people.
What Kant developed is what Tom Regan, a modern American philosopher, would term an extreme rights position. Regan moderates Kant's position by allowing some utilitarian concepts to enter the discussion, but he maintains that "rights are more basic than utility and independent of it." Regan argues that all individuals, including nonhuman animals (because we cannot distinguish all humans from all animals), are to be treated with respect. He then holds that nonhuman animals are of direct moral significance and that humans are morally responsible for their protection. The definition of sentience has vexed many rights philosophers, because many animals seem to possess a set of psychological characteristics such as memory, wants or desires, acting intentionally, and so forth. The issue of sentience and what beings inherently possess rights is unresolved. Many feel that the rights view can be expanded to include some idea of hierarchy, with humans deserving of more rights, and lower animals having far fewer. Elements of utilitarianism are incorporated into this version of the rights theory.
The rights ethical view accords with many people's view of animals, insofar as it explains why we should treat animals as individuals worthy of respect. Most people would agree that humans are not free to treat animals as if they were inanimate objects, because many animals do seem to possess psychological characteristics similar to humans. The rights view also
undergirds several principles that have been articulated to govern the use of animals in scientific research. Thus, the U.S. Government's Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training contains the statement (Principle IV) that "proper use of animals, including the avoidance or minimization of discomfort, distress, and pain when consistent with sound scientific practices, is imperative." Because individual animals are to be treated with respect, another of the government principles states, in effect, that the fewest possible animals should be used in research and that animals are not to be killed wantonly.
Many critics of the animal rights view espouse the idea of kinship, that is, our duty or obligation to our children, family, and country and, more broadly, to the human species. This accords with personal experience and even allows for special feeling toward our pets and domestic animals. The obvious drawback of this formulation is that it seems to be exclusionary, rather than impartial or fair. Finally, several modern philosophers have noted that all of the above theories fail to recognize that humans do not operate in a vacuum, but in fact are part of the whole of nature and must act accordingly. Often called moral ecology or deep ecology, this view talks about humans' obligations to treat the whole of nature with respect. Nature is viewed not as a "renewable resources" but rather as the context in which we exist, and it must be considered as a whole or unity. These ideas, which have been applied to the treatment of animals by Strachan Donnelley, a contemporary moral philosopher from the Hastings Center, seem to have much intuitive appeal because they incorporate elements of both utilitarianism and rights philosophies and recognize the complexity of life. This approach broadens the view from the individual to a whole community, including humans and animals, and recognizes that the complexity and richness of nature are characteristics that are due respect and protection. According to this view, endangered species are due more protection than a laboratory mouse.
History of Social Responses to Xenotransplantation
Physicians have been interested in using animal tissues to treat human disease for centuries (Lederer, 1995). In the seventeenth century, transfusion of blood from animals into people was abandoned after limited use. Midnineteenth century experiments by the English obstetrician James Blundell, transfusing sheep blood into dogs, suggested that human-to-human transplantation would be more effective than animal-to-human transplantation. Xenografting received more attention in the late nineteenth and early twentieth centuries. There were efforts to treat kidney failure by transplanting animal (sheep, goat, and pig) kidneys into patients. Skin grafting in patients with burns or trauma was attempted using both cadaver skin and skin from a wide variety of
animals; bone grafts from living dogs were also undertaken. Gonad transplants from goats and monkeys were tried as a treatment for impotence and lack of vitality. The role of antibodies and the immune system in rejection of either human or animal tissues was not recognized until the late 1940s, when modern immunological concepts began to be described. Some surgeons were encouraged by the development of immunosuppressive agents in the late 1950s and, in the early 1960s, attempted nearly 20 primate-to-human transplants of either kidneys or livers. Ultimately all of these organs were rejected or the patients died of infections that they were unable to combat due to immunosuppression. However, it should be noted that one patient, who received a chimpanzee kidney, survived nine months and died of infection, not organ failure.
The early twentieth century experiments in xenografting prompted considerable public comment and criticism. Surgeons who attempted xenotransplantation experienced intense media scrutiny and attacks from American antivivisectionists, who also criticized the short-lived enthusiasm and ambition of surgeons for new techniques. Depictions in popular magazines and films such as the 1932 movie Island of Lost Souls, based upon H. G. Wells' The Island of Doctor Moreau, and Disney Studios' cartoon The Mad Doctor suggest cultural unease about the dissolution of the border between animal and human and the medical hubris that produced it.
Further public reaction was engendered by Dr. Leonard Bailey's transplantation of a baboon heart into Baby Fae in 1984. Questions concerning both the scientific basis for this transplantation experiment and the ethics of research with human subjects were raised. At the time of this experiment it was known that the success of transplantation improved when there was major blood group compatibility, a compatibility that was lacking in this case. Further, this case raised issues of how to obtain adequate informed consent under life-and-death circumstances, particularly when a child is involved.
Two points that can be made regarding these survey comments include (1) the role of the media in disseminating information and in shaping the public's understanding and acceptance of new medical procedures, and (2) the public's concern for animals, which has changed not only over the past few centuries (see above), but also over the last few decades. A new social contract concerning the use of animals in xenotransplantation may have to be negotiated, the details of which will depend on the public's response to the issues.
A Moderate Ethical Perspective of Xenotransplantation
As knowledge about biology increases and the capacity to perform such things as transplanting animal organs into people improves, more attention needs to be given to the respective value of, and relationships between, animals and humans. Utilizing closeness to being human-like as a benchmark measure of value, James Walters, a contemporary philosopher, has developed ideas that grant highest moral status to those who are closest to qualities that most humans possess (Walters, 1995). These qualities connote self-consciousness and the capacity to experience the richness of life. For Walters, the idea of proximity includes potentiality for human existence, development toward that existence, and the bonding of persons with other beings.
Walter's approach enables him to value beings in terms of their proximity or likeness to persons, rather than their being members of a biological species. He thus holds that it may be more justifiable to use anencephalic infants as organ sources than to use chimpanzees. The intelligence and human-like behavior of other species are being recognized increasingly. The language capabilities of chimpanzees approximates those of two-and-a-half-year-old human children, and wolves have a well-developed social structure. So, according to Walter's analysis, these animals may have higher moral status than some humans. Finally, it is clear that humans, like other animals, are "speciesists," in that we display partiality to fellow human beings, even if some of them lack capabilities possessed by other species. However, humans are intelligent enough and powerful enough to impose their views on the whole of nature, which imposes special obligations and constraints on human actions. This consideration raises the same issues that are considered by moral ecologists (discussed earlier).
Application to Xenotransplantation
The above review shows that ethicists and philosophers, who have addressed the morality of xenotransplants, start with differing assumptions that lead them to draw different conclusions. Most people would appear to agree with the type of thinking voiced by Walters regarding the far greater value of persons over animals that lack human abilities and characteristics. Others assume that members of the human species have the highest priority and that appropriate, humane, and judicious use of animals is justified. Many are willing to extend some idea of kinship to certain, if not all, nonhuman primates, as well as to companion animals such as dogs and cats. For these people, the use of such animals must be justified by clear and well-articulated goals. Thus, many would not be willing to use chimpanzees as a source of organs, because they are quite human-like and because they are an endangered
species. Yet they would approve of the use of baboons if this were the only alternative to the death of the patient and if the transplantation had a reasonable chance of success. It almost goes without saying that if pigs could be developed as a source of organs, most people would not object because these animals are traditionally used as a source of food, are distant from humans phylogenetically, and fall much lower on the personhood scale. Thus, although no philosophical or ethical consensus has emerged, most people would favor proceeding with well-designed xenotransplantation experiments that begin with baboons, but would favor the use of swine if these animals prove to be a viable source of organs for humans. Such experiments would be subject to review by the Institutional Animal Care and Use Committee and must be performed in accord with all applicable regulations and local institutional policy (see section below on reviewing and monitoring xenotransplantation).
Economic Issues Regarding Xenotransplants
Forecasting the economic impact of xenotransplantation is difficult at the present stage of development and eventually must address both the expense of whole organ xenotransplants and the expense of cell and tissue xenotransplants, which are likely to be quite different. The section that follows provides background information about expenditures for allotransplantation in the aggregate and at the procedure level and describes the environment in which economic decisions about xenotransplantation are likely to be made. Allotransplants are quite expensive, but the scarcity of human organs has served to limit aggregate expenditures. Despite the difficulty in prediction, xenotransplantation will certainly result in increased overall expenditures simply because success will translate need into demand.
Aggregate Expenditures for Organ Transplantation
In 1994, aggregate expenditures for organ transplantation were approximately $4.1 billion (Evans, 1995b). This figure represents a small fraction of all national health care expenditures for that year, less than half a percent of a total of almost $1 trillion. The fact that annual expenditures for organ transplantation account for only a small proportion of national health care expenditures is explained by the relatively low number of transplanted and surviving patients. Were more organs available, the total expenditures would most certainly be higher. The transplant expenditures for 1994 were incurred by approximately 125,000 surviving recipients (i.e., patients who either had undergone the procedure during the year or were receiving posttransplant care
for an earlier year's transplant). The expenditures were further based on billed charges (the amount a patient or third-party payer was billed by a hospital), which are usually higher than the actual insurance reimbursement (Evans, 1995b).
Managed care, discussed more thoroughly at the end of this section, is having a profound impact on reducing organ transplant expenditures. Recent estimates indicate that, because of managed care contracting for transplant services, billed charges have decreased by approximately 34 percent (Table 4-1). So, for example, if all transplant patients were associated with a managed care network in 1994, total transplant-related expenditures would have been about $3.0 billion, rather than $4.0 billion. Thus, transplant expenditures would have represented an even lower share of national health care spending. Managed care aside, the aggregate expenditures for transplantation are relatively less visible and controversial than the high per capita expenses, that is, the expense associated with an individual.
Transplant Procedure Expenses
With annual per capita patient expenditures averaging about $33,000, 7 organ transplantation is among the most costly of medical treatments. The high expense is a function of both the surgery itself and continuing care, including immunosuppressive drugs. Organ transplants—including bone marrow transplants—consistently rank in the top ten most expensive medical treatments, according to data from the Mayo Clinic. Between 1990 and 1992, liver and heart transplants were ranked in either first or second place, based on the billing data for transplant procedures (Evans, 1993). Follow-up charges ranged between $10,000 and $21,000 in 1993. Such expenses, however, vary enormously depending on the organ (or tissue) transplanted, where the procedure is performed, and the health status of the patient before transplant. For example, average annual patient expenditures in 1994 were highest for lung transplants ($118,845) and lowest for kidney transplants ($19,195), including follow-up care (Figure 4-1). Lung transplants were the least commonly performed, accounting for only about 4 of every 10,000 transplant procedures undertaken in 1993. Kidney transplants, in contrast accounted for about 60 of every 100 transplant procedures (UNOS, 1994).
TABLE 4-1 Aggregate Expenditures for Transplantation Organ, 1994
|
Transplant |
Network Pricing |
Billed Charges ($ million) |
Network Effect (%) |
|
Kidney |
1,703.9 |
1,796.4 |
-5 |
|
Liver |
756.4 |
1,344.9 |
-78 |
|
Heart |
392.3 |
645.2 |
-64 |
|
Lung |
101.4 |
187.3 |
-85 |
|
Pancreas |
105.7 |
110.3 |
-4 |
|
Heart-lung |
11.0 |
20.3 |
-85 |
|
Total |
3,070.6 |
4,104.3 |
-3 |
|
SOURCE: Roger W. Evans, Ph.D., Section of Health Services Evaluation, Mayo Clinic, Rochester, Minnesota. |
|||
Ethics and Public Policy
Charges also vary depending on where the transplant is done. Figure 4-2 depicts the hospital charge per case and expected patient survival rates for liver transplants at different hospitals. The charges ranged from a low of $97,852 to a high of $457,522, a difference of more than 300 percent, with seemingly no effect on patient survival. The patient's health status prior to transplant also has a significant effect on charges. The poorer the health status, the higher were the charges for either heart or liver transplants. Patients on life support have the highest total charges, followed by hospitalized and then homebound patients. Not surprisingly, patients on life support before the transplant have the worst outcomes, as measured by one-year survival rates (Evans, 1993).
Expenditures for Xenotransplantation
The projected economic impact of xenotransplantation is complicated by uncertainties. These include the number of procedures that could be performed, as well as animal preparation, xenotransplant procedure, and patient posttransplant expenses. Other key variables include whether the procedure is a temporary measure to an allotransplant—and thus an additive expense—or whether the procedure is a permanent xenotransplant that might ultimately reduce the patient's long-term health care expenses. Some cellular xenotransplants, such as pancreatic islet cells or encapsulated cells, may even reduce some expenses by eliminating the need for long-term immunosuppression and its associated risks.
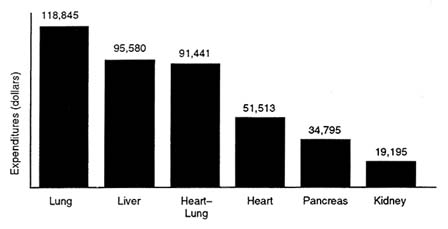
FIGURE 4-1 Average annual expenditures per surviving transplant recipient, 1994 (billed charges). SOURCE: Roger W. Evans, Ph.D., Section of Health Services Evaluation, Mayo Clinic, Rochester, Minn.
One of the greatest determinants of overall economic impact, however, is induced demand, that is, demand created by the availability of a procedure that is now rationed because of an inadequate organ supply. The demand will come from patients on the waiting list, patients who are not eligible for placement on the waiting list, or patient with conditions that may now benefit from xenotransplantation. It is estimated that nearly 124,000 patients, who could conceivably require a solid organ transplant, might benefit from xenotransplants. About 18,000 allotransplant procedures are now performed annually. Figure 4-3 presents an estimate of first-year expenditures with and without the availability of xenotransplants. If all those in need of a transplant receive an allo- or a xenotransplant, annual expenditures would rise from a conservative $2.9 billion to $20.3 billion, reflecting a change from less than half a percent of national health expenditures to more than 2 percent. It is important to note, however, that these estimates are quite conservative because they assume that the expense of xenotransplant procedures would be about the same as allotransplants. Further, the estimates are based strictly on the cost of the procedure (they exclude the expense associated with continuing care).
Insurance Coverage
The charges for allotransplants are covered to varying degrees by both public and private insurance, as long as the procedure is not considered
experimental. Private insurers account for the bulk of the payments, with the exception of payments for kidney transplants. Kidneys are the most frequently transplanted organ, primarily because of the success of the procedure and the availability of Medicare coverage. As a result of legislation enacted in 1972, Medicare has become the primary source of kidney transplant payments, covering about 90 percent of patients (Evans, 1993).
When Congress passed the 1972 Social Security Act Amendments, Medicare was extended to cover the disabled population under age 65. At the same time, the legislation deemed patients with chronic renal failure as automatically ''disabled," thus creating the first and only diagnosis-specific entitlement in the history of Medicare (IOM, 1991). Those insured under Social Security, their spouses, and dependents were thereby covered under Medicare for treatment of renal failure, including renal dialysis and transplantation. Over time, Medicare also has extended coverage to heart, liver, and bone marrow transplants, but not as an entitlement. Numerous restrictions apply. Medicaid, the joint federal-state public insurance program for certain people with low incomes, accounts for only a small proportion of organ transplants (Evans, 1993).
Private insurance usually pays for heart, liver, and bone marrow transplants, but less typically for heart, lung, and pancreas transplants, which are considered experimental by many insurers. The extent of reimbursement varies according to the policy and, as previously described, usually falls short of actual billed charges.
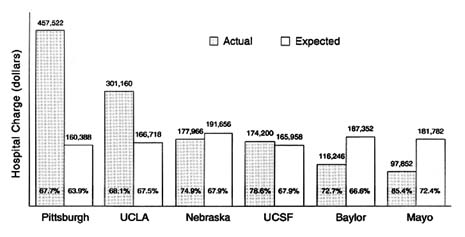
FIGURE 4-2 Liver transplantation total hospital charge per case and 3-year patient survival. SOURCE: Roger W. Evans, Ph.D., Section of Health Services Evaluation, Mayo Clinic, Rochester, Minn.
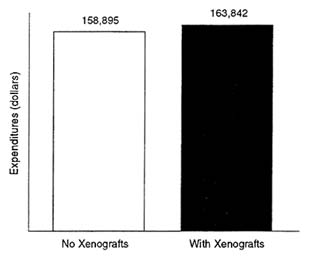
FIGURE 4-3 Impact of xenotransplantation on first-year transplantation procedure expenditures only, 1994. SOURCE: Roger W. Evans, Ph.D., Section of Health Services Evaluation, Mayo Clinic, Rochester, Minn.
Impact of Managed Care
The health care landscape is being transformed by managed care. Almost unheard of in 1980, when 3 percent of employees in medium-size and large establishments were enrolled, managed care enrollment had skyrocketed by 1993 to 49 percent of such employees (BLS, 1994).
Managed care refers mostly to health maintenance organizations (HMOs) and preferred provider organizations, which seek to provide health care in a manner that controls rising expenditures. Some of the most common cost control strategies are capitation, case management, quality reviews, and reliance on network providers who offer their services at discounted fees in exchange for a higher volume of services. Benefits typically emphasize those services that offer favorable health outcomes for the lowest expense. When two treatments are available for the same condition, managed care encourages (e.g., by offering financial incentives) or requires patients to select the least costly.
Managed care coverage of transplants is similar to that of traditional fee-for-service plans, although the data are somewhat sketchy. For example, surveys reveal that organ and tissue transplants (cornea and bone marrow) were covered by more than 90 percent of HMOs in 1992 (with the exception of heart-lung and pancreas transplants) (Evans, 1993). The major difference between managed care and fee-for-service plans is the price. Through
negotiated discounts with providers, managed care organizations have succeeded at reducing transplant fees by more than one-third (Figure 4-1). Ironically, these dramatic price reductions have served to "improve" the cost-effectiveness of transplantation, although without any apparent effect on medical outcome and survival.
The introduction of any new and expensive technology into managed care is likely to encounter resistance unless it is cost-effective. Managed care providers, and the employers who contract with them and dictate many of the coverage decisions, are wary of high-expense, low-benefit technologies. For xenotransplants to be covered by managed care, their cost-effectiveness will have to be demonstrated by carefully designed studies, as was done by comparing kidney allotransplants with dialysis. Most managed care providers and other insurers have decided to cover kidney transplants because research demonstrated that annual expenditures for a patient with a functioning transplant are far lower than those for renal dialysis (Evans, 1993). The final decision about whether to reimburse or cover xenotransplantation under private insurance ultimately resides in the hands of each insurer or employer, based on a host of financial and social considerations (discussed below). Xenotransplants for healthier patients would more likely be covered because they would have greater chances for success.
Pharmaceutical company investment in xenotransplant research has also come to reflect the changing health care marketplace. Therapeutic modalities, such as encapsulated cells and tissues, that hold the possibility of a cure without the need for expensive, long-term immunosuppression are more attractive to pharmaceutical investment because of their potential cost-effectiveness. Reimbursement would be almost guaranteed if cost-effectiveness could be demonstrated unambiguously.
Justice, Fairness, and the Ability to Pay
Should society pay for xenotransplants if they are found to be successful? This is a fundamental question of justice and fairness, the branch of ethics that is concerned with questions of macroallocation and microallocation of resources and whether these decisions fairly distribute societal benefits and harms. Macroallocation refers to how society allocates resources in the broadest of terms, such as whether it pays more for health care than for defense and education; microallocation refers to how society allocates resources on a lower level, such as whether an individual HMO covers xenotransplants instead of prenatal care (Evans, 1995a,b).
If whole organ xenotransplants are as expensive as allotransplants and their success at the experimental stage induces greater demand, the previous section has demonstrated that aggregate expenses will invariably rise—by
several hundred percent. If the past is any guide, Congress is likely to attempt to rein in Medicare and Medicaid expenditures, and private insurers are likely to decline coverage unless the xenotransplantation is actually cheaper than existing treatment. Should they?
The actions of Congress and private insurers are likely to reflect whether and to what extent the public is willing to pay. Congress will ask about the additional expenditures, the additional benefits (in terms of lives saved and the quality of life), whether to raise taxes to cover the expenditures, or whether to take resources from other programs. The public debates, which are likely to be no different from those surrounding other expensive health technologies, will involve compelling questions about how to quantify the benefits, including the quality and prolongation of life, increased productivity, and possible medical savings in other areas (Evans, 1993, 1995a,b). Similarly, private insurers will ask whether they must raise premiums, whether their subscribers will be willing to pay more, whether this will hurt their market position because their rates may no longer be competitive, or whether coverage of xenotransplantation should encourage them to forgo coverage of something else (Menzel, 1992, 1995). These are the kinds of questions that are exceptionally difficult to answer because they hinge on an even more fundamental, often unanswerable, question—how much is saving a life worth?
Reviewing And Monitoring Xenotransplantation
Experimental protocols describing research on either patients or animals are reviewed by committees at the institution where the work is to be performed. There are many similarities in the federal regulations that govern both committees. Key among these similarities is that the regulations call for the institution to assure the Office for Protection from Research Risks (OPRR) that the committees are complying with federal regulations. Thus, the regulatory framework is a self-assurance process, with annual reporting requirements and both random and for-cause audits of the activities of the committees by OPRR. A summary of the regulations pertaining to the committee that reviews research on patients is discussed first, followed by a description of the regulations for the committee that reviews the use of animals and a consideration of reviewing complex protocols such as xenotransplantation.
The institutional review board, sometimes known as the human subjects committee, is the committee that reviews protocols that describe proposed research involving patients. Regulations governing the IRB appear in the Code of Federal Regulations (45 CFR 46). The primary aim of the IRB is to protect patients who are the subjects of a research project. The committee is composed of both female and male scientists and clinicians, at least one nonscientist, and
at least one member representing the community, who is not affiliated with the institution in any way. Thus, committee membership is designed to include people with a diversity of expertise, backgrounds, and experiences. IRBs use a number of criteria in approving a research project: risks to subjects must be minimized; a risk–benefit ratio must be considered for the individual subject; selection of subjects must be equitable; informed consent must be obtained from the subject or a legally authorized representative; informed consent must be documented; safety of the subject must be ensured by the collection of appropriate monitoring data; the patient must be able to withdraw from the experiment at any time; privacy must be protected; and additional safeguards must be in place for the enrollment of prisoners, children, the mentally disabled, and others whose ability to give voluntary informed consent is uncertain or in question. Institutional officials cannot approve projects that have not been approved by the IRB. The IRB is authorized to suspend or terminate any research project. Regulations specify what must be addressed in the informed consent document. This includes full disclosure of the risks and benefits of the research, an explanation of the purpose of the research, an indication of whether medical treatment will be provided by the institution if a complication occurs, and a statement that consent is voluntary and a patient's refusal to participate in the research will not jeopardize her or his regular medical care. In addition to the regulations, OPRR issues guidelines from time to time that in practice have the force of regulations. 8 The IRB is required to maintain detailed records, which include copies of the protocol and supporting documents, minutes of all meetings, records of continuing reviews, copies of all correspondence with investigators, a membership list, and procedures followed in reviewing proposals.
The institutional animal care and use committee (IACUC) is the committee reviewing protocols for the use of animals (CFR 1–3). The overall aim of the IACUC is to promote the necessary and humane care and use of animals in research. The IACUC is composed of scientists, at least one veterinarian with expertise in laboratory animal science and medicine, at least one person who is a nonscientist, and a person who is not affiliated with the institution in any way. By regulation, the IACUC must review each proposal to use an animal in research to ensure that the protocol contains the following information: that methods described will be used to avoid or minimize pain and distress to the animal during research; that anesthetics and analgesics will be used when appropriate; that if the animal experiences severe pain or distress, it will be euthanized as promptly as possible; that animals will be well
cared for, with veterinary care available; that all personnel must be appropriately qualified and trained; and that methods of euthanasia are in accordance with guidelines. The IACUC's powers are similar to those of the IRB: institutional officials may not approve a project that has not been approved by the IACUC; the IACUC is authorized to suspend or terminate a project if it finds that the project is not being conducted as described in the protocol; and record-keeping requirements are similar to those for the IRB. In addition, an annual report must be sent to OPRR, giving the committee membership and dates on which semiannual inspections occurred. In contrast to the IRB, the IACUC has an additional obligation to review the program for the care and use of laboratory animals at least once every six months and to inspect all areas where animals are housed or used at least once every six months.
Two aspects of both IRBs and IUCACs are drawbacks when reviewing xenotransplant protocols. First, because of the broad range of scientific areas covered by protocols presented to a committee, it is unlikely that the members of the committee can include experts in all fields relevant to each protocol. Second, the committees are not constituted to protect the public health.
Absent broad scientific expertise, the committees may be unable to evaluate the scientific basis of the investigator's proposal. According to regulations, the committees can use consultants to assist in the review, although these consultants cannot vote on the proposal. In addition, many projects are funded by agencies that are equipped to provide a peer review of the quality of the scientific basis of the proposed research. Proposals that fail to receive extramural funding may still go forward, and no attempt is made to suspend projects that do not pass external scientific review. In the case of xenotransplantation protocols, which involve questions concerning the state of current immunological knowledge, human organ availability, technical feasibility, infectious disease risks, and ethical and societal issues, the committees may be very dependent on consultants or may require national review by a committee composed of experts in the relevant fields. Further, the committees would be materially aided by federally promulgated guidelines that outlined many of the issues that committee members should consider when reviewing and monitoring xenotransplantation research. Such guidelines were being prepared for public comment by the Centers for Disease Control and Prevention, National Institutes of Health, Food and Drug Administration, and other components of the United States Public Health Service as this report was being written.
A second issue is that these committees were created to protect individual subjects or an individual animal and are not constituted to protect public health. Protection of public health must be provided by a national or international body that has both the requisite breadth of view and the required knowledge. The public health issue in xenotransplantation centers on the possibility of a transmissible infectious agent being introduced into the patient
by animal organs, tissues, or cells. The potential threat to public health must be assessed, and measures to safeguard public health must be identified by properly constituted national bodies and written as a set of guidelines that the committees can use when reviewing and approving protocols.
The FDA is the agency that has the potential authority to regulate xenotransplantation. This agency, however, is accustomed to regulating manufacturers of drugs, medical devices, or biologics such as vaccines. To try to fit xenotransplantation into one of these categories may bring with it an already developed set of regulatory requirements, which might not be appropriate to the transplantation of animal organs into people. An example of a possible approach is that of recent regulations concerning human tissue banks. The need to regulate these banks grew out of the finding that tissues were a source of HIV infection for some recipients. The regulations require careful screening of the donor and extensive testing of the tissues. These regulations did not interfere with the surgical use of the tissue, relied heavily on standards already developed by the American Association of Tissue Banks, and included an important role for self-regulation. National guidelines, developed by the Public Health Service and implemented by local committees, may be a useful model for handling the potential risk to public health posed by infectious agents that may accompany the use of animal organs in xenotransplantation.
FDA Regulation of Xenotransplantation
The FDA plays a complex and evolving role in regulating human organ and tissue transplantation. This section describes FDA's current regulatory framework, which generally treats processed cells and tissues—of either allogeneic or xenogeneic origin—as biological products (FDA, 1993; Kessler et al., 1993). Given the pace and scope of innovation in the field, FDA and other federal agencies are building on this framework by developing guidelines for xenotransplantation, the subject of the second part of this section.
Current Regulatory Framework
The FDA's most stringent form of regulatory control—the requirement for premarket approval for safety and effectiveness—does not apply to whole organs, regardless of whether they are allogeneic or xenogeneic (Merrill, 1995). Organ transplantation is considered a surgical procedure conducted in the practice of medicine, an area historically outside the purview of FDA. Likewise, FDA does not require premarket approval of cells and tissues that are unmanipulated. However, the agency does require premarket approval of
cells and tissues if they are pretreated or manipulated. The cells and tissues can be of allogeneic, xenogeneic, or autologous (i.e., self) origin. FDA's overall regulatory framework and the critical role of tissue processing are presented in Table 4-2. (Although this table provides a general approach, it must be understood that clinical research protocols submitted to FDA are evaluated on a case-by-case basis.)
Manipulated cells and tissues are defined, for regulatory purposes, as undergoing propagation, expansion, selection, encapsulation, or other pharmacological treatment outside the body (ex vivo) (FDA, 1993). Any of these methods for ex vivo processing of cells and tissues are viewed by FDA as meeting the statutory definition of a biological product: "any virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative … applicable to the prevention, treatment, or cure of diseases or injuries of man" (Section 351(a) of the Public Health Service Act). Consequently, FDA requires manipulated cells and tissue transplants to undergo the same premarket approval requirements as other biological products. Some, however, dispute FDA's regulatory jurisdiction over manipulated cells and tissues. They argue that these do not meet the statutory definition of biological products and therefore ought not be subject to premarket approval and other legal requirements.
FDA's distinction between manipulated and unmanipulated tissues is best illustrated by bone marrow transplants. Conventional bone marrow transplants, of either allo- or xenogeneic origin, are not subject to premarket approval. However, bone marrow purged of T-cells is considered under the current regulatory framework to be manipulated. Premarket approval is therefore required. If the transplanted cells or tissues are nonliving, the regulatory approach is different. Table 4-2 shows that nonliving tissues and organs, such as pig heart valves and artificial hearts, are regulated as devices. Pig heart valves, commonly used in medical practice, are nonliving because they are pretreated with glutaraldehyde, which sterilizes the tissue and cross-links the DNA, but does not interfere with the valve's mechanical properties.
For combination products—such as encapsulated porcine islet cells—the primary mode of action dictates how the product is regulated. Although the encapsulant is considered a device, the primary mode of action is through replacement cell therapy. This means that encapsulated islets are evaluated as biological products by FDA's Center for Biologics Evaluation and Research, but input is sought about the encapsulant from medical device reviewers within another center of the agency.
What are the requirements for premarket approval of biological products? The process begins with the submission of an investigational new drug application (IND), an application for human clinical testing. The title is a misnomer for biologicals, including most xenotherapies, but it stems from
federal regulations that apply identical requirements for drugs and biologicals. The IND compiles all known pharmacological and toxicological information about the product, describes the general objectives of the research, and describes a specific protocol for early safety testing in humans, called a Phase I study.9 Several INDs already have been granted by the FDA for xenotherapies, but their content is proprietary. Because Phase I studies are designed to test for safety in a few human subjects, elaborate and expensive requirements (e.g., for "good manufacturing practices") do not apply. This has been a point of confusion with academic investigators, who erroneously think that the IND for Phase I testing in humans entails prohibitive expenditures. If the Phase I study is successful, investigators can seek FDA's approval to proceed with further clinical testing in humans in Phase II and Phase III studies—the prelude to formal licensing of the biological product. Far more stringent and expensive requirements pertain to these phases of human testing.
Genetic modifications to xenogeneic organs, tissues, and cells not only require premarket approval by the FDA but also are likely to require review by the NIH Recombinant DNA Advisory Committee (RAC). The latter body has oversight over gene therapy protocols that receive federal funding or are performed at institutions receiving federal funding. The FDA review is, by law, restricted to product safety and efficacy, whereas the RAC's review is much broader, delving into social and ethical concerns. The two reviews can proceed simultaneously.
TABLE 4-1 Current FDA Regulation of Organs, Tissues, and Cells
|
|
Unmanipulated |
Manipulated |
Nonliving |
|||||||
|
Source |
Allo organs |
Allo cells/tissues |
Xeno organs |
Allo cells/tissues |
Auto cells/tissues |
Encapsulated cells/tissues |
Transgenic cells/tissues |
Antibody-treated cells/tissues |
Pig heart valve |
Artificial heart |
|
Premarket approval |
Nonea |
Nonea |
Nonea |
Biologic |
Biologic |
Biologic |
Biologic |
Biologic |
Deviceb |
Device |
|
a Infectious disease and other requirements apply, but there is no requirement for premarket approval. Xenogeneic organs may be subject to Section 361 of the Public Health Service Act, according to proposals. This section provides authority to issue regulations to control communicable diseases. b Pretreated with glutaraldehyde or another agent that sterilizes or "fixes" the issue. SOURCE: FDA (1993) and Kessler et al. (1993). |
||||||||||



































