
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles
MATTHIAS J. SCHNELL, LINDA BUONOCORE, EVELYNE KRETZSCHMAR, ERIK JOHNSON, AND JOHN K. ROSE*
Departments of Pathology, Cell Biology, and Biology, Yale University School of Medicine, 310 Cedar Street, New Haven, CT 06510
ABSTRACT In a previous study we demonstrated that vesicular stomatitis virus (VSV) can be used as a vector to express a soluble protein in mammalian cells. Here we have generated VSV recombinants that express four different membrane proteins: the cellular CD4 protein, a CD4-G hybrid protein containing the ectodomain of CD4 and the transmembrane and cytoplasmic tail of the VSV glycoprotein (G), the measles virus hemagglutinin, or the measles virus fusion protein. The proteins were expressed at levels ranging from 23–62% that of VSV G protein and all were transported to the cell surface. In addition we found that all four proteins were incorporated into the membrane envelope of VSV along with the VSV G protein. The levels of incorporation of these proteins varied from 6–31% of that observed for VSV G. These results suggest that many different membrane proteins may be co-incorporated quite efficiently with VSV G protein into budding VSV virus particles and that specific signals are not required for this co-incorporation process. In fact, the CD4-G protein was incorporated with the same efficiency as wild type CD4. Electron microscopy of virions containing CD4 revealed that the CD4 molecules were dispersed throughout the virion envelope among the trimeric viral spike glycoproteins. The recombinant VSV-CD4 virus particles were about 18% longer than wild type virions, reflecting the additional length of the helical nucleocapsid containing the extra gene. Recombinant VSVs carrying foreign antigens on the surface of the virus particle may be useful for viral targeting, membrane protein purification, and for generation of immune responses.
Vesicular stomatitis virus (VSV) is the prototypic rhabdovirus and is among the simplest of the enveloped animal viruses (reviewed in ref. 1). VSV has been an important model system in part because it grows rapidly to very high titers and can be prepared in large quantities. The VSV genome is an 11, 161 nucleotide negative-strand RNA that is tightly encased in the nucleocapsid (N) protein. The nucleocapsid is packaged as a 35-turn helix within membrane-enveloped, bullet-shaped particles. Two virally encoded proteins, L and P, form a polymerase that is packaged in the virion. This polymerase is required for transcription of the genome to generate five subgenomic mRNAs encoding the five structural proteins and for replication of full-length genomes. The two other viral proteins are the matrix protein (M) and the glycoprotein (G). M protein probably bridges between the nucleocapsid and the short cytoplasmic domain of the transmembrane G protein. G protein forms trimeric spikes on the virion surface and is responsible for binding a cell surface receptor and for membrane fusion to initiate infection.
We reported recently that VSV can be used as a genetically stable, high-level expression vector (2). Here we have investigated VSVs ability to express cellular and viral membrane proteins and we have also examined the incorporation of these proteins, into the viral membrane. Incorporation of foreign viral spike glycoproteins into the envelope of recombinant VSV particles might allow development of a new generation of killed virus vaccines or retargeting of virus particles to specific cell types. Also, expression of other membrane proteins from the VSV genome and their subsequent incorporation into the virions could yield large quantities of the expressed protein in highly purified form for structural or functional studies. Because of these potential applications, we are especially interested in understanding the factors governing membrane protein incorporation into VSV particles. By generating VSV recombinants expressing foreign envelope proteins, we are able to achieve high-level expression of foreign membrane proteins at the same time that the VSV proteins are being made. This system allows us to examine the structural requirements for foreign protein incorporation into VSV particles.
Previous studies have shown that the envelope proteins of several other viruses can be incorporated into the envelope of VSV particles when VSV and a second virus are propagated in the same cells. This phenomenon of pseudotype formation is well known (reviewed in ref. 3), but the extent of foreign envelope protein incorporation into VSV has not been examined in detail. Also, the cellular CD4 protein expressed from a vaccinia vector was found to be incorporated into VSV particles at a low level of about 60 molecules per virion, and no preference was seen for a chimeric CD4 carrying the transmembrane and cytoplasmic domains of VSV G protein (4). In contrast, a study of HIV envelope protein incorporation into VSV particles lacking G protein indicated a requirement for the cytoplasmic tail of G on the HIV envelope protein (5). These studies complemented earlier studies showing a lower efficiency of incorporation of G proteins with truncated tails into the VSV G ts mutant lacking the G protein (6).
Here, using recombinant VSVs expressing very high levels of foreign membrane proteins, we have been able to quantitate both the expression levels of the foreign proteins and their incorporation into VSV particles. We conclude that there is significant extra space in the VSV envelope that can, in many cases, accommodate large amounts of foreign membrane proteins.
MATERIALS AND METHODS
Plasmid Construction. A plasmid expressing the positive strand RNA complement of the VSV genome with a site for foreign gene expression (pVSV-XN1) was described previ-
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: VSV, vesicular stomatitis virus; HIV, human immunodeficiency virus; wt, wild type; BHK cells, baby hamster kidney cells; N, nucleocapsid; M, matrix; G, glycoprotein; H, hemagglutinin; F, fusion.
|
* |
To whom reprint requests should be addressed. e-mail: jrose@biomed.med.vale.edu. |
ously (2). This plasmid contains unique XhoI and NheI sites flanked by VSV transcription start and stop signals. VSV constructs encoding the human CD4, human CD4 with the VSV G transmembrane and cytoplasmic domains (CD4G), measles virus fusion (F), and measles virus hemagglutinin (H) proteins, were generated as follows. Each gene was excised from the appropriate plasmids with XhoI and XbaI and ligated to pVSV-XN1 that had been cleaved with XhoI and NheI. The inserts were obtained from pBS-CD4 encoding CD4 (7) or pBS-CD4G encoding a chimeric molecule with the transmembrane and cytoplasmic domains of CD4 replaced with those from the VSV G protein. The measles F protein gene was subcloned from peΔ5F1 (8) and the measles H protein gene was subcloned from peH1 (8). The plasmids encoding the measles proteins were generously provided by Roberto Cattaneo (University of Zurich). The resulting plasmids were called pVSV-CD4, pVSV-CD4G, pVSV-MF, or pVSV-MH.
Recovery of VSV Recombinants. Baby hamster kidney cells (BHK-21, American Type Culture Collection) were maintained in DMEM supplemented with 5% fetal bovine serum. Cells on 10-cm dishes (≈80% confluent) were infected at a multiplicity of 10 with vTF7–3 (9). After 1 h, plasmids encoding the N, P, and L proteins and the appropriate recombinant antigenomic RNA were transfected into the cells using TransfectACE (10), as described (2). Plasmid amounts were 10 μg of the respective full length plasmid (VSV-CD4, VSV-CD4G, VSV-MF, VSV-MH), 3 μg pBS-N, 5 μg pBS-P, and 2 μg pBS-L. Subsequent steps were performed as described (2).
Preparation and Analysis of Protein from Recombinant VSV. For metabolic labeling of the VSV proteins, BHK cells on a 3.5-cm plate (≈80% confluent) were infected with recombinant or wild type (wt) VSV at a multiplicity of infection of ≈20. After 4 h cells were washed with DMEM— methionine and incubated for 1 h at 37°C in 1 ml of DMEM lacking unlabeled methionine and containing 100 μCi (1 Ci= 37 GBq) of [35S]methionine. For immunoprecipitation, SDS was added to a 0.2% final concentration in the cell lysates, and the medium samples were adjust to 1% Nonidet-40 and 0.2% SDS. Samples were incubated at 4°C overnight in the presence of specific antisera, and immune complexes were precipitated with excess fixed Staphylococcus aureus (Pansorbin; Calbiochem). Cell extracts (2% of the total) and radioimmunoprecipitation were analyzed by SDS/10% polyacrylamide gel and detected with a Phosphorlmager (Molecular Dynamics). For metabolic labeling of recombinant and wt VSV virions, a monolayer of BHK cells (≈80% confluent) on a 3.5-cm dish was infected with a multiplicity of infection of ≈20. After 2 h cells were washed with DMEM—methionine and incubated overnight at 37°C in 1.5 ml of DMEM +3% FCS lacking unlabeled methionine and containing 200 μCi of [35S]methionine. Cell debris and nuclei were removed by centrifugation at 1,250×g for 5 min, and virus was pelleted from the medium through 10% sucrose at 38,000 rpm in a Beckman SW41 rotor for 1 h. Virus pellets were resuspended in 50 μl or 10 mM Tris·HCl, pH 7.4 and 10 μl was analyzed by SDS/PAGE.
Immunofluorescence Microscopy. Cells on 3.5-cm plates were infected for 4 h, fixed in 3% paraformaldehyde, and stained with mAb I1 to the VSV GI protein (11) a mAb OKT4 (12) to CD4, a polyclonal rabbit antibody directed against the VSV G protein cytoplasmic domain (13), or polyclonal rabbit antibodies directed against the measles H or F proteins (8), followed by fluorescein isothiocyanate-conjugated goat antimouse antibody (Jackson ImmunoResearch) or by fluorescein isothiocyanate-conjugated goat anti-rabbit antibody (Jackson ImmunoResearch). Cells were examined by indirect immunofluorescence using a Nikon Microphot-FX microscope equipped with a 40× planapochromat objective.
Electron Microscopy and Immunogold Labeling. Virus particles from infections performed in 10-cm dishes were recovered from cultures by first pelleting cell debris at 1,500×g for 10 min. The virus was then concentrated and purified by centrifugation twice through 20% sucrose (20% sucrose in 1× TBE, pH 7.5) at 120,000×g for 60 min. Virus samples were absorbed onto carbon-coated grids for 5 min and then blocked with 1% BSA in PBS for 30 min at room temperature. The grids were then placed on a 50-μl drop of anti-VSV G mAb I1 (11) or anti-CD4 mAb OKT4 (12) diluted 1:50 in PBS containing 1% BSA. After 1 h excess antibody was removed by placing grids sequentially onto five 50-μl drops of 1% BSA in PBS for 2 min each time. The grids were then placed on a 50-μl drop of goat anti-mouse IgG (Fc) labeled with 15-nm gold particles (AuroProbe, Amersham). Unbound gold conjugates were removed by five sequential 2-min washes with PBS. The virus-Immunogold complexes were then negative stained by incubation the grids for 4 min on 50-μl drops of 2% phosphotungstic acid (pH 7.5). Excess stain was removed and the grids were air dried. Images of viruses were obtained with a Zeiss EM910 electron microscope.
RESULTS
We previously described the recovery of a recombinant VSV expressing the bacterial chloramphenicol acetyl transferase from a extra transcription unit inserted into an infectious clone of VSV. Chloramphenicol acetyl transferase expression employed the VSV vector pVSV-XN1 in which a new transcription unit and cloning site were inserted between the G and L genes of VSV (1). In the present study we have used this same vector to derive recombinant VSVs expressing four different membrane proteins that are transported to the plasma membrane and we have examined their incorporation into the membrane of particles budding from the cell surface.
Expression of CD4 and CD4G in VSV. In initial studies we used the gene encoding the cellular membrane protein CD4 (14) or a CD4G chimeric protein in which the transmembrane and cytoplasmic domains of CD4 were replaced with the corresponding domains of VSV G protein (Fig. 1). We cloned the sequences encoding the CD4 or CD4G proteins in the vector pVSV-XN1. Recovery of recombinant VSVs expressing the CD4 or CD4G proteins from plasmid DNA was obtained using published techniques (2). After initial recovery of recombinant viruses, expression of CD4 epitopes on the cell surface was verified by indirect immunofluorescence of infected cells for each recombinant (data not shown). We next determined the protein expression level in BHK cells infected with each recombinant. We infected BHK cells at a multiplicity of 20 for 4 h and labeled with [35S]methionine for 1 h. Because
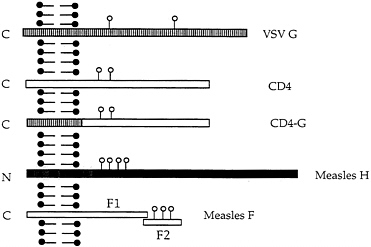
FIG. 1. Schematic diagram of VSV G protein and the other membrane proteins expressed from recombinant VSVs. VSV G, CD4, CD4 with the VSV G transmembrane domain and cytoplasmic tail, measles H protein, and measles F1 and F2 proteins are illustrated. The left side of the diagram represents the cytoplasmic side of the plasma membrane. Sites of N-linked glycan addition are indicated by ○.
VSV shuts down host cell protein synthesis, the viral and additional encoded proteins can be visualized without immunoprecipitation. The results in Fig. 2A show, that in addition to the VSV N, P, M, G, and L proteins, new proteins of the size expected for CD4 and CD4G were seen. Quantitation of protein expression (correcting for methionine content) showed that CD4, and CD4G were expressed at about 25–30% of the level of VSV G protein. The identities of the CD4 and CD4G proteins were confirmed by immunoprecipitation with antibodies directed against CD4 or against the VSV G tail. The CD4-specific antibody immunoprecipitated both the CD4 and CD4G proteins (Fig. 2B, lanes 1 and 2). The faster migrating CD4G band results from the shorter VSV G tail of 29 amino acids compared with the CD4 tail of 38 amino acids. The presence of the G tail in the CD4G construct was also confirmed with VSV G tail-specific antibody that immunoprecipitated the CD4G protein in addition to VSV G protein (Fig. 2B, lane 4), but not the CD4 protein (lane 3).
Both CD4 and CD4G are Incorporated at High Levels into VSV Particles. To determine if the CD4 and CD4G proteins were incorporated into VSV particles, we labeled infected cells with [35S]methionine, purified labeled VSV by sedimentation through 10% sucrose and then examined the proteins present by SDS/PAGE. The results in Fig. 2C show that both the CD4 and CD4G proteins were present in the purified virions. Quantitation using the PhosphorImager (correcting for the lower methionine content in CD4 and CD4G) showed that both CD4 and CD4G were present in virions at a level of 23–25% of VSV G. Therefore, the level of protein incorporation appears to correspond roughly to the level of synthesis relative to VSV G protein and no specific signals are required at least for coincorporation of CD4 into virions with VSV G protein. Quantitation from several experiments indicated that the ratios of G to N proteins in the CD4 or CD4G viruses were not decreased and in fact were slightly increased when compared with wt virus. Thus it appears that CD4 or CD4G proteins are not displacing G but might rather be filling in free space between the VSV G trimers. This quantitation was also verified using virus purified by rate zonal sedimentation (data not shown).
Electron Microscopy Shows CD4 and CD4G Distributed Over the Virion Surface. Although biochemical analysis of purified virions showed incorporation of CD4 and CD4G into VSV, it was unclear if every particle contained the foreign protein or if there were changes to the structure of VSV virions as a result of the inclusion of an additional gene and protein. To address these questions directly, purified wt VSV, VSV-CD4, and VSV-CD4G virions were examined by electron microscopy using Immunogold-coupled antibodies and negative stain (Fig. 3). A and B show wt VSV stained with anti-G antibodies and anti-CD4 antibodies respectively. The anti-G antibodies are clearly reacting with the virion surface while the anti-CD4 antibodies do not. B and C show the VSV-CD4 virus reacting with the anti-G or the anti-CD4 antibodies, and E and F show the same for the VSV-CD4G virus. These results confirm the incorporation of CD4 and CD4G into the virion membrane. In addition, CD4 and CD4G appeared to be present in all particles and in an even distribution over the virion surface. These results indicate that CD4 is inserted among the VSV G trimers and is not present in a subset of particles or clustered at one site on the virion.
VSV Recombinants are Longer. The length of the bullet-shaped VSV particles is known to be determined by the length of the helical nucleocapsid containing the RNA, since the shorter RNA genomes of defective interfering particles are packaged into truncated particles (15). As can be seen in Fig. 3, VSV-CD4 or VSV-CD4G virions were longer then wt VSV. In Fig. 4 the lengths of VSV and VSV-CD4 particles are compared using particles that have been aligned. VSV virions were found to be a uniform 170 nm long, while the VSV-CD4 virions were 200 nm long. This 18% increase in length corresponds reasonably well to the increase in genome size for the VSV-CD4 construct that has an insert of 1,800 nucleotides added to a genome that was originally 11,161 nucleotides.
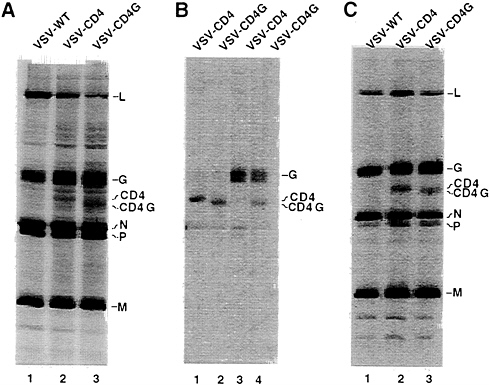
FIG. 2. Expression of CD4 and CD4G from recombinant VSVs and their incorporation into VSV virions. (A) Cells infected with VSV wt, VSV-CD4, or VSV-CD4G were labeled with [35S]methionine and cell lysates were fractionated directly by SDS/PAGE. Positions of the VSV proteins, CD4 and CD4G are indicated. (B) SDS/PAGE of immunoprecipitates of cell lysates prepared with antibodies directed against the extracellular domain of CD4 protein (lanes 1 and 2) or the VSV G protein cytoplasmic domain (lanes 3 and 4). (C) SDS/PAGE of purified virions labeled with [35S]methionine. Images of labeled proteins were generated by a PhosphorImager (Molecular Dynamics).
FIG. 3. Electron microscopy of Immunogold-labeled recombinant VSVs. Purified wt VSV (A and B), VSV-CD4 (C and D), or VSV-CD4G (E and F) particles were negative stained after labeling with a mAb directed against VSV G (A, C, and E) or CD4 (B, D, and F) followed by gold-labeled goat anti-mouse IgG particles. The bar represents 100 nm.
Reduced Titers and Virus Quantities of Recombinants Expressing CD4 Proteins. Titers of recombinant VSVs expressing CD4 or CD4G proteins were typically lower than wt titers by at least one log, while VSV-chloramphenicol acetyl transferase viruses grew to normal wt titers (1). To analyze the replication cycle of the recombinant CD4 and CD4G viruses more carefully, a one step growth curve was performed. BHK cells were infected simultaneously at a multiplicity of 20 to allow synchronous infection of all cells. After removing unadsorbed virus, virus growth was monitored by plaque assay (Fig. 5). Both recombinant viruses replicated at a rate similar to wt VSV, but in both cases the final titer was reduced ten-fold or more compared with wt. The total yield of virus protein from the same number of infected cells was lower by about a factor of three for the recombinants. The even greater reduction in titer suggested that particles released had a lower specific infectivity than wt virus.
The cloning strategy used to generate VSV-CD4G left an extra transcription termination signal UAUGAAAAAAA (16, 17) preceding the normal transcription stop-start signal just before L. If this upstream signal were used it might cause extensive termination without reinitiation and result in a strong inhibition of L protein expression. This could explain the slower growth of the CD4G virus compared with the CD4 virus as well as the reduced expression of L protein (Fig. 2A, lane 3).
Expression of Measles Virus Glycoproteins in VSV Recombinants. Because the cellular CD4 protein was incorporated well into VSV virions, we then wished to determine if glycoproteins from another virus could also be expressed in VSV recombinants and incorporated into the VSV viral envelope. For these experiments we chose measles virus, a negative strand RNA virus from the distantly related paramyxovirus group. Measles virus encodes two glycoproteins, H and F, that
FIG. 4. Size comparison of VSV wt and VSV-CD4 particles. Images of VSV or VSV-CD4 particles were excised from electron micrographs and displayed so that the sizes could be compared directly. Lengths including the surface spikes are shown by the bars.
are expressed from separate mRNAs and have molecular weights different from any VSV proteins permitting quantitation without immunoprecipitation. The H protein is a type II membrane protein (N terminus is cytoplasmic) with an apparent molecular mass of 80 kDa while the F protein is a type I protein that is cleaved from a precursor of 61 kDa to F1 and F2 proteins of 41 and 20 kDa. Stick diagrams of the proteins are shown in Fig. 1.
VSV recombinants expressing measles H and F proteins were generated with the same starting vector used for the CD4 and CD4G proteins. The VSV recombinant expressing measles H protein (VSV-MH) was recovered easily and grew to wt VSV titers, while the VSV expressing F protein (VSV-MF) grew slowly and to much lower titers. We do not have an explanation for the slow growth of this recombinant, but several independent isolates showed the same phenotype of
FIG. 5. One step growth curve of recombinant VSV. BHK cells were infected (multiplicity of infection=20) for 30 min with VSV wt, VSV-CD4 or VSV-CD4G and unabsorbed virus was removed by washing the cells twice with PBS. Complete medium was added and samples were collected at the indicated times postinfection. Virus titers were determined by plaque assays on BHK cells.
slow growth suggesting that it was due to the F gene and not to some secondary mutation picked up in the recovery.
Expression of the measles proteins by VSV-MH and VSV-MF was analyzed by infecting BHK cells with recombinants and labeling with [35S]methionine. As shown in Fig. 6, the VSV-MH virus generated a new protein band at 80 kDa and the VSV-MF virus encoded a new protein of about 41 kDa running just ahead of the VSV N protein (45 kDa). The H protein was synthesized at a level of 62% of G while the F1 protein was synthesized at a level of 46% of G (after correcting for methionine content). The identity of these proteins was verified by immunoprecipitation with antibodies specific for measles F and H proteins and with markers of measles proteins generated from HeLa cells infected with measles virus. Expression of both proteins on the cell surface was also verified by indirect immunofluorescence microscopy (data not shown). The F2 protein is typically a diffuse band of 18–20 kDa and was not visible on this gel. We also determined that both F and H proteins were found in purified virions (Fig. 6, lanes 1 and 2), with H present at level of 31% of G while F1 was present at 6% the level of G. The fact that the measles F protein was found in lower amounts in the virions compared with the other proteins may be explained by previous results showing that only a small fraction of F protein reaches the cell surface (8). Therefore the majority would not be available for inclusion in budding VSV virions.
DISCUSSION
Incorporation of one or more foreign glycoproteins into the envelope of a virus may potentially be useful for targeting viruses to specific cells and for the development of a new generation of vaccines. We have reported here that both viral and nonviral proteins can be incorporated at high levels along with the VSV glycoprotein into the envelope of VSV. This high level incorporation is achieved by expressing the foreign proteins from genes inserted directly into the VSV genome,
FIG. 6. Expression of measles virus glycoproteins from VSV recombinants and their incorporation into VSV particles. Lysates from [35S]methionine-labeled cells infected with VSV-MF (lane 3), or VSV-MH (lane 4) analyzed by SDS/PAGE. [35S]Methionine-labeled virions from VSV-MF (lane 1) or VSV-MH (lane 2) infected cells analyzed by SDS/PAGE. Positions of the VSV proteins, and measles F1 and H proteins are indicated. Images of labeled proteins were generated by a PhosphorImager (Molecular Dynamics).
and does not require the presence of specific signals on the foreign proteins. It is important to discuss these results in the context of the extensive literature on rhabdovirus assembly.
The budding of VSV particles at the plasma membrane involves an interaction of the helical nucleocapsid containing the genome, the matrix protein, and the membrane-spanning glycoprotein. The matrix protein is able to bind to the plasma membrane independently (18, 19), can cause vesiculation of the plasma membrane (20), and may serve as a bridge between the cytoplasmic tail of the glycoprotein and the helical nucleocapsid.
A temperature-sensitive mutant of the VSV glycoprotein is still able to bud particles lacking the G protein, but at a 10–20-fold reduced efficiency (21, 22), while temperature-sensitive mutations in the matrix protein block budding completely (21). A recombinant rabies virus (also in the rhabdovirus family) lacking the glycoprotein gene, buds bullet-shaped particles completely lacking G protein, but at a 30-fold reduced level (23). These results suggest that cooperative interactions of nucleocapsid, matrix, and glycoprotein are required to promote efficient rhabdovirus budding. Deletions of the cytoplasmic tail of the VSV G protein (6) or in the rabies G protein tail (23) reduce the efficiency of glycoprotein incorporation into the virus particle suggesting an important interaction occurring through the glycoprotein tail, presumably with the matrix protein.
The results presented here illustrate that VSV particles can incorporate several unrelated foreign membrane proteins into the viral envelope at levels up to 30% of G protein itself. In no case did incorporation of the foreign protein reduce the level of VSV G relative to nucleocapsid or matrix proteins, suggesting that there is extra space in the envelope for incorporation of the foreign proteins. In the case of CD4, electron microscopy showed that the foreign proteins are dispersed among the VSV spikes. Because both cellular and foreign viral proteins with unrelated sequences are incorporated into the VSV envelope, there cannot be specific signals involved in assembly of these foreign proteins into the VSV membrane. In fact, substitution of the CD4 transmembrane and cytoplasmic domains with those from VSV G did not increase the level of CD4 incorporation into the VSV particle. We therefore favor a model in which many proteins on the cell surface can be passively trapped in the budding VSV particle. Early studies showed that many cellular plasma membrane proteins are found at low levels in VSV particles, while others are excluded (24). Also the generation of VSV pseudotypes carrying foreign envelope proteins shows that some foreign proteins can be incorporated into the viral membrane (3). In our studies, the foreign proteins are being expressed at extremely high levels from the VSV genome and their high level of incorporation into particles is presumably the result of their presence at high concentrations on the plasma membrane.
The recombinant expressing the measles F protein deserves special comment because this virus grew slowly (maximal titers of 107/ml) and lost expression of the F protein after only three passages. We have not seen loss of foreign gene expression with the other VSV recombinants even after much more extensive passaging. With the VSV-MF recombinant there is apparently a strong selective pressure to eliminate the F protein expression, presumably because the protein interferes with some aspect of VSV replication. Mutations that eliminate F expression are apparently selected rapidly.
Plasma membrane proteins that are not incorporated into VSV particles are presumably excluded from regions of virus budding because of steric constraints such as large cytoplasmic domains, association with large cytoplasmic proteins, or association with domains enriched in specific lipids. Earlier results from our laboratory showed that the HIV envelope protein was largely excluded from budding VSV particles (5). Only an HIV envelope protein carrying the VSV G cytoplasmic domain was incorporated efficiently suggesting that the G domain was able to provide a specific assembly signal. We have confirmed and extended that result using a recombinant VSVs expressing the HIV envelope or an HIV envelope G chimeric protein from an extra gene (E.J. and J.K.R., unpublished results).
Although many foreign proteins can likely be incorporated into VSV particles, it is also likely that a significant amount of VSV G protein is required to promote particle formation efficiently. As a result, the amount of space available for foreign proteins may be limited. Preliminary results attempting to increase the level of CD4 incorporation using a new VSV vector in which CD4 is expressed at higher levels than VSV G have not resulted in increased CD4 incorporation into particles. In a study related to ours, it was reported that CD4 could be incorporated into avian leukosis virus particles at relatively high levels (25), although in that study it was found that CD4 bearing the cytoplasmic domain of the avian leukosis virus envelope proteins was not incorporated as well as CD4, apparently because of competition with the avian leukosis virus envelope protein itself (25).
One important remaining question is whether foreign proteins can ever completely substitute for VSV G in assembly. An earlier study on VSV (murine leukemia virus) pseudotype formation employing a temperature sensitive VSV G mutant concluded that this would not be possible because some VSV G protein appeared to be required to nucleate VSV assembly (26). However, results showing incorporation of the HIV-G chimeric protein into virions lacking VSV G suggest that this may be possible if the appropriate signals are appended to the proteins to be incorporated (5).
M.J.S. received support from an AIDS Infektionsforschungs fellowship from the Deutsches Krebsforschungszentrum. This work was also supported by National Institutes of Health Grant R37AI24345 to J.K.R.
1. Wagner, R.R. & Rose, J.K. (1996) in Rhabdoviridae: The Viruses and Their Replication, eds. Fields, B.N. & Knipe, D.M. (Lippincott-Raven, New York), Vol. 1, pp. 1121–1136.
2. Schnell, M.J., Buonocore, L., Whitt, M.A. & Rose, J.K. (1996) J. Virol. 70, 2318–2323.
3. Závada, J. (1982) J. Gen. Virol. 63, 15–24.
4. Schubert, M., Joshi, B., Blondel, D. & Harmison, G.G. (1992) J. Virol. 66, 1579–1589.
5. Owens, R.J. & Rose, J.K. (1993) J. Virol. 67, 360–365.
6. Whitt, M.A., Chong, L. & Rose, J.K. (1989) J. Virol. 63, 3569–3578.
7. Shaw, A.S., Amrein, K.E., Hammond, C., Stern, D.F., Sefton, B.M. & Rose, J.K. (1989) Cell 59, 627–636.
8. Cattaneo, R. & Rose, J.K. (1993) J. Virol. 67, 1493–1502.
9. Fuerst, T.R., Niles, E.G., Studier, F.W. & Moss, B. (1986) Proc. Natl. Acad. Sci. USA 83, 8122–8126.
10. Rose, J.K., Buonocore, L. & Whitt, M.A. (1991) BioTechniques 10, 520–525.
11. Lefrancois, L. & Lyles, D.S. (1982) Virology 121, 168–174.
12. Reinherz, E.L., Kung, P.C., Goldstein, G. & Schlossman, S.F. (1979) Proc. Natl. Acad. Sci. USA 76, 4061–4065.
13. Guan, J.L., Cao, H. & Rose, J.K. (1988) J. Biol. Chem. 263, 5306–5313.
14. Maddon, P.J., Dalgleish, A.G., McDougal, J.S., Clapham, P.R., Weiss, R.A. & Axel, R. (1986) Cell 47, 333–348.
15. Holland, J.J. (1990) in Defective Viral Genomes, eds. Fields, B.N. & Knipe, D.M. (Raven, New York), Vol. 1, pp. 151–166.
16. Rose, J.K. (1980) Cell 19, 415–421.
17. Rose, J.K. & Schubert, M. (1987) in Rhabdovirus Genomes and Their Products, ed. Wagner, R.R. (Plenum, New York), pp. 129–166.
18. Chong, L.D. & Rose, J.K. (1993) J. Virol. 67, 407–414.
19. Chong, L.D. & Rose, J.K. (1994) J. Virol. 68, 441–447.
20. Justice, P.A., Sun, W., Li, Y., Ye, Z., Grigera, P.R. & Wagner, R.R. (1995) J. Virol. 69, 3156–3160.
21. Knipe, D.M., Baltimore, D. & Lodish, H.F. (1977) J. Virol. 21, 1149–1158.
22. Schnitzer, T.J. & Lodish, H.F. (1979) J. Virol. 29, 443–447.
23. Mebatsion, T., Konig, M. & Conzelmann, K.K. (1996) Cell 84, 941–951.
24. Lodish, H.F. & Porter, M. (1980) Cell 19, 161–169.
25. Young, J.A., Bates, P., Willert, K. & Varmus, H.E. (1990) Science 250, 1421–1423.
26. Witte, O.N. & Baltimore, D. (1977) Cell 11, 505–511.







