
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes
(retrovirus vector/gene therapy/293 cells/transient transfection)
DANIEL S. ORY *, BEVERLY A. NEUGEBOREN †, AND RICHARD C. MULLIGAN †‡
Whitehead Institute for Biomedical Research, and Department Biology, Massachusetts Institute of Technology, Cambridge, MA 02142
ABSTRACT We have generated a human 293-derived retroviral packaging cell line (293GPG) capable of producing high titers of recombinant Moloney murine leukemia virus particles that have incorporated the vesicular stomatitis virus G (VSV-G) protein. To achieve expression of the retroviral gag-pol polyprotein, the precise coding sequences for gag-pol were introduced into a vector which utilizes totally nonretroviral signals for gene expression. Because constitutive expression of the VSV-G protein is toxic in 293 cells, we used the tetR/VP 16 transactivator and tet° minimal promoter system for inducible, tetracycline-regulatable expression of VSV-G. After stable transfection of the 293GPG packaging cell line with the MFG.SnlsLacZ retroviral vector construct, it was possible to readily isolate stable virus-producing cell lines with titers approaching 107colony-forming units/ml. Transient transfection of 293GPG cells using a modified version of MFG.SnlsLacZ, in which the cytomegalovirus IE promoter was used to drive transcription of the proviral genome, led to titers of ≈106colony-forming units/ml. The retroviral/VSV-G pseudotypes generated using 293GPG cells were significantly more resistant to human complement than commonly used amphotropic vectors and could be highly concentrated (>1000-fold). This new packaging cell line may prove to be particularly useful for assessing the potential use of retroviral vectors for direct in vivo gene transfer. The design of the cell line also provides at least theoretical advantages over existing cell lines with regard to the possible release of replication-competent virus.
Currently, retroviral-mediated gene transfer is widely utilized to obtain efficient transduction of mammalian cells in vitro, and to date, has been the gene transfer method of choice for clinical protocols aimed at the evaluation of ex vivo strategies for gene therapy (1). While standard murine-based retroviral vectors are well suited for use in such ex vivo applications, the vectors have found only limited use in strategies involving direct in vivo gene transfer (1). One major limitation of the commonly used vector/packaging cell systems is the inability to easily purify and concentrate the large amounts of virus often needed for direct in vivo gene transfer applications. A second limitation relates to the sensitivity of virus with amphotropic host range to inactivation by human serum (2–4). A final limitation of all murine-based vectors is their inability to integrate in quiescent cells (5, 6).
One recent advance that may prove to be important for the eventual use of retrovirus vectors for direct in vivo gene transfer was the demonstration that it is possible to generate retrovirus vector particles which have incorporated the vesic ular stomatitis virus G (VSV-G) protein (7). The resulting VSV-G/retroviral pseudotypes possessed the wide host range of VSV and could be highly concentrated without loss of biological activity (8). This finding follows very early studies which had demonstrated the capacity of retroviruses and VSV to form viral pseudotypes upon coinfection of cells with both viruses (9). In the recent work, the procedure used to generate virus involved the use of transient transfection techniques to express the VSV-G protein, since the constitutive expression of significant levels of VSV-G in most cells is toxic. However, this method of virus production significantly limits the evaluation of the potential applications of the viral pseudotypes, since only small amounts of virus can be easily produced. To overcome these difficulties, we have generated a stable human-derived cell line which constitutively expresses the necessary retroviral proteins for packaging and provides for large amounts of the VSV-G protein by inducible expression. We describe here the manner in which the cell line was constructed and some of the characteristics of the virus that is generated from the cells.
MATERIALS AND METHODS
Cell Lines and Drug Selections. Adenovirus 5-transformed human embryonic kidney 293 cells (10) were obtained from B. Panning (Whitehead Institute). The 293 cells were grown in 293 growth medium containing Dulbecco’s modified eagle medium (DMEM) (GIBCO/BRL), 10% (vol/vol) inactivated fetal bovine serum (IFS) (Sigma), 2 mM L-glutamine (GIBCO/BRL), and 50 units/ml penicillin and streptomycin (GIBCO/BRL). Drug selections in transfected 293 cells were performed at 2 μg/ml puromycin (Sigma), 0.3 mg/ml G418 (GIBCO/BRL) and 100 μg/ml Zeocin (Invitrogen). All growth media, except where noted, was supplemented with 1 μg/ml tetracycline. NIH 3T3 cells (ATCC CRL 1658) were grown in DMEM containing 10% (vol/vol) calf serum (Sigma), and 50 units/ml penicillin and streptomycin. Mus dunni cells were grown in DMEM containing 5% (vol/vol) calf serum (Sigma), and 50 units/ml penicillin and streptomycin.
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: VSV-G, vesicular stomatitis virus G; CMV, cytomegalovirus; HCMV, human CMV; cfu, colony-forming unit; RT, reverse transcriptase; IFS, inactivated fetal bovine serum; RCV, replication-competent virus; MuMLV, Moloney murine leukemia virus; IE, immediate early.
Plasmid Constructs. The plasmid pBC.tTA (see Fig. 1) was constructed from pBC12/cytomegalovirus (CMV)/interleukin 2 (11) by replacement of the interleukin 2 sequences (bp 756–1439) with the tet transctivator gene from pUHD10–1 (12). To construct pMDtet.G (Fig. 1), the 1.6-kb EcoRI fragment from pSVGL (13) containing the VSV-G gene was cloned into the EcoRI cloning site in pMD.tet which is within exon 3 of the genomic human β-globin sequence. pMDtet was constructed with a 0.47-kb XhoI-BamHI fragment from pUHC 13–3 (12), which contains the tet operator and minimal human cytomegalovirus (HCMV) enhancer-promoter sequences, a 1.34-kb BamHI-XbaI fragment from pUCMdβs(R)S (14) that includes the genomic human β-globin sequences from the BamHI site in exon 2 through 690 bp in the 3′ untranslated region, and a 3.06-kb XbaI-XhoI fragment from pSL301 (Invitrogen).
To construct pMD.gagpol (see Fig. 1), PCR was performed with pCRIPenv− (15) using the following pairs of primers: 5′-CGGAATTCATGGGCCAGACTGTTACC-3′ and 5′-AGCAACTGGCGATAGTGG-3′, 5′-CGGAATTCTTAGGGGGCCTCGCGG-3′ and 5′-ACTACATGCTGAACCGGG-3′. The PCR products were digested with EcoRI and XhoI and with EcoRI and HindIII, respectively, to generate 0.94-kb EcoRI-XhoI and 0.94-kb HindIII-EcoRI fragments. These fragments were ligated with the 3.3-kb XhoI-HindIII fragment from pCRIPenv− and with pUC19, which had been linearized with EcoRI and calf intestinal phosphatase treated, to produce pUC19.gagpol. The 5.2-kb EcoRI fragment from pUC19.gagpol was cloned into the EcoRI cloning site in pMD to yield pMD.gagpol. pMD was constructed with the 3.1-kb EcoRI-BamHI fragment from pBC12/CMV/interleukin 2 that includes the pXF3 backbone and HCMV enhancer-promoter region and the previously described 1.34-kb BamHIXbaI fragment derived from pUCMdβs(R)S. The 3.1-kb EcoRI-BamHI and 1.34-kb BamHI-XbaI fragments were ligated after the EcoRI and XbaI overhangs were blunt-ended by Klenow treatment.
The plasmids pJ6Ωpuro and pJ6Ωbleo conferring resistance to puromycin and bleomycin (and zeocin), respectively, were kindly provided by J.Morgenstern (16). The plasmid pSV2neo confers resistance to G418 (17).
Retroviral Vectors. MFG.SnlsLacZ (see Fig. 1) was kindly provided by O.Danos (18). This vector is a derivative of MFG (19) in which mutations have been introduced at nucleotides 412 (A to T), 429 (T to A), and 631 (C to T) [nucleotide 625 of the Moloney murine leukemia virus (MuMLV) sequence]. These substitutions produce the sequence, ATGGGCCCGGGGTAG, thereby preventing expression of the N-terminal portion of gag that would otherwise be expressed by the vector. The ΔU3nlslLacZ retroviral vector was constructed by precise replacement of the U3 region in the 5′ long-terminal repeat of MFG.SnlsLacZ with the HCMV enhancer-promotor (bp −671 to −2) (20). For the construction of ΔU3nlsLacZ, a 701-bp fragment encoding the HCMV promoter was generated by PCR with the pMD plasmid as the template with the pair of primers, 5′-GGGCCCAAGCTTCCCATTGCATACGTTGTATC-3′ and 5′-GGACTGGCGCCGGTTCACTAAACGAGCTC-3′, creating a 5′ HindIII site and a 3′ KasI site. The PCR product was digested with HindIII and KasI to yield a 677-bp fragment. The 91-bp KasI-StyI was isolated from the 3′ long-terminal repeat of MFG (19). The 253-bp StyI-EagI and the 4994-bp EagI-ScaI fragments were isolated from MFG.SnlsLacZ, and the backbone for ΔU3nlsLacZ is a 2.65-kb HindIII-SmaI fragment from pUC18.
DNA Transfection. Stable transfection of 293 cells was performed by the calcium phosphate precipitation method (22) with 5 μg pBC.tTA, 5 μg pMDtet.G, and 1 μg pJ6Ωpuro. For all stable and transient transfections, plasmid DNA was prepared by double banding on CsCl density gradients (23). Cells (1.5×106) were plated on 60-mm dishes in 4 ml 293 growth media the night before transfection. Chloroquine (final concentration, 25 μM) and tetracycline (final concentration, 1 μg/ml) were added to the media 5 min before transfection. The media was changed 7 h posttransfection. The transfected cells were plated 48 h posttransfection by limiting dilution in media containing puromycin and tetracycline and independent clones were isolated.
293 G cells were always grown in 293 growth medium supplemented with tetracycline and puromycin (293G growth medium). Stable transfection of the 293G cells was performed by the calcium phosphate precipitation method with 10 μg pMD.gagpol linearized with ScaI and 2 μg pSV2neo. Cells (2×106) were plated on 60-mm dishes in 4 ml 293G media the night before transfection. Chloroquine (final concentration, 25 μM) was added to the media 5 min before transfection. The media was changed 7 h posttransfection. The transfected 293G cells were plated by limiting dilution 48 h posttransfection in 293G growth medium supplemented with G418 and independent clones were isolated.
293GPG cells were grown in 293G growth medium supplemented with G418 (293GPG medium). Stable transfection of the 293GPG cells with MFG.SnlsLacZ was performed by the calcium phosphate precipitation method with 12.5 μg MFG.-SnlsLacZ linearized with AseI and 2.5 μg pJ6Ωbleo linearized with AflIII. Cells (4×106) were plated on 60-mm dishes in 4 ml 293GPG media the night before transfection. The media was changed 9 h posttransfection. The transfected 293GPG cells were plated by limiting dilution 48 h posttransfection in 293GPG media supplemented with zeocin and independent clones were isolated.
Transient transfections with 293GPG cells were performed on 60-mm dishes where 4–5×106 cells were plated the night before in 4 ml 293 growth medium. Four micrograms of ΔU3nlsLacZ was diluted into 300 μl OptiMEM (GIBCO/ BRL) and incubated at room temperature for 30 min with 25 μl lipofectamine (GIBCO/BRL) diluted into 300 μl OptiMEM. The DNA-lipofectamine mixture was diluted into 2.4 ml OptiMEM and layered on top of the 293GPG cells, which had been rinsed 30 min before transfection and had media replaced with 2 ml OptiMEM. Seven to 8 h posttransfection, 2 ml 293 media was added, and the media was changed at 24 h with 2.5 ml 293 media. The supernatant was harvested at 72 h and viral titers determined as described below.
Analysis of VSV-G Expression in Transfected Cells. The pMDtet.G and pBC.tTA cotransfected clones were screened for inducible VSV-G expression by plating each clone in parallel into two 35-mm tissue culture dishes at 50% confluence. The following day one plate was rinsed twice with 2 ml 293G media without tetracycline and maintained in this media. At 48 h the postnuclear cellular lysates were prepared and the paired samples run on a 7.5% SDS/PAGE under reducing conditions. The gels were transferred onto nitrocellulose (0.45 mm; Schleicher & Schuell) with a semidry electroblotter (Owl Scientific, Woburn, MA). Western blot analysis was performed by using a murine monoclonal anti-VSV-G IgG (Sigma) at a dilution of 1:800 and a peroxidase-conjugated F(ab′)2 fragment donkey anti-mouse IgG (Jackson ImmunoResearch Laboratories) was used at a dilution of 1:10,000. Detection by chemiluminescence was performed using commercially available reagents (Renaissance; New England Nuclear).
Assays For Reverse Transcriptase (RT) and β-Galactosidase Activity. 293G cells transfected with pMD.gagpol were screened for RT activity in the culture medium of subconfluent clones growing in 24-well culture dishes as described (24). Cells were stained for β-galactosidase activity as described (25).
Viral Titers, Virus Concentration, and Stability of Virus to Human Serum. To determine viral titers, NIH 3T3 cells were plated at 1×105 cells per well in 6-well culture dishes 16 h before infection and incubated for 24 h with serial dilutions of
viral supernatants containing 8 μg/ml polybrene (Sigma). Viral titer was determined as the average number of cells with blue nuclei (β-galactosidase-producing cells) per 20 1-mm2 fields (2–3×104 cells) multiplied by a factor to account for plate size, dilution of viral stock, and division of target cells in tissue culture wells. Viral supernatants harvested either from stable virus-producing cell lines or transiently transfected cells were concentrated by ultracentrifugation (8).
The stability of the GPGnlsLZ pseudotyped retrovirus and the ΨCRIPLacZ amphotropic retrovirus was determined in normal human serum. Twenty microliters virus harvest, which was diluted 1:5 in 20 mM Hepes buffer (pH 7.0), 10 μl of PBS, or 10 μl of Gal(α1–3)galactose (final concentration, 10 mg/ml) (Dextra Laboratories, Reading, U.K.), and 20 μl of fresh normal human serum, heat-inactivated human serum, or IFS were mixed on ice and then incubated at 37°C for 1 h (26). Heat inactivation of the human serum was carried out at 56°C for 1 h. The virus-serum mixture was diluted in 1.5 ml DMEM with 8 μg/ml polybrene. Serial dilutions of the virus-serum mixture were incubated with NIH 3T3 cells in 6-well dishes as described above to determine viral titers. Relative titers (percent) were determined for fresh and heat-inactivated human serum treatment as compared with IFS treatment.
Helper Virus Assay. Retroviral stocks were assayed for replication-competent virus (RCV) by a vector rescue assay (27). Mus dunni cells with a stably integrated MFGLacZ (MDZ), were plated at a 2×105 cells per 10-cm dish. The following day the MDZ cells were infected with viral stocks that were 0.45 μM filtered and to which 8 μg/ml polybrene was added. The media was changed at 24 h and again 3 days later. The 24-h MDZ supernatant was harvested and passed through a 0.45 μM filter, 8 μg/ml polybrene was added, and the supernatant was overlayed on naive Mus dunni cells, which were plated the day before at 4×105 cells per 10-cm dish. Twenty-four hours later the supernatant was removed and the cells supplied with normal media. The following day the cells were stained for β-galactosidase activity. The entire plate was scanned with a light microscope (×4 phase objective) for the presence or absence of LacZ expressing cells (blue cells). To determine the sensitivity of the assay, graded dilutions of a titered 4070A amphotropic virus stock (Tektagen, Malvern, PA) were used in place of the test virus. The assay was shown to be able to detect one particle of 4070A per 8 ml supernatant.
RESULTS
General Strategy for Construction of Packaging Cell Line. Although most previously described retroviral packaging cell lines have been derived from murine cell lines (15, 28–30), we chose to use the human-derived cell line 293 (10) as the parental cell line for our studies for several reasons. First, in contrast to murine cells, human cells do not harbor a large number of endogenous retroviral genomes nor express endogenous viral-like RNAs that may contribute to the generation of replication-competent virus through recombination events involving packaged vector sequences (31–33). In addition, retrovirus vectors produced from human cells have been shown to be resistant to the mechanisms of virus inactivation involving natural antibodies and complement that occur when virus derived from murine cells is exposed to human serum (2, 4). Finally, 293 cells can be transiently transfected at high efficiency, a property potentially useful for generating small quantities of high titer virus in a rapid fashion (22).
Because the high-level constitutive expression of VSV-G is toxic to cells, we employed the tetracycline-regulatable gene expression system of Gossen and Bujard (12) to provide for the inducible expression of VSV-G. The expression construct used to express the VSV-G protein, pMDtet.G (Fig. 1), contains a minimal CMV immediate early (IE) promoter to which seven tet operator sequences (12) are linked upstream, and an
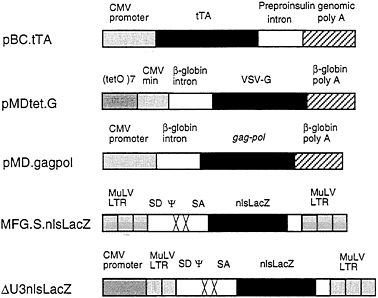
FIG. 1. Schematic diagrams of plasmid and retroviral constructs. The construction of pBC.tTA, pMDtet.G, and pMD.gagpol is detailed in Materials and Methods. The pBC.tTA construct encodes the VP16-tet transactivator fusion protein. In pMDtet.G expression of the VSV-G protein is under the control of the inducible tet°/CMV minimal promoter sequences. The pMD.gagpol construct encodes the MuLV gag-pol sequences. MFG.SnlsLacZ is a replication-defective retroviral vector with splice donor (SD) and splice acceptor (SA) sites in which a nuclear localizing LacZ (nlsLacZ) has been cloned into the ATG of the env gene (19). ΔU3nlsLacZ encodes a nuclear localizing β-galactosidase under the control of the HCMV enhancer-promoter.
intervening sequence and poly(A) site from the human β-globin gene. For expression of the tet/VP16 transactivator (12), we used the vector pBC.tTA, which utilizes the full CMV IE promoter and an intervening sequence and poly(A) signal from the rat insulin II gene (Fig. 1). The tet/VP16 transactivator binds to the tet operator sequences in the promoter region of pMDtet.G and activates transcription of VSV-G from the minimal CMV promoter (12). Transcription is suppressed in the presence of tetracycline and is activated when tetracycline is removed from the media.
For expression of the MuMLV gag-pol sequences, we used the expression vector pMD, which employs the CMV IE promoter and an intervening sequence and poly(A) site from the human β-globin gene for expression of inserted sequences (Fig. 1). Notably, this vector contains no retroviral sequences. A segment of the MuMLV genome that precisely encodes gag-pol was then inserted into the vector (see Fig. 1). These design features help to minimize the overlap of sequences between different vectors and the packaging cell sequences that can often occur, and which have been previously shown to contribute to the formation of RCV (34, 35) In comparison to pCRIPenv−, the construct used in the generation of ΨCRIP cells (15), pMD.gagpol generated significantly higher levels of RT activity during transient transfection of 293 cells (data not shown).
The overall scheme for generating the 293GPG packaging cells is shown in Fig. 2. The first step in the generation of the cells was the isolation of a 293-derived cell line that expressed VSV-G. Rather than sequentially introduce the pMDtet.G and pBC.tTA constructs into cells, we chose to simultaneously introduce both constructs via a tripartite cotransfection with a selectable marker in the hopes of providing a natural selection for integrants that express an acceptable level of VSV-G in the repressed state. Having identified a clone of cells that express high levels of VSV-G in the absence of tetracycline and low basal levels in the presence of tetracycline, the pMD.gagpol vector was then introduced by cotransfection into the cells. Candidate packaging cell lines were then screened for high RT levels in the media as described below.
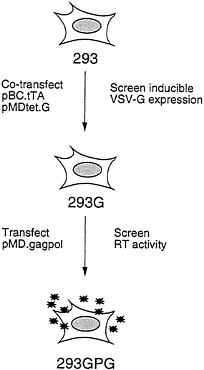
FIG. 2. Strategy for generation of pseudotyped packaging cell line. 293 cells were cotransfected with pBC.tTA and pMDtet.G. Clones were screened by Western blot analysis for induction of VSV-G expression. The 293G clone was transfected with pMD.gagpol and clones were screened by RT activity. The 293GPG clone demonstrated the highest level of RT activity and was used to generated stable producer cell lines.
Construction of a 293-Derived Cell Line Which Expresses VSV-G in an Inducible Fashion. To generate a cell line capable of expressing VSV-G, 293 cells were cotransfected by calcium phosphate precipitation techniques with equimolar amounts of pBC.tTA and pMDtet.G (Fig. 1) and with a plasmid encoding resistance to puromycin as a selectable marker. The pBC.tTA-and pMDtet.G-transfected cells were then cultured in media containing puromycin and 1 μg/ml tetracyline during selection (to prevent expression of the VSV-G). Seventy-two independent drug-resistant clones were subsequently isolated and screened by removal of tetracycline from the growth medium. Western blot analysis identified 12 clones that exhibited high levels of VSV-G expression in the absence of tetracycline, yet low or no detectable VSV-G expression in the presence of tetracycline (data not shown). The 293 clone chosen for further study, termed 293G, demonstrated particularly high levels of VSV-G expression per mg of cellular protein, comparable to twice the amount of VSV-G expressed after transient transfection with pMD.G (Fig. 3A). The two VSV-G bands detected in de-repressed 293G cells (Fig. 3A, lane 3) represent the completely glycosylated (upper band) and an incompletely glycosylated (lower band) form of VSV-G. Treatment of the postnuclear cellular lysate from the 293G cells with N-glycosidase F demonstrates a single unglycosylated VSV-G band (Fig. 3A, lane 9). The observation of incomplete glycosylation of VSV-G in the 293G cells suggests that the extremely high level of expression of VSV-G may overwhelm the capacity of the cells for glycosylation or that the large quantity of VSV-G protein may stimulate intracellular recycling with deglycosylation (36).
The high-level expression of VSV-G observed after transfection with the VSV-G expression constructs was consistently associated with significant morphologic changes in the cells. In transiently transfected 293 cells (data not shown) and in derepressed 293G cells, we observed formation of large multinucleated syncytia of cells, the appearance of which correlated precisely with VSV-G expression (Fig. 3B). The VSV-G protein has a putative fusagenic domain spanning amino acids 123
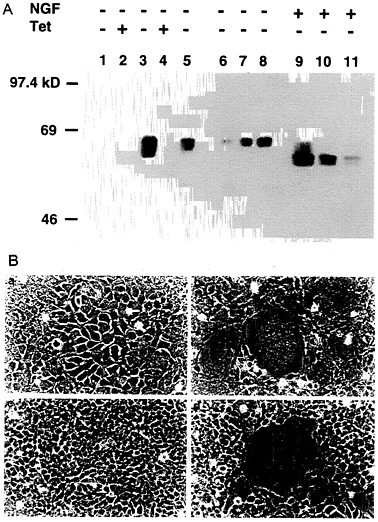
FIG. 3. Induction of VSV-G expression in 293G and 293GPG cell lines. (A) Western blot analysis of SDS/PAGE (7.5%) of cell lysates (10 μg/lane) from 293 cells (lane 1), 293G cells (lanes 2, 3, and 9), and 293GPG cells (lanes 4, 5, and 10). Lanes 6, 7, 8, and 11 show 5 μg, 10 μg, 20 μg, and 10 μg of cell lysate from 293 cells transiently transfected with pMD.G, respectively. Tet (+) indicates growth in the presence of tetracycline (1 μg/ml) and Tet (−) indicates growth for 48 hours in the absence of tetracycline. NGF (+) indicates treatment with 2 units N-glycosidase F (Boehringer Mannheim) for 24 h at 37°C before analysis. (B) Morphology (×100 field) of 293G (a and b) and 293GPG (c and d) cells grown in the presence of tetracycline (1 μg/ml) (a and c) and in the absence of tetracycline (b and d) for 72 h. Prominent syncytia formation is observed in 293G (b) and 293GPG (d) cells after induction of VSV-G expression.
to 137 (37), which facilitates fusion between the membrane of the enveloped virus and the plasma membrane of target cell. The high-level cell surface expression of VSV-G in our transient and stable cell lines may promote fusion of plasma membranes of adjacent cells in response to local pH changes (38).
Construction of a Packaging Cell Line Which Expresses Both VSV-G and MuMLV gag-pol. To generate a stable cell line which expresses both VSV-G and gag-pol, pMD.gagpol was linearized at the ScaI site and introduced into 293G cells along with a plasmid encoding resistance to neomycin, using calcium phosphate transfection techniques. Sixty-nine G418-resistant clones (15) were isolated, and each culture supernatant was screened for the level of RT activity. Twenty-four positive clones with RT activity equivalent to or greater than that of ΨCRE (15) were identified on an initial screen (data not shown). The clone selected for further study, 293GPG, released ≈25-fold more RT activity than that released by ΨCRE and ΨCRIP (15) and ≈10-fold more activity than that released by Anjou 65 cells (22) (Fig. 4). Removal of tetracycline from the growth medium of 293GPG cells demonstrated the continued presence of inducible VSV-G expression by Western

FIG. 4. RT activity of supernatant from 293GPG clone. 293GPG cells were grown to 75% confluence in 60-mm dishes, the media was changed, and supernatant was harvested 24 h later. RT assay was performed as described. Supernatants from RT reaction cocktail without supernatant (Blank) and 293 cells are negative controls. Supernatants from ΨCRE, ΨCRIP, and Anjou 65 cells are positive controls. Supernatant from the 293GPG cells was diluted 1:5, 1:10, 1:25, 1:50, and 1:100 as indicated.
blot analysis (Fig. 3A) and by the formation of syncytia (Fig. 3B).
Production and Characteristics of Recombinant Virus from 293GPG Cells. To examine the capacity of the 293GPG cells to produce high titers of recombinant retrovirus vectors, the cells were cotransfected with the retroviral vector MFG.-SnlsLacZ, linearized by AseI, and a plasmid encoding resistance to zeocin (16). Sixteen independent drug-resistant clones were isolated. The clones were then cultured in tetracyclinefree media and the Supernatants were harvested at 96 h and were used to infect NIH 3T3 cells for determination of viral titer by 5-bromo-4-chloro-3-indolyl β-D-galactoside (X-Gal) staining. Three of the clones, termed GPGnlsLZ2, GPGnlsLZ3, and GPGnlsLZ4, generated virus with titers of ≈107 colony forming units (cfu)/ml. To further determine the optimal time for virus harvest after removal of tetracycline, Supernatants from the three previously selected clones were collected serially at successive time points (Fig. 5). Maximal virus production per 24-h period was shown to occur between 48 and 96 h after removal of tetracycline.
Because 293 cells have been shown to yield efficient transient gene expression after transfection (22), we next examined the ability to harvest high titer virus after transient transfection of the cells with a retroviral vector. Although, as shown above, cells stably transfected with the MFG.S vector yielded high titers of virus, preliminary transient transfection studies with the MFG.S vector yielded titers of only ≈105 cfu/ml (data not shown). Based on the studies of others (39), which indicated the high transcriptional activity of the CMV IE promoter after transient transfection of 293 cells, we generated a derivative of MFG.S, termed ΔU3nlslLacZ, in which the enhancer-promoter region of the 5′ long-terminal repeat was replaced with the complete CMV IE promoter in such a way that transcription would initiate at the proper viral start site (Fig. 1). Using this construct in conjunction with lipofectamine transfection, we were able to obtain an average efficiency of transfection of the 293GPG cells of 40%. More importantly, when virus was harvested 24–72 h after transfection, titers exceeding 106 cfu/ml were obtained (Table 1).
To determine whether virus produced from 293GPG cells could be efficiently concentrated, a large amount of culture supernatant was generated from the GPGnlsLZ2 and GPGnlsLZ3 clones. The pseudotyped virus, initially possessing titers of 1.6×106 cfu/ml, could be concentrated by ultracentrifugation >1000-fold to achieve titers of >109 cfu/ml with 65–67% recovery of the infectious viral particles (Table 1).
Virus produced from the GPGnlsLZ cell lines were examined for the presence of RCV using a sensitive assay involving a Mus dunni cell line which harbors a highly transmissible retroviral genome encoding LacZ (21). Helper virus assays performed on unconcentrated viral supernatants as well as supernatants from transient transfections demonstrated that the retroviral stocks generated from the packaging cells were free of RCV (Table 1). It was not possible to perform cocultivation-based helper virus assays on the 293GPG-derived producer cell lines, since cocultivation of the cells with the Mus dunni indicator cells led to extensive cell fusion (data not shown).
Finally, because retroviruses produced from human cells are known to be resistant to inactivation by human serum (2, 4), we investigated the extent to which the viral pseudotypes produced from the 293GPG packaging cell line were resistant to inactivation by serum. Amphotropic virus from a ΨCRIPLZ producer clone and pseudotyped virus from the GPGnlsLZ4 clone were incubated with human serum, and the relative titers of each virus stock were then determined. The titer of the amphotropic virus was reduced by ≈250-fold after incubation with human serum, but not after incubation with heat-inactivated human serum (Fig. 6). In contrast, incubation of the VSV-G/retrovirus pseudotypes with human serum resulted in only a 5-fold reduction in titer. In light of the demonstrated role of natural human antibodies directed against proteins carrying Gal(α1–3)galactose terminal carbohydrates in the virus inactivation process (3, 4), we next
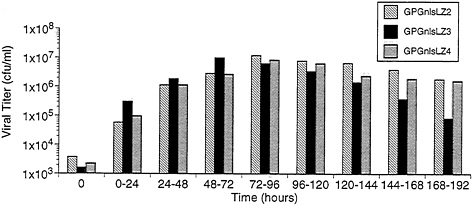
FIG. 5. Time course of VSV-G pseudotyped virus production by GPGnlsLZ clones. Clones GPGnlsLZ2, GPGnlsLZ3, and GPGnlsLZ4 were grown to 95% confluence in 100-mm dishes in tetracycline (1 μg/ml). Cells were rinsed and placed in 5 ml growth media without tetracycline. Supernatants were harvested and replaced with fresh media (no tetracycline) at 24 intervals. Supernatants were titered on NIH 3T3 cells. Titer is expressed as cfu/ml. Times indicate the period after tetracycline removal during which viral production was assessed.
Table 1. Viral titers from 293GPG and GPGnlsLZ producer cells
|
Producer cells |
Retroviral vector* |
Viral titer, units/ml† |
Helper virus assay‡ |
|
GPGnlsLZ2§ |
|
1.2×107 |
— |
|
GPGnlsLZ3 |
|
9.8×106 |
— |
|
GPGnlsLZ4 |
|
8.4×106 |
— |
|
Concentrated GPGnlsLZ2¶ |
|
5.4×109 |
ND |
|
Concentrated GPGnlsLZ3∥ |
|
3.4×109 |
ND |
|
ΔU3nlsLZ |
2.8×106 |
— |
|
|
293GPG (18) |
ΔU3nlsLZ |
3.0×106 |
— |
|
293GPG (20) |
ΔU3nlsLZ |
1.1×106 |
ND |
|
293GPG (20) |
ΔU3nlsLZ |
2.5×106 |
ND |
|
*nlsLacZ virus produced by transient transfection with ΔU3nlsLZ. †nlsLacZ virus titers determined on NIH 3T3 cells. ‡Helper virus assay performed using Mus dunni LacZ mobilization assay. Less than one amphotropic 4070A virus particle per milliliter was able to be detected by this method. ND, not determined. §Twenty-four hour viral supernatant harvest after removal of tetracycline from growth media (72- to 96-hr collection for GPGnlsLZ2, 48- to 72-hr collection for GPGnlsLZ3, and 72- to 96-hr collection for GPGnlsLZ4). ¶Unconcentrated titer was 1.6×106, virus was concentrated >3300-fold with 65% total virus recovery. ∥Unconcentrated titer was 1.6×106, virus was concentrated >3200-fold with 67% recovery of total virus. **Numbers in parentheses indicate cell passage at time of transfection. |
|||
examined the inactivation of virus exposed to human serum in the presence of an excess of Gal(α1–3)galactose. In the case of amphotropic virus, incubation with 10 mg/ml Gal(α1–3)galactose completely blocked virus inactivation, while in the case of the VSV-G/retroviral pseudotypes, the titer was unaffected. (Fig. 6).
DISCUSSION
The 293GPG packaging cell line described above possesses a number of features that should greatly facilitate the further evaluation of the potential applications of retroviral vectors. Most importantly, the cell line makes it possible both to generate stable virus-producing cell lines which produce very high titers of VSV-G/retroviral pseudotypes and to generate virus rapidly by transient transfection techniques. Because of the ability of the pseudotypes to be efficiently concentrated, it will now be feasible to generate the large amounts of extremely high titer (>109 cfu/ml) virus critical for examining the potential utility of retroviral vectors for in vivo infection and the ability to transduce cells refractory to infection by standard vectors. While the virus produced from 293GPG cells is not fully resistant to human serum, it is significantly more resistant than amphotropic virus generated from murine cells. Based on our Gal(α1–3)galactose blocking studies, we believe that the
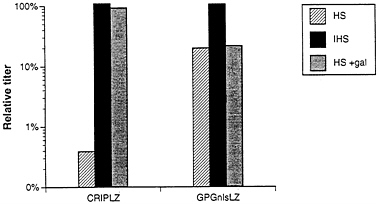
FIG. 6. Sensitivity to human serum of viruses produced by ΨCRIPLZ and GPGnlsLZ4 producer cell lines. Viruses produced from amphotropic ΨCRIPLZ producer cells and VSV-G pseudotyped GPGnlsLZ4 producer cells were incubated for 1 h at 37°C with fresh human serum (HS) and heat-inactivated human serum (IHS) in the presence and absence of 10 mg/ml Gal(α1–3)galactose. Surviving titers of LacZ virus were determined by infection of NIH 3T3 cells. The serum sensitivity assay was performed twice, and averaged relative titers for HS and IHS treatment versus IFS treatment are shown.
residual sensitivity of the virus pseudotypes to human serum may be due to natural IgM antibodies known to react against VSV (26). Cosset et al. (40) have recently described another human-derived packaging cell line capable of producing recombinant retrovirus that appears to be fully resistant to inactivation by human serum. In those studies, however, either the amphotropic envelope or an envelope derived from the virus RD 114 (41) was used rather than the VSV-G protein. Another potentially useful packaging cell line for the production of VSV G/retroviral pseudotypes has recently been described by Neinhuis and coworkers (42). That cell line makes use of murine cells and a strategy for the regulated expression of VSV-G similar to the one we have employed.
As a consequence of the use of the VSV-G protein as the determinant of the host range of virus produced by the 293GPG cell line, the packaging cell line exhibits several somewhat unusual properties relative to other packaging cell systems. First, while the expression of VSV-G in 293GPG cells is tightly regulated by tetracycline, the cells nevertheless express sufficient levels of VSV-G to yield titers of 103 cfu/ml in the presence of tetracycline. One concern that has not yet been fully evaluated is that the low levels of VSV-G expression in the presence of tetracycline may lead to the gradual loss of packaging function with the prolonged culture of the cells due to a growth selection against VSV-G-expressing cells. While we have observed that viral titers exceeding 106 cfu/ml can still be obtained by transient transfection after 20 passages of the cells, the prolonged passage of any working stock of the cells should probably be avoided. A second novel property of the cells is that, in addition to spontaneously fusing upon induction of VSV-G expression, the cells also promote the fusion of target cells with which they are cocultivated. While this property of the packaging cells makes the use of cocultivation techniques for achieving the highly efficient transduction of cells impractical, it is hopeful that the ability to generate highly concentrated virus stocks will compensate for this limitation. Another interesting property of 293GPG cells is the potential susceptibility of stable virus-producing cell lines generated from the cells to superinfection by the virus produced from the same cells. Although the titers of virus released from the cells in the presence of tetracycline are low, we do not yet know the extent to which reinfection of virus-producing cells may occur. Finally, because the packaging cells are routinely maintained in media containing tetracycline, puromycin, and G418, other selectable markers must be used for stably introducing vector constructs into the cells. In addition to zeocin, we have also
successfully used hygromycin and histidinol (43) selections with the 293G and 293GPG cells.
Most of the problems related to the generation of RCV by different retroviral packaging cell lines can be practically eliminated through the use of specific vectors (34, 35) and/or the practice of isolating clonal virus producing cell lines and screening the cell lines for helper. Nevertheless, several of the design features of the 293GPG packaging cell line may also provide at least some theoretical advantages over existing cell lines with regard to the possibility of release of RCV. One problem with all existing murine-based packaging cell lines is the presence of both endogenous retroviral DNA sequences (31, 33) and retroviral-like RNAs (44) that are efficiently packaged and transmitted to cells, and may contribute to the generation of helper virus under certain conditions. Another issue relates to the potential of different packaging cell lines to give rise to helper virus due to the overlap of sequences between the particular vector used and the precise packaging sequences present in the packaging cell line (34, 35). A more global inherent defect in the design of all murine and human-derived packaging cell lines is the ability of retroviral RNAs which lack packaging sequences to be packaged, albeit at low efficiency, and transmitted to cells (40). Even in the case of third generation packaging cells (15), it has been possible to observe the transmission of viral packaging functions to recipient cells. Cosset et al., for example, have recently documented and quantitated the transfer of both gag and env encoding genomes derived from both a packaging construct used to generate ΨCRIP cells (15) and a construct used in the generation of the human-derived FLY cell line (40), another third generation packaging cell line. In our laboratory’s unpublished studies with ΨCRE/ΨCRIP cells and the parental MFG vector, which contains an extended gag ORF (19), we have also obtained data consistent with the transfer of packaging functions and the possible emergence of helper virus in the context of high titer cross-infections employed to generate complex populations of virus-producing cells.
The design of the 293GPG cells may be relevant to each of the above issues. In the construction of the 293GPG cell line, we have used only the precise viral sequences necessary to encode gag-pol and an expression vector that utilizes totally nonretroviral sequences. We have also utilized totally nonretroviral sequences to provide for the host range of the virus produced from the cells rather than use conventional retroviral env gene expression constructs. Depending on the vector used in conjunction with 293GPG cells, this design feature of the cells may reduce the probability of undesirable recombination events. More significantly, it is hopeful that the removal of all extraneous viral sequences in the transcript used to express gag-pol and the use of nonretroviral transcripts encoding VSV-G will reduce the efficiency with which those transcripts can be packaged and transmitted to cells, relative to that which occurs with conventional third generation packaging cell transcripts. This hypothesis will need to be tested directly.
We thank Dr. Jean Schaffer for helpful discussions and critical review of this manuscript. D.S.O. is supported by a National Institutes of Health Physician Scientist Award (HL02910). Support for this work was also provided for by a Program of Excellence in Molecular Biology Grant from the National Heart, Lung, and Blood Institute (HL41484).
1. Mulligan, R.C. (1993) Science 260, 926–932.
2. Takeuchi, Y., Cosset, F.-L.C., Lachmann, P.J., Okada, H., Weiss, R.A. & Collins, M.K.L. (1994) J. Virol. 68, 8001–8007.
3. Takeuchi, Y., Porter, C.D., Strahan, K.M., Preece, A.F., Gustafasson, K., Cosset, F.-L., Weiss, R.A. & Collins, M.K.L. (1996) Nature (London) 379, 85–88.
4. Rother, R.P., Fodor, W.L., Springhorn, J.P., Birks, C.W., Setter, E., Sandrin, M.S., Squinto, S.P. & Rollins, S.A. (1995) J. Exp. Med. 182, 1345–1355.
5. Roe, T., Reynolds, T.C., Yu, G. & Brown, P.O. (1993) EMBO J. 12, 2099–2108.
6. Lewis, P.F. & Emerman, M. (1994) J. Virol. 68, 510–516.
7. Emi, N., Friedmann, T. & Yee, J.-K. (1991) J. Virol. 65, 1202– 1207.
8. Burns, J.C., Friedmann, T., Driever, W., Burrascano, M. & Yee, J.-K. (1993) Proc. Natl. Acad. Sci. USA 90, 8033–8037.
9. Zavada, J. (1972) J. Gen. Virol. 15, 183–191.
10. Grahm, F., Smiley, R. & Nairu, R. (1977) J. Gen. Virol. 36, 59.
11. Cullen, B.R. (1986) Cell 46, 973–982.
12. Gossen, M. & Bujard, H. (1992) Proc. Natl. Acad. Sci. USA 89, 5547–5551.
13. Rose, J.K. & Gallione, C. (1981) J. Virol. 39, 519–528.
14. Sadelain, M., Wang, C.H., Antoniou, M., Grosveld, F. & Mulligan, R.C. (1995) Proc. Natl. Acad. Sci. USA 92, 6728–6732.
15. Danos, O. & Mulligan, R.C. (1988) Proc. Natl. Acad. Sci. USA 85, 6460–6464.
16. Morgenstern, J.P. & Land, H. (1990) Nucleic Acids Res. 18, 1068.
17. Southern, P.J. & Berg, P. (1982) J. Mol. Appl. Genet. 1, 327–341.
18. Berns, A.J., Clift, S., Cohen, L.K., Donehower, R.C., Dranoff, G., Hauda, K.M., Jaffee, E.M., Lazenby, A.J., Levitsky, H.I. & Marshall, F.F. (1995) Hum. Gene Ther. 6, 347–368.
19. Riviere, I., Brose, K. & Mulligan, R.C. (1995) Proc. Natl. Acad. Sci. USA 92, 6733–6737.
20. Boshart, M., Weber, F., Jahn, G., Dorsch-Hasler, K., Fleckenstein, B. & Schaffner, W. (1985) Cell 41, 521–530.
21. Dranoff, G., Jaffee, E., Lazenby, A., Golumbek, P., Levitsky, H., Brose, K., Jackson, V., Hamada, H., Pardoll, D. & Mulligan, R.C. (1993) Proc. Natl. Acad. Sci. USA 90, 3539–3543.
22. Pear, W.S., Nolan, G.P., Scott, M.L. & Baltimore, D. (1993) Proc. Natl. Acad. Sci. USA 90, 8392–8396.
23. Sambrook, J., Fritsch, E.F. & Maniatis, T.T. (1989) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY), 2nd Ed.
24. Goff, S., Traktman, P. & Baltimore, D. (1981) J. Virol. 38, 239–248.
25. Lim, K. & Chae, C.-B. (1989) BioTechniques 7, 576–579.
26. Beebe, D.P. & Cooper, N.R. (1981) J. Immunol. 126, 1562–1568.
27. Osbourne, W.R.A., Hock, R.A., Kaleko, M. & Miller, A.D. (1990) Hum. Gene Ther. 4, 609–615.
28. Miller, A.D. & Buttimore, C. (1986) Mol. Cell. Biol. 6, 2895– 2902.
29. Mann, R., Mulligan, R.C. & Baltimore, D. (1983) Cell 33, 153–159.
30. Markowitz, D., Goff, S. & Bank, A. (1988) J. Virol. 62, 1120– 1124.
31. Scadden, D.T., Fuller, B. & Cunningham, J.M. (1990) J. Virol. 64, 424–427.
32. Cosset, F.L., Girod, A., Flamant, F., Dryand, A., Ronfort, C., Valsesia, S., Molina, R.M., Faure, C., Nigon, V.M. & Verdier, G. (1993) Virology 193, 385–395.
33. Purcell, D.F.J., Broscius, C.M., Vanin, E.F., Buckler, C.F., Neinhuis, A.W. & Martin, M.A. (1996) J. Virol. 70, 887–897.
34. Miller, A.D. & Rosman, G.J. (1989) BioTechniques 7, 982–990.
35. Miller, A.D. (1990) Hum. Gene Ther. 1, 5–14.
36. Hebert, D.N., Foellmer, B. & Helenius, A. (1995) Cell 81, 425–433.
37. Zhang, L. & Ghosh, H.P. (1994) J. Virol. 68, 2186–2193.
38. Blumenthal, R.A., Bali-Puri, A., Walter, A., Corell, D. & Eidelman, O. (1987) J. Biol. Chem. 262, 13614–13619.
39. Finer, M.H., Dull, T.J., Qin, L., Farson, D. & Roberts, M.R. (1994) Blood 83, 43–50.
40. Cosset, F.-L., Takeuchi, Y., Battini, J.-L., Weiss, R.A. & Collins, M.K.L. (1995) J. Virol. 69, 7430–7436.
41. Reeves, R.H. & O’Brien, S.J. (1984) J. Virol. 52, 164–171.
42. Yang, Y., Vanin, E.F., Whitt, M.A., Fornerod, M., Zwart, R., Scheiderman, R.D., Grosveld, G. & Neinhuis, A.W. (1995) Hum. Gene Ther. 6, 1203–1213.
43. Hartman, S.C. & Mulligan, R.C. (1988) Proc. Natl. Acad. Sci. USA 85, 8047–8051.
44. Torrent, C., Bordet, T. & Darlix, J.L. (1994) J. Mol. Biol. 240, 434–444.






