
2
Issues of Concern to NASA: Discussion and Conclusions
In attempting to assess and mitigate the health risks posed to spacecraft crews by radiation in space, numerous issues must be addressed and the physical and biological systems involved are complex. This chapter describes some of these issues and systems and discusses the most relevant problems associated with exposure to radiation in space. Where appropriate, some sections include a discussion of the general areas of research needed to characterize or reduce the health risks posed by space radiation. For clarity, risk is defined here as the likelihood of the occurrence of harmful effects resulting from exposure to radiation.
The potential risks to crew members' offspring as a consequence of crews' exposure to radiation in space are not large compared to the natural background mutation rate, and so the potential risk to the total human genetic pool is very small (see “Heritable Effects” below). However, crew members should be counseled about possible genetic effects of space travel. Although the possibility of catastrophic solar events, which could have short-term debilitating consequences despite the best countermeasures, cannot be excluded, observed solar events have not been in that category. Possible effects of exposure to radiation in space also include opacifications of the lens of the eye, and synergistic effects arising from the microgravity environment cannot be excluded, but there is no evidence that these types of biological effects represent risks comparable with that of carcinogenesis.
The most likely deleterious biological effect resulting from irradiation during interplanetary missions is late-occurring cancers. While other possible effects cannot be excluded, research for several decades has not produced evidence of a comparable risk from other biological sources or comparable risks from synergistic effects. The fundamental problem to be addressed in protecting against radiation is assessing the level of risk, specifically, determining the cancer types and the probability of their induction as a result of irradiation during a specific mission. Under the assumption that the biological information will exist, or can be modeled, what is needed in addition is adequate knowledge of the physical characteristics (the type of particles and their energies behind shielding) that can cause the risk to vary and of how the cancer types and induction probabilities vary as a function of these physical characteristics. Three sets of physical factors contribute to the variability of the risk itself and thus to uncertainty in determining risk: (1) the types of particles and their energies (radiation quality); (2) the amount of radiation; and (3) the extent and timing of exposure, i.e., acute exposure, protracted exposure, or a combination of both. (In fact, these three factors correspond to one probability distribution at the relevant biological sites as a function of particle type, energy, and time at the relevant biological sites.) A quantitative description of each of these three factors is already available to some degree—it is the variability of the distributions that remain in question.
Types of Particles and Their Energies
The two primary types of radiation, galactic cosmic rays (Figures 2.1 and 2.2) and solar radiation (Figure 2.3) vary according to their source and the event producing them. Both are altered substantially in particle type and energy as the primary galactic cosmic and solar particles traverse Earth's atmosphere and the primaries and secondaries are trapped in its magnetosphere as shown in Figure 2.4. The radiation quality likewise changes as the primary and secondary radiation particles traverse the spacecraft or the crew members themselves.1 Figure 2.5 shows the estimated change in the dose equivalent at various depths in different shielding materials in comparison to its value at zero shield thickness. The large changes as a function of depth and material arise because the primary particles not only are attenuated but also produce secondary particles with a range of types of energy. As a result, the radiation quality is modified and so is the equivalent dose. It is the combination of primary particles, attenuated primaries, and secondary particles at the biologically relevant site that determines the biological effects, not the primary spectrum per se.
Galactic Cosmic Rays
The major components of galactic cosmic radiation (GCR) are energetic protons and heavier ions with even atomic numbers, which are more abundant than those with odd numbers (see Figure 2.1). The relative abundance of each particle type generally decreases with increasing atomic number, although a significant increase occurs at iron-56 followed by a sharp decrease at higher numbers. There is a broad but consistent distribution in the energy per nucleon, with a peak in abundance in the vicinity of 1 GeV/nucleon (see Figure 2.2).
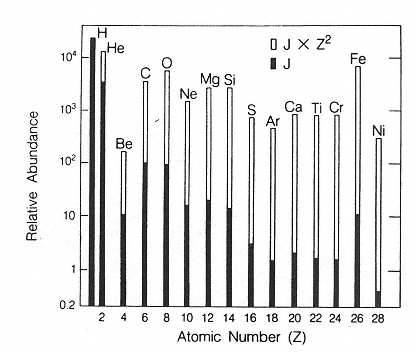
Figure 2.1
Histogram showing the relative abundances of the even-numbered galactic cosmic ray nuclei (solid bars) compared to their abundances weighted by the square of the particle's charge to give a measure of the “ionizing power” of each element (open bars).
SOURCE: Wefel, J.P. 1979. Instrumentation for radiation measurement in space. Pp. 117–183 in: Proceedings of the Workshop on the Radiation Environment of the Satellite Power System (SPS). (W. Schimmerling, and S.B. Curtis, eds.). U.S. DOE Report CONF-7809164. National Technical Information Service, Springfield, Va.
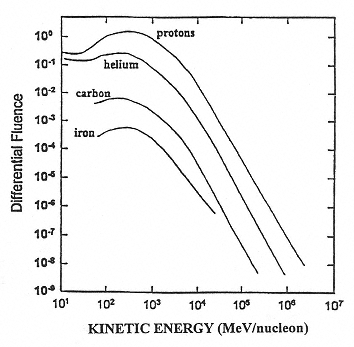
Figure 2.2
A representative fluence distribution as a function of the kinetic energy per nucleon. SOURCE: Simpson, J.A. 1983. Elemental and Isotopic Composition of the Galactic Cosmic Rays. Figure 5a in: Annual Review of Nuclear and Particle Science 33:323–381. Reproduced, with permission, from the Annual Review of Nuclear and Particle Science, Volume 33, © 1983 by Annual Reviews Inc.
Galactic cosmic rays are relatively constant in terms of distribution of particle types and energies over time, but they do decrease in intensity by roughly a factor of 10 during solar events because the increased energy emitted from the sun produces an increased interplanetary magnetic field that deflects a large fraction of the galactic cosmic rays. This change in intensity is a variation with time and does not reflect uncertainty in our knowledge of the spectrum. The uncertainty lies in our ability to predict the intensity over appropriate periods of time. The variability of the instantaneous galactic cosmic ray intensity is approximately a factor of 10, but the average variability is much less because solar events occur over a small fraction of time. Moreover, the probability of solar events occurring varies cyclically with the periodicity of the 11-year solar cycle, and thus planetary missions would be less exposed to galactic cosmic rays during solar maximum. Therefore, depending on when missions are flown, variation in cosmic ray intensity may not be a major factor in the uncertainty of risk estimates for radiation exposure. Nevertheless, the uncertainty in the absolute amount of particles and their energies is significant to factors of 2 to 4 at the modal energies, but with larger uncertainties at the lower particle energies.
Solar Particles.
Solar particle events (SPEs) produce substantial intensifications of the most energetic particles (including protons, heavier ions and electrons) emanating from the sun. The risk of harmful effects to space crews is generally assumed to be primarily from the protons and, to a lesser extent, the heavier particles, which are relatively less abundant than in the case of cosmic rays. The proton distributions as a function of energy have been measured extensively, and so the uncertainty in the distribution is small, but the intensity may vary by
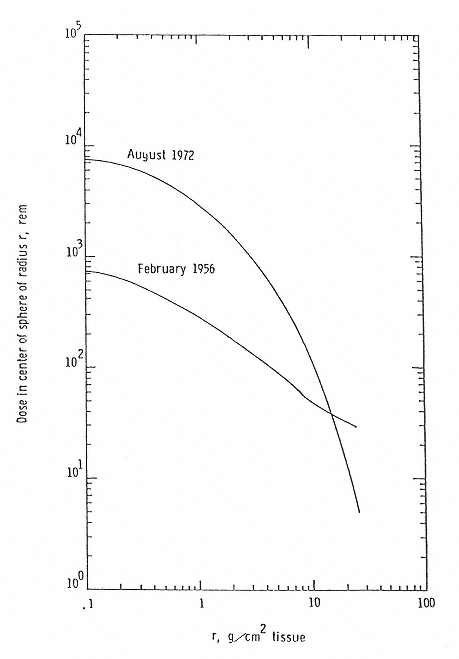
Figure 2.3
Dose Equivalent from two major solar events. SOURCE: Wilson, J.W., Townsend, L. W., Schimmerling, W., Khandelwald, G.S., Kahn, F., Nealy, J.E., Cucinotta, F.A., Simonsen, L.C., Shinn, J.L., and Norbury, J.W. 1991. Transport Methods and Interactions for Space Radiations. National Aeronautics and Space Administration Publication 1257. Available from the National Technical Information Service, Springfield, Va.
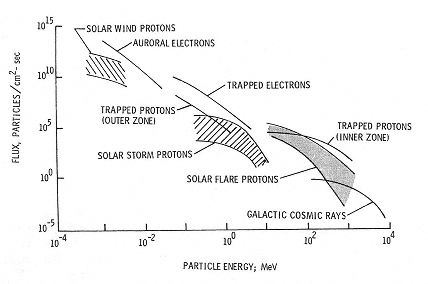
Figure 2.4
Space radiation environment. SOURCE: Wilson, J.W., Townsend, L.W., Schimmerling, W., Khandelwald, G.S., Kahn, F., Nealy, J.E., Cucinotta, F.A., Simonsen, L.C., Shinn, J.L., and Norbury, J.W. 1991. Transport Methods and Interactions for Space Radiations. National Aeronautics and Space Administration Publication 1257. Available from the National Technical Information Service, Springfield, Va.
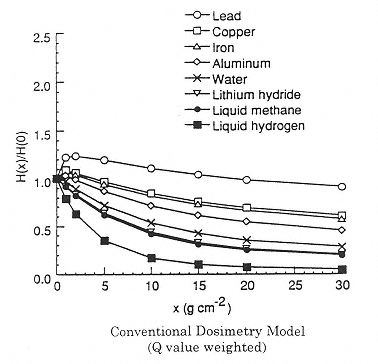
Figure 2.5
Calculated effect of shield material on the dose equivalent using the program NUCFRG2. Attenuation of dose equivalent in one-year exposure behind several shield materials. H(x)/H(0) is the normalized dose equivalent behind several shield materials. H(x) is the Sievert dose at a shield density x. H(0) is the Sievert dose without shielding (at the surface). SOURCE: Wilson, J.W., Kim, M., Schimmerling, W., Badavi, F.F., Thibeault, S.A., Cucinotta, F.A., Shinn, J.L., Kiefer, R. 1995. Issues in space radiation protection: Galactic cosmic rays. Health Phys. 68:50–58. Reproduced from the journal Health Physics with permission from the Health Physics Society.
several orders of magnitude. This variability in intensity translates to a significant uncertainty in risk, although that risk is reduced during solar minima. Conversely, the number of SPEs increase around solar maxima, increasing overall risk.
Simonsen et al. estimated the radiation doses and dose equivalents from the October 1989 SPE for various human organs as a function of thickness of water shielding.2 The doses to the skin, intestine, and bone marrow at water thickness of 0.5 cm were 7.21, 0.56, and 0.8 Gy, respectively, corresponding to 11.32, 0.75, and 1.07 Sv. At a water thickness of 10 cm, these values decreased to 0.35, 0.13, and 0.15 Gy or 0.46, 0.17, and 0.20 Sv, respectively. NASA's current lifetime limit for radiation exposure is 1 to 4 Sv, depending on age and gender.
Secondary Particles
The broad distribution of the primary background radiation in space by particle type and energy has significant uncertainties at the lower energies, and significant variability is contributed by the uncertainty in the timing and intensity of solar events. However, a major uncertainty in estimating space radiation's harmful effects for space crews is the uncertainty of the actual particle distribution at the point of exposure of crew members inside a spacecraft, inside a space suit for crew members conducting extravehicular activities, or actually at the sites of specific organs of crew members. The human body has an equivalent thickness of approximately 20 cm of unit density tissue for isotropically distributed high-atomic-number, high-energy (HZE) particles; the skin, although on the surface of the body, will be irradiated by a significant fraction of primary particles and the resultant secondary particles which have passed through the body. As the primary particles pass through the spacecraft and the bodies of the people themselves, secondary particles, including heavy secondaries and nuclear recoils, photons, electrons, neutrons, and even pions and muons, are produced in abundance. After only a centimeter or less of shielding material is traversed, the number of these secondary particles exceeds the number of the primary particles. Some of the secondaries, such as the low-energy nuclear recoils and secondaries, have linear energy transfers (LETs) greater than do most of the primary HZE particles. At the same time, the secondary electrons form a low-LET radiation background that also may have some biological significance for intracellular effects such as DNA damage and subsequently produced mutations.
Estimates of Uncertainty in Radiation Risk Factors
There are no rigorous estimates of the uncertainties associated with assessing health risks to crews in a radiation space environment. Curtis et al.3 provide estimates of the errors associated with values for the major contributors to a calculation of the risk from high-LET radiation, and these are shown below:
- 10 to 15% uncertainty in the initial charged-particle spectra;
- 50% uncertainty in the radiation transport calculation;
- 200 to 300% uncertainty in the risk coefficients for low-LET radiation;
- 200 to 500% uncertainty in the risk coefficients for high-LET radiation; and
- 400 to 1500% uncertainty in the overall risk.
Little information is provided to substantiate the values, and no comparisons are made with experimental data. As noted by the authors, the values should be treated as rough estimates.
Conclusions
- Current models of the GCR spectra may have reached a 10% root mean square accuracy.4 The enhancements needed in the models are the statistical uncertainties to be expected in the spectra due to the solar cycle and, for verification purposes, an accurate representation of the even-odd isotope abundance ratio. Distributions in energy and type of fragmentation products from particles with energies representing GCR
- need to be calculated at different times in the solar cycle and compared with subsequent laboratory measurements.
- Superficially, it appears that intensive research, both theoretical and experimental, ground-and space-based, over the last decade has altered by only about 25% the calculated distribution of primary particles as a function of their type and intensity. A review is needed to compare the physical data and theoretical methods available approximately one decade ago with those currently available in the three major areas (types of particles, their energies, and their quantity) specified in this report to see if the ~ 25% number is correct.
- Experimental measurements of particles emanating from a thick laminated shield need to be compared with calculations to benchmark the computer codes (modeling programs) and reduce the uncertainty in the shielding calculations. Once the cross section codes have been validated, the transport code itself must be validated. These measurements must include a “broad beam” geometry to assure that secondary particle products are fully accounted for.
- The risk of experiencing adverse biological effects in space depends on the length of a mission, not only because the dose received from GCR depends on mission length, but also because the probability of SPEs occurring increases with time as well. Specifically, it is necessary to know how much solar events contribute to total proton fluence over the period of the shortest anticipated mission. A better estimate is needed of the risk posed by a minimum-length mission compared to that by a 460-day mission.
Biological Effects of Radiation
There is extensive literature on what is currently known about the biological effects of radiation as summarized, for example, in the published deliberations of the NRC's Committee on the Biological Effects of Ionizing Radiations (BEIR) and the United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). This section introduces the various end points of concern for risk assessment and describes the potential magnitude of any radiation risks to crews during extended spaceflight. Based on proposed mechanisms of origin, the adverse health outcomes associated with exposure to radiation are considered under two broad categories: early and late effects, the former mostly deterministic, the latter mostly stochastic.
Early Effects
General Considerations.
Deterministic Effects The acute somatic effects of ionizing radiation are by nature deterministic (formerly known as nonstochastic) effects. They are mass phenomena involving depletion of cells in a given organ system or tissue; only after a significant number of cells have been killed is any clinical effect apparent. Thus, the dose-response relationship shows a threshold, with the threshold dose for most acute effects on the order of 1 Gy or more. (By contrast, stochastic effects, such as genetic alteration and cancer, can potentially arise from damage to single cells.) Thus, significant risks for experiencing early effects occur only with very high radiation fluences.
Radiation Environment Because of the deterministic nature of early effects, it is likely that radiation arising from HZE particles and background cosmic radiation would pose no significant risk of early deleterious biological effects for spacecraft crew members. The number of cells damaged by individual HZE particles would be too small to significantly affect organ function, and the doses arising from penetrating background cosmic radiation would be too low.
As previously discussed, the principal risk for experiencing early effects would thus be derived primarily from SPEs, which are associated with the release of high fluences of protons of varying energies. The doses of radiation associated with such events may be sufficiently high to produce acute effects. The primary danger would be to crew members who were outside the shielding afforded by the spacecraft for a sufficient length of time during one of these SPEs.
Relative Biological Effectiveness of Protons Studies conducted with a relatively broad range of proton energies indicate that the values for relative biological effectiveness (RBE) for acute effects are similar to those for 250-keV x rays;5 thus the risk per unit dose of early effects arising from exposure to protons from SPEs should be similar to that for effects arising from low-LET radiation such as electrons and gamma rays. Thus the risk estimates described below are derived from rather extensive data on effects of low-LET radiation, given that very few data are available on the incidence of such early effects following whole-body proton irradiation. It should be noted that there has been no evidence for new or qualitatively different early effects arising from proton irradiation per se that would lead to uncertainty in the prediction of early biological effects in space.
Early Systemic Effects
Prodromal Radiation Sickness Early prodromal effects of irradiation occur within a few hours of acute exposure and are characterized primarily by nausea and vomiting.6 The latter in particular can have serious consequences in space, particularly for individuals wearing helmets and space suits. The whole-body dose at which vomiting occurs in approximately 50% of individuals is in the range of 1.5 to 2.0 Gy for acute exposures.7 Nausea and perhaps vomiting may occur in a few individuals exposed to radiation at doses in the range of 0.5 to 1.0 Gy, but such symptoms would likely be mild and occur only 12 hours or longer after irradiation.8 On the other hand, nausea and vomiting would be expected in virtually all individuals receiving doses of 2.5 to 3.0 Gy, the severity increasing with the dose.9 These prodromal effects of radiation, which occur within 1 to 2 days after exposure and then subside,10 can be minimized by use of antinausea medications.
Total Body Irradiation—Acute Radiation Syndrome The clinical effects of acute, whole-body exposure to radiation involve primarily the hematopoietic system and are due in particular to depletion of circulating white blood cells (granulocytes) and platelets.11 Again, the threshold for the production of significant clinical effects is in the range of 1.5 to 2.0 Gy, although changes in white blood cell counts can be detected after exposure at substantially lower doses.12 The clinical effects occur 2 to 4 weeks after exposure when the granulocyte and platelet counts reach a minimum, and are characterized by infections and bleeding.13 If only minimal supportive care is available, mortality may occur following exposure at doses in the range of 3.0 to 4.0 Gy.14 Significantly higher doses would be required to produce the above effects if exposure to radiation were extended beyond one day.
Conclusion The probability that radiation fluxes within a spacecraft would be sufficient to cause early systemic effects is extremely low. The principal potential risk of these effects would be to crew members working outside the spacecraft for a prolonged period of time during an SPE.
Skin
Skin damage could be a potential problem for crew members working outside a space vehicle, where the skin would receive the highest dose of any organ system. The acute radiation dose necessary to produce erythema is of the order of 6.0 Gy, whereas that for moist desquamation is in the range of 15 to 20 Gy.15 The doses required to produce these effects will increase by a factor of 2 or more for protracted exposure. 16 Such single exposures would occur only in association with major SPEs. Transient epilation may also occur following acute doses of the order of 6.0 Gy, but hair regrowth always occurs after such doses. 17
Impairment of the normal healing of soft tissue injuries may occur following exposure to radiation. Studies with mice and rats indicate that the doses required for impairment of wound healing are high, 5.0 Gy or more, and thus within the range sufficient to produce erythema or skin damage.18 One study found that a reduction occurred in the rate of healing as measured by tensile strength, but the overall healing time and the final strength of the healed wound were not affected.19 Proper care of wounds to prevent infections and bleeding is essential.
Fertility
Two primary consequences of gonadal irradiation are (1) reduced fertility or transient or temporary sterility, which may last from several months to several years, and (2) permanent sterility. The nature of these effects and the doses required to produce them vary in males and females. For the male, the doses reported to cause temporary sterility generally fall in the range of 0.5 to 4.0 Gy for single acute exposures to low-LET radiation, although the threshold dose may be lower.20 A single acute dose as low as 0.15 Sv, about 0.15 Gy, has been reported to produce a decrease of the sperm count in some normal men.21 The duration of temporary sterility is dose-dependent and may last from 8 to 10 months up to several years.22 Permanent sterility has been reported following doses in the range of 2.5 to 4.0 Gy. An unresolved question involves the effect of dose rate on male fertility. Some data from canine experiments suggest that, as a consequence of the cyclic process of spermatogenesis, susceptibility to radiation-induced infertility may be enhanced by low-dose-rate, protracted exposure.23 Under conditions of space travel (assuming crews stay within NASA's present lifetime limit of 1 to 4 Sv), it is expected that the acute exposure of crew members (male and female) will be low enough so that any reduction in fertility should be minor and transient. Figure 2.3 gives doses (assuming little shielding) from an SPE. If a crew member were outside the spacecraft during a flare, there could be some early systemic effects as outlined above.
Doses of radiation necessary to sterilize most females fall in the range of 6.0 to 20 Gy, although a small percentage of women may be permanently sterilized by exposure at lower doses. Temporary sterility or reduced fertility may occur at doses as low as 1.25 Gy.24,25 Doses of 2 to 6.5 Gy are required to sterilize 5 percent of women for more than 5 years; protraction of exposure appears to reduce this effect. 26
Other Organ Systems
Damage to the epithelium of the gastrointestinal tract, particularly the small intestine and distal stomach, occurs in individuals receiving 4.0 to 5.0 Gy or more of whole-body radiation.27 These doses are presumed to be significantly higher than those that might be received by crew members during space travel.28 At doses below 4 Gy, however, transient early symptoms of nausea may occur within a few hours of irradiation.29 These symptoms can be expected to subside within 1 to 2 days, but their severity increases with dose. Individuals receiving doses of whole-body radiation sufficiently high to cause even mild intestinal damage (4.0 to 5.0 Gy) most likely will have incurred life-threatening damage to their hematopoietic systems.
Conclusions
Early effects of radiation in the major organ systems occur only following relatively high doses of radiation. Thus, with the possible exception of skin damage and a transient reduction in fertility, the early effects of irradiation are not likely to be a significant risk to spacecraft personnel. Skin damage would occur only in crew members working outside the spacecraft during an SPE.
Late Effects.
General Considerations
Potentially important late effects following exposure to radiation during spaceflight include induction of cancer and damage to the central nervous system (CNS). Uncertainties concerning the risk of cancer induction are related mainly to the quantification of these risks. With respect to assessing the potential for CNS damage, it is first necessary to establish, prior to conducting research directed toward quantitation, whether CNS damage is likely to occur. It is also necessary to consider the potential for an increased risk of cataract formation and to determine if there will be increased heritable effects leading to increases in the rates of mutation in the human population. The uncertainties in risk estimates are large (see above, “Estimates of Uncertainty in Radiation Risk Factors”) for the many reasons discussed below.
Cancer and Uncertainty in Estimates of Its Induction
As pointed out above, induction of cancer is generally considered the most significant deleterious biological effect of exposure to radiation in a space environment. Estimates of the risk of developing cancer as a result of spaceflight and the uncertainties in these estimates have been discussed in considerable detail in NCRP Report No. 98, Guidance on Radiation Received in Space Activities.30 Current understanding of the risk to humans of contracting cancer following exposure to radiation—whether in the terrestrial environment or in deep space—is founded on data from studies of atomic bomb survivors. Based on these data, risk estimates have been developed for both the incidence and mortality of leukemia and solid tumors in a number of organ sites31 following low-LET irradiation. Uncertainties in these estimates derive from several sources. First, because this is an ongoing study of a population, approximately 40% of which is still several sources. living, estimates are highly dependent on whether the models used to project lifetime risks are appropriate. Second, because the atomic bomb survivors in Japan were exposed at an acute high dose rate, principally to gamma rays, with a relatively minor component of the dose coming from fission neutrons, uncertainty in estimating the level of risk is increased when radiation is delivered at low dose rates or when the total dose delivered is protracted over a period from weeks to months. To correct for these differences, current risk estimates have incorporated a dose rate effectiveness factor (DREF) that reduces by a factor of 2 the estimated risk of contracting certain neoplasms under conditions of low dose rate or protracted exposure.32,33 The DREF is based on current models for mechanisms of radiation-induced carcinogenesis and on results derived from experimental studies.34,35 A third area of uncertainty in using existing data to assess risks from exposure to radiation in spaceflight is related to models used to extrapolate from risks estimated from a Japanese population irradiated in 1945, with specific and unique patterns of cancer incidence rates and age-specific mortality rates, to modern Western populations.
These sources of uncertainty in estimating the risk of cancer are generic to consideration of risk estimates in any exposed population. In addition, there are unique aspects of risks that are specific to radiation exposure in deep space. As described above (in the section titled “Types of Particles and Their Energies”), the radiation environment in deep space consists principally of galactic cosmic rays composed of protons, helium ions, and, to a lesser extent, heavy ions rather than the mainly low-LET radiation to which atomic bomb survivors were exposed. For the most part, radiation in space occurs at a low fluence rate. However, additional risks are associated with the higher dose and dose rate exposures from SPEs, the most important component of which is protons from a risk standpoint.
Since there are no epidemiological studies of humans exposed to the kinds of radiation that will be encountered in space, estimates of risks for biological effects induced by high-LET radiation are based on the risk estimates for exposure to low-LET radiation multiplied by weighting factors that express the effectiveness of an absorbed dose of such radiation in terms of equivalent doses (Table 2.1). The radiation weighting factor WR is used in radiation protection to weight the absorbed dose averaged over an organ to obtain the equivalent dose to that organ for the radiation quality of interest. The WR values, as well as derivation of the related quality factors (Q), are based on many experimental RBE values for stochastic effects, including those for cancer induction in animals and cancer-related end points such as mutations and chromosomal aberrations, and are selected by advisory groups such as the International Commission on Radiological Protection (ICRP).36 While more direct estimates of such risks would be preferred, use of these factors is state of the art, given current understanding of the mechanisms of cancer development and the role played by radiation in inducing carcinogenesis.
Reducing the uncertainties associated with the values of quality factors is necessary to improve risk estimates associated with space travel. Clearly, uncertainties in these quality factors translate directly to uncertainties in risk. These values are highly dependent on an improved understanding of RBE as a function of particle type and energy transferred for tumor induction over a range of LETs. Such an understanding is crucial to the development of appropriate quality factors for the range of radiation types encountered in deep space.
An additional source of uncertainty in risk that must be addressed relates to dose-response relationships for cancer induction and the influence of dose rate for protons such as encountered in deep space. Such information is required to derive estimates of risk at the low fluences that will exist in space. For the low
TABLE 2.1 Radiation Weighting Factors (WR)
|
Radiation Type and Energy Range |
WR |
|
Photons, all energies |
1 |
|
Electrons and muons, all energiesa |
1 |
|
Neutrons, energy < 10 keV |
5 |
|
10 keV to 100 keV |
10 |
|
100 keV to 2 MeV |
20 |
|
2 MeV to 20 MeV |
10 |
|
20 MeV |
5 |
|
Protons, other than recoil protons, energy > 2 MeV |
2b |
|
Alpha particles, fission fragments, heavy nuclei |
20 |
|
NOTE: All values relate to the radiation incident on the body or, for internal sources, emitted from the source. a Excluding auger electrons emitted from nuclei bound to DNA. b ICRP recommends a WR of 5 for protons, other than recoil protons, with energy >2 MeV (see International Commission on Radiological Protection. 1991. 1990 Recommendations of the International Commission on Radiological Protection. ICRP Publication 60. Annals of the ICRP 21. Pergamon Press, Elmsford, N.Y.). SOURCE: National Council on Radiation Protection and Measurements (NCRP). 1993. Limitation of Exposure to Ionizing Radiation. NCRP Report No. 116. National Council on Radiation Protection and Measurements, Bethesda, Md. |
|
dose rates expected from heavy ions, dose rate considerations should not be as important because the probability that two different heavy ions will traverse the same human cell is small.
Because essentially no data from human populations are available to allow investigators to make direct estimates of risk from exposure to these types of radiation, or which address the factors influencing sources of uncertainty in risk estimation, such estimates are heavily dependent on data from other studies. Hence, both an adequate understanding of the relationships between RBE and particle type and energy, as well as information on dose response and dose rate effects derived from experimental studies are essential to understanding the cancer risks associated with deep-space travel. Existing experimental data are inadequate.
Even in animal systems, data on tumor induction following exposure to protons and heavy ions are sparse. Critical data on cellular responses to irradiation, required to support the use of laboratory animal tumor data for estimating risks to humans, are also lacking in many instances. Cell survival studies, while not directly applicable to estimation of cancer risks, do permit comparisons of the effectiveness of different types and levels of radiation and determination of the repairability of induced DNA damage. Cellular studies of the induction of somatic mutations and chromosomal aberrations provide data that can be linked fairly directly to carcinogenic effects. Such studies, particularly in human cell systems, are important for understanding possible mechanisms of carcinogenicity and in the appropriate application of animal data to the estimation of risks to humans.
As described in Chapter 1, data for tumor induction following proton irradiation are available for only a few tumor types following acute exposure. The limited dose-response data that can be obtained from these studies suggest similarities to responses that would be seen after gamma ray irradiation.37–39 Only one study found evidence to support an RBE of greater than 1.40 Additional support for similarities in effects from exposure to proton and to low-LET radiation comes from the work of Burns et al., who have reported a curvilinear dose response for rat skin tumor induction similar to that occurring after exposure to electrons and a reduction in the carcinogenic effects of exposure to protons.41
Cellular studies have been conducted using protons of different energies to examine cell survival and induction of chromosomal aberrations. 42–45 Although the range of energies used is lower than that encountered in the space environment, these data also suggest similarities in effects between protons, gamma rays, and x rays. The dose responses tend to be linear/quadratic, and there is clear evidence for repair of proton-induced DNA damage.
While most data tend to support the view that the risks for carcinogenic effects, as a result of irradiation by high-energy protons, will be similar to those for low-LET radiation, additional studies of protons in the
range of energies relevant to those encountered in space, 0.1 GeV and higher (see Figure 2.2), could strengthen this conclusion considerably. The purpose of such experiments would be to determine whether biological effects of exposure to these higher-energy protons are qualitatively similar to those seen with exposure to low-LET radiation and to determine whether repair of proton-induced DNA damage can be observed.
Information on tumor induction following exposure to heavy ions is also limited. Burns et al. have conducted experiments on skin tumor induction in rats following argon irradiation (Figure 2.6).46 These data provide evidence of a linear dose response for tumor induction with this high-LET radiation and support the expectation of a relatively high RBE. However, because of the dominance of the dose-squared term (concave upward) in the low-LET dose response, the data do not allow for the estimation of a single RBE value that could be used in the determination of an appropriate weighting factor that is independent of dose. As stated previously, the determination of appropriate quality factors requires information on the relationship between LET and RBE for tumor induction. The only systematic study of such relationships was conducted for the Harderian gland in mice.47 The data show a rise in RBE with LET that peaks at an RBE of 30 in the 100- to 200-keV/µm range. Importantly, there was no clear-cut evidence of a decrease in RBE at LET of up to about 400 keV/µm as predicted by biophysical models and as observed for cell killing and mutation. Because the quality factor-versus-LET relationships adopted by ICRP incorporate a decrease at LET greater than 100 keV/µm, these Harderian gland data suggest possible important discrepancies that need to be explored with other tumor-induction models. Studies of Harderian gland tumorigenesis also suggest that the RBE values for fission spectrum neutrons are similar to those for 100- to 200-keV/µm heavy ions.48 If this is the case, there are dose-response and dose rate data for the induction of several tumors in mice after neutron irradiation that could be used in support of establishing a reliable quality factor for heavy ions in this energy range.49
While not able to be used directly for the derivation of quality factors, studies of cells have provided evidence that for high-LET radiation, linear dose-response relationships are only slightly influenced by fractionation or protraction.50–52 In addition, studies of mutagenesis and induction of chromosomal aberrations suggest possible qualitative as well as quantitative differences between high-and low-LET radiation and different particle types of the same LET that need to be examined further for their applicability to understanding cancer risks.53,54 Of these, it is important to note the recent observations of high-LET radiation effects on chromosome instability and the induction of delayed radiation damage leading to expression of damage in the progeny of surviving irradiated cells.55
Conclusions The present state of knowledge regarding cancer induction by irradiation, as described above, requires that additional research be directed in two areas. First, a pragmatic set of studies is needed to provide data necessary for the determination of appropriate quality factors that should be used in making risk calculations. These should be systematic studies of RBE as a function of particle type and energy for a select number of heavy ions and for protons using well-defined animal models for tumorigenesis. In addition, information on dose rate and fractionation effects for protons is also needed. Improvements in risk estimates beyond those attainable with these data require a more complete understanding of the mechanisms of tumor induction and of principles that will aid in using data, from experimental systems subjected to relatively high radiation doses, to estimate effects on humans exposed to low, protracted doses and in estimating risks across populations. These kinds of studies will require the development and exploration of new model systems and the application of developing technologies in cell and molecular biology.
Central Nervous System
Outside Earth's magnetic field, the fluence rates of the GCR are at the maximum during solar minimum: about 4 protons per cm-2 s-1, 0.4 helium ions per cm-2 s-1, and 0.04 HZE particles per cm-2 s-1. The number of particles traversing cell nuclei depends, of course, on the size of the nucleus (Figure 2.7),56 which in the CNS can vary from the small nuclei of microglia to the very large nuclei of motor neurons. Assuming a nuclear area of 100 µm2, Curtis et al. estimated that each cell nucleus in the body would be hit by a proton once every 3 days, by a helium ion once per month, and by higher weight atomic particles once per year.57,58 Estimates for strikes by the heavier particles, in particular, are strongly influenced by the degree of particle fragmentation occurring as the radiation traverses the shielding of the spacecraft.
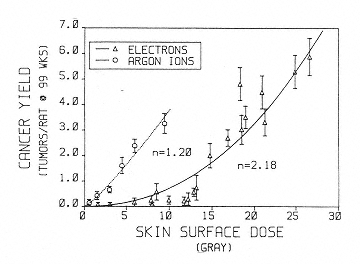
Figure 2.6
Cancer yield in rat skin as a function of surface dose (single dose, 3 to 5 Gy/min; 300 to 500 rad/min) in rats
exposed at 28 to 58 days of age. Errors are estimated from total number of tumors. The curves are least fit to the power function y = bn, where n = exponent of quadratic, y = y axis, and b = x axis. SOURCE: Burns, F.J., Hoselet, S., and Garte, S.J. 1989. Extrapolations of rat skin tumor incidence: Dose, fractionation and linear energy transfer. Pp. 571–582 in: Low Dose Radiation: Biological Basis of Risk Assessment (K.E. Baverstock and J.W. Stather, eds.). Taylor and Francis, London. Reprinted with permission from Taylor and Francis.
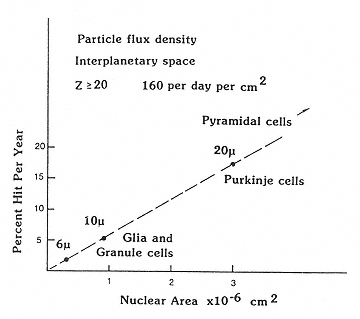
Figure 2.7
The dependence of size of the nuclei of neurons on the probability of traversal of heavy charged particles in space. SOURCE: Gauger, G.E., Tobias, C.A., Yang, T., and Whitney, M. 1986. The effect of space radiation on the nervous system. Adv. Space Res. 6:243–249. Reprinted from Advances in Space Research with kind permission from Elsevier Science Ltd., The Boulevard, Langford Lane, Kidlington OX5 1GB, UK.
It is probable that most of the damage done to the CNS by protons is repairable by DNA repair and regrowth of cells. Certainly, animals in experiments have been exposed to higher fluence rates than are likely to be encountered in space—and have not shown clinically detectable changes in the CNS. The main concern, however, is about HZE particles, particularly iron ions. The axons and dendrites of neurons are very radioresistant, and the cell nuclei, which do not undergo division in adult life, appear to be very resistant as well. They are not lost after irradiation in mitosis, as is usually the case with proliferating cells. What is inadequately known is whether any functional capability is diminished and, in particular, whether effects such as decreased DNA repair occur late in life long after exposure.
Lack of Data for Estimating Risks The reason for concern regarding the CNS is due to the fact that it cannot be stated with confidence what late effects, if any, might occur in the CNS of humans exposed to the various types of radiation in space such as heavy ions and secondaries of the more prevalent protons. There is evidence that in photoreceptors iron ions cause an increased loss of DNA.59 Whether a significant interaction occurs between aging and radiation-damaged cells (which is suggested by at least one investigator60) is also not known. Before investigators can conclude that the risks of late effects to the CNS are so improbable that they are not of concern, there have to be some data for relevant end points and doses.
Effects of HZE Particles The concern about HZE particles is that the energy deposition may be significantly different from that of radiation qualities for which we have some radiobiological understanding. One particle of very high Z and energy can traverse a number of contiguous cells. There is very dense ionization in the inner part or core of the particle track, with secondary particles and delta rays extending to neighboring cells. Although this pattern of potential damage raised concerns many years ago about the possibility of microlesions, the concerns have not yet been answered satisfactorily. While the lack of a solution to these concerns may seem surprising, the existence of the HZE particle component has been known only since 1948, and biological research using heavy ions has been restricted to a very small number of centers in the world with suitable accelerators. Furthermore, the critical experiments have proven difficult to carry out.
The effects of HZE particles on the CNS include (1) cellular effects, including biochemical changes; (2) functional changes; and (3) late effects, especially DNA repair deficiencies.
Cellular Effects High doses of low-LET radiation cause cellular changes and degeneration in neuronal tissues. Heavy ions are more effective in causing cellular damage, and the effects appear earlier than those appearing after exposure to low-LET radiation. In studies of the forebrain of rabbits, damage could be detected after exposure to 0.5 Gy of neon ions.61 Studies indicate that in the brains of fruit flies, swelling of neurons and changes in membranes could be detected with fluences that resulted in an average of less than one traversal per cell body by an argon particle.62 Dose-response data are lacking for clearly defined damage at either the cellular level or in specific areas of the brain. For example, what is the effect of various radiation fluences on the centers in the floor of the fourth ventricle, the site of a number of centers including the cardiac and respiratory centers, in which there are closely packed neuron cell bodies? If traversal of neurons by the various HZE particles in the GCR does in fact result in early- or late-occurring damage, the fourth ventricle is an area of the brain that could be at risk (there are also others). This example indicates how little we know about the potential effects of radiation on the CNS. Without such information it is impossible to assess the potential risk of clinically important damage to the CNS that might result from crew members' exposure to radiation during long-duration missions in deep space.
Functional Effects A number of studies have been carried out on the effect of HZE particles on the function of the CNS in rats and mice. Doses in the range of 0.5 Gy appear to impair the function of the neural networks involved in motor performance.63 Aging and exposure to radiation affect the CNS in similar ways and it is sometimes difficult to isolate the cause for affected functions, such as balance. Taste aversion has long been used as a test of behavioral and other changes induced by irradiation, and studies indicate that iron ions are more effective than lower-LET radiation in altering this particular type of behavior.64
Late Effects, Especially Repair Deficiencies Because many of the experiments concerning heavy ions have been associated with end points relevant to radiotherapy, questions about the effects on nonrenewing cell systems, especially the CNS, have remained unanswered. Differentiated cells such as those in the CNS or the liver can incur relatively large doses of radiation of different qualities and still retain their function. What is not known is whether untoward effects may appear later. The integrity of the transcribing regions of the genome must be reserved to ensure the fidelity of the RNA transcripts and also of the proteins, translated from RNA, that are necessary for the correct functioning of cells.
Changes with age in the retinal DNA of rabbits after irradiation have been studied by Lett and his coworkers.65–67 With both low-LET radiation and heavy ions, the evidence suggests that the initial radiation-induced DNA damage is repaired but that a subsequent breakdown of DNA occurs with age. The age at which secondary changes in the DNA of the photoreceptor occurred decreased with an increase in the LET of the radiation. The secondary changes in the DNA occurred earlier and were more marked with iron ions than with other heavy charged particles or with photons. Significant effects were noted after exposure at 2 to 3 Gy of iron ion radiation.
If it is assumed that the photoreceptors are a reasonable surrogate for neurons in the CNS, then the above results suggest that it is necessary to obtain adequate dose-response data using the most sensitive techniques for detecting DNA damage. Furthermore, it is mandatory to determine whether or not breakdown of DNA, which is an indication of impending cell death, occurs many years after exposure to radiation.
Conclusion Existing studies suggest that HZE particles may induce damage in the CNS. As yet, there are no complete data for RBE-LET relationships for the relevant end points for assessing the risk of radiation-induced damage. The results of studies with HZE particles suggest that it is not possible to predict the risk of CNS damage accurately from the effects of low-LET radiation.
Cataracts
Cataracts are considered a hazard of exposure to radiation, and limits for exposure are set for terrestrial workers who deal with radiation sources. The limits are based on estimates from studies of humans exposed to low-LET radiation. There is considered to be a threshold dose below which lenticular opacities of clinical importance do not occur. For this reason, cataract induction is considered a deterministic effect. However, the threshold is more a matter of the level of detection capable of detecting the beginning of cataractogenesis, and the most likely mechanism is consistent with a stochastic effect.
Assessing the risk of cataractogenesis from irradiation in space, in particular in deep-space missions, requires a knowledge of the associated RBE values of the various types of radiation. There are no data for induction of cataracts in humans exposed to HZE particles, and only sparse data for induction by protons. Thus, reliance on data from animal experiments is necessary.
The sensitivity of the lens of different species varies by more than an order of magnitude, decreasing with increasing size. In humans, a threshold dose for low-LET radiation of about 2.0 Gy has been considered reasonable. For the atomic bomb survivors, a somewhat lower threshold dose, 1.0 to 1.5 Gy, was derived by Otake and Schull.68 Since these results pertain to high-dose-rate exposures, it is important to know the reduction in effect that may result from fractionation or from lowering of the dose rate. Data from patients receiving radiotherapy or irradiation prior to bone marrow transplantation suggest a significant decrease, perhaps on the order of fivefold, in cataractogenic effects compared with the number induced by single high-dose-rate exposures. On the basis of experiments with rats,69 no such sparing would be predicted for the effects of exposure to very high LET radiation. Evidence from studies on monkeys indicates that the cataractogenic effect of protons will not be very different from that of gamma rays. 70 Therefore, exposure to protons on a Mars mission, unless there is an unexpectedly high exposure during SPEs, should not cause clinically significant opacities. The estimates of the risk of cataract induction from exposure to heavy ions are somewhat disparate,71,72 and until more definitive estimates are in hand, relatively high RBE values should be used in calculating the equivalent doses for estimating risk.
Heritable Effects
The great majority of data on the assessment of genetic or heritable effects in human populations following exposure to radiation has come from studies of the atomic bomb survivors. The following end points have been assessed: untoward pregnancy outcomes (major congenital malformation, stillbirth, neonatal death); sex of child; tumors with onset prior to the age of 20; death of liveborn infants through an average age of 26.2 years, exclusive of death from malignancy; growth and development of liveborn infants; cytogenetic abnormalities; and mutations altering the electrophoretic behavior or function of a selected battery of erythrocyte and blood plasma proteins. There were no significant increases in any of these indicators from a combined parental gonadal equivalent dose of 0.4 to 0.5 Sv.73
A recent study by Kodaira et al. examined variations in size of six minisatellite regions (see glossary) in the DNA of 64 children from 50 families in which one or both parents were exposed to the atomic bomb explosion and in 60 children from families in which neither parent was exposed.74 There was no difference in the frequency of change in the two groups. A similar result was reported by Satoh et al. for mutations detected by the denaturing gradient-gel electrophoretic method.75
UNSCEAR76 made estimates of the (unirradiated) background incidence of mutational effects per generation for the end points studied on the acutely exposed Japanese population. The values ranged from approximately 10-5 per locus for loci encoding proteins to 3 × 10-3 per locus for untoward pregnancy outcomes. UNSCEAR also estimated the acute dose that would, on average, double the background incidence—the doubling dose—as 1.7 to 2.2 Sv. Allowing for chronic exposure, a gonadal dose reduction factor was applied to give a minimal estimated doubling dose of 4 Sv for genetic effects. A complete description of the approach used may be found in UNSCEAR77 and in Neel and Schull.78 The present lifetime exposure limit for astronauts is < 4 Sv. Hence the actual increase above background in heritable effects per locus, depending on the particular locus, and the risk of heritable effects to individuals engaged in extended space travel will be low. In addition, because the number of individuals who might be exposed to ionizing radiation during long-range spaceflight will represent a very small fraction of the population, any genetic risk to the human gene pool would be negligible.
Variation in Susceptibility to Radiation Across Subject Types.
The rapid increase in knowledge of the mechanisms of tumor induction and heritable effects has led to a clear appreciation of the potential for a genetic predisposition to the induction of cancer by exogenous agents and endogenous processes and to induction of heritable changes. Such a predisposition might be specific for a single agent such as ionizing radiation (e.g., predisposition in ataxia telangiectasia heterozygotes) or it might involve sensitivity to a wide range of exogenous agents and endogenous processes (e.g., Li-Fraumeni syndrome (p53 heterozygosity)). Given that within the normal human population a range of risk exists for induction of cancer, it is difficult at this time to assign a value for increased risk owing to a single genetic susceptibility. In general, most of the genetic susceptibility or sensitivity factors that are common in the population tend to increase relative risk by small amounts. Those conferring high relative risk are present at a low frequency. The latter is particularly true for susceptibility for which background frequencies of cancer are high.
It has become increasingly apparent that the sensitivity of cells to radiation is controlled in part by the relationship of DNA repair kinetics to cell cycle progression. The quintessential example is the gene p53, which is involved in cell cycle control at the G1 checkpoint, the time point in the cell cycle at which DNA synthesis begins, and in the repair of DNA damage either directly or indirectly.79 A deficiency in p53 can both affect the efficiency of DNA repair and abrogate the G1 checkpoint, both of which can increase sensitivity to the induction of mutations and chromosomal aberrations. At G2, which is the time between the end of DNA synthesis and mitosis (the G2/M checkpoint), p53 appears to function in the direct repair of DNA damage, and not in control at the G2/M checkpoint.80 Mice that are homozygous or heterozygous for a knockout of the p53 gene are more susceptible to both spontaneous tumor formation, and tumor formation following exposure to a range of chemicals.81
The question of interest then is, What kinds of genotypes might elicit increases in sensitivity to radiation? For example, it is apparent that control of the cell cycle is a very complex process involving in part the interactions of cyclins, cyclin-dependent kinases, and cyclin-dependent kinase inhibitors. Alterations in any of these components could lead to abrogation in the cell cycle control, which would lead to abnormal responses to DNA damage and an increased sensitivity to genetic alteration. It remains of considerable importance to understand the mechanisms of genetic instability arising from abrogation of control at checkpoints in the cell cycle, and to determine the effects these mechanisms can have on radiosensitivity. Work in this area by the wider community of cancer investigators would lead to understanding of the role of genetic instability in cancer predisposition, and to development of assays for detecting individuals at increased risk.
While there will be a range of genotypes among individuals selected for long-range spaceflight, there is a very low probability that there will be highly sensitive individuals in the group. Very specific genotypes, such as those giving rise to ataxia telangiectasia and Li-Fraumeni syndrome, are obvious phenotypically, and such individuals would not constitute part of the selection pool. More subtle individual differences in sensitivity to ionizing radiation would not be detectable phenotypically. It could be argued that a radiosensitivity assay (G2 sensitivity) such as that described by Scott et al.82 and Jones et al.83 could be conducted on lymphocyte samples from potential crew members. However, such an assay is not, at present, directly predictive of an increased sensitivity to an adverse health outcome. Hence, it is appreciated by cancer investigators that a more complete assessment of the G2 sensitivity assay needs to be conducted in order to establish its range of sensitivity and possible predictive capability for cancer or heritable effects.
DNA Repair
The repair mechanisms utilized by the human body after exposure to radiation are an important part of any discussion of radiation effects, and the repair of damage to DNA is of obvious interest when considering late effects such as cancer. It has been known for a number of years that sophisticated and complex cellular processes exist for repairing all types of DNA damage: single-strand breaks, double-strand breaks, and a wide variety of types of base damage, all of which can result from exposure to radiation. It was also appreciated that the cell would be further protected if such repairs were completed prior to entry of the cell into the S-phase or mitosis/meiosis. If repair occurred at a later time, then there would be an increased probability of induction of point mutations and chromosomal aberrations from errors of replication on a damaged DNA template, errors of segregation, and/or loss of unrejoined chromosomal fragments. In the past 5 years, the repair processes for handling DNA damage have been largely characterized at the molecular level, and their complexity has been established. It is interesting to note that several of the repair processes are modifications of the functions of other cellular housekeeping proteins, such as transcription complexes or cell cycle control genes. For example, the nucleotide excision repair (NER) pathway involves at least 16 proteins, a number of which are components of the TFIIH/BTF2 complex that is a component of the RNA polymerase II transcription initiation complex. The very specific incisions required for removal of DNA damage are produced by enzymes of this complex. Reviews by Wood et al.84 and Sancar85 provide details of NER and the effect that mutations in this pathway can have, as illustrated by xeroderma pigmentosum, Cockayne's syndrome, and trichothiodystrophy. While the actual process of excising damaged nucleotides by NER is quite well worked out, the cellular control and damage recognition processes are still the subject of extensive research efforts.
Repair of Oxidative Damage and Double-Strand Breaks
More recently, an understanding of repair of DNA damage induced by ionizing radiation has emerged. Two recent reviews, one on the repair of oxidative damage86 and a second on double-strand break repair,87 describe the current level of knowledge. The enzymology of repair of some damaged bases and sugars has been quite thoroughly described in bacterial systems and to a lesser extent in S. cerevisiae, and is mostly inferred for mammalian systems.88 Although there is a very broad range of base damage, it appears that several of the repair activities recognize a range of substrates, thereby leading to the requirement for rela-
tively few enzymes to repair the bulk of DNA and sugar lesions. In broad terms, the process of base and sugar damage repair involves damage recognition, base excision of purine or pyrimidines, site incision, and fragment release. Clearly, variations in efficiency among cell types or species, or within a population, can occur at any one of these steps, each of which is under genetic control. At this time, however, no human syndrome has been identified that results in a sensitivity to ionizing radiation attributable to a deficiency in the repair of oxidative damage.
The understanding of the mechanism of repair of DNA double-strand breaks has taken several significant steps forward recently. Studies have demonstrated that there is a close association between the repair of site-specific double-strand breaks introduced during V(D)J recombination and those generated by DNA-damaging agents.89 V(D)J recombination allows for the creation of the diversity in immunoglobulin and T cell receptor genes by reassorting variable (V), joining (J), and diversity (D) elements into single exons by recombination.90 In addition, a range of mammalian cell mutants that are sensitive to low-LET radiation are deficient in V(D)J recombination. This association was shown to be through the DNA-dependent protein kinase (DNA-PK) that is involved in the rejoining of double-strand breaks. The significant activity of DNA-PK in this regard is that it binds to, and is activated by, DNA double-stranded ends.
DNA-PK consists of two polypeptides, Ku80 and Ku70, with the former being a DNA binding protein and the latter having an unknown function, and DNA-PKcs, a catalytic subunit that contains a serine-threonine kinase domain.91 The kinase activity is limited to the situation when DNA-PK is bound to DNA. DNA-PK can phosphorylate a number of DNA binding proteins in vitro, including transcription factors such as Sp1, c-Jun, and p53. It appears that DNA-PK enters the DNA at one end and can move along the molecule.
It has been shown that several radiosensitive mammalian mutants are defective in Ku80, that cells from severe combined immunodeficient (scid) mice have a DNA repair defect, and that additional radiosensitive cell lines have a deficiency in DNA-PKcs.92 Thus, it appears that repair of ionizing radiation-induced double-strand breaks is performed in part by DNA-PK. It has been suggested that there could be a link between double-strand break repair machinery and transcription, as has been described for NER.93 A preferential repair of ionizing radiation-induced DNA damage on the transcribed strand has recently been described, 94 and DNA-PK is a potent inhibitor of transcription by RNA polymerase I.95 On the basis of these mechanistic studies, it is predicted that there will be a range of individual sensitivities to ionizing radiation that is, in part, dependent on the efficiency of the repair processes for double-strand breaks. To date, no human syndromes that are characterized by defects in DNA-PK have been identified, although the DNA-PKcs gene maps to the same human chromosome region as the one for the human gene that complements scid.96
Other Studies
A good deal has been learned about repair mechanisms by studying the human syndrome ataxia telangiectasia (AT), which is characterized by a sensitivity to cell killing and mutation induction in cells in vitro as a result of exposure to low-LET x rays, and, in some cases, by a loss of x-ray-induced inhibition of initiation of DNA synthesis. It was presumed by investigators that these phenotypes were the consequence of a DNA repair defect, and that different steps or components were controlled by genes in the four different complementation groups, all of which map to a single chromosome region. However, the recent cloning of the AT mutated gene (ATM)97 and additional characterization of homologous genes in yeast98,99 have shown that the defect in AT cells is not the result of a repair defect but results from an altered cell cycle control, and perhaps an inability to activate damage-inducible DNA repair.100 All four complementation groups appear to involve the same ATM gene.
The radiosensitivity and cancer susceptibility of ATM homozygotes are well established and very clear-cut. On the other hand, whether or not there is increased sensitivity in ATM heterozygotes is less clear. It has been reported that there is an increased breast cancer risk for ATM heterozygotes,101 although this remains equivocal. Recently, Scott et al.102 demonstrated that lymphocytes from obligate AT heterozygotes had an increased sensitivity to x-ray-induced chromosomal aberrations in G2. In addition, they showed that about 42% of women with breast cancer showed a sensitivity that was similar to that of obligate AT heterozy-
gotes.103 This does not mean that these persons were all AT heterozygotes, or that AT heterozygosity predisposes to breast cancer, but rather that altered DNA damage-processing (including repair) genes are more likely to be present in breast cancer patients, and could be partially causative. Thus, heterozygosity for DNA repair genes, where the phenotype is not immediately apparent, could be a marker for susceptibility to cancer, particularly following exposure to ionizing radiation. It is expected that screening for ATM heterozygosity will soon be possible based on recent reports of the genomic organization and gene sequence.104,105
Conclusion
A growing understanding of the various mechanisms of repair of ionizing radiation-induced DNA damage, and of the effects of mutations in genes involved in the repair itself or in its control, is likely to greatly aid in predicting the risk of adverse biological effects arising from exposure to radiation, and eventually in identifying individuals at increased risk.
Loss of Research Programs
Over many years, NASA maintained only a very small radiation health program because of the responsibility, mandated to the Department of Energy (DOE) and its predecessors, for radiation studies. Recently, the funding for NASA radiation research has increased. However, although the percentage increase in funding has been large, the budget in years past was small. Moreover, DOE has significantly reduced its funding for radiation studies in the last few years. Major radiation and animal facilities have been closed, including the high-LET radiation sources for experimental studies at Oak Ridge and Argonne national laboratories. DOE funding of the important facility at Columbia University has been terminated, and the future of the radiation facilities at the Armed Forces Radiobiology Research Institute is now threatened. BEVALAC, the only facility in the United States that was producing beams of heavy ion spectra of energy and LET suitable for cellular and animal studies, as well as for investigations of fragmentation and aspects important for dosimetry, was closed by DOE in 1993. As noted by several previous advisory groups (see, for example, Appendix D), this closure has had very serious consequences for efforts to estimate risks from exposure to radiation in deep space.
Two accelerators, one in Germany and one in Japan, have been developed for heavy ion radiotherapy (see Appendix C) and could be of use in the NASA program. There is no question that international collaboration involving accelerators (with guarantees of appropriate particles and beam time), personnel (to operate and use the facilities), and the necessary financial commitments would be of help in carrying out the priority experiments outlined in Chapter 4.
Although sources of heavy ions exist in the United States and other countries (Appendix C), it is essential to have not only facilities that provide beams for the required range of ions of various energies up to 1 GeV/nucleon, but also laboratory and animal facilities that are readily accessible to U.S. investigators. The present U.S. source of 1 GeV/nucleon heavy ions, the Brookhaven Alternating Gradient Synchrotron, is now used by NASA for only about 100 hours per year. At this rate of utilization, it would take more than 20 years to obtain the physical and biological data needed to make rational decisions about the shielding needed to protect space crews from the biological effects of radiation in space (see Chapter 4).
Collaborative efforts cannot involve transfer of animals among international sites because of strict national quarantine restrictions aimed at reducing the spread of potentially hazardous microorganisms and viruses. Moreover, the travel of animals over a number of time zones would force a resetting of their biological clocks, during which time they would be physiologically and psychologically altered and not useful for controlled experiments. Hence, a requirement for international efforts is the establishment of identical animal colonies at international sites so as to eliminate scientific and legalistic impediments and any effects of biological variability in experimental results. All animal colonies, for example, would have to conform to international accreditation standards, and animal experiments would have to be approved by local institutional review boards and by the board of a collaborating investigator's institution.
References.
1. Lemaignen, L. 1988. Study of Biological Effects and Radiation Protection to Future European Manned Space Flights. Document No. DDT 33061. European Space Agency, ESA Publications Division, ESTEC, Noordwijk, The Netherlands.
2. Simonsen, L.C., Cucinotta, F.A., Atwell, W., and Nealy, J.E. 1993. Temporal analysis of the October 1989 proton flare using computerized anatomical models. Radiat. Res. 133: 1–11.
3. Curtis, S.B., Nealy, J.E., and Wilson, J.W. 1995. Risk cross sections and their application to risk estimation in the galactic cosmic-ray environment . Radiat. Res. 141: 57–65.
4. Badhwar, G.M., and O'Neill, P.M. 1996. Galactic cosmic radiation model and its applications. Adv. Space Res. 17: 7–17.
5. National Council on Radiation Protection and Measurements (NCRP). 1989. Guidance on Radiation Received in Space Activities. Recommendations of the National Council on Radiation Protection and Measurements. NCRP Report No. 98. National Council on Radiation Protection and Measurements, Bethesda, Md.
6. Conklin, J.J., and Walker, R.I., eds. 1987. Military Radiobiology. Academic Press, Orlando, Fla.
7. NCRP, 1989, Guidance on Radiation Received in Space Activities.
8. NCRP, 1989, Guidance on Radiation Received in Space Activities.
9. NCRP, 1989, Guidance on Radiation Received in Space Activities. See also Conklin and Walker, eds., 1987, Military Radiobiology.
10. NCRP, 1989, Guidance on Radiation Received in Space Activities.
11. Conklin and Walker, eds., 1987, Military Radiobiology.
12. NCRP, 1989, Guidance on Radiation Received in Space Activities; Conklin and Walker, eds., 1987, Military Radiobiology.
13. Conklin and Walker, eds., 1987, Military Radiobiology.
14. NCRP, 1989, Guidance on Radiation Received in Space Activities; Conklin and Walker, eds., 1987, Military Radiobiology.
15. NCRP, 1989, Guidance on Radiation Received in Space Activities.
16. NCRP, 1989. Guidance on Radiation Received in Space Activities.
17. NCRP, 1989, Guidance on Radiation Received in Space Activities.
18. Radakovich, M., Dutton, A.M., and Schelling, J.A. 1954. The effect of total body irradiation on wound closure. Ann. Surg. 139: 186–194. See also Raventos, A. 1954. Wound healing and mortality after total body exposure to ionizing radiation . Proc. Soc. Exp. Biol. Med. 87: 165–167.
19. Raventos, 1954, Wound healing and mortality after total body exposure to ionizing radiation. See also Lawrence, W., Jr., Nickerson, J.J., and Warshaw, L.M. 1953. Roentgen rays and wound healing: experimental study. Surgery 33: 376–384.
20. Meistrich, M.L., and Van Beck, M.E.A.B. 1990. Radiation sensitivity of the human testes. Adv. Radiat. Biol. 14: 227–268.
21. Hahn, E.W., Feingold, S.M., Simpson, L., and Batata, M. 1982. Recovery from aspermia induced by low-dose radiation in seminoma patients. Cancer 50: 337–340.
22. Meistrich and Van Beck, 1990, Radiation sensitivity of the human testes.
23. Lushbaugh, C.C., and Cassarett, G.W. 1976. Effects of gonadal irradiation in clinical radiation therapy: A review. Cancer 37: 1111–1125.
24. Ash, P. 1980. The influence of radiation on fertility in man. Br. J. Radiol. 53: 271–278.
25. Damewood, M.D., and Grochow, L.B. 1986. Prospects for fertility after chemotherapy or radiation for neoplastic disease. Fertil. Steril. 45: 443–459.
26. United Nations Scientific Committee on the Effects of Atomic Radiation. 1962. Report of the United Nations Scientific Committee on the Effects of Atomic Radiation. General Assembly Official Records: 17th Session. Supplement No. 16 (A/5216). United Nations, New York.
27. Conklin and Walker, eds., 1987, Military Radiobiology.
28. Simonsen, L.C., Cucinotta, F.A., Atwell, W., and Nealy, J.E.. 1993. Temporal analysis of the October 1989 proton flare using computerized anatomical models. Radiat. Res. 133: 1–11.
29. NCRP, 1989, Guidance on Radiation Received in Space Activities.
30. NCRP, 1989, Guidance on Radiation Received in Space Activities.
31. International Commission on Radiological Protection (ICRP). 1991. 1990 Recommendations of the International Commission on Radiological Protection. ICRP Publication 60. Annals of the ICRP 21. Pergamon Press, Elmsford, N.Y.
32. ICRP, 1991, 1990 Recommendations of the International Commission on Radiological Protection.
33. United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). 1993. Annex F: Influence of dose and dose rate on stochastic effects of radiation. Pp. 619–728 in: Sources and Effects of Ionizing Radiation: United Nations Committee on the Effects of Atomic Radiation: UNSCEAR 1993 Report to the General Assembly, with scientific annexes. United Nations, New York.
34. ICRP, 1991, 1990 Recommendations of the International Commission on Radiological Protection.
35. UNSCEAR, 1993, Annex F: Influence of dose and dose rate on stochastic effects of radiation.
36. National Council on Radiation Protection and Measurements (NCRP). 1993. Risk Estimates for Radiation Protection. NCRP Report No. 115. National Council on Radiation Protection and Measurements, Bethesda, Md.
37. Yochmowitz, M.G., Wood, D.M., and Salmon, Y.L. 1985. Seventeen-year mortality experience of proton radiation in Macaca mulatta. Radiat. Res. 102: 14–34.
38. Clapp, N.K., Darden, E.B., Jr., and Jernigan, M.C. 1974. Relative effects of whole-body sublethal doses of 60-MeV protons and 300 kVp x rays on disease incidence in RF mice. Radiat. Res. 57: 158–186.
39. Burns, F.J., Hoselet, S., and Garte, S.J. 1989. Extrapolations of rat skin tumor incidence: Dose, fractionation and linear energy transfer. Pp. 571–582 in: Low Dose Radiation: Biological Basis of Risk Assessment (K.E. Baverstock and J.W. Stather, eds.). Taylor and Francis, London.
40. Burns et al., 1989, Extrapolations of rat skin tumor incidence.
41. Burns, F.J., Albert, R.E., Vanderlaan, M., and Strickland, P. 1975. The dose-response curve for tumor induction with single and split doses of 10 MeV protons. Radiat. Res. 62: 598 (abstract).
42. Hall, E.J., Kellerer, A.M., Rossi, H.H. and Lam, Y.M.P. 1978. The relative biological effectiveness of 160 MeV protons. Int. J. Radiat. Oncol. Biol. Phys. 4: 1009.
43. Raju, M.R., Bain, E., Carpenter, S.G., Cox, R.A., and Robertson, J.B. 1978. A heavy particle comparative study. Part II: Cell survival versus depth. Br. J. Radiol. 51: 704–711.
44. Todorov, S.L., Grigor'ev, Y.G., Rizhov, N.I., Ivanov, B.A., Malyutina, T.S., and Micleva, M.S. 1972. Dose response relationship for chromosomal aberrations induced by x rays or 50 MeV protons in human peripheral lymphocytes. Mutat. Res. 15: 215.
45. Edwards, A.A., Lloyd, D.C., Prosser, J.S., Finnon, P., and Moquet, J.E. 1986. Chromosome aberrations in human lymphocytes by 8.7 MeV protons and 23.5 MeV helium-3 ions. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 50: 137–145.
46. Burns et al., 1989, Extrapolations of rat skin tumor incidence.
47. Alpen, E.L., Powers-Risius, P., Curtis, S.B., DeGuzman, R., and Fry, R.J.M. 1994. Fluence based relative biological effectiveness for charged particle carcinogenesis in mouse Harderian gland. Adv. Space. Res. 14: 573, 581.
48. National Council on Radiation Protection and Measurements (NCRP). 1990. The Relative Biological Effectiveness of Radiations of Different Quality. NCRP Report No. 104. NCRP, Bethesda, Md.
49. NCRP, 1990, The Relative Biological Effectiveness of Radiations of Different Quality.
50. NCRP, 1990, The Relative Biological Effectiveness of Radiations of Different Quality.
51. Blakely, E.A., Ngo, F.Q.H., Curtis, S.B., and Tobias, C.A. 1984. Heavy ion radiobiology: Cellular studies. Adv. Radiat. Biol. 11: 295.
52. Raju, M.R. 1980. Heavy Particle Radiotherapy. Academic Press, New York.
53. Kronenberg, A. 1995. NASA space radiation health program: Ground based radiobiology research program. Presentation to the Task Group on the Biological Effects of Space Radiation, Committee on Space Biology and Medicine, National Research Council, Washington, D.C., November 13, 1995.
54. Kadhim, M.A., MacDonald, D.A., Goodhead, D.T., Lorimore, S.A., Marsden, S.J., and Wright, E.G. 1992. Transmission of chromosomal instability after plutonium alpha-particle irradiation. Nature (London) 355: 738–740.
55. Kadhim et al., 1992, Transmission of chromosomal instability after plutonium alpha-particle irradiation.
56. Gauger, G.E., Tobias, C.A., Yang, T., and Whitney, M. 1986. The effect of space radiation on the nervous system. Adv. Space Res. 6: 243–249.
57. Curtis, S.B., and Letaw, J.R., 1989. Galactic cosmic rays and cell-hit frequencies outside the magnetosphere. Adv. Space Res. 9: 293–298.
58. Curtis, S.B. 1992. Relating space radiation environments to risk estimates. In: Biological Effects and Physics of Solar and Galactic Radiation (C.E. Swenberg, G. Horneck, and E.G. Starsinopoulos, eds.). Plenum Press, New York.
59. Williams, G.R., and Lett, J.T. 1995. Damage to the photoreceptor cells of the rabbit retina from 56Fe ions: Effect of age at exposure. Adv. Space Res. 18: 55–58. See also Williams, G.R., and Lett, J.T. 1994. Effects of 40Al and 56Fe ions on retinal photoreceptor cells of the rabbit: Implications for manned missions to Mars. Adv. Space Res. 1: 217–220.
60. Williams and Lett, 1995, Damage to the photoreceptor cells of the rabbit retina from 56Fe ions.
61. Lett, J.T., Cox, A.B., Keng, P.C., Lee, A.C., Su, C.M., and Bergtold, D.S. 1980. Late degeneration in rabbit tissues after irradiation by heavy ions. Pp. 131–142 in: Life Sciences and Space Research, Volume XVIII (R. Holmquist, ed.). Pergamon Press, Oxford.
62. Philpott, D.E., Sapp, J., Miquel, J., Kato, K., Corbett, R., Stevenson, J., Black, S., Lindseth, K.A., and Benton, E.V. 1985. The effect of high energy (HZE) particle radiation (40Ar) on aging parameters of mouse hippocampus and retina. Scanning Electron Microsc. Part 3: 1177–1182.
63. Joseph, J.A., Hunt, W.A., Rabin, B.M., and Dalton, T.K. 1992. Possible “accelerated striatal aging” induced by 56Fe heavy-particle irradiation: Implications for manned space flights. Radiat. Res. 130: 88–93.
64. Rabin, B.M., Hunt, W.A., and Joseph, J.A. 1989. An assessment of the behavioral toxicity of high-energy iron particles compared to other qualities of radiation. Radiat. Res. 119: 113–122.
65. Lett, J.T., Keng, P.C., Bergtold, D.S., and Howard J. 1987. Effects of heavy ion on rabbit tissues: Induction of DNA stand breaks in retinal photoreceptor cells by high doses of radiation. Radiat. Environ. Biophys. 26: 23–26.
66. Williams and Lett, 1994, Effects of 40Al and 56Fe ions on retinal photoreceptor cells of the rabbit.
67. Williams and Lett, 1995, Damage to the photoreceptor cells of the rabbit retina from 56Fe ions.
68. Otake, M., and Schull, M. 1990. Radiation-related posterior lenticular opacities in Hiroshima and Nagasaki atomic bomb survivors based on the DS86 dosimetry system. Radiat. Res. 121: 3–13.
69. Worgul, B.Y., Merriam, Jr., G.R., Medvedovsky, C., and Brennen, D.J. 1989. Accelerated heavy particles and the lens. III. Cataract enhancement by dose fractionation. Radiat. Res. 118: 93–100.
70. Lett, J.T., Less, A.C., and Cox, A.B. 1991. Late cataractogenesis in Rhesus monkeys irradiated with protons and radiogenic cataract in other species. Radiat. Res. 126: 147–156.
71. Lett et al., 1991, Late cataractogenesis in Rhesus monkeys irradiated with protons and radiogenic cataract in other species.
72. Wu, B., Medvedovsky, C., and Worgul, B.V. 1994. Non-subjective cataract analysis and its application in space radiation risk assessment. Adv. Space Res. 14: 493–500.
73. Neel, J.V., and Schull, W.J., eds. 1991. The Children of Atomic Bomb Survivors: A Genetic Study. National Academy Press. Washington, D.C.
74. Kodaira, M., Satoh, C., Hiyama, K., and Toyama, K. 1995. Lack of effects of atomic bomb radiation on genetic instability of tandem-repetitive elements in human germ cells. Am. J. Hum. Genet. 57: 1275–1283.
75. Satoh, C., Takahashi, N., Asakawa, J., Hiyama, K., and Kodaira, M. 1993. Variations among Japanese of the factor IX gene (f9) detected by PCR-denaturing gradient gel electrophoresis. Am. J. Hum. Genet. 52: 167–175.
76. United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). 1993. Sources and Effects of Ionizing Radiation: United Nations Committee on the Effects of Atomic Radiation: UNSCEAR 1993 Report to the General Assembly, with scientific annexes. United Nations, New York. Pp. 754–757.
77. UNSCEAR, 1993, Sources and Effects of Ionizing Radiation.
78. Neel, J.V., and Schull, W.J., eds. 1991. The Children of Atomic Bomb Survivors: A Genetic Study. National Academy Press. Washington, D.C.
79. Cox, L.S., and Lane, D.P. 1995. Tumour suppressors, kinases and changes: How p53 regulates the cell cycle in response to DNA damage. Bioessays 17: 501–508.
80. Donner, E.M., and Preston, R.J. 1996. The relationship between p53 status, DNA repair and chromatid aberration induction in G2 mouse embryo fibroblast cells treated with bleomycin. Carcinogenesis 17: 1161–1165.
81. Harvey, M., McArthur, M.J., Montgomery, C.A., Butel, J.S., Bradley, A., and Donehower, L.A. 1993. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat. Genet. 5: 225–229.
82. Scott, D., Spreadborough, A., Levine, E., and Roberts, S.A. 1994. Genetic predisposition in breast cancer. Lancet 344: 1444.
83. Jones, L.A., Scott, D., Cowan, R., and Roberts, S.A. 1995. Abnormal radiosensitivity of lymphocyte from breast cancer patients with excessive normal tissue damage after radiotherapy: Chromosome aberrations after low dose-rate irradiation. Int. J. Radiat. Biol. 67: 519–528.
84. Wood, R.D., Aboussekhra, A., Biggerstaff, M., Jones, C.J., O'Donovan, A., Shivji, M.K.K., and Syzmkowski, D.E. 1993. Nucleotide excision repair of DNA by mammalian cell extracts and purified proteins. Pp. 625–632 in: DNA and Chromosomes. Vol. LVIII. Cold Spring Harbor Press, Cold Spring Harbor, N.Y.
85. Sancar, A. 1995. Excision repair in mammalian cells. J. Biol. Chem. 270: 15915–15918.
86. Demple, B., and Harrison, L. 1994. Repair of oxidative damage to DNA: Enzymology and biology. Annu. Rev. Biochem. 63: 915–948.
87. Jeggo, P.,39 A., Taccioli, G.E., and Jackson, S.P. 1995. Menage à trois: Double strand break repair, V(D)J recombination and DNA-PK. Bioessays 17: 949–957.
88. Demple and Harrison, 1994, Repair of oxidative damage to DNA.
89. Taccioli, G.E., Rathburn, G., Oltz, E., Stamato, T., Jeggo, P.A., and Alt., F. 1993. Impairment of V(D)J recombination in double-strand break repair mutants. Science 260: 207–210.
90. Alt, F.W., Oltz, E.M., Young, F., Gorman, J., Taccioli, G., and Chen, J. 1992. VDJ recombination. Immunol. Today 13: 306–314.
91. Anderson, C.W. 1993. DNA damage and the DNA-activated protein kinase. Trends Biochem. Sci. 18: 433–437.
92. Jackson, S.P. 1996. The recognition of DNA damage. Current Opinion in Genetics and Development 6:19–25.
93. Jackson, 1996, The recognition of DNA damage.
94. Leadon, S.A., Barbee, S.L., and Dunn, A.B. 1995. The yeast RAD2, but not RAD1, gene is involved in the transcription-coupled repair of thymine glycols. Mutat. Res. 337: 169–178.
95. Kuhn, A., Gottlieb, T.M., Jackson, S.P., and Grummt, I. 1995. DNA-dependent protein kinase: A potent inhibitor of transcription by RNA polymerase I. Genes Dev. 9: 193–203.
96. Blunt, T., Finnie, N.J., Taccioli, G.E., Smith, G.C.M., Demengeot, J., Gottlieb, T.M., Mizuta, R., Varghese, A.J., Alt, F.W., Jeggo, P.A., and Jackson, S.P. 1995. Defective DNA-dependent protein kinase activity linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell 80: 813–823.
97. Savitsky, K., Sfez, S., Tagle, D.A., Ziv, Y., Sartiel, A., Collins, F.S., Shiloh, Y., and Rotman, G. 1995. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum. Molec. Genet. 4: 2025–2032.
98. Morrow, D.M., Tagle, D.A., Shiloh, Y., Collins, F.S., and Hieter, P. 1995. TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell 82: 831–840.
99. Greenwell, P.W., Kronmal, S.L., Porter, S.E., Gassenhuber, J., Overmaier, B., and Petes, T.P. 1995. TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell 82: 823–829.
100. Meyn, M.S. 1995. Ataxia telangiectasia and cellular responses to DNA damage. Cancer Res. 55: 5991–6001.
101. Swift, M., Morrell, D., Massey, R.B., and Chase, C.L. 1991. Incidence of cancer in 161 families affected by ataxia telangiectasia. New Engl. J. Med. 325: 1831–1836.
102. Scott et al., 1994, Genetic predisposition in breast cancer.
103. Jones, L.A., Scott, D., Cowan, R., and Roberts, S.A. 1995. Abnormal radiosensitivity of lymphocyte from breast cancer patients with excessive normal tissue damage after radiotherapy: Chromosome aberrations after low dose-rate irradiation. Int. J. Radiat. Biol. 67: 519-28.
104. Rasio, D., Negrini, M., and Croce, C.M. 1995. Genomic organization of the ATM locus involved in ataxia telangiectasia. Cancer Res. 55: 6053–6057.
105. Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle, D.A., Smith, S., Uziel, T., Sfez, S., Ashkenazi, M., Pecker, I., Frydman, M., Harnik, R., Patanjali, S.R., Simmons, A., Clines, G.A., Sartiel, A., Gatti, R.A., Chessa, L., Sanal, O., Lavin, M.F., Jaspers, N.J., Taylor, A.R., Arlett, C.F., Miki, T., Weissman, S.M., Lovett, M., Collins, F.S., and Shiloh, Y. 1995. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268: 1749–1753.






















