
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Specific infection of CD4+target cells by recombinant rabies virus pseudotypes carrying the HIV-1 envelope spike protein
TESHOME MEBATSION AND KARL-KLAUS CONZELMANN *
Department of Clinical Virology, Federal Research Centre for Virus Diseases of Animals, Paul-Ehrlich-Strasse 28, D-72076 Tübingen, Germany
ABSTRACT A recombinant rabies virus (RV) mutant deficient for the surface spike glycoprotein (G) gene was used to study the incorporation of envelope proteins from HIV-1 expressed from transfected plasmids. A hybrid HIV-1 protein in which the cytoplasmic domain was replaced with that of RV G was incorporated into the virus envelope and rescued the infectivity of the RV mutant. The RV(HIV-1) pseudotype viruses could infect only CD4+cells, and their infectivity was neutralized specifically by anti-HIV-1 sera. In contrast to the chimeric protein, wild-type HIV-1 envelope protein or mutants with truncated cytoplasmic domains failed to produce pseudotyped particles. This indicates the presence of a specific signal in the RV G cytoplasmic domain, allowing correct incorporation of a spike protein into the envelope of rhabdovirus particles. The possibility of directing the cell tropism of RV by replacement of the RV G with proteins of defined receptor specificity should prove useful for future development of targetable gene delivery vectors.
The host range of viruses is determined primarily by the interaction of viral envelope glycoproteins with specific proteins on the host cell surface that act as receptors. This interaction is very often species-specific and, in some cases, even tissue-specific. As a result, some viruses, like retroviruses, have a quite limited host range, whereas others, such as rhabdoviruses, can infect a variety of cell types including in many cases cells of human, animal, and insect origin. Developing means for redefining the receptor specificity of viral particles would have numerous applications in basic research and clinical application. On the one hand, this might allow better infection of cells of therapeutic interest that are poorly amenable to retrovirus infection, such as hepatocytes or early hematopoietic progenitors. On the other hand, this would also allow targeting of specific cell types in complex cell populations. In the in vivo situation, the availability of targeting methods should, for example, permit the development of new gene therapy models.
Alteration of the tropism of viruses by incorporation of a foreign envelope protein, however, is hampered in most virus systems by the fact that the presence of the viral spike protein(s) is required also to drive the viral assembly and budding process (for review, see ref. 1). Retroviruses represent one example where this does not apply (2, 3). In the past, retroviral vectors deficient for the spike glycoprotein gene (env) have been used to generate pseudotype viruses carrying foreign proteins in their envelopes, predominantly the vesicular stomatitis rhabdovirus (VSV) G protein. As expected, the pseudotype viruses exhibit the expanded host range of the foreign glycoprotein (4–6). Since incorporation of the heterologous spike proteins is apparently nonspecific, low titers of infectious pseudotype viruses are usually obtained, representing the major drawback in clinical application.
Another group of viruses that are able to bud in the absence of their viral spike glycoprotein are the rhabdoviruses (7) which include VSV and rabies virus (RV). Rhabdoviruses have a single negative strand RNA genome of 11,000–12,000 nt and replicate in the cytoplasm (8). Virus assembly and budding takes place at the cell membrane where the viral ribonucleoprotein complex is enwrapped into an envelope containing an internal matrix protein and the single transmembrane spike glycoprotein (G) (9). By applying a system that allows genetic engineering of RV (10, 11), we could recently recover a recombinant RV mutant deficient for the G gene and demonstrate that, in the absence of G protein, bullet-shaped spikeless rhabdovirus particles are released from the infected cells (7). In the same study, we could also confirm that, similar to VSV G (12) and in contrast to retroviruses (13), the cytoplasmic domain of the RV G protein contributes considerably to sorting and incorporation of the protein into the viral envelope. Moreover, previous studies confined to a temperature-sensitive G protein mutant of VSV (tsO45) have indicated that the cytoplasmic domain of VSV G protein is sufficient for directing a foreign protein, the HIV-1Env, into VSV (14).
In the present study, we exploited the G deficiency of a recombinant RV mutant to confirm that the RV G cytoplasmic tail provides a signal allowing specific incorporation of HIV-1 Env, expressed from transfected plasmids, into the envelope of virus particles and to verify that no residual G protein is required to initiate rhabdovirus pseudotype formation. The generated RV(HIV) pseudotype particles possess the tropism of HIV-1 in that they can infect HeLa cells expressing the major HIV receptor CD4, but not CD4− HeLa cells. The transient G-deficient RV mutant system provides a versatile and safe tool to rapidly analyze incorporation and function of a wide range of surface protein constructs. Moreover, since foreign genes in RV and VSV genomes are highly stable (15, 16), recombinant rhabdoviruses represent promising candidates for future development of targetable, nonintegrative viral vectors for delivery of therapeutic or protective genes.
MATERIALS AND METHODS
Cells, Viruses, cDNA, and Antibodies. The following reagents were obtained through the AIDS Research and Reference Reagent Program (National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda). HeLa CD4+ and HeLa CD8+ cells were from Richard Axel (17), and human anti-HIV-1 immune globulin was from Alfred Prince (18, 19). The recombinant vaccinia virus vTF73, ex-
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: RV, rabies virus; SAD, Street-Alabama-Dufferin strain of RV; VSV, vesicular stomatitis virus; G, glycoprotein.
|
* |
To whom reprint requests should be addressed, e-mail: klaus. conzelmann@tue.bfav.de. |
pressing T7 RNA polymerase (20), was kindly provided by T. Fuerst and B.Moss, and the HIV-1 (NL-43) gp160 cDNA was from W.Garten and H.-D.Klenk (Institute of Virology, University of Marburg, Germany). The G-deficient Street-Alabama-Dufferin RV mutant, SAD ΔG, was recovered and propagated after deletion of the entire G protein coding region from a full-length RV cDNA in cells expressing RV G from a transfected plasmid in the vaccinia/T7 polymerase system (7).
Construction of Expression Plasmids. An expression plasmid encoding wild-type HIV-1 Env (pEnv) was generated by insertion of the BbsI-XhoI-Klenow fragment spanning the entire gp160 coding region from the HIV NL-43 cDNA in the EcoRV site of pT7T (21). To replace the HIV cytoplasmic domain, an HpaI site (underlined) was introduced by PCR using a primer (5′-GCTCTAGACTAGTTAACTATAGAAAGTACAGC-3′) overlapping the transmembrane/ cytoplasmic domain-encoding region and a second primer (5′-AACAATTACACAAGCTTAAT-3′) spanning an upstream unique HindIII site (underlined) of the Env cDNA. An HpaI site was also introduced by PCR (5′-AAGTCGACCGTTAACAGAAGAGTCAATCGATCA-3′) upstream of the pT7T-G sequence encoding the RV SAD B19 G cytoplasmic tail. After ligation of the HIV-derived HindIII-HpaI fragment and the G-derived HpaI-PstI fragment, the construct was used to replace the sequence downstream of HindIII in pEnv to yield pEnv-RVG. The removal of an HpaI fragment of pEnv resulted in pEnv-Δ107; the encoded protein possessed a deletion of 107 amino acids adjacent to the transmembrane domain (see Fig. 1). The Env construct possessing a carboxyl-terminal truncation (pEnv-44) was generated by introduction of a translational stop codon (complementary sequence underlined) into the cytoplasmic tail-encoding sequence of pHIV-Env by PCR mutagenesis using a primer with the sequence 5′-ACGAATTCATTAGTTCACTAATCGAATG-3′.
Expression of Envelope Proteins. BSR cells were first infected with vTF7–3 at a multiplicity of infection of 5. After 1 hr of infection, cells were transfected with CsCl-purified plasmids by using the Stratagene mammalian transfection kit as described (21). To adjust the level of cell surface expression, various concentrations of plasmid DNA were used for transfection (see below). After incubation for 16 hr, cells were fixed with 4% paraformaldehyde and incubated at 4°C for 30 min with human anti-HIV-1 IgG (1:500 dilution) or a monoclonal antibody directed against RV G protein (diluted 1:100). Cells were stained with fluorescein isothiocyanate-conjugated goat anti-human IgG or goat anti-mouse IgG (Dianova, Hamburg, Germany). For double staining, rhodamine-conjugated goat anti-mouse IgG (Dianova) was used in combination with fluorescein isothiocyanate-conjugated goat anti-human IgG.
Complementation of SAD ΔG with Envelope Proteins. BSR cells were infected at a multiplicity of infection of 1 with SAD ΔG phenotypically complemented with RV G protein and were then superinfected with vTF7–3. Plasmids encoding the spike proteins, pT7T-G, pEnv, pEnv-Δ107, pEnv-44, and pEnv-RVG (1, 7.5, 5, 3, and 3 μg, respectively) were then transfected as described above. After incubation at 37°C for 24 hr, cell culture media were harvested and cleared of cell debris. To determine the infectious titers of pseudotype viruses, supernatants were serially diluted and used for inoculation of confluent monolayers of CD4+ or CD8+ HeLa cells. After 24 hr of infection cells were fixed with 80% acetone and expression of RV nucleoprotein was examined by direct immunofluorescence after staining with an fluorescein isothiocyanateconjugate directed against RV nucleoprotein (Centocor).
Sucrose Gradients and Western Blotting. Supernatants from complementation experiments (approximately 6×106 cells) were harvested 24 hr after transfection, and virions were pelleted through a 10% sucrose cushion at 19,000 rpm in a Beckman SW 41 rotor. Pellets were resuspended in TEN buffer (10 mM Tris, pH 7.4/50 mM NaCl/1 mM EDTA), layered on continuous 10–50% sucrose gradients, and centrifuged at 27,000 rpm in an SW 41 rotor for 1 hr. Virus proteins from 12 equal gradient fractions were resolved by SDS/PAGE and transferred to nitrocellulose membranes using a semidry transfer apparatus (Hoefer). After incubation with a blocking solution (2.5% dry milk/0.05% Tween 20 in PBS), the blot was incubated overnight with a mixture of human HIV-1 IgG (1:10,000) and rabbit sera raised against purified RV G protein (S72, 1:20,000), RV ribonucleoprotein complex (S50, 1:20,000) or a peptide deduced from the the RV matrix protein sequence (M1–B4, 1:10,000) in PBST. After three successive washes in PBST, the blot was incubated for 2 hr with a mixture of peroxidase-conjugated goat anti-human and goat anti-rabbit IgG (Dianova) diluted 1:10,000 in PBST. The blot was washed as above, stained with an Enhanced Chemiluminescence Western blot detection kit (Amersham) for 1 min, and exposed to x-ray films (Biomax MR, Kodak).
RESULTS
The HIV-1 env protein is synthesized as a precursor (gp160) that is cleaved during transport to the plasma membrane into two noncovalently associated subunits, gp120, which is involved in binding to the HIV receptor, and gp41, which contains the membrane anchor and a carboxyl-terminal cytoplasmic domain of 150 amino acids (22, 23). To determine the influence of both sequence and length of the cytoplasmic domain on formation of RV(HIV) pseudotypes, various cDNA constructs resulting in altered cytoplasmic domains were prepared in the expression plasmid pT7T (21), which is under the control of the T7 RNA polymerase promoter. Starting from a plasmid encoding the authentic HIV-1 NL-43 Env protein (pEnv), we prepared a construct that encodes a protein in which the entire Env cytoplasmic domain is replaced by the 44 amino acid cytoplasmic domain of RV G (EnvRVG). To investigate whether the considerable length of the HIV Env cytoplasmic domain may affect incorporation into RV particles, two other plasmids encoding proteins with short Env cytoplasmic domains, similar in length to that of RV G, were constructed. Env-44 represented a protein with a carboxyl-terminally truncated cytoplasmic domain, whereas in pEnv-Δ107 an internal deletion removed the membrane-proximal 107 amino acids and fused the carboxyl-terminal 43 residues to the membrane anchor domain (Fig. 1).
Transient Expression of Glycoproteins. To analyze expression of the recombinant glycoproteins on the cell surface, which represents a prerequisite for incorporation into RV particles, plasmids were transfected into BSR cells that had been infected with the recombinant vaccinia virus vTF7–3 providing T7 RNA polymerase (20). Surface expression was compared by indirect immunofluorescence with human anti-HIV IgG (18) after transfection of equal amounts of protein-encoding plasmids. Interestingly, compared with the authentic HIV-Env, the level of cell surface expression was higher for all Env constructs with modified cytoplasmic domains (data not shown). To obtain similar levels of expression, the amounts of the plasmids in transfection experiments were therefore adjusted. After transfection of 7.5 μg pT7T-Env and of 5, 3, and 3 μg of pEnv-Δ107, pEnv-44, and pEnv-RVG, respectively, surface fluorescence intensity and the number of expressing cells were similar. Representative micrographs are shown in Fig. 2. By double labeling cells expressing both RV G and each of the plasmids encoding an Env protein, no differences in the distribution of proteins on the cell surface were observed (data not shown).
We next determined whether the introduced mutations affected the function of the HIV envelope proteins in attachment to the HIV receptor and induction of membrane fusion. The adjusted amounts of plasmids were transfected into HeLa

FIG. 1. Organization of wild-type and hybrid envelope proteins. The amino acid sequence of part of the transmembrane domains (underlined) and of the entire cytoplasmic domains of mutants and wild-type proteins are shown.
cells constitutively expressing the major HIV receptor, CD4 (CD4+ HeLa, ref. 17), and that had been infected with vTF7–3. At 14 hr after transfection, each mutant caused formation of syncytia equivalent to that produced by the HIV-Env protein (Fig. 3). In addition, analysis of 35S-labeled proteins from cell extracts and media had revealed correct proteolytic cleavage of mutant precursors to generate gp120 (data not shown). Taken together, these results indicated correct transport to the cell surface, proteolytic processing, and fusogenicity of the mutants.
Rescue of SAD ΔG by Engineered Env Protein Mutants. The construction and recovery of a G-deficient RV mutant lacking the entire G gene (SAD ΔG) were described previously (7). Stocks of phenotypically complemented SAD ΔG were produced in BSR cells expressing RV G from transfected pT7T-G. Upon inoculation of noncomplementing BSR cell cultures, infection was restricted to the cells initially infected by the G-complemented particles and was not able to spread through the monolayers. The supernatants obtained from such cultures contained spikeless rhabdovirus particles that could not infect fresh BSR cells (7). As for BSR cells, infection of HeLa cells was found to require the presence of the viral spike protein.
To determine whether any of the mutant Env proteins could rescue the infectivity of SAD ΔG, BSR cells were infected with both G-complemented SAD ΔG and vTF7–3 and were then transfected with plasmids encoding wild-type or hybrid Env
FIG. 2. Surface expression of envelope protein constructs. Cells were infected with vTF7–3 and transfected with plasmids encoding Env (A), Env-Δ107 (B), Env-44 (C), Env-RVG (D), or RV G (E) (7.5, 5, 3, 3, and 1 μg, respectively). (F) Mock-transfected cells. Sixteen hours after transfection, cells were processed for indirect immunofluorescence as described.
proteins or, as a control, were transfected with RV G. After incubation for 24 hr, the supernatants were collected and the infectivity of virus particles was determined on HeLa CD4+ and HeLa CD8+ cells by direct immunofluorescence. RV G protein rescued the infectivity of SAD ΔG for both cell lines, regardless of the presence of CD4 at the cell surface (Fig. 4 A and B). In addition, rescue was observed in one of the four Env protein constructs, Env-RVG, which is the Env protein possessing the cytoplasmic tail of RV G. In contrast to RV G, however, particles complemented with Env-RVG could only infect CD4+ HeLa cells (Fig. 4 E and F) and their infectivity could be specifically neutralized by anti-HIV-1 serum (data not shown). This indicates that infection is mediated by the hybrid Env protein. Compared with RV G, the infectious titers obtained with Env-RVG were in average 25 times lower (Table 1). No fluorescent cells were observed after complementation of SAD ΔG with the wild-type Env or the constructs possessing the short gp41 cytoplasmic tails, indicating that the presence of defined sequences of the RV G tail is required for specific incorporation into the viral envelope.
Protein Composition of RV(HIV) Pseudotype Viruses. To verify the incorporation of the Env-RVG protein into the envelope of SAD ΔG particles, virions were purified by velocity centrifugation in continuous 10–50% sucrose gradients. The protein composition of gradient fractions was analyzed by Western blotting with a combination of sera directed against
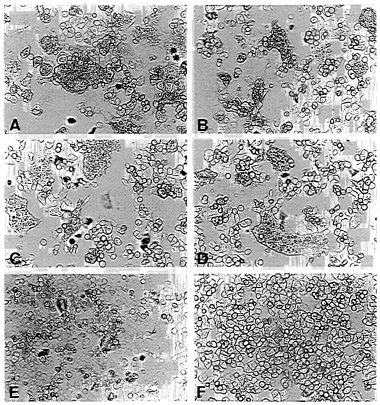
FIG. 3. Fusion activity of envelope protein constructs. CD4+ HeLa cells were infected with vTF7–3 and transfected with pEnv (A), pEnv-Δ107 (B), pEnv-44 (C), pEnv-RVG (D), and pT7T-G (E) by using the amounts of plasmids determined to yield similar surface expression of proteins (see Fig. 2). (F) Mock-transfected cells. Syncytium formation is shown after an incubation of 14 hr at 37°C.
FIG. 4. Rescue of the infectivity of SAD ΔG by engineered Env proteins. BSR cells were infected with G-complemented SAD ΔG and vTF7–3 and transfected with Env protein-coding plasmids. At 24 hr after transfection, supernatants were used to inoculate monolayers of CD4+ and CD8+ HeLa cells. Cells were then fixed after 24 hr of incubation and examined by direct immunofluorescence with a conjugate directed against RV nucleoprotein. (A and B) RV G. (C and D) HIV-Env. (E and F) Env-RVG. For A and B, 100 μl and for C-F 1 ml of the respective supernatant from 106 transfected BSR cells was used for inoculation.
RV proteins and anti-HIV IgG. Both the peak of infectivity and the majority of virus proteins were found in fractions 6 and 7 for particles pseudotyped with RV G or Env-RVG (Fig. 5). In addition to RV nucleoprotein, phosphoprotein, and matrix protein, RV(Env-RVG) pseudotype particles contained a protein reacting with the anti-HIV serum and migrating at ≈37 kDa. This corresponds well to the size predicted for the hybrid subunit composed of the HIV gp41 extracellular and transmembrane domains fused to the RV G cytoplasmic tail. In addition, a faint band of high molecular weight, which should correspond to the gp120 subunit, was identified. Since shedding of the gp120 subunit from virus particles during sucrose gradient centrifugation is apparently notorious (24), this finding was not unexpected. In contrast to Env-RVG, which was the only construct able to rescue infectivity of SAD ΔG, no HIV-related proteins could be detected in virions generated in the presence of the wild-type or the other mutant
Table 1. Rescue of SAD ΔG by recombinant spike proteins
|
|
Titers of infectious pseudotype particles per ml* |
|
|
Spike protein |
CD4+ HeLa cells |
CD8+ HeLa cells |
|
Env |
0 |
0 |
|
Env-Δ107 |
0 |
0 |
|
Env-44 |
0 |
0 |
|
Env-RVG |
1×103−4×103 |
0 |
|
RV G |
2×104−1×105 |
3×104−2×105 |
|
Mock |
0 |
0 |
|
*Titers were determined by counting fluorescing cells, assuming that nucleoprotein-expressing cells result from infection with one SAD ΔG particle. Titer ranges result from four independent experiments. |
||
FIG. 5. Protein composition of pseudotype particles. SAD ΔG particles phenotypically complemented with RV G protein or EnvRVG were analyzed by velocity centrifugation in 10–50% sucrose gradients and Western blotting. Infectivity and the majority of virus proteins are located in gradient fractions 6 and 7. In addition to the RV nucleoprotein (N), phosphoprotein (P), and matrix protein (M), RV(HIV) pseudotype viruses contain a gp41-derived chimeric transmembrane subunit protein of ≈37 kDa (“gp41”) and the gp120 subunit.
Env proteins (data not shown). Taken together, these results confirmed true incorporation of the hybrid Env-RVG protein and revealed the requirement of the RV G cytoplasmic tail to promote this process.
DISCUSSION
The possibility of generating recombinant RV pseudotype viruses with altered cell specificity was suggested by several previous observations. The recent availability of a defined, recombinant RV mutant lacking the entire G gene revealed that the only viral surface protein, or part of it, is not required to drive budding of particles from the cell surface membrane. In addition, the cytoplasmic tail of RV G was found to considerably contribute to sorting of G spikes into the virions (7). Finally, a temperature-sensitive VSV mutant containing at nonpermissive temperature only carboxyl-terminal VSV G protein fragments (25) was successfully rescued by a chimeric HIV Env protein possessing the VSV G cytoplasmic tail. This indicated that the G cytoplasmic domain sequence might be even sufficient to direct foreign proteins into virions (14).
The data presented here demonstrate that the cytoplasmic tail of the RV G contains a signal sufficient and necessary to direct a chimeric HIV Env spike protein, Env-RVG, into the envelope of “rabies” virus particles in the absence of G or of parts of it. In contrast to RV, which is able to infect all culture cells analyzed so far, the tropism of the pseudotype particles is restricted and is obviously determined by the specificity of the incorporated spike protein and its interaction with the HIV-1 CD4 receptor complex. This implies that, in contrast to RV, which enters cells after receptor-mediated endocytosis and fusion of membranes in acidic lysosomes, RV(HIV) pseudotype membranes should fuse with the cell surface membrane at neutral pH. Subsequent successful viral replication and protein expression could be monitored directly. As a consequence of the genetic deficiency for a spike protein, infection was restricted to the primarily infected cells, allowing direct determination of the yield of infectious pseudotype viruses.
In contrast to Env-RVG, wild-type HIV-1 Env or the two constructs with Env-derived cytoplasmic tails identical or similar in length to that of G were not capable to rescue the infectivity of SAD ΔG particles, although they were proteolytically processed and expressed at the cell surface and also
induced syncytium formation in CD4+ HeLa cells in a level comparable to that of Env-RVG. Thus, rather than the length of ≈44 amino acids, the specific sequence of the RV cytoplasmic tail is required to promote or to allow incorporation of spikes into the viral envelope. This is strongly supported by preliminary data on an additional Env mutant capable of rescuing the infectivity of SAD ΔG. The mutant has a cytoplasmic tail of 65 amino acids, the carboxyl terminus of which corresponds to the sequence of the RV G tail. As suggested by the ability of the G protein from another lyssavirus serotype to replace RV G (26) and by comparison of available lyssavirus G sequences, the critical motif might be located in the carboxyl-terminal region of the tail in which 7 of 14 residues are conserved with regard to other lyssaviruses (unpublished data).
As demonstrated recently, similar to VSV G (27), RV G protein possesses an independent exocytosis activity (7) and, in the presence of G, the yield of rhabdovirus particles is augmented. As estimated from the gradient analyses, an approximately 5-fold higher amount of particles complemented with G compared with RV(HIV) pseudotype particles was observed. Although substantial amounts of Env-RVG spikes were incorporated into the particles, as shown directly by the presence of the anchored, gp41-derived chimeric 37 kDa subunit (Fig. 5), the infectious titers differed by a factor of 20–25. Since the infectivity of viruses is largely governed by the features of their spike proteins and the interaction with receptors, the discrepancy in infectious titers may not directly reflect the efficiency by which the hybrid protein can substitute for RV G in formation of infectious virions.
It is not known so far whether oligomerization has an influence on the putative interaction of the cytoplasmic tail with internal virus proteins. Similar to the RV spike (28), HIV-Env may probably form trimers (29). The successful incorporation of Env-RVG suggests that this protein may present the tail(s) in an appropriate configuration. Incorporation of the monomeric cellular glycoprotein CD4 into VSV tsO45 has been reported previously (30). However, particles containing CD4, or a chimeric CD4 with the VSV cytoplasmic tail, were observed only at permissive temperature, suggesting that VSV G protein is required for incorporation of CD4 into VSV.
Our results verified that a recombinant rhabdovirus can be generated whose tropism is exclusively determined by a foreign surface glycoprotein incorporated specifically into the viral envelope. Due to the transient nature of the assay, the described system should provide a versatile and, most importantly, a safe tool in determining whether other glycoproteins might be able to direct rhabdovirus vectors to specific target cells.
We thank Veronika Schlatt and Karin Kegreiss for perfect technical assistance. This work was supported by Grants BEO 21/0310118A and 0311171 from the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie.
1. Simons, K. & Garoff, H. (1980) J. Gen. Virol. 50, 1–21.
2. Delchambre, M., Gheysen, D., Thines, D., Thiriart, C., Jacobs, E., Verdin, E., Horth, M., Burny, A. & Bex, F. (1989) EMBO J. 8, 2653–2660.
3. Gheysen, D., Jacobs, E., deForesta, F., Thiriart, C., Francotte, M., Thines, D. & DeWilde, M. (1989) Cell 59, 103–112.
4. Dong, J., Roth, M.G. & Hunter, E. (1992) J. Virol. 66, 7374– 7382.
5. Burns, J.C., Friedmann, T., Driever, W., Burrascano, M. & Yee, J.-K. (1993) Proc.Natl. Acad. Sci. USA 90, 8033–8037.
6. Yee, J.K., Miyanohara, A., LaPorte, P., Bouic, K., Burns, J. & Friedman, T. (1994) Proc. Natl. Acad. Sci. USA 91, 9564–9568.
7. Mebatsion, T, König, M. & Conzelmann, K.-K. (1996) Cell 84, 941–951.
8. Rose, J.K. & Schubert, M. (1987) in The Rhabdoviruses, ed. Wagner, R.R. (Plenum, New York), pp. 129–166.
9. Wagner, R.R. & Rose, J.K. (1996) in Fields Virology, eds. Fields, B.N., Knipe, D.M., Howley, P.M., Chomock, R.M., Melnick, J.L., Monath, T.P., Roizman, B. & Straus, S.E. (Lippincott-Raven, Philadelphia), pp. 1121–1135.
10. Schnell, M.J., Mebatsion, T. & Conzelmann, K.-K. (1994) EMBO J. 13, 4195–4203.
11. Conzelmann, K.-K. (1996) J. Gen. Virol. 77, 381–389.
12. Whitt, M.A., Chong, L. & Rose, J.K. (1989) J. Virol. 63, 3569–3578.
13. Hunter, E. (1994) Semin. Virol. 5, 71–83.
14. Owens, R.J. & Rose, J.K. (1993) J. Virol. 67, 360–365.
15. Schnell, M.J., Buonocore, L., Whitt, M.A. & Rose, J.K. (1996) J. Virol. 70, 2318–2323.
16. Mebatsion, T., Schnell, M.J., Cox, J.H., Finke, S. & Conzelmann, K.-K. (1996) Proc. Natl. Acad. Sci. USA 93, 7310–7314.
17. Maddon, P.J., Dalgleish, A.G., McDougal, J.S., Clapham, P.R., Weiss, R.A. & Axel, R. (1986) Cell 47, 333–348.
18. Prince, A.M., Reesink, H., Pascual, D., Horowitz, B., Hewlett, I., Murthy, K.K., Cobb, K.E. & Eichberg, J.W. (1991) AIDS Res. Hum. Retroviruses 7, 971–973.
19. Prince, A.M., Horowitz, B., Baker, L., Shulman, R.W., Ralph, H., et al. (1988) Proc. Natl. Acad. Sci. USA 85, 6944–6948.
20. Fuerst, T.R., Niles, E.G., Studier, F.W. & Moss, B. (1986) Proc. Natl. Acad. Sci. USA 83, 8122–8126.
21. Conzelmann, K.-K. & Schnell, M.J. (1994) J. Virol. 68, 713–719.
22. Wain-Hobson, S., Sonigo, P., Danos, O., Cole, S. & Alizon, M. (1985) Cell 40, 9–17.
23. Willey, R.L., Bonifacino, J.S., Potts, B.J., Martin, M.A. & Klausner, R.D. (1988) Proc. Natl. Acad. Sci. USA 85, 9580–9584.
24. Schneider, J., Kaaden, O., Copeland, T.D., Orozlan, S. & Hunsmann, G. (1986) J. Gen. Virol. 67, 2533–2538.
25. Metsikkö, K. & Simons, K. (1986) EMBO J. 5, 1913–1920.
26. Mebatsion, T., Schnell, M.J. & Conzelmann, K.-K. (1995) J. Virol. 69, 1444–1451.
27. Rolls, M.M., Webster, P., Balba, N.H. & Rose, J.K. (1994) Cell 79, 497–506.
28. Gaudin, Y., Tuffereau, C., Segretain, D., Knossow, M. & Flamand, A. (1992) Virology 187, 627–632.
29. Gelderblom, H.R. (1991) AIDS 5, 617–638.
30. Schubert, M., Joshi, B., Blondel, D. & Harmison, G.G. (1992) J. Virol. 66, 1579–1589.





