
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese, Co-chairs, held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Development of HIV vectors for anti-HIV gene therapy
(lentivirus/T cell/packaging cell line/vesicular stomatitis virus glycoprotein G)
ERIC POESCHLA*, PIERRE CORBEAU*, AND FLOSSIE WONG-STAAL*†‡
Departments of *Medicine and †Biology, Mail Code 0665, University of California at San Diego, 9500 Gilman Drive, La Jolla, CA 92093–0665
ABSTRACT Current gene therapy protocols for HIV infection use transfection or murine retrovirus mediated transfer of antiviral genes into CD4+ T cells or CD34+ progenitor cells ex vivo, followed by infusion of the gene altered cells into autologous or syngeneic/allogeneic recipients. While these studies are essential for safety and feasibility testing, several limitations remain: long-term reconstitution of the immune system is not effected for lack of access to the macrophage reservoir or the pluripotent stem cell population, which is usually quiescent, and ex vivo manipulation of the target cells will be too expensive and impractical for global application. In these regards, the lentivirus-specific biologic properties of the HIVs, which underlie their pathogenetic mechanisms, are also advantageous as vectors for gene therapy. The ability of HIV to specifically target CD4+ cells, as well as non-cycling cells, makes it a promising candidate for in vivo gene transfer vector on one hand, and for transduction of non-cycling stem cells on the other. Here we report the use of replication-defective vectors and stable vector packaging cell lines derived from both HIV-1 and HIV-2. Both HIV envelopes and vesicular stomatitis virus glycoprotein G were effective in mediating high-titer gene transfer, and an HIV-2 vector could be cross-packaged by HIV-1. Both HIV-1 and HIV-2 vectors were able to transduce primary human macrophages, a property not shared by murine retroviruses. Vesicular stomatitis virus glycoprotein G-pseudotyped HIV vectors have the potential to mediate gene transfer into non-cycling hematopoietic stem cells. If so, HIV or other lentivirus-based vectors will have applications beyond HIV infection.
Quantitative Aspects of HIV Pathogenesis and the Strategy of Gene Therapy
A remarkably quantitative model of the natural history of HIV infection has now coalesced from recent studies (1–10). Although a complete picture of pathogenesis is not yet at hand, there is every reason to believe that continuous, high-level viral replication is central to disease causation. Much insight has been gained from the ability to directly quantitate the virion genome itself in blood (1–4, 8, 9) and in tissues (5, 6), as opposed to following indirect surrogate markers or outgrowth of virus in culture. These studies have consolidated a new paradigm: Before the recent application of newer assays with higher sensitivity and dynamic range (e.g., branched-chain DNA, quantitative reverse transcription-PCR, in situ PCR), a model of long-term true microbiologic latency espousing little in vivo replication still held considerable sway.
The new estimates reveal a furiously destructive process behind a facade of apparent clinical latency: approximately 1010 virions produced per day, 140 viral generations per year, a t1/2 for productively infected T cells of 1.6 days and for virions of about 6 hr, and a daily turnover of 109 infected CD4+ T cells; the latter rate estimate may exceed the normal turnover by several logs (1, 3, 4, 7). The numbers tie replication directly to pathogenesis and fit well with our understanding of quasi-species diversity in the lentivirinae, the subgroup of the family Retroviridae to which HIV-1 and HIV-2 belong. Characteristic properties of lentiviruses include high genetic complexity and incubation periods of months to many years before disease development (11). However, the two most singular features of lentiviral infections compared with those caused by other retroviruses are extent of replication within the host (1, 3, 4, 12) and the capacity to infect non-dividing or even post-mitotic cells. For example, all lentiviruses infect terminally differentiated macrophages in vivo.
HIV-1 viral RNA load, even a single initial measurement, has now been shown to correlate well with the prognosis for subsequent CD4+ lymphocyte depletion and disease development (2). It is thus reasonable to surmise that inhibition of viral replication can delay or prevent disease development. Emerging data with presently available combinations of antiretroviral agents targeted at the viral reverse transcriptase and protease suggest utility in some patients (13). Long-term outcomes with respect to viral load, development of multiply resistant virus, or disease status, however, have not yet been ascertained with these combinations. There is a general consensus that it is desirable to initiate therapy early, after infection, to forestall irreversible damage of virus to the immune system, e.g., by depleting the repertoire of immune cells. However, because therapy likely needs to be sustained for the life of the individual, strategies that confer long-term antiviral effects, such as gene therapy, would be important therapeutic options. Antiviral gene therapy, also termed intracellular immunization, aims to reconstitute the immune system with genetically altered cells that resist infection. Two key determinations are the choice of antiviral genes and the choice of gene delivery vehicles into relevant target cells.
Antiviral Genes
The emergence of resistance mutants during antiviral drug monotherapy arises from three ineluctable realities: (i) the large number of HIV virions produced daily (1, 3, 4), (ii) the inherent variability yielded by RNA virus replication (11), and (iii) the inability of available drugs to completely suppress
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: VSV-G, vesicular stomatitis virus glycoprotein G; GFP, green fluorescent protein; LTR, long terminal repeat.
|
‡ |
To whom reprint requests should be addressed, e-mail: fwongstaal@ucsd.edu. |
replication (14). When viewed in the context of the quantitative properties of HIV infection in vivo, the apparently inevitable development of resistance following drug monotherapy illustrates a potential advantage for gene therapy of HIV disease. By virtue of their action through Watson-Crick base pairing, only nucleic acids (antisense molecules and ribozymes) can presently be prospectively targeted at specific sites within the approximately 10-kb HIV genome, which are widely or universally conserved in natural isolates (15, 16). Such regions are likely to be less susceptible to escape mutations that simultaneously preserve viability. Furthermore, genes that target these multiple sites can be combined to virtually eliminate the possibility of virus escape, akin to the concept of multidrug combinations, but still delivered in a single vector. Basic science investigations of steric interaction are also the fundamental source for protease inhibitors, the most potent anti-HIV drugs yet, which are among the first clinically effective drugs developed from precise knowledge of three-dimensional protein structure. Molecular prediction is, however, more versatile and more specific with nucleic acid-based therapies.
Ribozymes are small, catalytic antisense RNAs that bind and cleave specific sites in target RNAs (17, 18). Cleavage, a cis reaction in the natural setting, can be engineered to occur in trans and results through the action of a central region containing secondary structure that is not base-paired with the substrate. The cleavage products are rapidly degraded in cells. The catalytic mechanism (one ribozyme molecule can cleave many substrate molecules in succession) may provide an advantage over antisense approaches. Our laboratory has concentrated on the hairpin ribozyme (19–27); other groups have employed hammerhead ribozymes for antiviral studies (28–31). Using Moloney murine leukemia virus-based retroviral vectors for delivery, hairpin ribozymes have been shown to confer protection from HIV-1 infection of T-cell lines, primary T cells, and macrophage-like progeny of CD34+ hematopoietic progenitor cells (19–25). The use of two ribozymes targeting the long terminal repeat (LTR) and env genes of HIV-1, each fused to an RNA decoy [the RRE (rev response) element), resulted in a potent antiviral vector that effectively inhibits replication of diverse clades of HIV-1 (F.W.-S. and A.Gervaix, unpublished data). Recently, a ribozyme-mediated inhibition of SIVmac was demonstrated in tissue culture (26). Furthermore, transduction of Rhesus macaque cord blood-derived CD34+ cells with this ribozyme conferred viral resistance to both the T cells and macrophage progenies (F.W.-S., M.Heush, G.Kraus, M.Rosenzweig, and P.Johnson, unpublished data). Application of this ribozyme to the SIVmac model, currently the most relevant animal model of AIDS pathogenesis, may allow testing of antiviral efficacy in vivo. In addition, a phase I trial for use of autologous T cells transduced with two hairpin ribozymes that cleave conserved sites in the HIV-1 LTR and pol has received FDA approval to enroll patients.
Gene Transfer Options
This and other ongoing gene therapy trials using anti-HIV molecules (32) entail the relatively cumbersome and expensive procedures of T-cell leukapheresis, ex vivo transduction with the Moloney murine leukemia virus vector, ex vivo expansion, and infusion of transduced cells (Fig. 1). The approach is feasible and currently necessary for proof-of-concept studies, but is not likely to be practical or comprehensive enough for routine use. In particular, it does not access the macrophage reservoir, an important component of the in vivo burden. While reconstitution with CD34+ hematopoietic progenitor cells has obvious advantages, the ability to reconstitute HIV-1-infected individuals, who exhibit complex derangements of hematopoiesis, remains uncertain.
If stem cell therapy proves workable for HIV disease, transduction of the most primitive precursor, probably a subset within the CD34+, CD38− population, has the most chance of success. Targeting this subset and converting from cumbersome ex vivo transduction processes to direct in vivo gene delivery are central goals.
Lentiviral Vectors
Lentiviral vectors have attracted interest with respect to both of these aims. The capacity of lentiviruses to infect non-cycling cells probably resides in the ability of the lentiviral preintegration complex to traverse an intact nuclear envelope through the nuclear targeting properties of both the p17Gag protein and the accessory protein Vpr (33, 34). This property, which is not shared by oncoretroviruses or Moloney murine leukemia virus-derived retroviral vectors, has spurred efforts to develop lentiviral-based gene therapy vectors. The practical goal to which such investigations aspire is stable transfer of genes to rare (and rarely dividing) stem cells and to postmitotic cells in the hematopoietic, nervous, and other body systems.
Other properties of HIV vectors may be particularly desirable for treatment of HIV infection. First, vector systems employing an HIV envelope may allow direct lineage-specific targeting to CD4+ T cells and to non-cycling macrophages and glial cells in vivo. Second, rescue of the vector in vivo by patients’ HIV-1 may result in an effective amplfication of the vector through several cycles before lack of selection pressure results in reverse transcription-derived mutations. Third, the tat and rev regulatory cycles may be exploited to achieve inducible expression of delivered genes. These combined features could elevate gene transfer efficiency to the realm of in vivo therapy.
Notable progress has recently been made with an HIV-1-based system employing vesicular stomatitis virus G protein (VSV-G)-pseudotyped HIV-1 vectors (35); titers exceeding 105/ml and delivery of a lacZ marker gene to post-mitotic cells (neurons) in rodent brain were reported. This system relies upon transient transfection to generate the vector because expression of VSV-G lyses the producer cells.
Although other gene transfer vectors can transduce nondividing cells (e.g., adenovirus vectors), other limitations, chiefly the lack of a stable, consistent genomic integration mechanism, limits their applicability. Adeno-associated virus has been reported to integrate at a specific locus in chromosome 19, but proof of integration and stable gene transfer by engineered vectors in non-dividing cells remains elusive (36).
Other lentiviral vector systems have been studied (37–44). All are derived from HIV-1. Several use wild-type replication-competent helper virus as the source of virion proteins, and some represent simple pseudotyping of an env gene-mutated full-length provirus by VSV-G (37). In general, two problems have been troublesome in this field: (i) vector titers, with the exception of Naldini et al. (35), have been low (101–103) or not reported and (ii) stable packaging lines have been difficult to develop for these viruses, which have more genes and much more complex genetic regulation schemes than the simple retroviruses such as Moloney murine leukemia virus. Carrol et al. (42) reported the first packaging cell lines derived from HIV-1. However, the HXBc2-derived packaging construct expressed a defective Vpr protein, which may interfere with the normal karyophilic properties of the HIV pre-integration complex (33, 34), and the lines expressed predominantly unprocessed gag/pol precursor.
Our focus with lentiviral vectors has been 3-fold. First, we are experimenting with both the native HIV envelopes for lineage-specific gene delivery and with pseudotyped particles because of their higher stability and potential to transduce CD4-negative stem cells. Second, we have concentrated on
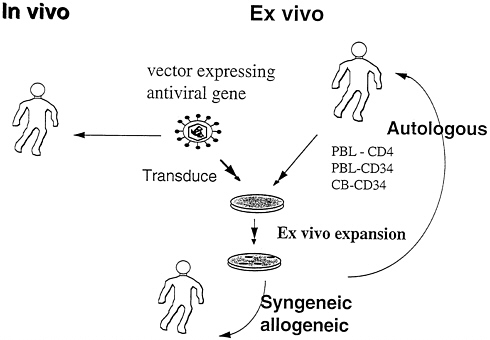
FIG. 1. Gene transfer options for HIV gene therapy.
HIV-2, as well as HIV-1. Vector systems derived from HIV-1 and HIV-2 inevitably raise safety concerns. However, HIV-1 displays nearly uniform lethality in humans, whereas HIV-2 is now documented to be less pathogenic (45). In addition, for delivery of anti-HIV-1 genes to treat HIV, a system based solely upon HIV-1 will be self-inactivating in direct proportion to the efficacy of the antiviral gene. HIV-2KR, the infectious molecular clone from which we have derived components for both HIV-1 and HIV-2-based gene transfer systems, was cloned from the human clinical isolate HIV-2PEI (46). HIV-2KR replicates to high titers in T-cell lymphoblastoid lines, primary human macrophages and peripheral blood lymphocytes. KR is able to infect pig-tailed macaques in vivo and induce transient viremia, as well as protective immunity against disease causation by a highly pathogenic HIV-2 strain (HIV-2EHO), but has itself proven completely apathogenic, as well as unrecoverable by culture at more than 2 years of follow-up following high-dose intravenous challenge (D. Looney, G.Kraus, W.Morton, F.W.-S., J.McClure and S.L. Hu, unpublished data).
Third, we have explored the stable expression in trans of HIV-1 and HIV-2 structural and accessory proteins to develop packaging cell lines from these viruses. Stable expression of viral structural proteins has proven considerably more elusive for HIV than in murine retroviral systems, presumably because of the complexity of HIV genetic regulation (e.g., the Tat and Rev axes) and because of the toxicity of the HIV proteins. The HIV envelopes (the duotropic HIV-1MN has been used for the HIV-1 system) can be used for CD4-specific targeting from these lines. For broad target-cell specificity, the VSV-G protein, recently shown to have numerous advantages for virion particle stability and extended host range (35, 47), can be used in transient packaging.
This paper describes our progress in developing stable cell lines capable of expressing the full complement of HIV-1 and HIV-2 proteins in trans and the development of HIV-based vectors that can be packaged in these lines or by pseudotyping with VSV-G.
Packaging Cell Lines
To devise HIV-1MN and HIV-2KR packaging constructs to supply HIV proteins in trans, deletions of 37 and 61 bp respectively were made in the regions between the major 5′ splice donor and the gag gene initiation codon (Fig. 2A). These deletions alone rendered both HIV-1MN and HIV-2KR proviruses replication-defective but able to express wild-type levels of structural proteins. A stable HIV-1MN packaging cell line was then derived from HeLa cells by cotransfecting the 37-bp psi-deleted provirus with a neo-R containing plasmid and selection in G418. A single cell clone that produced a high level (approximately 20 ng/ml) of p24, designated Ψ422, was isolated and further characterized. For HIV-2KR, a plasmid modified by the 61-bp psi-deletion, replacement of the 3′ LTR with the bovine growth hormone polyadenylylation signal and inclusion of a downstream neoR expression cassette was transfected into COS-1 cells. After selection in G418, clones producing 300–700 ng/ml of p26 were isolated. Both the HIV-1 and HIV-2 packaging cell lines produced no replicating virus as measured by long-term cocultivation with permissive T-cell lines and transfer of supernatant to LTR β-galactosidase indicator cells; reverse-transcription-PCR and RNAase protection assays showed that the psi-deleted proviruses were not packaged efficiently into particles. However, expression of a high level of viral proteins was maintained through over 6 months in culture. Electron microscopy revealed production of viral particles with fully mature lentiviral morphology for both HIV-1 and HIV-2 packaging lines. Details of these results will be reported elsewhere.
Vectors
HIV-1 and HIV-2 based lentiviral vectors were constructed according to the scheme illustrated in Fig. 2B. The 5′ LTR was
Table 1. Transduction capacity of HIV-1MN packaging cell lines expressing HIV-1 and HIV-2 gpt vectors
|
Cell supernatant |
Target cell |
Titer,* transducing units/ml |
|
Ψ422 (packaging line) |
HeLa-T4 |
0 |
|
Vector-transfected HeLa |
HeLa-T4 |
0 |
|
Clone 1 (HIV-1 vector) |
HeLa-T4 |
2.3×104 |
|
|
HeLa |
0 |
|
Clone 2 (HIV-1 vector) |
HeLa-T4 |
5.4×104 |
|
|
HeLa |
0 |
|
Clone a (HIV-2 vector) |
HeLa-T4 |
1.2×104 |
|
|
HeLa |
0 |
|
Clone b (HIV-2 vector) |
HeLa-T4 |
1.1×104 |
|
|
HeLa |
|
|
*Average titer from three experiments. |
||
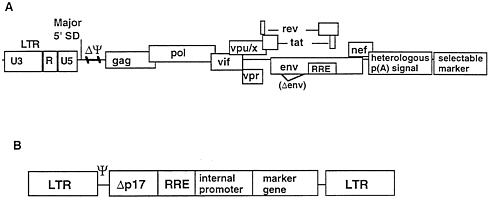
FIG. 2. General scheme for construction of packaging and vector plasmids.
linked sequentially to the 5′ leader including the psi region, a short portion of the p17 region of the gag gene, the respective rev response element (RRE), an internally promoted marker gene and the 3′ LTR. Marker genes used in these studies included xanthine-guanine phosphoriboxyl transferase (gpt), neomycin phosphotransferase (neoR), Escherichia coli β-galactosidase (lacZ) green fluorescent protein (gfp) (48, 49), and the Streptomyces hindustanus phleomycin resistance gene (she ble). In some vectors an element from Mason-Pfizer monkey virus previously shown to substitute for Rev-RRE-mediated activity in HIV mRNAs nuclear transport was included instead of the RRE (50).
Vector Packaging and Transduction
HIV-1 and HIV-2 vectors were transfected into the producer cell lines and the cells were doubly selected for stable expression of viral proteins and vectors. Clones of these stable cell lines were also generated, and titers of the transducing vector in the supernatants were measured. As shown in Table 1, both HIV-1 and HIV-2 vectors were packaged in the HIV-1MN packaging cell line Ψ422. Titers of 10e4 to 10e5 were achievable. These vectors were able to transduce terminally differentiated primary macrophages, in contrast to murine retrovirus vectors, which failed to do so (P.C.G.Kraus, F.W.-S., unpublished work). With the HIV-2KR packaging line, transfection of an HIV-2 neoR vector yielded titers of 10e3 to 10e4 (Table 2). These titers are two to three logs higher than previously reported values of the stable HIV-1 packaging line. It is not clear whether it is the choice of the packaging constructs (both HIV-1MN and HIV-2KR contain coding sequences for all of the accessory genes, and both are duotropic for T cells and monocytes) or that of the producer cells which allowed expression of high titers of infectious vectors. Pseudotyped HIV-2 vectors were generated by transient triple cotransfection of a packaging construct with an additional deletion in the env gene, the vector plasmid, and a plasmid encoding VSV-G under control of the hCMV promoter (kindly supplied by T.Friedman, Uuniversity of California, San Diego). Production of 10e5 or higher titers of the pseudotyped vectors was observed. A vector expressing GFP as a reporter gene gave similar titers (E.P. and F.W.S., unpublished work).
Table 2. Transducing titer of HIV-2 neoR vectors produced from HIV-2KR packaging cell clone on U937 cells
|
Exp. |
Titer, transducing units/ml |
Mean |
|
1 |
1.3×10e4 |
1.8×10e4 |
|
2 |
8.5×10e3 |
|
|
3 |
3.2×10e4 |
|
Experiments to determine if these vectors can transduce non-cycling CD34+/CD38− cells in culture, or long-term repopulating cells in in vivo animal models are in progress.
Perspectives
Our current understanding of AIDS pathogenesis affirms the central role of HIV in both disease initiation and progression. Recent studies on virus dynamics in patients under chemotherapy (1), as well as long-term prospective studies of plasma viral burden in patients that progress to disease at different rates (2) support a virus threshold hypothesis for disease progression. It is now also recognized that insidious damage inflicted by the virus upon the immune system occurs from the onset of infection, underscoring the importance of early intervention in infected individuals. Although recent clinical results from trials of combinations of antiviral agents, including the potent protease inhibitors, have been encouraging, whether such therapy can be sustained lifelong without recument problems of toxicity, viral resistance, and economics is unclear. Gene therapy has been considered by many to be an attractive strategy for conferring long-term therapeutic benefits.
Gene therapy for HIV infection, however, faces experimental obstacles common to gene therapy and genes that are intrinsic to the nature of HIV infection in particular. The extreme inefficiency of transducing hematopoietic progenitor cells that would give rise to long-term repopulation of multilineage progeny cells in animals is a general frustration. For HIV infection, the need to access the nonproliferative macrophage target cell reservoir, the uncertainty of whether bone marrow derived hematopoiesis may be impaired in adult AIDS patients, and the lack of high-titer vectors that allow in vivo targeting are additional concerns. The ability of HIV vectors to both target CD4+ cells in vivo and transduce non-cycling cells may help resolve some of these issues.
E.P. is a recipient of a National Institutes of Health Physician-Scientist Award. The work described here is supported by the National Institutes of Health SPIRAT award to F.W.-S. and the University of California, San Diego Center for AIDS Research.
1. Perelson, A.S., Neumann, A.S., Markowitz, M., Leonard, J.M. & Ho, D.D. (1996) Science 271, 1582–1586.
2. Mellors, J.W., Rinaldo, C.R., Gupta, P., White, M.R., Todd, J.A. & Kingsley, L.A. (1996) Science 272, 1167–1170.
3. Ho, D.D., Neumann, A.U., Perelson, A.S., Chen, W., Leonard, J.M. & Markowitz, M. (1995) Nature (London) 373, 123–126.
4. Wei, X.P., Ghosh, S.K., Taylor, M.E., Johnson, V.A., Emini, E. A., et al. (1995) Nature (London) 373, 117–123.
5. Pantaleo, G., Graziosi, C., Demarest, J.F., Butini, L., Montroni, M., et al. (1993) Nature (London) 362, 355–358.
6. Embretson, J., Zupanic, M., Ribas, J.L., Burke, A., Racz, P., et al. (1993) Nature (London) 362, 359–362.
7. Coffin, J. (1995) Science 267, 483.
8. Piatak, M., Saag, M.S., Yang, L.C., Clark, S.J., Kappes, J.C., et al. (1993) Science 259, 1749–1754.
9. Ho, D.D., Moudgil, T. & Alam, M. (1989) N. Engl. J. Med. 321, 1621–1625.
10. Ho, D.D. (1996) Science 272, 1124–1125.
11. Coffin, J.M. (1992) Curr. Top. Microbiol. Immunol. 176, 143–164.
12. Coffin, J.M. (1993) in Reverse Transcriptase, eds. Skalka, A. & Goff, S. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 445–479.
13. Saag, M.S., Holodniy, M., Kuritzkes, D.R., O’Brien, W.A., Coombs, R., et al. (1996) Nat. Med. 2, 625–629.
14. Richman, D.D. (1994) AIDS Res. Hum. Retroviruses 10, 901–905.
15. Poeschla, E.M. & Wong-Staal, F. (1995) in AIDS Clinical Review 1995/96, eds. Volberding, P. & Jacobsen, M.A. (Dekker, New York), pp. 1–45.
16. Yu, M., Poeschla, E. & Wong-Staal, F. (1994) Gene Ther. 1, 13–26.
17. Symonds, R.H. (1992) Annu. Rev. Biochem. 61, 641–671.
18. Poeschla, E. & Wong-Staal, F. (1994) Curr. Opin. Oncol. 6, 601–606.
19. Ojwang, J., Hampel, A., Looney, D., Wong-Staal, F. & Rappaport, J. (1992) Proc. Natl. Acad. Sci. USA 89, 10802–10806.
20. Yu, M., Ojwang, J., Yamada, O., Hampel, A., Rappaport, J., Looney, D. & Wong-Staal, F. (1993) Proc. Natl. Acad. Sci. USA 90, 6340–6344.
21. Yamada, O., Yu, M., Yee, J.-K., Kraus, G., Looney, D. & Wong-Staal, F. (1994) Gene Ther. 1, 38–45.
22. Yu, M., Leavitt, M., Maruyama, M., Yamada, O., Young, D., Ho, A. & Wong-Staal, F. (1995) Proc. Natl. Acad. Sci. USA 92, 699–703.
23. Leavitt, M.C., Yu, M., Yamada, O., Kraus, G., Looney, D., Poeschla, E. & Wong-Staal, F. (1994) Hum. Gene Ther. 5, 1115–1120.
24. Yu, M., Poeschla, E.M., Yamada, O., De Grandis, P., Leavitt, M.C., Heusch, M., Yee, J.-K., Wong-Staal, F. & Hampel, A. (1995) Virology 206, 381–386.
25. Yamada, O., Leavitt, C., Yu, M., Kraus, G. & Wong-Staal, F. (1994) Virology 205, 121–126.
26. Heusch, M., Kraus, G., Johnson, P. & Wong-Staal, F. (1996) Virology 216, 241–244.
27. Leavitt, M.C., Yu, M., Wong-Staal, F. & Looney, D. (1996) Gene Ther., 3, 599–606.
28. Sarver, N., Cantin, E.M., Chang, P.S., Zaia, J.A., Ladne, P.A., Stephens, D.A. & Rossi, J.J. (1990) Science 247, 1222–1225.
29. Weerasinghe, M., Liem, S.E., Asad, S., Read, S.E. & Joshi, S. (1991) J. Virol. 65, 5531–5534.
30. Dropulic, B., Lin, N.H., Martin, M.A. & Jeang, K.T. (1992) J. Virol. 66, 1432–1441.
31. Rossi, J., Elkins, D., Zaia, J. & Sullivan, S. (1992) AIDS Res. Hum. Retroviruses 8, 183–189.
32. Nabel, G., Fox, B., Post, L., Thompson, C. & Woffendin, C. (1994) Hum. Gene Ther. 5, 79–92.
33. Heinzinger, N.K., Bukinsky, M.I., Haggerty, S.A., Ragland, A.M., Kewalramani, V., Lee, M.A., Gendelman, H.E., Ratner, L., Stevenson, M. & Emerman, M. (1994) Proc. Natl. Acad. Sci. USA 91, 7311–7315.
34. Stevenson, M., Brichacek, B., Heinzinger, N., Swindells, S., Pirruccello, S., Janoff, E. & Emerman, M. (1995) Adv. Exp. Med. Biol. 374, 33–45.
35. Naldini, L., Blömer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F.H., Verma, I.M. & Trono, D. (1996) Science 272, 263–266.
36. Goodman, S., Xiao, X., Donahue, R.E., Moulton, A., Miller, J., Walsh, C., Young, N.S., Samulski, R.J. & Nienhuis, A.W. (1994) Blood 84, 1492–1500.
37. Akkina, R., Walton, R., Chen, M.-C., Li, Q.-X., Planelles, V. & Chen, I. (1996) J. Virol. 70, 2581–2585.
38. Poznansky, M., Lever, A., Bergeron, L., Haseltine, W. & Sodroski, J. (1991) J. Virol. 65, 532–536.
39. Parolin, C., Dorfman, T., Palu, G., Gottlinger, H. & Sodroski, J. (1994) J. Virol. 68, 3888–3895.
40. Richardson, J.H., Kaye, J.F., Child, L.A. & Lever, A.M. (1995) J. Gen. Virol. 76, 691–696.
41. Buchschacher, G.L., Jr., & Panganiban, A.T. (1992) J. Virol. 66, 2731–2739.
42. Carrol, R., Lin, J.-T., Dacquel, E.J., Mosca, J.D., Burke, D.S. & St. Louis, D.C. (1994) J. Virol. 68, 6047–6051.
43. Rizvi, T.A. & Panganiban, A.T. (1993) J. Virol. 67, 2681–2688.
44. Shimada, T., Fuji, H., Mitsuya, H. & Nienhuis, A.W. (1991) J. Clin. Invest. 88, 1043–1047.
45. Marlink, R., Kanki, P., Thior, I., et al. (1994) Science 265, 1587–1590.
46. Talbott, R., Kraus, G., Looney, D. & Wong-Staal, F. (1993) Proc. Natl. Acad. Sci. USA 90, 4226–4230.
47. Lin, S., Gaiano, N., Culp, P., Burns, J.C., Friedmann, T., Yee, J.K. & Hopkins, N. (1994) Science 265, 666–669.
48. Chalfie, M., Tu, Y., Euskirchen, G., Ward, W.W. & Prasher, D.C. (1994) Science 263, 802–805.
49. Heim, R., Cubitt, A.B. & Tsien, R.Y. (1995) Nature (London) 373, 663–664.
50. Bray, M., Prasad, S., Dubay, J.W., Hunter, E., Jeang, K.T., Rekosh, D. & Hammarskjold, M.L. (1994) Proc. Natl. Acad. Sci. USA 91, 1256–1260.





