
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Oncogenic potential of the adenovirus E4orf6 protein
MARY MOORE, NOBUO HORIKOSHI, AND THOMAS SHENK *
Howard Hughes Medical Institute, Department of Molecular Biology, Princeton University, Princeton, NJ 08544–1014
ABSTRACT The group C adenovirus E4orf6 protein has previously been shown to bind to the p53 cellular tumor suppressor protein and block its ability to activate transcription. Here we show that the E4orf6 protein blocks the induction of p53-mediated apoptosis when AT6 cells, which harbor a temperature-sensitive p53, are shifted to the permissive temperature. The E4orf6 protein does not, however, prevent the induction of apoptosis in p53-deficient H1299 cells by treatment with tumor necrosis factor α and cycloheximide. The E4orf6 protein also cooperates with the adenovirus E1A protein to transform primary baby rat kidney cells, and it cooperates with the adenovirus E1A plus E1B 19-kDa and E1B 55-kDa proteins to increase the number of baby rat kidney cell transformants and enhance the rate at which they arise. The level of p53 is substantially reduced in transformed cells expressing the E4orf6 protein in comparison to adenovirus transformants lacking it. The E4orf6 gene also accelerates tumor formation when transformed baby rat kidney cells are injected subcutaneously into the nude mouse, and it converts human 293 cells from nontumorigenic to tumorigenic in nude mice. In addition to the well-studied E1A and E1B oncogenes, group C adenoviruses harbor a third oncogene, E4orf6, which functions in some respects similarly to the E1B oncogene.
Although adenoviruses are not known to be associated with tumorigenesis in humans, some human adenovirus serotypes can directly induce tumors in rats or hamsters, and all serotypes tested can transform cultured rodent cells (reviewed in refs. 1 and 2). Candidate viral oncogenes were first identified as the genes that are always retained in cells transformed by group C adenoviruses. Most of the viral genome is lost from cells transformed by these viruses, which include adenovirus types 2 and 5; only the E1A and E1B genes are consistently retained. The E1A and E1B genes were subsequently confirmed to be both necessary and sufficient for transformation by mutational analysis of the viral genome and transfection experiments employing the cloned genes.
More recent work has provided a detailed mechanistic explanation for the transforming ability of these viral genes (reviewed in refs. 1 and 2). The E1A proteins bind to a number of cellular growth-regulatory proteins and modulate their function. Most notably, E1A proteins bind to the retinoblastoma tumor suppressor protein and its family members (3), freeing the cellular S phase-specific transcription factor E2F (4) and deregulating the control of cell cycle progression (5). The E1A proteins generally stabilize p53 and induce apoptosis when introduced into cells (6). The E1B proteins cooperate with E1A to transform cells at least in part by preventing the apoptotic response (7). The E1B 55-kDa protein binds to p53 and interferes with its ability to activate transcription (8–10), presumably blocking its ability to induce apoptosis. The E1B 19-kDa protein, which is related to the Bcl-2 family of cellular proteins (11), prevents the induction of apoptosis by a variety of inducers, including p53 (7, 12). Either one of the two E1B proteins is sufficient to cooperate with E1A to transform cells (12).
We have recently shown that the adenovirus E4orf6 protein, like the E1B 55-kDa protein, can bind to p53 both in vitro and in extracts from infected cells (13). Whereas the E1B 55-kDa protein binds to the amino-terminal activation domain of p53, the E4orf6 protein binds near the carboxyl terminus of the protein, close to its oligomerization domain. Nevertheless, like the E1B 55-kDa protein, the E4orf6 protein efficiently blocks the ability of p53 to activate transcription (13). p53 activates transcription, at least in part, by contacting a constituent of the basal transcriptional machinery, TAFII31, through its aminoterminal transcriptional activation domain (14, 15). The E4orf6 protein blocks the interaction of p53 with TAFII31, even though it does not appear to directly contact the aminoterminal activation domain of p53 (13).
Here we describe several biological consequences of the E4orf6 interaction with p53. E4orf6 protein can block the induction of apoptosis by p53, but it does not exhibit transforming activity when expressed in the absence of other adenovirus proteins in rat cells. It can, however, cooperate with the E1A proteins to transform rat cells, it can enhance transformation by the E1A plus E1B proteins, and it can enhance the oncogenicity of virus-transformed cells in nude mice. Thus, group C adenoviruses contain a third oncogene that appears to function in some respects similarly to the E1B gene.
MATERIALS AND METHODS
Plasmids. The cytomegalovirus immediate early promoter was utilized to express the E1A (pCMVE1A), E1B 19-kDa (pCMV19K), or E1B 55-kDa (pCMV55K) coding region; each of these constructs has been described elsewhere (16, 17). The simian virus 40 early promoter/enhancer was utilized to express the E1A coding region in pSVE1A (18). The plasmid pXhoI-C (19) contains the leftmost 15.5% of the adenovirus 5 genome, including the E1A and E1B genes with their endogenous promoters. An E4orf6-specific cDNA was prepared from mRNA by reverse transcription, amplified by PCR, cloned, and sequenced. The E4orf6 coding region was expressed either from the cytomegalovirus immediate early promoter (pCMV34K) (13) or from the mouse mammary tumor virus long terminal repeat (MMTV LTR) (pMMTVE4orf6). The E2A coding region was also expressed from the MMTV LTR (pMMTVE2A).
Apoptosis Assay. Cells were transfected with the pCMVE4orf6 expression plasmid (13) using Lipofectamine (GIBCO/BRL) 24 hr before inducing apoptosis. H1299 cells (20), which express no p53, were treated with human tumor
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: TNF, tumor necrosis factor; TUNEL, terminal deoxynucleotidyltransferase-mediated UTP end labeling.
|
* |
To whom reprint requests should be addressed. e-mail: tshenk@molbiol.princeton.edu. |
necrosis factor (TNF)-α (10 ng/ml) and cycloheximide (30 μg/ml) for 7 hr. AT6 cells (21), which express a temperature-sensitive p53 allele, were placed at 32°C for 3 days for the induction of apoptosis. Cells were then harvested, apoptotic cells were identified by terminal deoxynucleotidyltransferase-mediated UTP end labeling (TUNEL) assay (22, 23), and expression of the E4orf6 protein was detected by immunofluorescence using the E4orf6-specific RSA3 antibody and Texas red-coupled donkey anti-mouse IgG.
Transformation Assay. Primary Fischer baby rat kidney cells were prepared as described (17). Cells were transfected by the calcium phosphate technique (24) and maintained in medium with 5% fetal calf serum. Transformed cultures were stained with Giemsa stain, and foci that were 4 mm or greater in diameter were counted at 30 days after transfection. Three independent experiments were performed for each plasmid combination. Five pCMVE1A/pCMVE4orf6 transformants, five pXhoC/MMTVE4orf6 transformants, and five pXhoC/ CMVE4orf6 transformants were cloned and maintained as continuous cell lines.
Selection of 293 Cell Lines Expressing E4orf6 Protein. The 293 human embryonic kidney cell line expresses the adenovirus E1A and E1B genes (25). The 293 cells were transfected with pMMTVE4orf6 and pMMTVE2A together with the pBabePuro puromycin-resistance marker (26) by the calcium phosphate technique. Clones were selected and maintained in medium containing 10% delipidated calf serum plus puromycin at 1 μg/ml.
Protein Analysis. For analysis of proteins by immunoprecipitation followed by Western blotting, cells were lysed in RIPA buffer (50 mM Tris·HCl, pH 7.4/150 mM NaCl/1% Triton X-100/0.1% SDS/1% sodium deoxycholate) and normalized for protein concentration, and specific proteins were immunoprecipitated with RSA3 (E4orf6-specific monoclonal antibody, ref. 27), 421 (p53-specific monoclonal antibody, ref. 28), or 2A6 (E1B 55-kDa-specific monoclonal antibody, ref. 29). The immunoprecipitates were subjected to electrophoresis in an SDS-containing polyacrylamide gel, and proteins were transferred to an Immobilon membrane (Millipore), which was then incubated with E4orf6- or p53-specific monoclonal antibody. Reactive protein species were detected by enhanced chemiluminescence (ECL; Amersham).
To assay proteins by immunofluorescence, cells were grown on coverslips. The coverslips were washed in phosphate-buffered saline and fixed in 100% methanol. The fixed calls were then incubated with an E4orf6-, E1B 55-kDa-, or p53-specific antibody followed by reaction with a secondary antibody conjugated with either fluorescein isothiocyanate (FITC) or tetramethylrhodamine B isothiocyanate (TRITC) and were examined with a confocal microscope.
The half-life of p53 in transformed baby rat kidney cells was determined by pulse-chase analysis (30).
PCR Amplification of cDNAs. Total cell RNA was prepared from transformed baby rat kidney cells, treated with RNase-free DNase I (1 unit/μg of RNA) (GIBCO/BRL), and subjected to reverse transcription with Superscript II polymerase (GIBCO/BRL) using a 3′-E4orf6-specific primer (P3, 5′-AATCCCACACTGCAGGGA-3′). The cDNA was then amplified with KlenTaq DNA polymerase (CLONTECH) in a PCR using P3 primer and a 5′-E4orf6-specific primer (P2, 5′-CGGCGCACTCCGTACAGT-3′). To control for the presence of DNA that might have survived treatment of the RNA preparations with DNase I, a second amplification was performed on each sample with a primer from the promoter region of the plasmid used to express the E4orf6 mRNA (P1, 5′-CGGTAGGCGTGTACG-3′) and the P3 primer.
Tumor Induction. Transformed cells were injected subcutaneously into 6- to 8-week-old female Swiss 3T3 nude mice (Taconic Laboratories) and assayed by palpation until a tumor was detected. Animals that did not develop a tumor were sacrificed on day 110.
RESULTS
The Adenovirus E4orf6 Protein Blocks p53-Induced Apoptosis. Since the adenovirus E4orf6 protein can bind to p53 and block its ability to activate transcription (13), we tested whether the E4orf6 protein could prevent apoptosis induced by p53. We employed AT6 cells for the assay. These cells lack an endogenous p53 gene and harbor an ectopic temperature-sensitive p53 allele (21). They grow normally when maintained at the nonpermissive temperature but undergo apoptosis when p53 function is restored by shifting to the permissive temperature. AT6 cells were transfected with a plasmid expressing the E4orf6 gene under control of the cytomegalovirus immediate early promoter; 24 hr later the cultures were shifted to the permissive temperature, and the cultures were assayed for apoptosis after incubation for 72 hr at 32°C. Transfected cells expressing the E4orf6 protein were identified by immunofluorescence using an E4-specific monoclonal antibody, and apoptotic cells were identified by using the TUNEL assay (22, 23). In this assay, chromatin is treated in situ with terminal deoxynucleotidyltransferase to label 3′-OH ends of DNA with biotin-dUTP, followed by reaction with fluorescein-labeled avidin to identify cells with fragmented DNA. In the confocal micrograph displayed in Fig. 1A, E4orf6-specific immunofluorescence is represented by the red signal and DNA fragmentation is monitored with the green signal. The red nuclei from transfected cells expressing the E4orf6 protein display little yellow signal (yellow results from the overlap of green and red signals), whereas the nuclei of cells that do not express the E4orf6 protein display a green signal indicative of DNA fragmentation, a hallmark of apoptosis. Expression of the E4orf6 protein (red signal) and expression of DNA fragmentation (green signal) are mutually exclusive, indicating that the viral protein protects against p53-induced apoptosis in AT6 cells.
It is noteworthy that the green apoptotic nuclei in Fig. 1A appear to be much smaller than the red E4orf6-expressing nuclei that were protected from apoptosis. However, examination of cells by phase-contrast microscopy (data not shown) revealed that nuclei undergoing apoptosis had not shrunk to a considerable extent. Rather, the green signal indicative of DNA fragmentation was limited to subdomains within apoptotic nuclei. Presumably the nuclear shrinkage that is characteristic of apoptosis would be observed at later times after the induction of p53.
We also tested the ability of the E4or6 protein to protect against the induction of apoptosis in H1299 cells that do not express p53. H1299 cells were transfected with the E4orf6-expressing plasmid, and 24 hr later the cultures were treated for 7 hr with TNF-α (10 ng/ml) and cycloheximide (30 μg/ml) to induce apoptosis. In the confocal micrograph shown in Fig. 1B, the E4orf6 protein is identified by immunofluorescence (red signal) and DNA fragmentation is marked by the TUNEL assay (green signal). In this case, overlapping green and red signals (yellow signal) are evident, indicating that the E4orf6 protein does not protect cells from the induction of p53-independent apoptosis by TNF-α. As noted above, nuclei undergoing apoptosis have not shrunk, and the yellow signal generated by DNA fragmentation was limited to subdomains within the nuclei whose boundaries are demarcated by the E4orf6-specific immunofluorescence (red signal).
E4orf6 Cooperates with E1A and E1B to Transform Rat Cells. The E1B proteins cooperate with E1A proteins to transform rodent cells at least in part by blocking apoptosis (7). Since the E4orf6 protein, like E1B proteins, can block p53-mediated apoptosis, we explored the possibility that it might contribute to oncogenesis. Primary baby rat kidney cells were

FIG. 1. The adenovirus E4orf6 protein prevents p53-dependent apoptosis. An E4orf6 expression plasmid was transfected into H1299 cells (A) or AT6 cells (B). H1299 cells were treated with human TNF-α and AT6 cells were shifted to 32°C to induce apoptosis. Apoptotic cells were detected by assaying DNA fragmentation with the TUNEL assay (green signal), and expression of the E4orf6 protein was detected by indirect immunofluorescence (red signal). The yellow signal marks nuclei that are positive for expression of E4orf6 protein and for DNA fragmentation. (×250.)
used for transformation assays. In the first assay (Fig. 2A), expression of the E4orf6 gene was controlled by the mouse mammary tumor virus promoter; and in the subsequent four assays (Fig. 2B), E4orf6 expression was controlled by the cytomegalovirus major immediate early promoter. All assays produced the same result. The E4orf6 protein alone was unable to induce the formation of transformed colonies, and the E1A proteins alone produced either no foci (Fig. 2A) or a limited number of foci (Fig. 2B) that were commonly flat in appearance and generally could not be cloned (data not shown). Rare E1A transformants that can be propagated have been shown to contain mutant p53 genes (31) that can cooperate with E1A to transform baby rat kidney cells by blocking the induction of apoptosis by wild-type p53 (7). Transfection with plasmids expressing the E1A plus E4orf6 proteins produced severalfold more colonies than E1A alone. Many of these foci were multilayered, and these cells could be cloned and propagated. E1A/E4orf6 transformants grew somewhat more slowly but were morphologically indistinguishable from E1A/E1B transformants. When assayed by Western blotting, E1A/E4orf6 transformants contained very low to non-detectable levels of E4orf6 protein (Fig. 3A, lanes 1–3), but E4orf6 mRNA was detected in four additional clones of E1A/E4orf6-transformed cells when assayed by reverse transcription followed by PCR amplification (Fig. 3B). Furthermore, the E1A/E4orf6 transformant that was examined con-
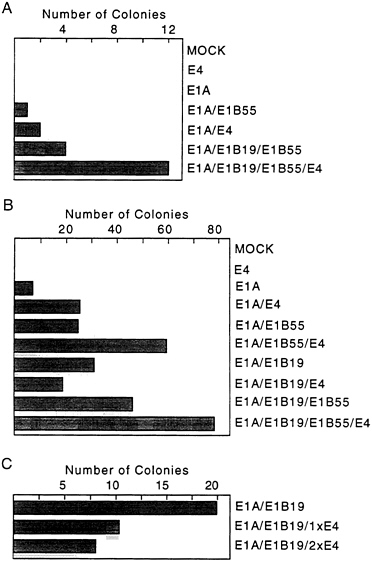
FIG. 2. The E4orf6 protein is an oncoprotein. Primary baby rat kidney cells were transfected with plasmids expressing the indicated adenovirus proteins and assayed for the formation of foci 30 days later. Cells receiving both E1B proteins were transfected with a plasmid containing the intact E1B transcription unit controlled by its own promoter, while cells receiving only the E1B 19-kDa or E1B 55-kDa protein received plasmids carrying cDNAs controlled by the cytomegalovirus major immediate early promoter. (A) Two 10-cm plates of cells were assayed for colony formation in response to the indicated adenovirus proteins. Expression of the E4orf6 protein was controlled by the mouse mammary tumor virus promoter, and E1A expression was controlled by the simian virus 40 early promoter. (B) Eight 10-cm plates in four independent experiments were assayed for colony formation. Expression of the E4orf6 and E1A proteins was controlled by the cytomegalovirus major immediate early promoter. (C) Three 10-cm plates of cells were assayed for colony formation. Cells received either the same amount (1xE4) or twice as much E4 as E1B expression vector (2xE4). All expression plasmids contained the cytomegalovirus major immediate early promoter.
tained substantially less p53 than cells transformed with the E1A plus E1B 19-kDa and E1B 55-kDa (E1B19,55) proteins (Fig. 3C, lanes 2 and 3).
The E4orf6 protein enhanced transformation by the E1A plus E1B 55-kDa proteins or by E1A plus E1B 19-kDa and E1B 55-kDa (E1B19,55) proteins (Fig. 2). The total number of colonies was increased by a factor of about 1.5 to 2.5; but, more significantly, colonies arose and grew more rapidly. For example, on day 10 after transfection, E1A/E1B 19,55 transformants averaged <0.5 mm in diameter, whereas E1A/ E1B19,55/E4orf6 transformants averaged 1.5 mm in diameter. At 14 days, transformants lacking E4orf6 protein averaged 2.0 mm in diameter and those containing it averaged 3.5 mm in diameter. Further, E4orf6 protein-containing transformants generally produced multilayered colonies that grew markedly
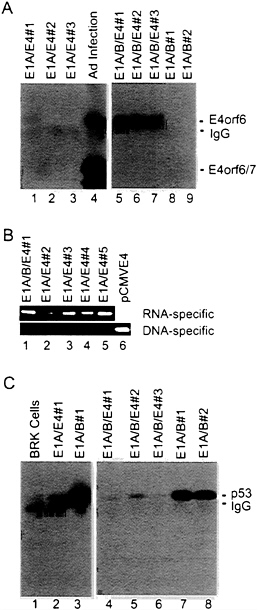
FIG. 3. The steady-state level of p53 in E1A/E1B-transformed baby rat kidney cells is substantially reduced when the E4orf6 protein is present. (A) Extracts were prepared from E1A/E4orf6 transformants (lanes 1–3), E1A/E1B19,55/E4orf6 transformants (lanes 5–7), E1A/E1B19,55 transformants (lanes 8 and 9), and adenovirus-infected 293 cells (lane 4). Immunoprecipitations were performed with antibody to E4orf6 protein (RSA3), and E4orf6 protein was detected in immunoprecipitates by Western blot assay using the same antibody. Bands corresponding to the E4orf6 protein, E4orf6/7 protein (which is also recognized by the RSA3 antibody), and the immunoglobulin light chain (IgG) are identified. (B) Total cell RNA was prepared from four E1A/E4orf6 transformants (lanes 2–5) that did not contain detectable amounts of E4orf6 protein and from one E1A/ E1B19,55/E4 transformant (lane 1) that expressed an easily detectable level of the E4orf6 protein. RNA samples were treated with DNase I, and cDNAs were prepared and amplified by PCR using primers that could detect E4-specific RNA (upper gel) or primers that were specific for DNA that might have survived the DNase I treatment (lower gel). (C) Extracts were prepared from baby rat kidney cells (BRK, lane 1), an E1A/E4orf6 transformant (lane 2), E1A/E1B19,55 transformants (lanes 3, 7, and 8), and E1A/E1B19,55/E4orf6 transformants (lanes 4–6). Immunoprecipitations were performed with antibody to p53 (421), and p53 was detected in immunoprecipitates by Western blot assay using the same antibody. Bands corresponding to the p53 and the immunoglobulin heavy chain (IgG) are identified.
taller—i.e., had more layers of cells—than colonies lacking it. E1A/E1B19,55/E4orf6 transformants contained substantial quantities of E4orf6 protein (Fig. 3A, lanes 5–7), considerably more than was found in E1A/E4orf6 transformants (Fig. 3A, lanes 1–3). But, like E1A/E4orf6 transformants, E1A/ E1B19,55/E4orf6 transformants exhibited substantially reduced levels of p53 in comparison with E1A/E1B19,55 transformants (Fig. 3C, lanes 4–8).
Curiously, the E4orf6 protein did not cooperate with the E1A plus E1B 19-kDa proteins in the transformation assay. Rather, the E4orf6 protein somewhat reduced the number of foci produced by E1A plus E1B 19-kDa protein (Fig. 2 B and C). The inhibition was not due to promoter competition, since the total amount of cytomegalovirus major immediate early promoter was held constant by inclusion of a plasmid containing the promoter with no insert in transfections that did not receive the E4orf6 expression plasmid in the experiment displayed in Fig. 2C. The lack of cooperation suggests that the E1B 19-kDa and E4orf6 proteins might function similarly in the transformation assay—i.e., block E1A-induced apoptosis; but the reason for the apparent interference by the E4orf6 protein in the assays receiving E1A and E1B 19-kDa proteins is unclear.
Two E1A/E1B19,55 baby rat kidney cell transformants and two E1A/E1B19,55/E4 transformants were tested for their ability to induce tumors in athymic Swiss nude mice (Table 1). Both transformants expressing the E4orf6 protein produced tumors at the site of injection that were detected by palpation on day 28. One of the cell lines lacking the E4orf6 protein produced detectable tumors after an extended delay (84 days), and the other did not generate tumors during the period of observation (110 days). In similar experiments, the tumorigenic potential of a derivative of human 293 cells that contained the adenovirus E1A/E1B19,55/E2A genes was compared with derivatives of 293 cells that also contained the E4orf6 gene (Table 1). As had been reported for 293 cells previously (32), injection of 5×106 cells lacking E4orf6 protein failed to induce tumors during a 110-day period that animals were monitored. In contrast, three independently derived 293 cell lines containing the E4orf6 protein produced tumors at the site of injection. Four solid tumors were subjected to histopathological examination, and as expected, they were composed of poorly differentiated epithelial cells.
E4orf6 protein could be detected by Western blot assay in the 293 cell derivatives containing the E4orf6 gene (Fig. 4A, lanes 2 and 3); and, as was found for rat cell transformants, the level of p53 was substantially reduced in cells containing the E4orf6 protein (Fig. 4B). The association of E4orf6 protein with other proteins in these cells was examined by immunoprecipitation from a cell extract using E4orf6-, E1B-55-kDa-, or p53-specific monoclonal antibodies followed by Western
Table 1. Tumor induction in Swiss nude mice
blot assay of the immunoprecipitate using an E4orf6-specific antibody (Fig. 4C). The E4 protein was co-immunoprecipitated by the E1B 55-kDa-specific antibody, consistent with earlier reports that the two proteins exist in a complex (33). The E4orf6 protein was also co-immunoprecipitated with the p53-specific antibody, as predicted by our earlier work (13). Curiously, while protein immunoprecipitated by the E4orf6-specific antibody migrated as a single band, the protein interacting with the E4orf6 antibody in the Western blot that was co-immunoprecipitated with either the E1B 55-kDa protein or p53 migrated as a doublet (Fig. 4C, lanes 1 and 2). The doublet was consistently seen in multiple experiments; and, while direct immunoprecipitation with E4orf6-specific antibody produced only the slower-migrating component of the doublet (Fig. 4C, lane 3), the faster-migrating band was consistently the major species when assayed by co-immunoprecipitation with p53. Further, the faster-migrating species was the major E4orf6-specific product present in tumor
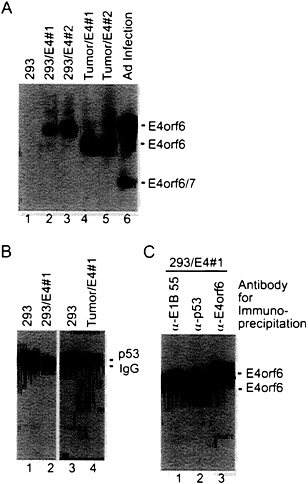
FIG. 4. Expression of E4orf6 protein reduces the steady-state level of p53 in 293 cells. (A) Extracts were prepared from 293 cell derivatives containing the E4orf6 gene (lanes 2 and 3), tumors produced in nude mice by E4orf6-containing cells (lanes 4 and 5), and adenovirus-infected 293 cells (lane 6). Immunoprecipitations were performed with antibody to E4orf6 protein (RSA3), and E4orf6 protein was detected in immunoprecipitates by Western blot assay using the same antibody. Bands corresponding to the E4orf6 protein and E4orf6/7 protein (which is also recognized by the RSA3 antibody) are identified. (B) Extracts were prepared from a 293 cell derivative expressing the E4orf6 protein (lane 2) and from a tumor produced in a nude mouse by the E4orf6-expressing cell line (lane 4). Immunoprecipitations were performed with antibody to p53 (421), and p53 was detected in immunoprecipitates by Western blot assay using the same antibody. Bands corresponding to the p53 and the immunoglobulin heavy chain (IgG) are identified. (C) An extract was prepared from a 293 cell derivative expressing the E4orf6 protein. Aliquots of the extract were immunoprecipitated with antibody to the E1B 55-kDa protein (α-E1B 55), antibody to p53 (α-p53), or antibody to the E4orf6 protein (α-E4orf6), and E4orf6 protein was detected in immunoprecipitates by Western blot assay using RSA3 antibody.
cells (Fig. 4A, lanes 4 and 5), raising the possibility that this species of E4orf6 protein is selected during tumorigenesis. The doublet does not result from differential phosphorylation of the E4orf6 protein (data not shown). As yet we do not understand the basis for the differential migration of the E4orf6 species. Possibly the doublet is produced by proteolysis within the cell extract; alternatively, it could be a variant of the E4orf6 protein that contributes to its oncogenic potential.
The Half-Life of p53 and Its Intracellular Location Are Altered in the Presence of the E4orf6 Protein. Normally, wild-type p53 is localized to the nucleus and has a half-life of 15–30 min (30, 34), whereas p53 is excluded from the nucleus in cells transformed by group C adenoviruses, and it is stabilized so that it has a half-life of about 10 hr (35). The stabilization of p53 results from the action of the E1A protein, and it can occur in the absence of the E1B 55-kDa protein when E1A cooperates with the H-ras oncogene to transform cells (6). The relatively low steady-state level of p53 in E1A/ E1B19,55/E4orf6 transformants as compared with E1A/ E1B19,55 transformants (Figs. 3C and 4B) suggested that p53 was not stabilized in the presence of the E4orf6 protein. Therefore, we performed a pulse-chase analysis to compare the half-life of p53 in transformed baby rat kidney cells in the presence and absence of E4orf6 protein (Fig. 5). The half-life of p53 was greater than 2 hr in the absence of E4orf6 protein. In the transformants containing the E4orf6 gene, the cells appeared to contain two pools of p53 with different stabilities. The major p53 species had a half-life of about 5 min, and the minor p53 species had a half-life of greater than 2 hr.
In addition to reducing the stability of p53, the E4orf6 protein altered the localization of p53 in transformed cells. In 293 cells lacking the E4orf6 protein, p53 was localized predominantly in discrete, intensely fluorescent, cytoplasmic bodies adjacent to the nucleus (Fig. 6A), as has been described previously for cells transformed by group C adenoviruses (35–37). The dense cytoplasmic bodies are located in close proximity to the centrosome in interphase cells (38). Many p53-containing cytoplasmic bodies were observed in 293 cells (Fig. 6A), and many fewer cytoplasmic bodies were evident in cells containing the E4orf6 protein. In these cells p53 was substantially localized to the nucleus, where it exhibited an uneven, spotty fluorescence (Fig. 6C). However, some cells in
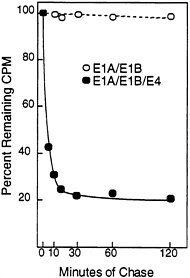
FIG. 5. The E4orf6 protein reduces the half-life of p53 in adenovirus-transformed cells. E1A/E1B-transformed (○) and E1A/E1B/ E4-transformed (●) baby rat kidney cells were labeled for 60 min with [35S]methionine followed by chase periods, after which immunoprecipitations were performed with the p53-specific monoclonal antibody 421. After electrophoresis, radioactivity in p53-specific bands was quantified using a PhosphorImager. At the start of the chase (time 0), E1A/E1B and E1A/E1B/E4 transformants contained 1.36×106 cpm and 2.66×106 cpm, respectively, in p53-specific bands.
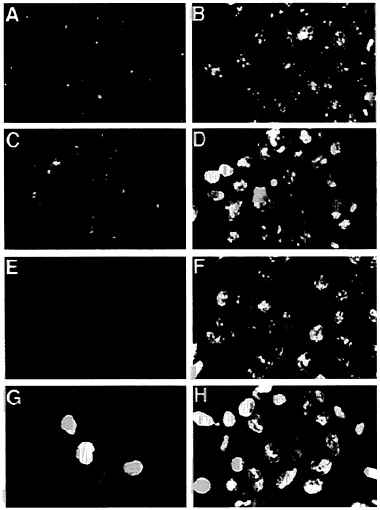
FIG. 6. The E4orf6 protein alters the location of p53 in 293 cells. E1A/E1B/E2A-containing 293 cells (A, B, E, and F) were compared with E1A/E1B/E2A/E4-containing 293 cells (C, D, G, and H). These cell lines were tested for tumorigenicity, and the results are shown in Table 1, where the E4orf6-containing cells are designated cell line #1. Indirect immunofluorescent staining with the monoclonal antibody 421 identified p53 (A and C) and E4orf6 protein was localized with the monoclonal antibody RSA3 (E and G). A fluorescent, nucleic acid-binding dye (Yo-yo) was used to counterstain nuclei (B, D, F, and H) that are examined by immunofluorescence. (×600.)
clonal populations of transformed cells containing the E4orf6 gene continued to exhibit cytoplasmic bodies containing p53, and we believe this is due to the fact that not all cells in these cultures express the E4orf6 protein at a high level (Fig. 6G). This could also explain the two populations of p53 observed in the half-life analysis (Fig. 5). The p53 with the short half-life could reside in cells with a high level of E4orf6 protein, whereas the long-lived p53 could be located in cells with less E4orf6 protein. We do not yet know the reason why individual cells in clonal populations appear to accumulate quite different amounts of nuclear E4orf6 protein. The E1B 55-kDa protein, which binds to p53, exhibited the same change in localization within 293 cells—i.e., it moved from the cytoplasmic body in the absence of E4orf6 protein to the nucleus in its presence (data not shown).
DISCUSSION
Our experiments demonstrate that the adenovirus E4orf6 protein has oncogenic potential. It can cooperate with the adenovirus E1A protein to transform baby rat kidney cells, and it can enhance transformation by the E1A protein plus E1B proteins (Fig. 2). Dobner and colleagues (M.Nevels, S. Rubenwolf, H.Schuett, T.Sprussl, H.Wolf, and T.Dobner, personal communication) have observed similar effects of the E4orf6 protein in transformation experiments. Further, the E4orf6 protein can accelerate tumor formation when transformed baby rat kidney cells are injected subcutaneously into the nude mouse, and it converts human 293 cells from nontumorigenic to tumorigenic in nude mice (Table 1).
A role for the E4orf6 protein in oncogenesis was missed in early studies with group C adenoviruses that identified the E1A and E1B genes as the viral oncogenes. Although several early studies described the presence of E4-specific mRNAs in addition to the E1A and E1B species in adenovirus-transformed rat and hamster cells (e.g., refs. 39–41; reviewed in ref. 42), a role for E4 in transformation was discounted because it was not always present in cells transformed by adenovirus types 2 and 5 and because cloned E1A and E1B genes were sufficient for transformation. Our results indicate that, although it is not essential for transformation by adenovirus, the E4orf6 protein contributes to the transformed phenotype of a cell when it is present.
Since the E4orf6 protein can cooperate with E1A proteins to transform cells (Fig. 2), one might ask why E1A/E4orf6 transformants, lacking an E1B gene, are not obtained when rodent cells are transformed by adenoviruses. The relative locations of the E1A, E1B, and E4 genes on the viral chromosome might favor retention of the adjacent E1A and E1B genes and loss of the E4 gene, which resides at the other end of the linear adenovirus chromosome. There is evidence suggesting that the viral genome is circularized during lytic replication (43), and circularization or oligomerization of the viral chromosome by recombination prior to integration could link the E1A gene as tightly to the E4orf6 as to the E1B gene. Nevertheless, it remains possible that the relative positions of the three oncogenes on the viral chromosome favor retention of the E1A and E1B genes. Alternatively, the E1A proteins might cooperate more efficiently with E1B proteins than with the E4orf6 protein in transformation assays. E1A/E4orf6 transformants produced smaller colonies than E1A/E1B transformants; and, when they were cloned, E1A/E4orf6-transformed rat cells initially grew more slowly than E1A/ E1B-transformed rat cells. With continued passage, both gene combinations produced cell lines with similar growth characteristics, but the cells with the E1A plus E4orf6 genes have presumably undergone selection for mutations in cellular genes that result in more rapid growth. The initially slow growth of E1A/E4orf6 transformants and the potential to more efficiently coselect E1A and E1B genes probably explain why E1A/E1B transformants, rather than E1A/E4orf6 transformants, are routinely isolated after infection of rodent cells.
The E4orf6 protein very likely cooperates with the E1A proteins to transform cells through its ability to bind p53 and alter its function (13). The E4orf6 protein blocks the ability of p53 to activate transcription, at least in part by interfering with its ability to bind to the TAFII31 subunit of transcription factor TFIID, and the physical interaction is presumably also responsible for the ability of the E4orf6 protein to block p53-mediated apoptosis (Fig. 1). The ability to block transcriptional activation and apoptosis mediated by p53 can explain how the E4orf6 protein, like the E1B proteins, cooperates with the E1A protein to transform cells. However, since the E1B proteins and the E4orf6 protein appear to function similarly, antagonizing the activity of p53, it is not clear why the E4orf6 protein enhances transformation by E1A plus E1B (Fig. 2) or why it influences tumorigenesis in nude mice (Table 1). Perhaps the E1B proteins do not completely neutralize the activity of p53, and the enhancing effects of the E4orf6 protein result from its ability to dramatically lower the level of p53 in comparison with the levels present in E1A/E1B transformants (Figs. 3 and 4), an effect that is consistent with the earlier observation that p53 levels are enhanced when cells are infected with a mutant adenovirus that is unable to produce the E4orf6 protein (44). As yet, we do not know how the E4orf6 protein influences p53 levels. The protein might displace E1B from p53, but p53 levels appear to be regulated by
the E1A protein and not the association of p53 with the E1B 55-kDa protein (6).
Since the E1B and E4orf6 proteins bind at different sites on p53, the E1B 55-kDa and the E4orf6 proteins might interact simultaneously with the residual low level of p53 that accumulates in the presence of the E4orf6 protein. The combination of adenovirus proteins might more completely inactivate p53 than either viral gene product alone. It is also possible that the altered localization of p53 in the presence of E4orf6 (Fig. 6) modifies the oncogenic properties of adenovirus-transformed cells. It has been shown that E1A/E1B-transformed 3Y1 rat cells containing a low steady-state level of the E1B 55-kDa protein form tumors more rapidly in nude mice than transformants with high levels of the transforming protein (36). 3Y1 transformants with relatively low levels of the E1B 55-kDa protein do not accumulate p53 in cytoplasmic bodies; rather, they contain nuclear p53 (36), just as we have observed for transformants containing the E4orf6 protein (Fig. 6).
It seems contradictory that the E4orf6 protein can cooperate with the E1A protein to transform cells (Fig. 2), presumably by blocking E1A-induced apoptosis (Fig. 1), while cells infected with mutant viruses lacking only the E1B 19-kDa protein undergo extensive apoptosis (45–47). The mutant viruses contain wild-type E4 genes and should express the E4orf6 protein in the cells undergoing apoptosis. However, the E1A protein has been shown to induce apoptosis through a p53-independent pathway (48, 49) in addition to the p53-dependent pathway. The p53-independent apoptosis might be induced indirectly by the E1A protein; that is, it might be caused by another adenovirus gene product whose expression is activated by the E1A protein. The E1B 19-kDa protein can block both p53-dependent and p53-independent apoptosis induced by the E1A protein. In contrast, the E4orf6 protein can prevent p53-induced apoptosis, but not apoptosis mediated by TNF-α in the absence of p53 (Fig. 1). So, if the p53-independent apoptosis seen in infected cells is due to a viral protein whose expression is induced by E1A protein, then one can propose an explanation for the inability of the E4orf6 protein to prevent apoptosis in virus-infected cells. Presumably, the E4orf6 protein antagonizes p53-dependent apoptosis induced directly by E1A, but it does not prevent p53-independent apoptosis induced indirectly by the E1A protein when it activates another apoptosis-promoting viral gene in adenovirus-infected cells.
Many adenovirus vectors that are being considered for gene delivery in humans contain the E4orf6 coding region. Given the ability of this protein to alter p53 function (13) and its oncogenic potential demonstrated here, it would be prudent to remove this coding region from gene transfer vectors.
We thank D.A.Haber for AT6 cells, A.Teresky for instruction and help in mouse injections, J.Goodhouse and H.Zhu for assistance with confocal microscopy, and Y.Shen for helpful discussions on transformation assays. This work was supported by grants from the National Cancer Institute (CA41086) and the Cystic Fibrosis Foundation (Z998). T.S. is an American Cancer Society Professor and an Investigator of the Howard Hughes Medical Institute.
1. Nevins, J.R. & Vogt, P.K. (1996) in Fields Virology, eds. Fields, B.N., Knipe, D.M. & Howley, P.M. (Lippincott-Raven, New York), pp. 310–343.
2. Shenk, T. (1996) in Fields Virology, eds. Fields, B.N., Knipe, D.M. & Howley, P.M. (Lippincott-Raven, New York), pp. 2111–2148.
3. Whyte, P.Buchkovich, K.J., Horowitz, J.M., Friend, S.H., Raybuck, M., Weinberg, R.A. & Harlow, E. (1988) Nature (London) 334, 124–129.
4. Bagchi, S., Raychaudhuri, P. & Nevins, J.R. (1990) Cell 62, 659–669.
5. Nevins, J.R. (1992) Science 258, 424–429.
6. Lowe, S.W. & Ruley, H.E. (1993) Genes Dev. 7, 535–545.
7. Debbas, M. & White, E. (1993) Genes Dev. 7, 546–554.
8. Sarnow, P., Ho, Y.-S., Williams, J. & Levine, A.J. (1982) Cell 28, 387–394.
9. Yew, P.R. & Berk, A.J. (1992) Nature (London) 357, 82–85.
10. Yew, P.R., Liu, X. & Berk, A.J. (1994) Genes Dev. 8, 190–202.
11. Chiou, S.-K., Tseng, C.-C., Rao, L. & White, E. (1994) J. Virol. 68, 6553–6566.
12. Rao, L., Debbas, M., Sabbatini, P., Hockenbery, D., Korsmeyer, S. & White, E. (1992) Proc. Natl. Acad. Sci. USA 89, 7742–7746.
13. Dobner, T., Horikoshi, N., Rubenwolf, S. & Shenk, T. (1996) Science 272, 1470–1473.
14. Lu, H. & Levine, A.J. (1995) Proc. Natl. Acad. Sci. USA 92, 5154–5158.
15. Thut, C.J., Chen, J.-L., Klemm, R. & Tjian, R. (1995) Science 267, 100–104.
16. Horikoshi, N., Usheva, A., Chen, J., Levine, A.J., Weinmann, R. & Shenk, T. (1995) Mol. Cell. Biol. 15, 227–234.
17. White, E. & Cipriani, R. (1990) Mol. Cell. Biol. 10, 120–130.
18. Findlay, C.A., Hinds, P.W. & Levine, A.J. (1989) Cell 57, 1083–1093.
19. Logan, J., Pilder, S. & Shenk, T. (1984) Cancer Cells 2, 527–532.
20. Brower, M., Carney, D.N., Oie, H.K., Gazdar, A.F. & Minna, J.D. (1986) Cancer Res. 46, 798–806.
21. Maheswaran, S., Englert, C., Bennett, P., Heinrich, G. & Haber, D.A. (1995) Genes Dev. 9, 2143–2156.
22. Gavrieli, Y., Sherman, Y. & Ben-Sasson, S.A. (1993) J. Cell Biol. 119, 493–501.
23. Zhu, H., Shen, Y. & Shenk, T. (1995) J. Virol. 69, 7960–7970.
24. Wigler, M., Pellicer, A., Silverstein, S., Axel, R., Urlaub, G. & Chasin, L. (1979) Proc. Natl. Acad. Sci. USA 76, 1373–1376.
25. Graham, F.L., Smiley, J., Russel, W.C. & Nairn, R. (1977) J. Gen. Virol. 36, 59–72.
26. Morgenstern, J.P. & Land, H. (1990) Nucleic Acids Res. 18, 3587–3596.
27. Ornelles, D.A. & Shenk, T. (1991) J. Virol. 65, 424–439.
28. Harlow, E., Pim, D.C. & Crawford, L.V. (1981) J. Virol. 37, 564–573.
29. Sarnow, P., Sullivan, C.A. & Levine, A.J. (1982) Virology 120, 510–517.
30. Oren, M., Maltzman, W. & Levine, A.J. (1981) Mol. Cell. Biol. 1, 101–110.
31. White, E. (1993) Genes Dev. 7, 2277–2284.
32. Gallimore, P.H., McDougall, J.K. & Chen, L.B. (1977) Cell 10, 669–678.
33. Sarnow, P., Hearing, P., Anderson, C.W., Halbert, D.N., Shenk, T. & Levine, A.J. (1984) J. Virol. 49, 692–700.
34. Reich, N.C., Oren, M. & Levine, A.J. (1983) Mol. Cell. Biol. 3, 2143–2150.
35. Zantema, A., Schrier, P.I., Davis-Olivier, A., van Laar, T., Vaessen, R.T.M.J. & van der Eb, A.J. (1985) Mol. Cell. Biol. 5, 3084–3091.
36. van den Heuvel, S.J.L., van Laar, T., Kast, W.M., Melief, C.J. M., Zantema, A. & van der Eb, A.J. (1990) EMBO J. 9, 2621–2629.
37. Zajdel, M.E.B. & Blair, G.E. (1988) Oncogene 2, 579–584.
38. Brown, C.R., Doxsey, S.J., White, E. & Welch, W.J. (1994) J. Cell. Physiol. 160, 47–60.
39. Esche, H. (1982) J. Virol. 41, 1076–1082.
40. Flint, S.J., Gallimore, P.H. & Sharp, P.A. (1975) J. Mol. Biol. 96, 47–68.
41. Flint, S.J. & Sharp, P.A. (1976) J. Mol. Biol. 106, 749–771.
42. Flint, S.J. (1980) in DNA Tumor Viruses, ed. Tooze, J. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 547–575.
43. Weiden, M.D. & Ginsberg, H.S. (1994) Proc. Natl. Acad. Sci. USA 91, 153–157.
44. Grand, R.J., Grant, M.L. & Gallimore, P.H. (1994) Virology 203, 229–240.
45. Pilder, S., Logan, J. & Shenk, T. (1984) J. Virol. 52, 664–671.
46. Subramanian, T., Kuppuswamy, M., Gysbers, J., Mak, S. & Chinnadurai, G. (1984) J. Biol. Chem. 259, 11777–11783.
47. White, E., Grodzicker, T. & Stillman, B.W. (1984) J. Virol. 52, 410–419.
48. Subramanian, T., Tarodi, B. & Chinnadurai, G. (1995) Cell Growth Diff. 6, 131–137.
49. Teodoro, J.G., Shore, G.C. & Branton, P.E. (1995) Oncogene 11, 467–474.







