
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
The function of herpes simplex virus genes: A primer for genetic engineering of novel vectors
BERNARD ROIZMAN
The Marjorie B.Kovler Viral Oncology Laboratories, The University of Chicago, 910 East 58th Street, Chicago, IL 60637
ABSTRACT Herpes simplex virus vectors are being developed for delivery and expression of human genes to the central nervous system, selective destruction of cancer cells, and as carriers for genes encoding antigens that induce protective immunity against infectious agents. Vectors constructed to meet these objectives must differ from wild-type virus with respect to host range, reactivation from latency, and expression of viral genes. The vectors currently being developed are (i) helper free amplicons, (ii) replication defective viruses, and (iii) genetically engineered replication competent viruses with restricted host range. Whereas the former two types of vectors require stable, continuous cell lines expressing viral genes for their replication, the replication competent viruses will replicate on approved primary human cell strains.
Herpes simplex viruses (HSV) and particularly HSV-1 are potential vectors for several applications in human health. These include (i) delivery and expression of human genes to central nervous system (CNS) cells, (ii) selective destruction of cancer cells, and (iii) prophylaxis against infections with HSV and other infectious agents. The properties of wild-type virus are fundamentally antithetical to such applications. HSV-1 is highly destructive to infected cells. In addition, HSV-1 is generally spread by contact of the tissues containing virus of one individual with mucous membranes of an uninfected individual. The virus multiplies at the portal of entry, infects sensory nerve endings innervating the site of multiplication, and is transported retrograde to the nucleus of sensory neurons. The sequence of events beyond this point are less well known. In experimental animal systems, the virus multiplies in some neurons but establishes a latent state in others. In a fraction of those infected, the virus periodically reactivates from latent state. In these neurons the newly replicated virus is transported anterograde, usually to a site at or near the portal of entry into the body, where it may cause a localized lesion. In immunosuppressed individuals, the lesions caused both by initial infection and recrudescences tend to be more extensive and persist longer than in immunocompetent individuals (reviewed in refs. 1 and 2). To serve as vectors, the viral genotype must be extensively altered to fit the objective of the vector. For example, to deliver and express human genes in the CNS, the desirable properties of HSV are its ability to establish latent infections and the huge coding capacity of the viral genome. The undesirable property is the capacity of the virus to commit the cell to destruction very early in infection. For selective destruction of cancer cells, the desirable property is the capacity of the virus to destroy cells. The undesirable properties are the wide host range of the wild-type virus and the capacity of the virus to reactivate from latent state. Each application therefore requires a different kind of vector and, in
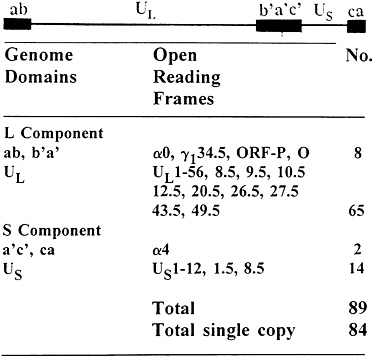
FIG. 1. Sequence arrangement in HSV DNA and distribution of viral genes in the HSV genome. The filled quadrangles represent terminal sequence ab and ca inverted and repeated internally to yield b′a′c′. See ref. 1 and Table 1 for details of genome structure and gene function. The latency associated transcripts (LATs) map within inverted repeats flanking UL.
principle, different kinds of genetic engineering. The purpose of this report is to summarize our knowledge of the molecular biology of HSV-1 relevant to experimental design of viral vectors.
Genome Structure and Gene Content
The genome structure and current gene content of HSV-1 are summarized in Fig. 1. Exclusive of the variable number of repeats of the terminal a sequence, the HSV-1 genome is approximately 152 kbp in size (3, 4). The genome consists of two long stretches of quasi-unique sequences, unique long sequence (UL) and unique short sequence (US), flanked by inverted repeats. UL is flanked by the sequence ab and its inversion b′a′, approximately 9 kbp each, whereas US is flanked by the sequence a′c′ and its inversion ca, 6.5 kbp each (5, 6). Thus, the HSV genome contains 15 kbp of DNA sequences (b′a′c′), which represent inverted repeats of terminal regions inserted between UL and US domains. The a sequence varies in size and may be present in multiple copies adjacent to the ba sequence, but only in a single copy at the
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: HSV-1, -2, herpes simplex virus 1 and 2; ICP, infected cell protein; gB, gC, gD, etc., glycoproteins B, C, D, etc.; UL, unique long sequence; US, unique short sequence; LAT, latency associated transcripts; ORF, open reading frame; CNS, central nervous system.
Table 1. The function of herpes simplex virus genes
|
Gene |
Product |
Dispensable in cell culture |
Regulation |
Function of gene product |
|
γ134.5 |
ICP34.5 |
Y |
γ1 |
Null mutants are attenuated and fail to block phosphorylation of eIF-2α by activated protein kinase RNA-dependent kinase; carboxyl terminus homologous to the corresponding domain of the GADD34 proteins. |
|
ORF-P |
ORF-P |
Y |
pre α |
ORF is antisense to the γ134.5 gene and repressed by binding of ICP4 to cap site. Proteins interact with p32, a component of SF2/ASF splicing factor. |
|
ORF-O |
ORF-O |
Y |
pre α |
Overlaps with ORF P, a protein made by frameshift from ORF-P. |
|
α0 |
ICP0 |
Y |
α |
Promiscuous transactivator, requires ICP4 for optimal activity; nucleotidylylated, phosphorylated by UL13, nuclear (early) and cytoplasmic (late) phases. Null mutants debilitated at low multiplicities of infection. |
|
UL1 |
gL |
N |
γ |
Complex with gH required for transport of both proteins to plasma membrane and for viral entry mediated by gH. |
|
UL2 |
|
Y |
β |
Uracil DNA glycosylase. |
|
UL3 |
|
Y |
γ2 |
Nuclear phosphoprotein of unknown function. Reported to localize to perinuclear region early and to the nucleus late in infection. |
|
UL4 |
|
Y |
Unknown |
Unknown. |
|
UL5 |
|
N |
β |
Forms complex with UL8 and UL52 proteins. |
|
UL6 |
|
N |
Unknown |
Virion protein; required for DNA cleavage and packaging. |
|
UL7 |
|
N |
Unknown |
Unknown. |
|
UL8 |
|
N |
β |
Forms complex with UL5 and UL52 (helicase/primase complex). Stabilizes interaction between primers and DNA template. |
|
UL9 |
|
N |
γ(?) |
Binds to origins of DNA synthesis in sequence-specific (origin) fashion; carries out helicase and ATPase activities. |
|
UL10 |
gM |
Y |
γ |
Glycoprotein present in virions and plasma membranes. |
|
UL10.5 |
|
? |
Unknown |
Unknown. |
|
UL11 |
|
Y |
γ(?) |
Myristoylated protein; necessary for efficient capsid envelopment and exocytosis. |
|
UL12 |
|
Y |
β |
Exonuclease (DNase) involved in viral nucleic acid metabolism; reported to localize in nucleoli and in virally induced nuclear dense bodies and to bind to a sequence along with other unidentified proteins. Complex may be involved in cleavage/packaging of viral DNA. |
|
UL12.5 |
|
Y |
Unknown |
Nuclease-associated with capsids. |
|
UL13 |
|
Y |
γ |
Virion (nuclear) protein kinase; substrates include ICP0, ICP22, vhs, UL3, UL49, etc. |
|
UL14 |
|
N |
Unknown |
Unknown. |
|
UL15 |
|
N |
γ |
ts mutant DNA+. Two exons; protein required for cleavage/packaging of DNA. |
|
UL16 |
|
Y |
Unknown |
Virion protein; gene located within intron of UL15. |
|
UL17 |
|
N |
γ |
Located within intron of UL15. |
|
UL18 |
VP23 |
N |
γ |
Protein required for capsid formation and cleavage/packaging of DNA. |
|
UL19 |
VP5, ICP5 |
N |
γ1 |
Major capsid protein. |
|
UL20 |
|
Y |
γ |
Membrane protein, associates with nuclear membranes, Golgi stacks, etc. Essential for viral exocytosis. |
|
UL20.5 |
|
|
γ2 |
Unknown. |
|
UL21 |
|
Y |
Unknown |
Nucleotidylylated phosphoprotein; unknown function. |
|
UL22 |
gH |
N |
γ2 |
Forms complex with gL (see above). Required for entry, egress, and cell-cell spread. |
|
UL23 |
ICP36 |
Y |
β |
Thymidine (nucleoside) kinase. |
|
UL24 |
|
Y |
γ |
Syn− locus; membrane-associated protein? |
|
UL25 |
|
N |
γ |
Virion protein reported to be required for packaging of cleaved viral DNA. |
|
UL26 |
|
N |
γ |
Serine protease; substrates are UL26 protein and UL26.5 (IC35). VP21 (C portion of UL26), VP24 (N terminus of protease) are products of the self-cleavage of UL26. |
|
UL26.5 |
ICP35 |
N |
γ |
Substrate of UL26 protease unique to B capsids and forms inner core or scaffolding; the precursor, ICP35b,c is cleaved to e, f. On packaging of DNA it is removed from capsid shell. |
|
UL27 |
gB, VP7 |
N |
γ1 |
Glycoprotein required for viral entry; forms a dimer and induces neutralizing antibody. A syn− locus maps to the carboxyl terminus. |
|
UL27.5 |
|
? |
Unknown |
Unknown, antisense to gB. |
|
UL28 |
ICP18.5 |
N |
γ |
Mr 87–95 K protein required for DNA cleavage/packaging. |
|
UL29 |
ICP8 |
N |
β |
Binds single-stranded DNA cooperatively, required for viral DNA replication: forms complex with DNA polymerase and UL42. ts mutants are DNA− and hence expression of early and late genes may be affected positively or negatively by ICP8. Because ICP8 denatures DNA, it affects renaturation of complementary strands of DNA and affects homologous pairing and strand transfer. |
|
UL30 |
|
N |
β |
DNA polymerase; forms complex with ICP8 and C terminal 247 amino acids of UL42. |
|
UL31 |
|
N |
γ2 |
Nucleotidylylated phosphoprotein, cofractionates with nuclear matrix. |
|
UL32 |
|
N |
γ2 |
Cytoplasmic/nuclear protein required for DNA cleavage/packaging. |
|
UL33 |
|
N |
Unknown |
DNA packaging; necessary for assembly of capsids containing DNA. |
|
UL34 |
|
N |
Unknown |
Abundant nonglycosylated, membrane-associated, virion protein phosphorylated by US3. |
|
UL35 |
VP26 |
N |
γ2 |
Basic phosphorylated capsid protein. |
|
UL36 |
ICP1–2 |
N |
γ2 |
Tegument phosphoprotein. DNA is not released from capsids at nuclear pores in cells infected with ts mutant. Reported to form complex with a Mr 140 K protein that binds a sequence DNA. |
|
Gene |
Product |
Dispensable in cell culture |
Regulation |
Function of gene product |
|
UL37 |
ICP32 |
N |
γ |
Cytoplasmic phosphoprotein; in presence of ICP8 it is transported to nucleus and associates with DNA, but phosphorylation is not dependent on ICP8. Required for maturation of virions. |
|
UL38 |
VP19C |
N |
γ2 |
Capsid assembly protein, binds DNA and may be involved in anchoring DNA in the capsid. |
|
UL39 |
ICP6 |
Y |
β |
Large subunit of ribonucleotide reductase. Autophosphorylates via unique N terminus but does not trans-phosphorylate. |
|
UL40 |
|
Y |
β |
Small subunit of ribonucleotide reductase. |
|
UL41 |
VHS |
Y |
γ |
Causes nonspecific degradation of mRNA after infection; shuts off host protein synthesis, enables sequential synthesis of viral proteins. |
|
UL42 |
|
N |
β |
Double-stranded DNA-binding protein, binds to and increases processivity of DNA polymerase. |
|
UL43 |
|
Y |
Unknown |
Amino acid sequence predicts membrane-associated protein. |
|
UL43.5 |
|
Y |
|
Antisense to UL43; low abundance nuclear protein; accumulates in assemblons. |
|
UL44 |
gC, VP7.5 |
Y |
γ2 |
Glycoprotein involved in cell attachment; required for attachment to the apical surface of polarized MDCK cells. |
|
UL45 |
|
Y |
γ2 |
Encodes a Mr 18 K protein of unknown function. |
|
UL46 |
VP11/12 |
Y |
γ |
Tegument phosphoprotein reported to modulate the activity of UL48 (aTIF). |
|
UL47 |
VP13/14 |
Y |
γ2 |
Nucleotidylylated tegument phosphoprotein modulates the activity of UL48 (αTIF). |
|
UL48 |
VP16, ICP25, αTIF |
N |
γ |
Tegument protein, induces α genes by interacting with OctI. The complex binds to specific sequences with the consensus GyATGnTAATGArATTCyTTGnGGG-NC. |
|
UL49 |
VP22 |
N |
γ |
Nucleotidylylated, mono(ADP-ribosyl)ated tegument phosphoprotein. |
|
UL49.5 |
|
N |
γ2 |
Sequence predicts a Mr 12,000 membrane-associated protein. |
|
UL50 |
|
Y |
β |
dUTPase. |
|
UL51 |
|
Y |
γ |
Unknown. |
|
UL52 |
|
N |
β |
Component of the helicase/primase complex. |
|
UL53 |
gK |
Y |
γ |
Glycoprotein required for efficient viral exocytosis; contains syn− locus. |
|
α27 |
ICP27 |
N |
α |
Nucleotidylylated multifunctional regulatory protein; causes redistribution of snRNPs, inhibits RNA splicing. It is required for late gene expression, and negatively regulates early genes. |
|
UL55 |
|
Y |
Unknown |
Unknown. |
|
UL56 |
|
Y |
Unknown |
Nuclear, virion-associated protein of unknown function. |
|
α4 |
ICP4 |
N |
α |
Nucleotidylylated, poly(ADP-ribosyl)ated phosphoprotein; regulates positively most β and γ genes and negatively itself, ORF-P and the α0 gene; blocks apoptosis. Binds to DNA in sequence specific fashion. |
|
α22 |
ICP22 |
Y |
α |
Nucleotidylylated regulatory protein, phosphorylated by UL13 and US3 protein kinases, required for optimal expression of ICP0 and of a subset of γ proteins. |
|
US1.5 |
US1.5 |
Y |
α |
Regulatory protein; extent to which it shares function with ICP22 not known. |
|
US2 |
|
Y |
Unknown |
Unknown. |
|
US3 |
|
Y |
β |
Protein kinase; major substrate is UL34 protein. |
|
US4 |
gG |
Y |
γ |
Glycoprotein involved in entry, egress, and spread from cell to cell. |
|
US5 |
gJ(?) |
Y |
Unknown |
Sequence predicts glycoprotein. |
|
US6 |
gD VP17/18 |
N |
γ1 |
Glycoprotein required for post-attachment entry of virus into cells. |
|
US7 |
gI |
Y |
γ |
gI and gE glycoproteins form complex for transport to plasma membrane and also to constitute a high-affinity Fc receptor. gI is required for basolateral spread of virus in polarized cells. |
|
US8 |
gE |
Y |
γ2 |
FC receptor; involved in basolateral spread of virus in polarized cells. |
|
US8.5 |
|
Y |
β or γ1 |
Unknown. |
|
US9 |
|
Y |
Unknown |
Tegument protein phosphorylated by UL13. |
|
US10 |
|
Y |
Unknown |
Tegument protein. |
|
US11 |
|
Y |
γ2 |
Tegument protein binds to UL34 mRNA in sequence- and conformation-specific fashion; binds to the 60S ribosomal subunit and localizes in the nucleolus. |
|
α47 |
ICP47 |
Y |
α |
Binds to TAP1/TAP2 and to block antigen presentation to CD8+ cells. |
|
OriSTU |
OriSRNA |
Y |
γ2 |
RNA transcribed across S origins of DNA synthesis. Function is not known. |
|
LATU |
LATs |
Y |
pre α? |
Transcripts, found in latently infected nerons. Function is not known. |
|
OriSTU is the transcriptional unit across the origin of DNA synthesis in the S component. LATTU is the transcriptional unit expressed in latently infected sensory neurons. This table was updated and modified from Ref. 1. Additional references are as follows: UL10.5, ref. 7; UL12.5, ref. 15; UL20.5, P.J.Ward and B.R. (unpublished data); UL27.5, Y.Chang, G.Campadelli-Fiume, and B.R. (unpublished work); UL43.5, ref. 14; US1.5, ref. 9; ORF-O, G.Randall and B.R. (unpublished work); US9, R.Brandimasti and B.R. (unpublished work). |
||||
terminus of the genome next to the c sequence and contains signals for cleavage of unit length DNA from concatemers and packaging of the DNA in preformed capsids (reviewed in ref. 1). HSV-1 is known to express at least 84 different polypeptides whose open reading frames (ORFs) are distributed as indicated in Fig. 1 (refs. 4 and 7–15; P.L.Ward and B.R., unpublished data; Y.Chang, G.Campadelli-Fiume, and B.R., unpublished data). Of this number, five ORFs, mapping in the inverted repeats, are present in two copies per viral genome. In addition to the ORFs listed in Fig. 1, infected cells contain
transcripts from genome domains not known to specify proteins. These include the LATs discussed below and an RNA (OriSRNA) derived by transcription of the two of the three origins of viral DNA synthesis mapping in inverted repeats (16, 17). The ORFs form several groups whose expression is coordinately regulated in a cascade fashion. The α genes are expressed first, functional α proteins are required for the expression of β genes, and both functional α proteins and viral DNA synthesis mediated by β proteins are required for (γ2), or enhance (γ1), the expression of late or γ genes (18, 19). Whereas α proteins perform regulatory functions or prevent a host response to infection, the function of β proteins is the management of the nucleic acid metabolism and viral DNA synthesis in the infected cell, as well as posttranslational modification of proteins made earlier and later in infection. The γ proteins are largely the structural components of the virions (reviewed in ref 1).
Since 1982 (20, 21) techniques have been available to delete or insert DNA sequences at specific sites. These studies have revealed the existence of ORFs that are expressed and the rather unexpected finding that 45 of the 83 ORFs specifying diverse proteins are dispensable for viral replication in at least some cells in culture. A list of the ORFs and the functions expressed by the gene products are shown in Table 1. The 38 ORFs that cannot be deleted without ablating the capacity of the virus to replicate include four genes specifying surface glycoproteins, two regulatory proteins [infected cell proteins no. 4 (ICP4) and no. 27 (ICP27)], seven proteins required for the synthesis of viral DNA, proteins required for assembly of the capsid, structural proteins, and proteins whose functions are not yet known. The 45 accessory ORFs, which are not required for viral replication in cells in culture, specify 11 proteins involved in entry, sorting, and exocytosis of virus (glycoproteins C, E, G, I, J, K, M; membrane proteins UL11, UL20, UL24, UL43), 2 protein kinases (UL13, US3), 2 proteins that preclude host response to infection (α47 and γ134.5), 3 regulatory proteins (α0, α22, US1.5), 5 proteins that augment the nucleotide triphosphate pool or repair DNA (thymidine kinase, dUTPase, ribonucleotide reductase, DNase, uracil glycosylase), 1 protein that causes the degradation of mRNA after infection (UL41), and numerous other proteins whose functions are not known (detailed references in ref. 1).
The Role of Selected Viral Genes in Viral Replication
The reproductive cycle of HSV has been described in detail elsewhere (1). The objective of viral replication is efficient, rapid synthesis and dissemination of viral progeny. In the process, the infected cell dies. Viral replication consists of a series of events very tightly regulated both positively and negatively. To accomplish its objectives, the virus brings into the newly infected cell several proteins packaged in the virion tegument (a layer of proteins located between the capsid and the envelope; see ref. 22), whose functions are best described as creating the environment for initiation of viral replication). One, designated as VP16 or α gene trans-inducing factor (αTIF) induces the transcription of α genes by cellular RNA pol II and accessory factors, whereas another encoded by UL41 causes the degradation of cytoplasmic RNAs (23–25). The major regulatory protein ICP4 made after infection acts both negatively by binding to high-affinity sites on viral DNA and positively by an as yet unknown mechanism (reviewed in ref. 1). The hypothesis that ICP4 is directly involved in transcription is based on reports that it binds TATA box-binding protein and transcription factor IIB, and on the evidence that after the onset of DNA synthesis, it is a component of γ-transcriptons— nuclear stuctures containing newly synthesized viral DNA, RNA polymerase II, ICP22, and a cellular protein known as L22 or EAP and that is normally present in nucleoli and ribosomes and binds small RNA molecules (ref. 26; R.Leopardi, P.L.Ward, W.Ogle, and B.R., unpublished work). ICP27 has multiple functions, but primarily it regulates posttranscriptional processing of RNA (27). ICP22 also appears to be a transcriptional factor; it is required for the expression of the α0 gene and also of a subset of γ genes and is a component of the γ-transcriptons (ref. 28; R.Leopardi, P.L.Ward, W.Ogle, and B.R., unpublished work). Among other proteins that regulate the replicative cycle is UL13, a protein kinase known to mediate the phosphorylation of ICP0, ICP22, and other proteins (ref. 28; W.Ogle, K.Carter, and B.R., unpublished work). The function of another viral protein kinase, US3, is less clear (1).
The functions of γ134.5 and α47 genes are of particular interest. γ134.5 appears to have at least two functions. One function of γ134.5 is to preclude the shutoff of protein synthesis caused by activation of the protein kinase RNA-dependent kinase and, ultimately, by the phosphorylation of the α subunit of the translation initiation factor eIF-2 (29). The carboxyl-terminal domain of γ134.5 required for this function is homologous to the corresponding domain of the mammalian protein GADD34—one of a set of proteins induced in growth arrest as a consequence of differentiation, serum deprivation, or DNA damage. Human GADD-34, or a chimeric gene consisting of the amino terminal domain of γ134.5 and the carboxyl terminus of GADD-34, effectively replaces the γ134.5 gene in the context of the viral genome (30). The second function of the γ134.5 enables the virus to multiply efficiently in a number of tissues, but particularly in the CNS of experimental animal systems (31, 32). The argument that this function of γ134.5 is independent of the function of the protein to preclude the phosphorylation of eIF-2α is based on the observation that viruses carrying GADD-34 in place of γ134.5 are not blocked in protein synthesis; they are nevertheless attenuated (32).
α47 binds the complex of TAP1/2 and thereby precludes the transport of peptides for presentation to CD8+ cells (33).
The Function of Viral Genes in Latency
To date the only domain of the viral genome shown to be expressed during latency maps in the inverted repeats flanking UL (16). The RNAs described to date consist of two populations. The low abundance population arises from an 8.3-kbp domain. The two abundant RNAs, of 2 and 1.5 kb respectively, and known by the acronym LAT, appear to be stable introns that accumulate in abundant amounts in nuclei of neurons harboring latent virus. Deletion of the upstream promoter or of the sequences encoding LATs has little effect on the establishment or maintenance of the latent state, but reduces the efficiency of latent virus to reactivate (1). LATs may be harbingers of neurons capable of reactivating than viral products required for establishment of latency. Given the multitude of viral accessory genes whose function is to render viral replication and dissemination more efficient, the notion that the virus depends solely on the cellular factors for dissemination seems unlikely. Recent studies have shown that the genome domain transcribed during latency contains ORF-0 and ORF-P, whose expression is repressed by ICP4, inasmuch as mutagenesis of the high-affinity binding site at the transcription initiation site of ORF-P led to the derepression of both genes (12). The virus carrying the derepressed gene is attenuated in experimental animal systems (mice) and underexpresses α0 and α22 proteins (34, 45). In addition, ORF-P protein colocalizes and binds to a protein (p34), which is a component of the SF2/ASF splicing factor (45). The role of ORF-P in latency is not known.
Genetic Engineering of Novel Viral Genomes
The two major techniques for construction of novel viruses depend on genetic recombination in infected or transfected cells. The first technique was based on the observation that transfection of cells with intact viral DNA and mutated fragment will result in a small fraction of the progeny carrying the mutated sequence (20, 21). To specifically select this population, the procedure first involved the insertion at or near the target for deletion by recombination through flanking sequences of the viral thymidine kinase as a selectable marker. Only the progeny of transfection, which carries the viral thymidine kinase gene, would multiply in thymidine kinase minus cells overlaid with appropriate medium. In the second step, the inserted thymidine kinase was deleted along with adjacent target sequences by recombination through flanking sequences with a mutated DNA sequence. In this instance, only thymidine kinase minus progeny would break through efficiently in cells overlaid with medium containing bromouracil deoxyriboside. A minor variant of this technique based on gene inactivation by random insertion of the mini-μu phage has the disadvantage in that the mini-μu DNA sequence is quite large and troublesome to remove (21).
The second procedure involves transfection of cells with overlapping cosmids containing appropriate insertions or deletions. Expression of genes contained in cosmids leads eventually through recombination to the reconstruction of full-length viral genomes. This procedure is less efficient, but the progeny of transfection need not be subjected to selection for the isolation of the desired genotype (35)
Both procedures suffer from gene rearrangements as a result of transfection. To link a specific genotype to the observed phenotype, it is essential to determine whether the wild-type phenotype is restored by the repair of the deleted sequences with a small DNA fragment. It is estimated that as much as 30% of the recombinants made by the techniques described above contain additional mutations detected only after the restoration of the missing sequence.
Requirements and Design of Viral Vectors
Vectors for delivery of cellular genes to CNS must not express viral genes that cause the infected cell to make cytotoxic viral proteins or that induce an immune response, which may damage the recipient cells. A huge literature describes attempts to obtain long-term expression of reporter genes in experimental animals, particularly mice. A potentially suitable vector for this type of application is based on construction of defective genomes, i.e., genomes that are unable to replicate in the recipient cells. In recent years, two different types of defective recombinant viruses have emerged. The first is based on defective HSV genomes, which arise spontaneously by recombination and are amplified during serial passage at high multiplicities (36). The defective genome subunit (the amplicon) consists minimally of the terminal a sequence and an origin of viral DNA synthesis. In virions, these unit are arranged head-to-tail. HSV-1 amplicons have long been shown to express efficiently cellular genes incorporated into them (37). Amplicons do not encode viral proteins and must therefore be supplied with both viral structural proteins and proteins required for viral DNA synthesis and exocytosis in order to be made. In theory, amplicons could accommodate as much as 150 kbp of DNA. In practice, three problems exist. First, until recently, amplicons were contaminated with helper DNA. Second, the amounts of amplicons made are not readily controllable and the usual yields are several orders of magnitude lower than those of wild-type infectious virus. Third, amplicons tend to be unstable on serial passage because the smaller the amplicon, the greater is its selective advantage (38). Of these problems, only the first one has been solved, since a helper virus incapable of packaging has been constructed (39). A significant potential problem is the rescue of the helper virus by recombination with the amplicon, which would enable the helper to package.
The second approach is based on construction of viruses lacking essential genes. The essential genes deleted singly or in combination include α4, α22, α27, UL48 (α gene transinducing factor or VP16), and UL41 (40). Deletion of the inverted repeats (b′a′c′) and of stretches of genes not essential for viral replication in cell culture (e.g., US except for US6, α22, and α47), and large stretches of UL (UL2, UL3, UL4, UL10, UL11, UL43, UL43.5, UL45, UL46, UL47, UL55, UL56) could make space for insertion of at least 40 kbp of DNA. By necessity, the debilitated recombinant virus must be grown in cell lines expressing the deleted viral genes. This type of vector also presents several problems. Foremost is that some of the selected genes targeted for deletion perform a multiplicity of functions. For example, they may be both transactivators and repressors, and they normally block the cells from programmed cell death triggered by viral gene expression (41). It could be predicted therefore that sooner or later the recipient cell will cease functioning because of slow but ultimately fatal expression of viral genes. Other problems, perhaps more readily surmountable, include potential for recombination between the defective viral genomes and viral genes resident in the cell genome and the stability of the cell lines. In neither model is reactivation from latency a problem inasmuch as the viruses are unable to replicate.
Whereas gene therapy may require delivery of therapeutic genes to a significant fraction of cells that normally express them or could serve as surrogate expressors, cancer therapy requires complete destruction of cancer cells. Theoretically, for selective destruction of cancer cells, it should be sufficient to introduce a defective viral genome expressing a factor that is excreted in abundant amounts and toxic only for cancer cells. Theory not withstanding, viruses that infect or at least selectively multiply and destroy tumor cells are likely to have a therapeutic advantage. In experimental animal systems several genetically engineered viruses appear to have met at least the initial requirements for further development (32). The mutations that render the viruses attenuated fall into three categories. The first set encodes enzymes (e.g., thymidine kinase, ribonucleotide reductase, etc.) involved in nucleic acid metabolism and that would not be available in neurons but would be available to the virus in dividing tumor cells. The second category are genes encoding proteins (e.g., γ134.5), which disable the capacity of the virus to replicate in CNS for reasons not well understood, as described earlier in the text (reviewed in ref. 32). Lastly, deletion of inverted repeats (b′a′c′) in itself grossly debilitates HSV-1 (42, 43). Irrespective of the set of genes involved for attenuation, it will be virtually impossible to introduce virus in all cancer cells. As in the case of defective viruses, even replicating viruses will have to carry and express factors that induce immune response or activate pro-drugs selectively in cancer cells.
The use of recombinant viruses for prophylaxis against viral infection requires mutants that are genetically stable, incapable of replicating in CNS, incapable of spreading in immunocompromised individuals, unable to reactivate, not transmissible from immunized individual to contacts, but immunogenic and protective against disease caused by subsequent infection. A large number of defective viruses have been proposed for immunization on the grounds that although they cannot produce infectious progeny and fundamentally make proteins only in the set of initially infected cells, the presentation to the immune system of viral antigens made in these cells is superior to that of subunit vaccines (e.g. ref. 44). The central issue is the immunogenic mass required for effective immunity and whether such a mass could be achieved by progeny of a replication-defective virus. Secondary issues are the stability of
the cell lines expressing the complementing viral genes, rescue of the defective virus by recombination, and qualification of transformed cell lines for administration of virus along with cellular DNA to healthy individuals. The construction of replicating virus is not less daunting, and parallels in many respects the construction of recombinant viruses for cancer therapy.
Conclusions
Development of genetic engineering evolved pari passu with our knowledge of viral gene content and function and is at a point where, subject to constraints imposed by the size of the virion, the construction of virtually any vector is feasible. The outstanding issues are not the construction of viral vectors, but rather (i) delivery of recombinant viruses to appropriate cells, (ii) regulation of expression of the gene carried by the viral vector, and (iii) regulatory issues related to qualification of continuous cell lines, which express complementing viral genes for replication of defective vectors. Theoretically, it should be possible to create viruses that carry surface protein destined for receptors present only on a specific set of cells, but this is not yet feasible. The problems associated with regulation of gene expression are particularly vexing because the available size for packaging genes in the viral genome is too small to accommodate the genomic versions rather than the cDNA version of most cellular genes of interest. As a consequence, alternative splicing to produce isoforms of cellular proteins and natural regulation of gene expression may not be feasible. The third issue arises from the difficulty of purifying virus away from cell debris which contains cellular DNA. Since only continuous cell lines are likely to express stably viral genes complementing replication defective viruses, it would be necessary to define the requirements for the qualification of such cell lines for production of viruses intended for human use. Considering the progress of the past 10 years, the solution of these problems should not be far behind.
I thank P.L.Ward, N.Markovitz, and A.E.Sears for their advise and assistance in the generation of Table 1. These studies were aided by grants from the National Cancer Institute (CA47451) and the National Institute for Allergy and Infectious Diseases (AI124009), and the U.S. Public Health Service.
1. Roizman, B. & Sears, A.E. (1996) in Fields Virology, eds. Fields, B.N., Knipe, D.M., Howley, P., Chanock, R.M., Hirsch, M.S., Melnick, J.L., Monath, T.P. & Roizman, B. (Lippincott-Raven, Philadelphia), 3rd Ed., pp. 2231–2295.
2. Whitley. R.J. (1996) in Fields Virology, eds. Fields, B.N., Knipe, D.M., Howley, P., Chanock, R.M., Hirsch, M.S., Melnick, J.L., Monath, T.P. & Roizman, B. (Lippincott-Raven, Philadelphia), 3rd Ed., pp. 2297–2342.
3. Kieff, E.D., Bachenheimer, S.L. & Roizman, B. (1971) J. Virol. 8, 125–132.
4. McGeoch, D.J., Dalrymple, M.A., Davison, A.J., Dolan, A., Frame, M.C., McNab, D., Perry, L.J., Scott, J.E. & Taylor, P. (1988) J. Gen. Virol. 69, 1531–1574.
5. Sheldrick, P. & Berthelot, N. (1975) Cold Spring Harbor Symp. Quant. Biol. 39, 667–678.
6. Wadsworth, S., Jacob, R.J. & Roizman, B. (1975) J. Virol. 15, 1487–1497.
7. Baradaran, K., Dabrowski, C.E. & Schaffer, P.A. (1994) J. Virol. 68, 4251–4261.
8. Barker, D.E. & Roizman, B. (1992) J. Virol. 66, 562–566.
9. Carter, K.L. & Roizman, B. (1996) J. Virol. 70, 172–178.
10. Cho, J. & Roizman, B. (1986) J. Virol. 57, 629–637.
11. Georgopoulou, U., Michaelidou, A., Roizman B. & Mavromara-Nazos, P. (1993) J. Virol. 67, 3961–3968.
12. Lagunoff, M. & Roizman, B. (1994) J. Virol. 68, 6021–6028.
13. Liu, F. & Roizman, B. (1991) J. Virol. 65, 206–212.
14. Ward, P.L., Barker, D.E. & Roizman, B. (1996) J. Virol. 70, 2684–2690.
15. Martinez, R., Shao, L., Bronstein, J.C., Weber, P.C. & Weller, S.K. (1996) Virology 215 152–164.
16. Stevens, J.G., Wagner, E.K., Devi-Rao, G.B., Cook M.L. & Feldman, L.T. (1987) Science 235, 1056–1059.
17. Hubenthal-Voss, J., Starr, L. & Roizman, B. (1987) J. Virol. 61, 3349–3355.
18. Honess, R.W. & Roizman, B. (1974) J. Virol. 14, 8–19.
19. Honess, R.W. & Roizman, B. (1975) Proc. Natl. Acad. Sci. USA 72, 1276–1280.
20. Post, L.E. & Roizman, B. (1981) Cell 25, 227–232.
21. Roizman, B. & Jenkins, F.J. (1985) Science 129, 1208–1218.
22. Roizman, B. & Furlong, D. (1974) in Comprehensive Virology, eds. Fraenkel-Conrat, H. & Wagner, R.R. (Plenum, New York), Vol. 3, pp. 229–403.
23. Batterson, W. & Roizman, B. (1983) J. Virol. 46, 371–377.
24. McKnight, J.L.C., Kristie, T.M. & Roizman, B. (1987) Proc. Natl. Acad. Sci. USA 84, 7061–7065.
25. Read, G.S. & Frenkel, N.J. (1993) Virology 46, 498–512.
26. Smith, C.A., Bates, P., Rivera-Gonzalez, R., Gu, B. & DeLuca, N.A. (1993) J. Virol. 67, 4676–4687.
27. Smith, I.L., Hardwicke, M.A. & Sandri-Goldin, R.M. (1992) Virology 186, 74–85.
28. Purves, F.C., Ogle, W.O. & Roizman, B. (1993) Proc. Natl. Acad. Sci. USA 90, 6701–6705.
29. Chou, J., Chen, J.-J., Gross, M. & Roizman, B. (1995) Proc. Natl. Acad. Sci. USA 92, 10516–10520.
30. He, B., Chou, J., Liebermann, D.A., Hoffman, B. & Roizman, B. (1996) J. Virol. 70, 84–90.
31. Chou, J., Kern, E.R., Whitley, R.J. & Roizman, B. (1990) Science 252, 1262–1266.
32. Andreansky, S.S., Bin, H., Gillespie, G.Y., Soroceanu, L., Markert, J., Roizman, B. & Whitley, A.J. (1996) Proc. Natl. Acad. Sci. USA 93, 11313–11318.
33. Hill, A., Jugovic, P., York, I., Russ, G., Bennink, J., Yewdell, J., Ploegh, H. & Johnson, D. (1995) Nature (London) 375, 411–415.
34. Lagunoff, M., Randall, G. & Roizman, B. (1996) J. Virol. 70, 1810–1817.
35. van Zijl, M., Quint, W., Braire, J., de Rover, T., Gielkens, A. & Berns, A. (1988) J. Virol. 62, 2191–2195.
36. Frenkel, N., Jacob, R.J., Honess, R.W., Hayward, G.S., Locker, H. & Roizman, B. (1975) J. Virol. 16, 153–167.
37. Kwong, A.D. & Frenkel, N. (1985) Virology 142, 421–425.
38. Kwong, A.D. & Frenkel, N. (1984) J. Virol. 51, 595–603.
39. Fraefel, C., Song, S., Lim, I., Lang, P., Yu, L., Wang, Y., Wild, P. & Geller, A.I. (1996) J. Virol., in press.
40. Marconi, P., Krisky, D., Oligino, T., Poloani, P.L., Ramakrishnan, R., Goins, W.F., Fink, D.J. & Glorioso, J.C. (1996) Proc. Natl. Acad. Sci. USA 93, this issue.
41. Leopardi, R. & Roizman, B. (1996) Proc. Natl. Acad. Sci. USA 93, 9583–9587.
42. Meignier, B., Longnecker, R. & Roizman, B. (1988) J. Infect. Dis. 158, 602–614.
43. Meignier, B., Martin, B., Whitley, R.J. & Roizman, B. (1990) J. Infect. Dis. 162, 313–322.
44. Nguyen, L.H., Knipe, D.M. & Finberg, R.W. (1992) J. Virol. 66, 7067–7072.
45. Bruni, R. & Roizman, B. (1996) Proc. Natl. Acad. Sci. USA 93, 10423–10427.






