
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector
LUIGI NALDINI *, ULRIKE BLÖMER, FRED H. GAGE, DIDIER TRONO †, AND INDER M. VERMA
The Salk Institute for Biological Studies, P.O. Box 85800, San Diego, CA 92186–5800
ABSTRACT We describe the construction of a safe, replication-defective and efficient lentiviral vector suitable for in vivo gene delivery. The reverse transcription of the vector was found to be a rate-limiting step; therefore, promoting the reaction inside the vector particles before delivery significantly enhanced the efficiency of gene transfer. After injection into the brain of adult rats, sustained long-term expression of the transgene was obtained in the absence of detectable pathology. A high proportion of the neurons in the areas surrounding the injection sites of the vector expressed the transduced β-galactosidase gene. This pattern was invariant in animals sacrificed several months after a single administration of the vector. Transduction occurs by integration of the vector genome, as it was abolished by a single amino acid substitution in the catalytic site of the integrase protein incorporated in the vector. Development of clinically acceptable derivatives of the lentiviral vector may thus enable the sustained delivery of significant amounts of a therapeutic gene product in a wide variety of somatic tissues.
Gene therapy is a promising new form of medicine because of its potential to reverse the genetic causes of several innate and acquired diseases (1, 2). The currently available methods of gene delivery suffer from several major limitations that curtail the realization of these high expectations. Nonviral methods are inefficient and only attain a transient expression of the transgene, while no viral vector yet offers a satisfactory combination of efficacy of gene transfer, sustained transgene expression, and biosafety (3, 4). Adenoviral vectors allow highly efficient delivery of the transgene in most tissues in vivo, but its expression is transient. This is mostly due to the immune response against the transduced cells, which also express a low level of viral proteins (5–9). Vectors derived from oncoretroviruses, such as the Moloney leukemia virus (MLV), integrate the transgene in the genome of the target cells without transferring any viral gene, two properties considered crucial for the sustained expression of the transgene (10). These prototypic retroviral vectors, however, are severely restricted in their potential targets, as they only transduce cells that divide shortly after infection (11). Consequently, they are most often employed in demanding ex vivo protocols of gene transfer (12–16). Furthermore, transcriptional shutoff of the transgene after reimplantation in vivo of the transduced cells is frequently observed (17, 18).
We have previously described a human lentivirus (HIV)-based vector that can transduce nondividing cells. As the particles are pseudotyped with the envelope of the vesicular stomatitis virus (VSV), the vector can serve to introduce genes into a broad range of tissues and can be used in vivo (19). Furthermore, we demonstrate that in vivo gene transfer is dependent on a functional integrase protein and that transgene expression is sustained for several months without detectable pathology. These characteristics suggest that lentiviral vectors could play a major role in the arena of gene therapy.
MATERIALS AND METHODS
Plasmid Construction. The construction of the HIV-derived plasmids pCMVΔR9, pHR′-CMVLacZ, and pHR′-CMVLucif has been described (19). Plasmid pCMVΔR8.2 was derived from pCMVΔR9 by substituting, for a 2.7-kbp SalI-BamHI fragment, a 0.5-kbp SalI-NotI fragment (obtained by a PCR that added to the proviral HIV-1 NL4–3 DNA sequence, at the end of the vpu gene, a stop codon in the env reading frame followed by a NotI site) and a 0.85-kbp NotI-BamHI fragment from pHR′, containing a NotI linker introduced at the BglII site 7620 in the HIV-1 HXB2D sequence, the Rev responsive element and the splice acceptor site for the second exon of the tat and rev genes. The construction deletes nucleotides 6308–7611 of the HIV-1 NL4–3 genome, encompassing a large portion of the env coding sequence, from the packaging plasmid. Its remaining HIV-1 sequences are derived from plasmid pR7 (20) [nucleotides 708–1506 and 7620–9416 in the HXB2D sequence (21)] and NL4.3 (nucleotides 1507– 6307). Plasmid pMD.G drives the expression of the VSV.G reading frame from the human cytomegalovirus immediateearly promoter (hCMV) and contains β-globin sequences upstream (exons 2 and 3, intervening sequence 2) and downstream (polyadenylylation site) of it. All plasmids contain the simian virus 40 origin of replication in the backbone.
Production and Assays of Vectors. Human kidney 293T cells (1.5×106) were plated on 10-cm plates and transfected the following day with 15 μg of pCMVΔR8.2, 20 μg of either pHR′ plasmid, and 5 μg of pMD.G by calcium phosphate DNA precipitation (22). Conditioned medium was harvested 62 hr after transfection, cleared of debris by low-speed centrifugation, filtered through 0.45-μm filters, and assayed for p24 Gag antigen by ELISA (DuPont). For transduction of rat 208F fibroblasts, cells were infected overnight with serial dilutions of vector stock in culture medium supplemented with 8 μg of polybrene per ml. After medium replacement, the cells were further incubated for 36 hr, and expression of β-galactosidase (β-gal) was scored by 5-bromo-4-chloro-3-indolyl β-D-galac-
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: MLV, Moloney leukemia virus; VSV, vesicular stomatitis virus; hCMV, human cytomegalovirus immediate-early promoter; β-gal, β-galactosidase.
|
* |
Present address: Somatix Therapy Corporation, 850 Marina Village Parkway, Alameda, CA 94501. |
|
† |
To whom reprint requests should be addressed, e-mail: didier_trono@qm.salk.edu |
toside (X-Gal) staining. Titers were calculated by counting the number of foci of blue cells per well and dividing it by the dilution factor. Expression of luciferase was assayed by washing the cultures twice with TBS (50 mM Tris·HCl, pH 7.8/130 mM NaCl/10 mM KCl/5 mM MgCl2), extracting them with 0.5% Nonidet P-40 in TBS containing 1 mM dithiotreithol, and measuring luminescence in a luminometer. Concentrated vector stocks were prepared by ultracentrifugation of conditioned medium at 50,000×g for 90 mm, resuspension of the pellets in a small volume (half percent of the starting volume of medium) of TBS containing 10 mM MgCl2, pooling, and incubation with or without 0.1 mM of each of the four deoxynucleotides (dNTPs), 3 mM spermine, and 0.3 mM spermidine for 2 hr at 37°C. After dilution in TBS, the vector particles were concentrated by a second ultracentrifugation, and the final pellet was resuspended in a very small volume (half thousandth of the starting volume of medium) of sterile saline containing 4 μg of polybrene per ml. Resuspension of the second pellet required prolonged incubation and pipetting. Stocks were stored frozen at −80°C and titered before and after freezing. MLV-based β-gal vector was similarly produced by the transient transfection into 293T cells of a plasmid driving the MLV gag and pol genes from the hCMV promoter (the vector pLNL-CMVLacZ, which carries the same β-gal expression cassette as the HIV-based vector) and the pMD.G plasmid. Before injection, all batches of vector were tested for the absence of replication-competent virus by infecting HeLa cells at high multiplicity of infection (HeLa P4 cells, which express CD4 and contain an integrated LacZ gene driven by the HIV long terminal repeat), obtained from the American Type Culture Collection stock (23), and HeLa cells previously transduced with a lentiviral vector carrying a different reporter gene and selected for its expression. The transduced cells were passaged two to three times, and the conditioned medium was tested for transfer of the markers to virgin 208F fibroblasts or for β-gal-inducing activity on P4 cells.
In Vivo Delivery of Vectors and Immunostaining of Sections from the Injected Brains. All animal procedures were performed according to an institution-approved protocol and under strict biological containment. Adult female Fischer 344 rats were anesthetized (ketamine, 44 mg/kg; acepromazine, 0.75 mg/kg; and xylazine, 4 mg/kg, in 0.9% NaCl i.p.) and positioned in a stereotactic head frame. After midline incision of the skin, holes were drilled in the appropriate locations in the skull with a dental bur, and a 5-μl Hamilton syringe with needle was used to slowly inject 2 μl of vector suspension in sterile saline into the striatum (AP +0.2, ML±3.5, DV −4.5) and hippocampus (AP −3.5, ML 3.0, DV −4.0) bilaterally. Holes were then filled with bone wax, the incision was sutured, and the animals were returned to their cages. For assessing transduction, the rats were deeply anesthetized and perfused with 4% cold paraformaldehyde and 0.2% glutaraldehyde intracardially. The brains were removed, post-fixed at 24 hr, saturated in 30% sucrose, and sectioned on a freezing microtome (50-μm serial sections). For light microscopy, sections were incubated with rabbit anti-β-gal antibodies (1:1000; Cortex Pharmaceuticals, Irvine, CA) and stained using avidinbiotin-peroxidase (Vectastain ABC Elite; Vector Laboratories) and diaminobenzidine. For immunofluorescence labeling, mouse monoclonal anti-NeuN and guinea pig anti-GFAP antibodies, secondary antibodies coupled to the fluorescent markers CY5, dichlorotriazinyl amino fluorescein, and Texas Red were also used, and the mounted sections were analyzed by confocal scanning laser microscopy (Bio-Rad model MRTC600). Fluorescent signals were collected, digitally color enhanced, and superimposed.
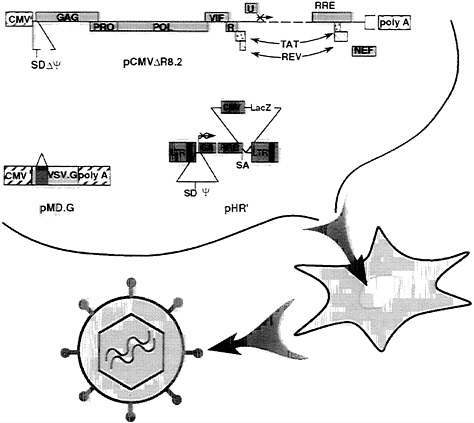
FIG. 1. Schematic of the generation of lentiviral vector. The relevant portions of the three plasmids cotransfected into 293T cells are depicted at the top; their contribution to the vector particles harvested in the transfectant conditioned medium are depicted at the bottom. pCMVΔR8.2, the packaging construct, provides all vector proteins but the envelope. The viral genes are expressed from the hCMV promoter and the insulin polyadenylylation site. Proviral 5′ leader and Ψ sequences have been deleted together with a large portion of the env gene. A translation stop codon was inserted upstream of the remaining env segment. The major 5′ splice donor site (SD) has been conserved. pHR′, the transducing vector, provides the vector genome. The viral long terminal repeats (LTRs) and the Ψ sequence are indicated. The gag gene is truncated after 350 bp and is out of frame (×), and it follows the Rev responsive element (RRE) and a splice acceptor site (SA). The position of a hCMV-driven expression cassette for the β-gal cDNA (LacZ) is shown. pMD.G encodes the heterologous VSV envelope that pseudotypes the vector. The hCMV promoter drives the VSV.G reading frame, which has β-globin sequences upstream (exons 2 and 3, intervening sequence 2) and downstream [poly(A) site].
RESULTS
Generation of High-Titer Lentiviral Vector with Improved Biosafety. The lentiviral vector is produced by the transient transfection of three plasmids into human kidney 293T cells (Fig. 1). Two complementary constructs were derived from the HIV-1 proviral DNA. The packaging construct expresses all HIV trans-acting proteins but the envelope from heterologous transcription signals. The transducing vector, pHR′, retains all HIV cis-acting sequences required for its transfer to the target cell, now framing an expression cassette for the transgene. The Escherichia coli β-gal gene and the firefly luciferase gene driven by the hCMV promoter were used as reporter genes in this study. A novel packaging plasmid, pCMVΔR8.2, was constructed to improve the biosafety of the vector. It was derived from the previously described plasmid pCMVΔR9 by deletion of 1.4 kbp from the env gene sequence, downstream of the (functional) vpu gene, and substitution with an inframe stop codon. The deletion of env sequences did not affect the yield or the transduction efficiency of the vector particles. The third plasmid, pMD.G, encodes the heterologous VSV envelope and is used for pseudotyping the particles generated by the other two constructs.
High-titer stocks were obtained by pelleting the vectors from the transfectants conditioned medium by two rounds of ultracentrifugation at 50,000×g for 90 min, as described for VSV.G-pseudotyped MLV vectors (24). Vector yield averaged 50% for each centrifugation step, both measured as p24 Gag equivalent and as transducing units in 208F rat fibroblasts. The overall yield of the protocol was ≈25%, with an increase in transducing titer of three orders of magnitude. As the transfectant conditioned medium contained, on average, 4×105 transducing units/ml, titers of 2–4×108 transducing units/ml were routinely achieved. When normalized to the content of p24 Gag antigen, the transducing activity of the vector was not affected by the centrifugation steps, averaging 4500 transducing units per ng of p24 in 208F fibroblasts. The absence of replication-competent virus from all vector stocks was proven by the lack of spreading or mobilization of reporter genes from transduced cells (see Materials and Methods).
Transduction Is Enhanced by Promoting Intraparticle Reverse Transcription. We previously showed that the HIV-based vector is less efficient in cells arrested in G0 than in cells growing or arrested in G1/S or G2. This became more marked the longer the culture had been in G0 and correlated with a progressive block in reverse transcription of the vector genome (19). We decided to test whether promotion of the reaction inside the vector particle, as described by Zhang et al. (25) for the HIV virus, would enhance its transducing activity. Upon incubation with a mixture of the four dNTPs and the polyamine spermine and spermidine for 2 hr at 37°C, the transduction of a luciferase reporter gene increased 2-fold in growing 208F fibroblasts and up to 5-fold in G0-arrested cells, becoming independent of the length of growth-arrest of the culture (Fig. 2). We then tested whether a similar effect was observed in the transduction of terminally differentiated cells in vivo.
High Efficiency of Transduction by the Lentiviral Vector of Rat Brain Neurons in Vivo. Stocks of lentiviral β-Gal vector, matched for the content of p24 Gag antigen, were incubated
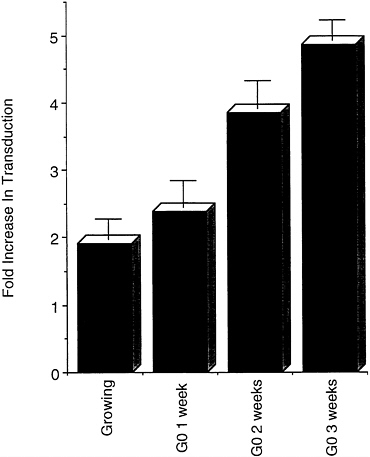
FIG. 2. Transduction is enhanced by promoting reverse transcription inside the lentiviral vector before infection. Rat 208F cells were plated at low density and either infected the following day (Growing) or grown to confluence, switched to medium containing 5% calf serum and 2 μM dexamethasone (11), and further incubated for the indicated number of weeks before infection (G0 x weeks, where x=number of weeks) with lentiviral luciferase vector preincubated with or without dNTPs and polyamines. Transduction was scored by comparing luminescence in cell extracts 48 hr after infection with both vectors. Plotted is the mean±SEM increase in transduction by the treated vector over the level induced by the untreated vector, calculated from infections performed at three different multiplicities of infection over a hundred-fold range in a typical experiment. In the cells arrested in G0, the transduction by the untreated vector was 28% (G0 1 week), 18% (G0 2 weeks), and 13% (G0 3 weeks) of that scored in growing cells.
with or without dNTPs and polyamines for 2 hr at 37°C before injection. β-Gal MLV-based vector, pseudotyped with the VSV envelope, was also treated with the dNTPs and matched for transducing activity on 208F cells. Vector suspension (2 μl) was injected into the corpus striatum and hippocampus of both sides of the brain of anesthetized adult female Fisher rats. Groups of three animals each were sacrificed at increasingly longer time intervals (2 weeks, 6 weeks, 3 months) from a single vector administration. Transduction rate was assessed for each injected brain by serial cryostatic sectioning, and immunostaining of each sixth section for β-gal. The relative proportion of transduced cell types was estimated by immunofluorescence costaining of representative sections with antibodies directed against β-gal and cell type-specific markers and confocal microscope analysis.
FIG. 3 . Immunohistochemical staining for β-gal of sections from the corpus striatum (Left) or the hippocampus (Right) of rat brains injected with a single dose of β-gal vector 2 weeks or 3 months before. The viral parent of the injected vector is indicated on the left, as also is the pretreatment of vector with dNTPs and polyamines. The HIV integrase mutant carries a single amino acid replacement in the catalytic site (D64V), which severely reduces the activity of the enzyme incorporated in the vector. Note the lower magnification in the three bottom rows. The lentiviral vector achieves an efficient gene transfer into cells of typical neuronal morphology; the transduction is significantly enhanced by the pretreatment with dNTPs and is abolished by the integrase mutation. No change in the pattern and density of β-gal-expressing cells is observed even after 3 months. In contrast, the MLV-derived vector shows a poor gene transfer, exclusively in cells of glial morphology, and no expression is detected after 3 months. The sections shown are representative of those obtained in the proximity of each injected site in all animals in the experimental group. One of every six serial sections from the injected brains was stained, and transduced cells could be detected in up to six stained sections, for the lentiviral vector treated with dNTPs.
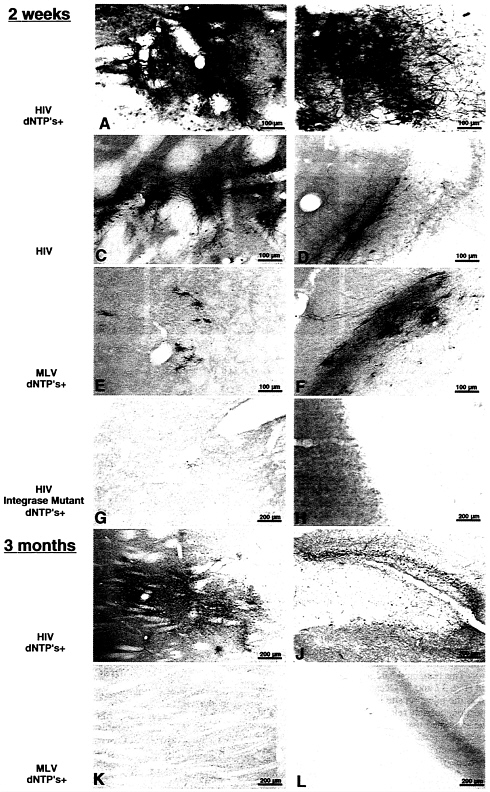

FIG. 4. Immunofluorescence staining for β-gal and the neuronal marker NeuN of sections from the corpus striatum (Upper) and the hippocampus (Lower) of a rat brain injected 6 weeks earlier with lentiviral β-gal vector pretreated with dNTPs. Shown are confocal microscope images obtained from each individual staining (Middle and Right) and their overlap (Left). A significant fraction of the neurons in the area surrounding the injection site expresses the transgene.
A conspicuous fraction of cells was reproducibly transduced by the lentiviral vector around the injection site, and cells expressing β-gal could be detected up to several millimeters away from it, as shown in Fig. 3 A, B, I, and J. The majority of transduced cells showed neuronal morphology and, when costained for immunofluorescence, expressed the neuronal marker protein NeuN (ref. 26; Fig. 4). Treatment of the lentiviral vector with dNTPs before delivery enhanced significantly its transduction rate in vivo (Fig. 3, compare A and B with C and D). An estimate of the average density of transduced cells indicated at least a 2-fold increase (data not shown). The MLV-based vector displayed a comparatively poor transduction rate (Fig. 3 E and F). Cells transduced by the MLV vector were smaller and different from those predominantly stained in lentiviral-injected brains and looked like oligodendrocytes and astrocytes. As expected, none could be found by immunofluorescence analysis that expressed β-gal together with the neuronal marker NeuN (data not shown).
Long-Term Expression of the Transgene in Vivo with No Detectable Pathology. The pattern and the estimated density of cells expressing β-gal in the injected areas did not change appreciably with the time elapsed since administration of the lentiviral vector. This remained true for 3 months, the longest time examined (Fig. 3 I and J). Occasionally, some detectable tissue damage in the injection site was observed at the earlier times after injection, possibly related to local bleeding, but the majority of the examined tissues were remarkably unaffected by the delivery and expression of the transgene, as also seen by hematoxylin and eosin staining (data not shown). Furthermore, the distribution of glial cells was not altered in the injected areas, as documented by immunofluorescent staining for the expression of the astrocytic marker glial fibrillary acidic protein (data not shown). In brains injected with the MLV-based vector and examined at late times after administration, almost no cells were detected that still expressed the transgene (Fig. 3 K and L).
Transduction in Vivo Occurs by Integration of the Transgene. To verify whether transduction in vivo occurs by integration of the transgene, we used a β-gal vector incorporating a mutant form of integrase carrying a single amino acid substitution (D64V) in the catalytic site. The generation and biological properties of this mutation, both in the context of the HIV virus and the lentiviral vector, have been described (27),(19). While the mutation severely decreases the activity of the enzyme in vitro and in vivo, it has no detectable effect on the preceding steps of the infection pathway, including particle budding, entry into the target cell, reverse transcription, and nuclear import. It did, though, reduce transduction by the β-gal vector in vitro to a residual activity <2% of that of the wild-type. For testing its effect in vivo, a concentrated stock of vector incorporating the mutant enzyme was prepared as above, matched to the wild-type for content of p24 Gag antigen, and injected into the brain of three rats. In all injected sites, examined either after 2 or 6 weeks, from none to a couple of rare cells could be detected expressing β-gal, providing genetic evidence that expression of the transgene in vivo depends on its integration (Fig. 3 G and H).
DISCUSSION
The design of a viral vector system relies upon the segregation in the viral genome of cis-acting sequences involved in its transfer to target cells from trans-acting sequences encoding the viral proteins. The prototype vector particle is assembled by viral proteins expressed from constructs stripped of all cis-acting sequences. These sequences are instead used to frame the expression cassette for the transgene driven by an heterologous promoter. As the particle will transfer only the latter construct, the infection process is limited to a single round without spreading. The safety and efficiency of an actual vector system depends on the extent to which this ideal, complete segregation of cis- and trans-acting functions is obtained.
In the case of HIV, several sequences have been implicated by deletion studies in the encapsidation and dimerization of viral RNA. In contrast to MLV, where the Ψ sequence, located in the 5′-untranslated leader downstream of the major splice donor site, strongly contributes to RNA packaging, the role of the corresponding region in the HIV genome seems more to discriminate genomic from spliced transcripts than to promote efficient encapsidation (28–30). Additional, and possibly more important, sequences have been identified in the transcribed long terminal repeats and 5′ leader sequences upstream of the major splice donor site (31–35). A major caveat, however, is that reconstitution of the packaging function has not yet been reported by juxtaposition of the identified sequence(s) to heterologous RNA (36). Thus, the packaging signal of HIV is either highly sequence-specific or, more likely, multipartite and distributed over a rather large stretch of its 5′ sequence (37).
Several features of the HIV-derived packaging plasmid described here prevent its transfer to the target cells. The combined modifications of the 5′ end delete or disrupt all structural motifs to date implicated in RNA encapsidation and dimerization, with the possible exception of the 5′ portion of the gag sequence (30, 38, 39). Recently, McBride and Panganiban (35) reported the encapsidation efficiency of HIV-1 transcripts carrying deletions of the 5′ leader sequence; relative to the wild-type counterpart, it was reduced to <0.1 for transcripts derived from a construct comparable to pCMVΔR8.2 and to <0.02 in the presence of competing wild-type RNA (35). Furthermore, the deletion of both long terminal repeats and of the primer binding site from the packaging plasmid would prevent reverse transcription and integration of any encapsidated transcript not recombined with the vector RNA. The transducing vector, on the other hand, is endowed with a full complement of the cis-acting sequences not identified until now, which allows its proficient transfer to the target cell.
It is well recognized that the retroviral infection is an inefficient process. Once the content of virions is delivered inside the target cell, uncoating, reverse transcription, interaction with cytoplasmic chaperones and the nuclear import machinery, and maturation to an integration-competent complex take place. These events, the mechanism of which is still poorly understood, can result in degradation and arrest at a stable intermediate, as well as integration of the viral genome (40). Partial reverse transcripts have been detected in HIV and MLV virions (41–43). Recently, it was shown that viral DNA synthesis can be promoted inside intact HIV-1 particles by exposure to dNTPs and magnesium chloride and that the efficiency of the reaction can be increased by the addition of the polyamine spermine and spermidine (25, 44, 45). The resulting HIV-1 virions exhibit an increased infectivity in primary T lymphocytes infected before activation, a setting in which reverse transcription was previously demonstrated to be a rate-limiting step (46–48). The stimulation of reverse transcription inside virions was also shown to increase the transduction efficiency of MLV-based retroviral vectors in dividing targets (49). Here, we find that it significantly augments the efficacy of gene transfer mediated by the lentiviral vector. This effect was most pronounced in nondividing cells and could also be observed in vivo. Performing such in vitro reverse transcription reactions before injecting the vector may be critical for some nonproliferating targets that maintain low cytoplasmic pools of dNTPs (50, 51).
The crucial advantage of the lentiviral vector is its integration in the genome of nondividing cells. This was proven here by the dependence of the transduction on the incorporation of a functional integrase in the vector. At least in the case of the brain, the only tissue studied so far, this provides for long-term sustained expression of the transgene. No decrease in the extent of β-gal immunoreactivity was observed even 3 months after a single vector administration. Given the recent report of predominant transgene-directed immune responses in animals transduced with adenoviral vectors (52), it remains to be determined whether the brain represents an immune haven, as long suspected, or whether the adenoviral proteins expressed by the transduced cells played a critical adjuvant role.
Retroviruses are thought to select, through poorly understood mechanisms, active chromatin sites for the integration of their genome (40). This may explain why gene delivery methods based on MLV-derived vectors often suffer from the transcriptional shutoff of the transgene, as was observed in this study, when the transduced cells return to a nonproliferating status and presumably revise their pattern of chromatin expression. The ability of the lentiviral vector to integrate in nondividing cells may allow for the selection of stably open chromatin sites, thus ensuring against the transcriptional silencing of the transgene.
The high prevalence of neurons observed among the transduced cell types in the brain may be due both to the neurotropism of the envelope of the VSV, a rhabdovirus (53), and to a preferential expression of the hCMV promoter in neurons, as recently observed with transgenic animals (54). It may also reflect preferential long-term expression in nondividing cells, for the reasons discussed above.
A major issue now concerns the biosafety of the lentiviral vector. The novel feature of the packaging plasmid described in this paper precludes the generation of wild-type HIV viruses, even by unlikely rearrangement and recombination events, given the actual absence of most of HIV env sequences in all three plasmids. In the previously described plasmid pCMVΔR9, the env reading frame was blocked by insertion of a linker containing multiple stop codons. The use of a separate plasmid encoding a heterologous envelope makes it extremely unlikely that a replication-competent recombinant be generated. This would require multiple recombination events between different plasmids and/or endogenous retroviral sequences, including recombination between nonhomologous sequences. Careful scrutiny and improvement of the vector-producing system, including evaluation of the minimal set of viral genes required for efficient packaging of the vector and generation of stable packaging systems better amenable to monitoring, are now required.
We are extremely grateful to R.Pomerantz and colleagues for communicating their results before publication. We thank members of the Verma, Gage, and Trono laboratories for helpful suggestions. L.N. was supported by the American-Italian Cancer Foundation, and U.B. was supported by the Deutsche Forschungsgemeinschaft (DFG). This work is supported by grants from the National Institutes of Health and the American Cancer Society (I.M.V.), U.S. Public Health Service Grant AI37510 (D.T.); National Institutes of Health Grants AG10435 and 08514 and Hollfelder Foundation (F.H.G.); and the H.N. and Frances Berger Foundation (D.T., I.M.V., and F.H.G.). I.M.V. is an American Cancer Society Professor of Molecular Biology and D.T. is a Pew Scholar.
1. Mulligan, R.C. (1993) Science 260, 926–932.
2. Verma, I. (1994) Mol. Med. 1, 2–3.
3. Crystal, R.G. (1995) Science 270, 404–410.
4. Leiden, J.M. (1995) New Engl. J. Med. 333, 871–873.
5. Yang, Y., Li, Q., Ertl, J. & Wilson, J.M. (1994) Proc. Natl. Acad. Sci. USA 91, 4407–4411.
6. Yang, Y., Ertl, J. & Wilson, J.M. (1994) Immunity 1, 433–442.
7. Dai, Y., Schwarz, E.M., Gu, D., Zhang, W.-W., Sarvetnick, N. & Verma, I.M. (1995) Proc. Natl. Acad. Sci. USA 92, 1401–1405.
8. Knowles, R.M., Hohneker, K.W., Zhou, Z., Olsen, J.C., Noah, T.L., Hu, P.-C., Leigh, M., Engelhardt, J.F., Edwards, L.J., Jones, K.R., Grossman, M., Wilson, J.M., Johnson, L.G. & Boucher, R.C. (1995) New Engl. J. Med. 333, 823–831.
9. Kremer, E.J. & Perricaudet, M. (1995) Br. Med. Bull. 51, 31–44.
10. Miller, A.D., Miller, D.G., Garcia, J.V. & Lynch, C.M. (1993) Methods Enzymol. 217, 581–599.
11. Miller, D.G., Adam, M.A. & Miller, A.D. (1990) Mol. Cell. Biol. 10, 4239–4242.
12. Dai, Y., Roman, M., Naviaux, R.K. & Verma, I.M. (1992) Proc. Natl. Acad. Sci. USA 89, 10892–10895.
13. Blaese, R.M., Culver, K.W., Miller, A.D., Carter, C.S., Fleisher, T., Clerici, M., Shearer, G., Chang, L., Chiang, Y., Tolstoshev, P., Greenblatt, J.J., Rosenberg, S.A., Klein, H., Berger, M., Mullen, C.A., Ramsey, W.J., Muul, L., Morgan, R.A. & Anderson, W.F. (1995) Science 270, 475–480.
14. Bordignon, C., Notarangelo, L.D., Nobili, N., Ferrari, G., Casorati, G., Panina, P., Mazzolari, E., Maggioni, D., Rossi, C., Servida, P., Ugazio, A.G. & Mavilio, F. (1995) Science 270, 470–475.
15. Kohn, D.B., Weinberg, K.J., Nolta, J.A., Heiss, L.N., Lenarski, C., et al. (1995) Nat. Med. 1, 1017–1023.
16. Grossman, M., Radar, D.J., Muller, D.W.M., Kolansky, D.M., Kozarsky, K., Clark, B.J.I., Stein, E.A., Lupien, P.J., Brewer, H.B., Raper, S.E. & Wilson, J.M. (1995) Nat. Med. 1, 1148– 1154.
17. Scharfmann, R., Axelrod, J.H. & Verma, I.M. (1991) Proc. Natl. Acad. Sci. USA 88, 4626–4630.
18. Palmer, T.D., Rosman, G.J., Osborne, W.R.A. & Miller, A.D. (1991) Proc. Natl. Acad. Sci. USA 88, 1330–1334.
19. Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F.H., Verma, I.M. & Trono, D. (1996) Science 272, 263–267.
20. Trono, D., Feinberg, M.B. & Baltimore, D. (1989) Cell 59, 113–120.
21. Ratner, L., Haseltine, W., Patarca, R., Livak, K.J., Starcich, B., Josephs, S.F., Donan, E.R., Rafalski, J.A., Whitehorn, E.A., Baumeister, K., Ivanoff, L., Petteway, S.R., Pearson, M.L., Lautenberger, J.A., Papas, T.S., Gharayeb, J., Chang, N.T., Gaho, R.C. & Wong-Staal, F. (1985) Nature (London) 313, 277–284.
22. Chen, C. & Okayama, H. (1987) Mol. Cell. Biol. 7, 2745–2752.
23. Charneau, P., Mirambeau, G., Roux, P., Paulous, S., Buc, H. & Clavel, F. (1994) J. Mol. Biol. 241, 651–662.
24. Burns, J.C., Friedman, T., Driever, W., Burrascano, M. & Yii, J.-K. (1993) Proc. Natl. Acad. Sci. USA 90, 8033–8037.
25. Zhang, H., Dornadula, G. & Pomerantz, R.J. (1996) J. Virology 70, 2809–2824.
26. Mullen, R.J., Buck, C.R. & Smith, A.M. (1992) Development (Cambridge, U.K.) 116, 201–211.
27. Leavitt, A.D., Robles, G., Alesandro, N. & Varmus, H.E. (1996) J. Virol. 70, 721–728.
28. Lever, A., Gottlinger, H., Haseltine, W. & Sodroski, J. (1989) J. Virol. 63, 4085–4087.
29. Aldovini, A. & Young, R.A. (1990) J. Virol. 64, 1920–1926.
30. Luban, J. & Goff, S.P. (1994) J. Virol. 68, 3784–3793.
31. Kim, H.-J., Lee, K. & O’Rear, J.J. (1994) Virology 198, 336–340.
32. Vicenzi, E., Dimitrov, D.S., Engelman, A., Migone, T.-S., Purcell, D.F.J., Leonard, J., Englund, G. & Martin, M.A. (1994) J. Virol. 68, 7879–7890.
33. Geigenmuller, U. & Linial, M.L. (1996) J. Virol. 70, 667–671.
34. Paillart, J.-C., Skripkin, E., Ehresmann, B., Ehresmann, C. & Marquet, R. (1996) Proc. Natl. Acad. Sci. USA 93, 5572–5577.
35. McBride, M.S. & Panganiban, A. (1996) J. Virol. 70, 2963–2973.
36. Berkowitz, R.D., Hammarskjöld, M.-L., Helga-Maria, C., Rekosh, D. & Goff, S.P. (1995) Virology 212, 718–723.
37. Kaye, J.F., Richardson, J.H. & Lever, A.M.L. (1995) J. Virol. 69, 6588–6592.
38. Buchschacher, G.L.J. & Panganiban, A.T. (1992) J. Virol. 66, 2731–2739.
39. Parolin, C., Dorfman, T., Palú, G., Göttlinger, H. & Sodroski, J. (1994) J. Virol. 68, 3888–3895.
40. Coffin, J.M. (1990) in Virology, eds. B.N.Fields & D.M.Knipe (Raven, New York), pp. 1437–1500.
41. Trono, D. (1992) J. Virol. 66, 4893–4900.
42. Lori, R., di Marzo Veronese, F., De Vico, A.L., Lusso, P., Reitz, M.S., Jr., & Gallo, R.C. (1992) J. Virol. 66, 5067–5074.
43. Zhu, J. & Cunningham, J.M. (1993) J. Virol. 67, 2385–2388.
44. Zhang, H., Zhang, Y., Spicer, T.P., Abbott, L.Z., Abbott, M. & Poiesz, B.J. (1993) AIDS Res. Hum. Retroviruses 9, 1287–1296.
45. Zhang, H., Zhang, Y., Spicer, T., Henrard, D. & Poiesz, B. (1995) J. Virol. 69, 3675–3682.
46. Zack, J.A., Haislip, A.M., Krogstad, P. & Chen, I.S.Y. (1992) J. Virol. 66, 1717–1725.
47. Gao, W.-Y., Cara, A., Gallo, R.C. & Lori, F. (1993) Proc. Natl. Acad. Sci. USA 90, 8925–8928.
48. Meyerhans, A., Vartanian, J.-P., Hultgren, C., Plikat, U., Karlsson, A., Wang, L., Eriksson, S. & Wain-Hobson, S. (1994) J. Virol. 68, 535–540.
49. Zhang, H., Duan, L.-X., Dornadula, G. & Pomerantz, R.J. (1995) J. Virol. 69, 3929–3932.
50. Cohen, A., Barankiewica, J., Lederman, H.M. & Gelfand, E.W. (1983) J. Biol. Chem. 258, 12334–12340.
51. McCormick, P.J., Danhauser, L.L., Rustum, Y.M. & Bertram, J.S. (1983) Biochem. Biophys. Res. Comm. 755, 36–40.
52. Tripathy, S.K., Black, H.B., Goldwasser, E. & Leiden, J.M. (1996) Nat. Med. 2, 545–550.
53. Wagner, R.R. (1987) in Rhabdovirus Biology and Infection: An Overview, ed. Wagner, R.R. (Plenum, New York), pp. 9–74.
54. Baskar, J.F., Smith, P.P., Nilaver, G., Jupp, R.A., Hoffmann, S., Peffer, N.J., Tenney, D.J., Colberg-Poley, A.M., Ghazal, P. & Nelson, J.A. (1996) J. Virol. 70, 3207–3214.







