
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and of Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA
Cell-surface receptors for retroviruses and implications for gene transfer
A. DUSTY MILLER
Fred Hutchinson Cancer Research Center, 1100 Fairview Avenue North, Room C2–023, Seattle, WA 98109
ABSTRACT Retroviruses can utilize a variety of cell-surface proteins for binding and entry into cells, and the cloning of several of these viral receptors has allowed refinement of models to explain retrovirus tropism. A single receptor appears to be necessary and sufficient for entry of many retroviruses, but exceptions to this simple model are accumulating. For example, HIV requires two proteins for cell entry, neither of which alone is sufficient; 10A1 murine leukemia virus can enter cells by using either of two distinct receptors; two retroviruses can use different receptors in some cells but use the same receptor for entry into other cells; and posttranslational protein modifications and secreted factors can dramatically influence virus entry. These findings greatly complicate the rules governing retrovirus tropism. The mechanism underlying retrovirus evolution to use many receptors for cell entry is not clear, although some evidence supports a mutational model for the evolution of new receptor specificities. Further study of factors that govern retrovirus entry into cells are important for achieving high-efficiency gene transduction to specific cells and for the design of retroviral vectors to target additional receptors for cell entry.
Many features make retrovirus vectors a good choice for gene transfer into animal cells. Most importantly, these vectors integrate efficiently into the target cell genome to promote stable gene transfer, and integration is precise with respect to the virus genome, resulting in unrearranged transfer of the desired genes. The only other integrating vector is derived from adeno-associated virus, but integration is inefficient (1) and appears not to be precise with respect to the viral genome (2). In addition, retroviral vectors can transduce both dividing and non-dividing cells, although this is true of vectors derived from HIV (3) and not the commonly used vectors derived from murine leukemia viruses, which require cell division (4). Furthermore, retrovirus vectors can be designed to eliminate all viral protein coding regions without affecting gene transfer rates, and can be made in the absence of replication-competent virus by using retrovirus packaging cell lines, which supply all of the viral proteins required for vector transmission. Gene transfer and expression mediated by such replication-incompetent vectors is called transduction to differentiate this process from virus infection followed by further virus replication.
A key consideration in retroviral vector design is the source of the viral envelope (Env) protein present on vector virions, because this protein binds to specific cell-surface proteins and is the primary determinant of the range of cells that can be transduced by the vector. The name of the virus or the virus group from which the Env protein was derived will be referred to as the pseudotype of the vector. Naturally occurring retroviruses can use a variety of different proteins for cell entry, although in general individual retroviruses appear to recognize a single receptor. Utilization of additional cell-surface proteins for vector entry has been achieved by incorporation of polypeptides into the Env protein to alter its receptor binding properties or by replacement of the retroviral Env protein with surface proteins from other viruses. These alterations can allow targeting of particular cells that express specific proteins or an expansion of the range of cells that can be transduced by targeting broadly expressed proteins. In this paper I will review the factors that govern retrovirus binding and entry into cells and implications for the design of retroviral vectors.
Virus Interference
Early evidence that retroviruses use multiple receptors for cell entry came from studies of virus interference. Infection of a cell by a replication-competent retrovirus results in synthesis of a retroviral Env protein that binds to the receptor used for virus entry. This effectively blocks entry of the original virus and other retroviruses that target the same receptor, whereas entry of retroviruses that use different receptors is unaffected. Interference between retroviruses has been shown to occur at the level of virus entry into cells and not at some other step in the virus life cycle. By interference analysis, retroviruses that infect human cells have been assigned to eight groups that use different receptors on human cells (Table 1). The genes encoding these receptors are scattered on different chromosomes (Table 1), indicating that the receptors are different proteins.
Cloned Retrovirus Receptors
In 1984 CD4 (previously called T4) was identified as a receptor for HIV-1, and became the first known retrovirus receptor (12, 13). Since then, six additional retrovirus receptors have been identified and their cDNAs cloned (Table 2). All except CD4 appear to be sufficient for entry of the corresponding retroviruses by the criteria that expression of these receptors in nonpermissive cells renders the cells susceptible to infection. In contrast, CD4 transfer into nonpermissive mouse cells does not allow infection by HIV. HIV binds to all cells that express CD4, but another factor is required for HIV entry. Recently, a coreceptor for T-cell tropic HIV-1 strains has been found and was named fusin to indicate its presumed role in virus entry following HIV-1 binding to CD4 (14). Expression of the human CD4 and fusin proteins in mouse cells renders the cells susceptible to HIV-1 infection, whereas either protein alone is insufficient. Even
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: MLV, murine leukemia virus; AM-MLV, amphotropic MLV; MoMLV, Moloney MLV; CHO, Chinese hamster ovary; GALV, gibbon ape leukemia virus; FeLV, feline leukemia virus.
Table 1. Retrovirus interference groups in human cells
more recently, a second protein related to fusin and previously named CC-CKR-5 has been found to be a coreceptor for macrophage-tropic HIV-1 strains (15, 16).
These results, showing that two proteins are required for HIV-1 entry, raise the possibility that coreceptors are required for entry of other retroviruses. However, their detection will require the identification of nonpermissive cells for which transfer of the known receptors does not render the cells susceptible to infection. Some retroviruses have a very wide host range; thus, if other proteins are required for entry of these viruses, functional homologs of these coreceptors must be widely distributed in cells from many species.
Two of the cloned retrovirus receptors, Ram1 and Glvr1, are closely related at the protein sequence level (21, 22, 24), and both are sodium-dependent phosphate transporters (23). These proteins are members of a large family of known and presumptive phosphate transporters from many organisms (Fig. 1). However, Ram1 and Glvr1 are clearly distinct since the genes encoding these proteins are located on different chromosomes in humans and mice (8, 9, 30, 31) and they show very different patterns of expression in animal tissues (23). In addition, these proteins serve as receptors for distinct groups of viruses in human cells (Table 1).
The 10A1 Retrovirus Can Use Either of Two Receptors for Cell Entry
Studies of cloned retrovirus receptors and most virus interference data suggested that individual retroviruses bind to a single protein for entry into cells. When different viruses bind to the same receptor, they typically show reciprocal interference; that is, infection of cells by either virus blocks entry by the other virus. The finding of nonreciprocal interference between some retroviruses complicated this picture. In the example shown (Table 3), transduction by a vector with an amphotropic, a 10A1, or an ecotropic pseudotype was measured in NIH 3T3 mouse cells infected with amphotropic MLV (AM-MLV), 10A1 MLV, Moloney MLV, or no virus. A typical pattern of interference for viruses that use different receptors for cell entry is shown by the amphotropic and ecotropic viruses, where ecotropic vector transduction is blocked by the presence of ecotropic MoMLV in the target cells, but is unaffected by the presence of amphotropic virus, and ampho-
Table 2. Cloned retrovirus receptors
|
Retrovirus |
Receptor |
Type* |
Function |
Refs. |
|
Human immunodeficiency virus |
CD4 |
TM1 |
Immune recognition |
|
|
|
Fusin, CC-CKR-5 (coreceptors) |
TM7 |
G protein-coupled chemokine receptors |
|
|
Simian immunodeficiency virus |
CD4 |
TM1 |
Immune recognition |
|
|
Murine ecotropic retrovirus |
Rec1 |
TM14 |
Basic amino acid transport |
|
|
Murine amphotropic retrovirus |
Ram1 |
TM10–13 |
Phosphate transport |
|
|
Gibbon ape leukemia virus |
Glvr1 |
TM10–13 |
Phosphate transport |
|
|
Bovine leukemia virus |
Blvr |
TM1 |
ND |
|
|
Avian leukosis virus type A |
Tva |
TM1 |
LDL receptor-like protein |
|
|
Feline immunodeficiency virus |
CD9 |
TM4 |
Signaling protein? |
|
|
ND, not determined; LDL, low density lipoprotein. *TM followed by a number indicates the number of transmembrane domains in the protein. |
||||
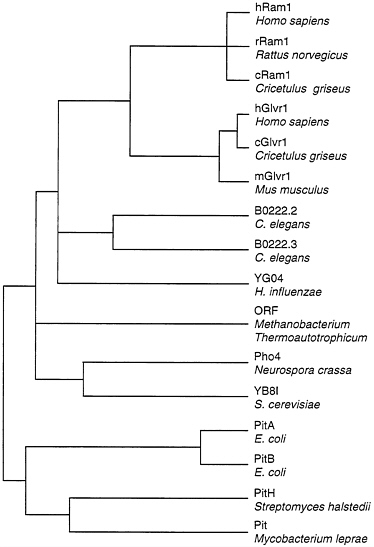
FIG. 1. Dendrogram of amino acid sequence similarities among phosphate transporters. Distances between sequences were computed by the Genetics Computer Group program PILEUP. Overall sequence identity for the branch point at the far left is about 21%. Sequences were obtained from GenBank: hRam1, L20852; rRam1, L19931; cRam1, U13945; hGlvr1, L20859; cGlvr1, U13946; mGlvr1, M73696; B0222.2 and B0222.3, U50312; YG04, P45268; M. thermoauto trophicum ORF, S08522; Pho4, M31364; YB8I, P38361; PitA, P37308; PitB, P43676; PitH, P41132; Pit, U15187.
tropic vector transduction is blocked by the presence of AM-MLV, but is unaffected by the presence of ecotropic MoMLV. Nonreciprocal interference is displayed by the amphotropic and 10A1 viruses, where 10A1-pseudotype vector transduction is unaffected by the presence of amphotropic virus in the target cells, but amphotropic vector transduction is blocked by the presence of 10A1 virus. These data suggested that 10A1 virus can enter cells by using a different receptor than that used by amphotropic virus, and that 10A1 virus can also bind to the amphotropic receptor and block amphotropic virus entry (Fig. 2).
Given that 10A1 virus can bind to Ram1, we tested the ability of Ram1 to mediate entry of the 10A1-pseudotype LAPSN vector. We also tested Glvr1 due to its similarity to Ram1 and thus the possibility that Glvr1 was the alternative receptor for entry of 10A1 virus. We found that expression of human Ram1, rat Ram1, human Glvr1, or mouse Glvr1 rendered Chinese hamster ovary (CHO) cells susceptible to 10A1-pseudotype LAPSN vector transduction (Table 4). Thus, 10A1 virus can bind and enter cells by using either of two different retrovirus receptors. Amphotropic virus can enter CHO cells expressing human or rat Ram1, but not those expressing human or mouse Glvr1 (data not shown). These results explain the nonreciprocal interference observed between 10A1 and amphotropic retroviruses.
Table 3. Nonreciprocal interference between 10A1 and amphotropic retroviruses
|
LAPSN pseudotype |
Vector titer, FFU/ml |
|||
|
3T3 |
3T3+ AM-MLV |
3T3+ 10A1 |
3T3+ MoMLV |
|
|
Amphotropic |
5×106 |
40 |
3 |
4×106 |
|
10A1 |
7×106 |
6×106 |
2×102 |
6×106 |
|
Ecotropic |
3×106 |
2×106 |
2×106 |
40 |
|
The LAPSN vector encodes alkaline phosphatase and neomycin phosphotransferase. LAPSN vector with an amphotropic, 10A1, or ecotropic pseudotype was made by using PA317 retrovirus packaging cells, wild-type 10A1 virus, or PE501 packaging cells, respectively. Transduction was measured by staining cells for alkaline phosphatase 2 days after exposure to the vectors. Data are from Miller and Chen (32). |
||||
Some Receptors Can Promote Entry of Retroviruses That Normally Utilize Independent Receptors
In human cells, gibbon ape leukemia virus (GALV) exclusively uses Glvr1 for entry and amphotropic retrovirus exclusively uses Ram1. These facts are reflected in the assignment of these viruses to separate interference groups for human cells (Table 1). However, analysis of the hamster homolog of Ram1 shows that it can function as a receptor for GALV or amphotropic retrovirus (34). In addition, certain chimeric receptors made between Ram1 and Glvr1 can also function as receptors for both viruses (Fig. 3). In this example, the hybrid receptor RRG promotes transduction by GALV or amphotropic pseudotype vectors with efficiencies similar to those found for GALV vector transduction of cells expressing the normal human Glvr1 receptor (GGG) or amphotropic vector transduction of cells expressing the normal rat Ram1 (RRR) receptor. Thus, the ability of retroviruses to utilize certain receptor homologs for entry, and therefore the interference pattern of these

FIG. 2. Nonreciprocal interference between 10A1 and amphotropic retroviruses.
Table 4. A 10A1-pseudotype retroviral vector can utilize Ram1 or Glvr1 for cell entry
|
Receptor |
Species |
Vector titer, FFU/ml |
|
Ram-1 |
Human |
1×106 |
|
|
Rat |
3×105 |
|
Glvr-1 |
Human |
6×105 |
|
|
Mouse |
7×105 |
|
None |
|
<500 |
|
Retrovirus receptor cDNAs were expressed by using the retroviral vector LXSN. CHO cells were seeded at 2×104 per well (d=3.5 cm) in multiwell dishes on day 1. On day 2, cells were cotransfected with 2.5 μg of β-galactosidase expression plasmid and 2.5 μg of the receptor expression construct. On day 3, one set of dishes was stained for β-galactosidase to assess transfection efficiency, whereas the other set was infected with 2 μl of the LAPSN vector pseudotyped with the 10A1 retrovirus in the presence of 50% medium conditioned by CHO cells. On day 4, cells were stained for alkaline phosphatase and foci of stained cells were counted. Transfection efficiencies were similar for all constructs, as measured by β-galactosidase staining. Data are from Miller and Miller (33). |
||
retroviruses, will depend on the specific receptors expressed in the target cells.
Retrovirus Interactions with Homologous Receptor Proteins from Other Species Are Complex
Limitations to retrovirus entry into cells from different species are due to variable expression of the receptor or its homologs, or to amino acid sequence differences or posttranslational modifications in the receptor homologs in different species that inhibit virus binding or entry. The former mechanism for virus resistance is primarily applicable to different cells from the same organisms that express variable levels of the receptor. An example of this is provided by HIV, which for entry requires the CD4 receptor that is found primarily on T lymphocytes and not on cells from many other tissues. Many examples of the latter mechanism of virus resistance have been found for cells from different species, in which a receptor homolog is expressed but is nonfunctional. For example, ecotropic retroviruses infect rodent cells, but do not infect human cells, even though human cells express a homolog of the murine ecotropic receptor that is 87% identical to the mouse protein. In this case, only two amino acid changes are required to convert the human protein into a functional receptor, or to render the mouse protein nonfunctional as an ecotropic retrovirus receptor (35, 36).
Other examples of virus restriction in different species are provided by viruses that utilize Pit receptor family members for entry. A simple example is the restriction of GALV entry into mouse cells, which, like the restriction to ecotropic virus infection of human cells, is not due to lack of receptor homolog expression, but to minor changes in mouse Glvr1 compared with human Glvr1.
A more complicated example is provided by 10A1 receptor usage in different species. As noted above, 10A1-pseudotype virus can use human Ram1 or human Glvr1 to enter CHO cells (Table 4) and to enter human cells (32). However, in rat cells 10A1 virus infection does not block GALV-pseudotype vector transduction (Table 5), showing that 10A1 cannot bind to or enter cells by using the rat GALV receptor. Indeed, 10A1 has the same interference properties as amphotropic retrovirus in rat cells, and thus uses the amphotropic virus receptor for entry. Thus, 10A1 virus behaves like an amphotropic virus in rat cells, but like a combination of an amphotropic virus and GALV in human cells. Therefore, the interference and receptor utilization properties of 10A1 virus depends on the cell type used for the analysis.
Env Amino Acid Sequence Is Not Predictive of Receptor Utilization
It is not clear how retroviruses have evolved to utilize such a diverse group of receptors for entry into cells. One possibility is that mutations in the Env protein promote weak binding and entry through interaction with new receptors, and if the new receptor specificity is beneficial for virus survival, selective pressure favors further mutations that promote more efficient
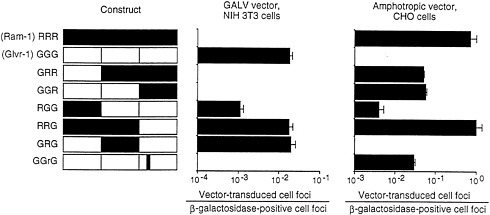
FIG. 3. GALV- and amphotropic-pseudotype vector transduction of cells expressing hybrid Ram-1/Glvr-1 receptors. CHO or NIH 3T3 cells were seeded in 3.5-cm dishes at 2×104 cells per dish. On day 2, hybrid constructs cloned in the retroviral vector LXSN were cotransfected (1:1) with a plasmid encoding β-galactosidase (2.5 pig each). Parallel dishes were stained for β-galactosidase, whereas the other set was infected with the retroviral vector LAPSN pseudotyped by amphotropic Env [LAPSN(PA317)] or a GALV Env [LAPSN(PG13)] at a multiplicity of infection of about 2. Cells were stained for alkaline phosphatase activity on day 5. Values are the number of vector-transduced (alkaline phosphatase-positive) foci divided by the number of β-galactosidase positive foci. Results are averages of duplicate dishes. The experiment was performed three times with similar results. Data are from Miller and Miller (33).
Table 5. 10A1-pseudotype LAPSN vector can use the amphotropic receptor, but not the GALV receptor, for entry into 208F rat cells
|
LAPSN pseudotype |
Vector titer, FFU/ml |
||
|
208F |
208F+AM-MLV |
208F+GALV |
|
|
10A1 |
1×107 |
500 |
1×107 |
|
Amphotropic |
2×106 |
200 |
2×106 |
|
GALV |
2×105 |
2×105 |
100 |
|
LAPSN vector with a 10A1, amphotropic, or GALV pseudotype was made by using wild-type 10A1 virus, PA317 retrovirus packaging cells, or PG13 packaging cells, respectively. Transduction was measured by staining cells for alkaline phosphatase 2 days after exposure to the vectors. Data are from Miller and Chen (32). |
|||
utilization of the new receptor. Another model involves replacement of portions of the retroviral Env protein with portions of cellular proteins that recognize other cell-surface proteins. For example, incorporation of a portion of the erythropoietin protein into an existing Env protein might allow the erythropoietin receptor to function as a new target for retrovirus binding and entry. This model for acquisition of new receptor specificities parallels the process of retrovirus acquisition of cellular oncogenes. Just as oncogenes can improve retrovirus replication and survival, the ability to utilize new receptors for cell entry should improve the survival potential of a retrovirus. In addition, the existence of cellular proteins that bind cell-surface molecules with high affinity seems a more likely source for the development of radically altered retrovirus receptor specificities compared with random Env mutations.
Predictions of the model for alteration of retrovirus receptor utilization by incorporation of cellular genes is that retroviruses that utilize similar receptors should contain more closely related Env proteins than retroviruses that use other receptors, and Env proteins should contain regions of similarity with cellular proteins. A comparison of Env proteins from several retroviruses shows that retroviruses that use different receptors can be more highly related than those that use the same receptor (Fig. 4). For example, GALV, 10A1, and subgroup B feline leukemia virus (FeLV-B) all can use Glvr1 as a receptor, but FeLV-B is more closely related to FeLV-A and FAIDS, which use different receptors, than it is to GALV or the 10A1 virus. Likewise, the 10A1 virus is more closely related to the polytropic (MoMCF; Moloney mink cell focus-forming virus), xenotropic (NZB), and ecotropic (FrMLV, MoMLV, and AKV) retroviruses than to GALV or FeLV-B. The same overall dendrogram is obtained even if one compares only the 200 amino acids at the amino termini of the processed Env proteins that are directly involved in receptor binding (not shown). Comparison of these amino terminal receptor-binding regions of the Env proteins from viruses in the FeLV, MLV, and GALV groups reveals a similar amino acid framework surrounding two variable regions, with no common features in the variable regions that would predict the receptor utilization pattern (37). In addition, no similarities have been found between regions of retroviral Env proteins and already sequenced cellular proteins. Thus, the data to date favor a mutational origin for new receptor specificities rather than a model involving acquisition of cellular proteins that can bind new cell-surface receptors. The mutational model can explain the diversity in Env sequences among viruses that recognize the same receptor as the result of convergent evolution of different parental retroviruses.
Another argument in favor of a mutational model for acquisition of new receptor specificities is the finding that small changes in a virus Env protein can result in a new receptor specificity. For example, no more than six amino acid changes are required to convert an amphotropic Env, which targets Ram1, to one having the receptor utilization properties of
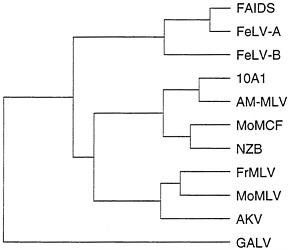
FIG. 4. Dendrogram of amino acid similarities between different retroviral Env proteins. Distances between sequences were computed by the Genetics Computer Group program PILEUP. Overall sequence identity for the branch point at the far left is about 42%. Sequences were obtained from GenBank: FAIDS, feline AIDS virus, M18247; FeLV-A, feline leukemia virus subgroup A, M12500; FeLV-B, feline leukemia virus subgroup B, X00188; 10A1, 10A1 MLV, M33470; AM-MLV, M33469; MoMCF, Moloney mink cell focus-forming virus, J02254; NZB, NZB MLV, K02730; FrMLV, Friend MLV, Z11128; MoMLV, J02255; AKV, AKV MLV, J01998; and GALV, M26927.
10A1 virus, which targets Ram1 or Glvr1 for entry (32, 38). However, because Ram1 and Glvr1 are related proteins, this change does not represent a dramatic switch in receptor specificity, and it will be interesting to see if minor amino acid changes in Env proteins can result in more dramatic changes in receptor utilization.
Endogenous Synthesis of Env Protein Can Block Retrovirus Entry
Retrovirus receptors can be rendered nonfunctional due to blockade by Env protein synthesized by a replication-competent retrovirus. This is the basis for the virus interference discussed above. Interference with receptor function can also result from synthesis of Env proteins from endogenous retroviruses or fragments of retroviruses that are inherited in animals. A well-documented example of this phenomenon involves the Fv-4 locus in mice (39), the phenotype of which is due to a truncated endogenous ecotropic retrovirus that is missing the gag and part of the pol genes, but which contains an intact env gene. Synthesis of this endogenous env gene product in mouse tissues blocks infection and leukemia caused by ecotropic retroviruses by blocking the ecotropic retrovirus receptor. Other examples of this phenomenon have been found for avian leukosis viruses in chickens (40) and for MCF viruses in mice (41).
Hamster Cells Secrete a Factor That Blocks Retrovirus Infection and a Similar Factor Is Found in Hamster Serum
CHO cells are resistant to infection by many retroviruses. In most cases, this resistance can be abrogated by prior treatment of the cells with the glycosylation inhibitor tunicamycin. The resistance to GALV and amphotropic retrovirus infection is due to a secreted protein factor that blocks infection (42). Thus, addition of CHO cell-conditioned medium to CHO cells that have been made susceptible to infection by treatment with tunicamycin blocks infection by GALV and amphotropic viruses. In contrast, addition of the conditioned medium does not block infection of tunicamycin-treated CHO cells by an ecotropic retrovirus, showing that the medium is not simply toxic, and that the effect is specific for retroviruses with particular Env proteins. Interestingly, the CHO cell-
conditioned medium does not block amphotropic vector transduction of human or mouse cells, nor does it block transduction of CHO cells made susceptible to amphotropic vector transduction by prior introduction of genes expressing human or rat Ram1 (21, 22), indicating that the factor can bind to and block the hamster receptor but not the human or rat receptors.
Hamster serum contains a similar factor that can block retrovirus infection of tunicamycin-treated CHO cells (Table 6). Addition of 5% serum from Chinese hamsters completely blocked transduction by an amphotropic vector, and 12.5% serum from Syrian hamsters also significantly inhibited transduction. In contrast, addition of 25% fetal bovine serum had no effect on transduction of tunicamycin-treated CHO cells by the amphotropic vector. Like the CHO cell-conditioned medium, the hamster sera had no effect on amphotropic vector infection of HeLa human cells (Table 6), indicating a species specificity for the factor.
Thus, hamster serum contains a similar, potentially identical, inhibitor of retrovirus infection to that secreted from CHO cells. Based on the principle of virus interference, the factor could be a fragment of an Env protein that is secreted from cells and blocks infection by binding to the virus receptor. Alternatively, it could be a normal cellular protein that naturally interacts with the phosphate transporter that serves as a receptor for GALV and amphotropic viruses resulting in a block to infection.
Receptor Glycosylation Can Affect Retrovirus Entry
Retroviral interference can by reversed by inhibitors of glycosylation that affect Env processing and subsequent binding to virus receptors (44). In addition, inhibitors of glycosylation can have direct effects on a retrovirus receptor to modulate infection. For example, the ecotropic retrovirus receptor homolog on Mus dunni cells functions as a receptor for most ecotropic retroviruses with the exception of MoMLV. Tunicamycin treatment renders the cells susceptible to infection by MoMLV, and alteration of a single amino acid in the Mus dunni receptor to prevent glycosylation at that site results in a receptor that promotes MoMLV infection (45). Thus, subtle changes in receptor glycosylation can have a major effect on the ability of a retrovirus to utilize the receptor for cell entry.
Vector Pseudotypes Available for Gene Transfer Applications
Given the complicated factors that govern retrovirus entry into cells from different tissues and different species, it is helpful that there are a wide range of retrovirus packaging cell lines that are available for production of retroviral vectors with
Table 6. Hamster sera inhibit amphotropic vector infection of tunicamycin-treated CHO cells but not HeLa cells
|
Target cells |
Additional serum |
Vector titer, CFU/ml |
Inhibition, % |
|
CHO |
None |
1×103 |
|
|
|
Chinese hamster (5%) |
<10 |
>99 |
|
|
Syrian hamster (12.5%) |
35 |
97 |
|
|
Fetal bovine (25%) |
2×103 |
|
|
HeLa |
None |
3×105 |
|
|
|
Chinese hamster (5%) |
2×105 |
|
|
|
Syrian hamster (12.5%) |
3×105 |
|
|
The indicated target cells were plated at 105 per 6-cm dish on day 1, infected with an amphotropic-pseudotype vector carrying the neo gene on day 2 in the presence of culture medium containing 5% FBS (no additional serum), 5% FBS plus 5% Chinese hamster serum, 5% FBS plus 12.5% Syrian hamster serum, or 5% FBS plus 25% additional FBS, and G418-resistant colony formation was measured. Inhibition is reported only when >50%. Data are from Miller and Miller (43). |
|||
different pseudotypes. Approximate host ranges of packaging cells derived using mammalian retroviruses are shown in Table 7. A listing of specific packaging cell lines can be found in ref. 46. The best vector pseudotype for a given application will be further influenced by the specific target tissue and the expression of suitable levels of receptors with proper posttranslational modifications to allow efficient virus entry. For example, Glvr1 is overexpressed compared with Ram1 in hematopoietic cells (23), and vectors with a GALV pseudotype have been found to transduce hematopoietic cells more efficiently than the same vectors with an amphotropic pseudotype (47, 48).
Conclusions
Retroviruses utilize a diverse set of proteins for cell entry. Single proteins are apparently required for binding and entry of most retroviruses, although two proteins are required for HIV. Although virus entry is dependent on the level of receptor expression in particular cells, there are many other factors that govern utilization of a receptor or its homologs in different species. Subtle alterations in the amino acid sequence of receptor homologs in different species can dramatically affect virus entry, either as a direct result of changes in the primary amino acid sequence or as an indirect result of altered protein modifications such as glycosylation. Indeed, restricted virus host range is not generally due to a lack of expression of homologous receptor proteins, but is more often related to minor alterations in these proteins. In addition, soluble proteins secreted by some cells and present in some animals, and retroviral Env proteins synthesized from replication-competent viruses or from endogenous virus sequences, can block receptor utilization. These are all important considerations in the design of retroviral vectors for gene transfer in cultured cells and in animals.
Recently it has become clear that certain retroviruses can use more than one receptor for entry into some cell types, and some receptors can promote entry of retroviruses that normally utilize different receptors in other cells. These results seriously complicate attempts to classify retroviruses into groups based on receptor utilization, as determined by interference analysis, because these groupings depend on the particular receptors expressed on the cell type used for the analysis. In fact, this problem was appreciated long before the molecular basis for this phenomenon was determined (49).
Further development of retroviral vectors for gene transfer applications has involved the incorporation of Env proteins
Table 7. Host range of selected retrovirus packaging cells
from other virus families, such as the vesicular stomatitis virus G protein (3, 50) and efforts to alter the receptor specificity of existing retroviral Env proteins by the incorporation of peptide or antibody domains that can bind to other cell-surface proteins (51, 52). An understanding of the principles governing cell entry by naturally occurring retroviruses will help in the design and application of these strategies.
A fascinating aspect of retroviruses is their utilization of diverse proteins for cell entry. The analysis presented here favors a mutational basis for retrovirus evolution to utilize new receptors, rather than acquisition and expression of cellular proteins that naturally bind to cell-surface receptors, but more information is needed to resolve this issue. Perhaps analysis of additional naturally occurring retroviruses and their receptors will reveal a clear example of acquisition of a cellular gene that enables utilization of a new cell-surface receptor for entry. Answers to these questions have important implications for the design of retroviral vectors with novel receptor specificities, and for the evolution of retroviruses, which are important agents of disease in humans and in animals.
I thank Michael Emerman and Greg Wolgamot for comments on this manuscript. This work was supported by grants from the National Heart, Lung, and Blood Institute and the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health.
1. Halbert, C.L., Alexander, I.E., Wolgamot, G.M. & Miller, A.D. (1995) J. Virol. 69, 1473–1479.
2. Kotin, R.M. & Berns, K.I. (1989) Virology 170, 460–467.
3. Naldini, L., Blomer, U., Gallay, P., Ory, D., Mulligan, R., Gage, F.H., Verma, I.M. & Trono, D. (1996) Science 272, 263–267.
4. Miller, D.G., Adam, M.A. & Miller, A.D. (1990) Mol. Cell. Biol. 10, 4239–4242.
5. Sommerfelt, M.A. & Weiss, R.A. (1990) Virology 176, 58–69.
6. Kewalramani, V.N., Panganiban, A.T. & Emerman, M. (1992) J. Virol. 66, 3026–3031.
7. Sommerfelt, M.A., Williams, B.P., McKnight, A., Goodfellow, P.N. & Weiss, R.A. (1990) J. Virol. 64, 6214–6220.
8. Garcia, J.V., Jones, C. & Miller, A.D. (1991) J. Virol. 65, 6316–6319.
9. Kaelbling, M., Eddy, R., Shows, T.B., Copeland, N.G., Gilbert, D.J., Jenkins, N.A., Klinger, H.P. & O’Hara, B. (1991) J. Virol. 65, 1743–1747.
10. Sommerfelt, M.A., Williams, B.P., Clapham, P.R., Solomon, E., Goodfellow, P.N. & Weiss, R.A. (1988) Science 242, 1557–1559.
11. Isobe, M., Huebner, K., Maddon, P.J., Littman, D.R., Axel, R. & Croce, C.M. (1986) Proc. Natl. Acad. Sci. USA 83, 4399–4402.
12. Dalgleish, A.G., Beverley, P.C., Clapham, P.R., Crawford, D.H., Greaves, M.F. & Weiss, R.A. (1984) Nature (London) 312, 763–767.
13. Klatzmann, D., Champagne, E., Chamaret, S., Gruest, J., Guetard, D., Hercend, T., Gluckman, J.C. & Montagnier, L. (1984) Nature (London) 312, 767–768.
14. Feng, Y., Broder, C.C., Kennedy, P.E. & Berger, E.A. (1996) Science 272, 872–877.
15. Deng, H., Liu, R., Ellmeier, W., Choe, S., Unutmaz, D., Burkhart, M., Di Marzio, P., Marmon, S., Sutton, R.E., Hill, C.M., Davis, C.B., Peiper, S.C., Schall, T.J., Littman, D.R. & Landau, N.R. (1996) Nature (London) 381, 661–666.
16. Dragic, T., Litwin, V., Allaway, G.P., Martin, S.R., Huang, Y., Nagashima, K.A., Cayanan, C., Maddon, P.J., Koup, R.A., Moore, J.P. & Paxton, W.A. (1996) Nature (London) 381, 667–673.
17. Sattentau, Q.J., Clapham, P.R., Weiss, R.A., Beverley, P.C., Montagnier, L., Alhalabi, M.F., Gluckman, J.C. & Klatzmann, D. (1988) AIDS 2, 101–105.
18. Albritton, L.M., Tseng, L., Scadden, D. & Cunningham, J.M. (1989) Cell 57, 659–666.
19. Kim, J.W., Closs, E.I., Albritton, L.M. & Cunningham, J.M. (1991) Nature (London) 352, 725–728.
20. Wang, H., Kavanaugh, M.P., North, R.A. & Kabat, D. (1991) Nature (London) 352, 729–731.
21. Miller, D.G., Edwards, R.H. & Miller, A.D. (1994) Proc. Natl. Acad. Sci. USA 91, 78–82.
22. van Zeijl, M., Johann, S.V., Closs, E., Cunningham, J., Eddy, R., Shows, T.B. & O’Hara, B. (1994) Proc. Natl. Acad. Sci. USA 91, 1168–1172.
23. Kavanaugh, M.P., Miller, D.G., Zhang, W., Law, W., Kozak, S.L., Kabat, D. & Miller, A.D. (1994) Proc. Natl. Acad. Sci. USA 91, 7071–7075.
24. O’Hara, B., Johann, S.V., Klinger, H.P., Blair, D.G., Rubinson, H. , Dunn, K.J., Sass, P., Vitek, S.M. & Robins, T. (1990) Cell Growth Differ. 1, 119–127.
25. Ban, J., Portetelle, D., Altaner, C., Horion, B., Milan, D., Krchnak, V., Burny, A. & Kettmann, R. (1993) J. Virol. 67, 1050–1057.
26. Ban, J., Truong, A.T., Horion, B., Altaner, C., Burny, A., Portetelle, D. & Kettmann, R. (1994) Arch. Virol. 138, 379–383.
27. Bates, P., Young, J.A. & Varmus, H.E. (1993) Cell 74, 1043– 1051.
28. Hosie, M.J., Willett, B.J., Dunsford, T.H., Jarrett, O. & Neil, J.C. (1993) J. Virol. 67, 1667–1671.
29. Willett, B.J., Hosie, M.J., Jarrett, O. & Neil, J.C. (1994) Immunology 81, 228–233.
30. Gazdar, A.F., Oie, H., Lalley, P., Moss, W.W. & Minna, J.D. (1977) Cell 11, 949–956.
31. Adamson, M.C., Silver, J. & Kozak, C.A. (1991) Virology 183, 778–781.
32. Miller, A.D. & Chen, F.C. (1996) J. Virol. 70, 5564–5571.
33. Miller, D.G. & Miller, A.D. (1994) J. Virol. 68, 8270–8276.
34. Wilson, C.A., Farrell, K.B. & Eiden, M.V. (1994) J. Virol. 68, 7697–7703.
35. Albritton, L.M., Kim, J.W., Tseng, L. & Cunningham, J.M. (1993) J. Virol. 67, 2091–2096.
36. Yoshimoto, T., Yoshimoto, E. & Meruelo, D. (1993) J. Virol. 67, 1310–1314.
37. Battini, J.L., Heard, J.M. & Danos, O. (1992) J. Virol. 66, 1468–1475.
38. Ott, D. & Rein, A. (1992) J. Virol. 66, 4632–4638.
39. Gardner, M.B., Kozak, C.A. & O’Brien, S.J. (1991) Trends Genet. 7, 22–27.
40. Robinson, H.L., Astrin, S.M., Senior, A.M. & Salazar, F.H. (1981) J. Virol. 40, 745–751.
41. Bassin, R.H., Ruscetti, S., Ali, I., Haapala, D.K. & Rein, A. (1982) Virology 123, 139–151.
42. Miller, D.G. & Miller, A.D. (1992) J. Virol. 66, 78–84.
43. Miller, D.G. & Miller, A.D. (1993) J. Virol. 67, 5346–5352.
44. Rein, A., Schultz, A.M., Bader, J.P. & Bassin, R.H. (1982) Virology 119, 185–192.
45. Eiden, M.V., Farrell, K. & Wilson, C.A. (1994) J. Virol. 68, 626–631.
46. Miller, A.D., Miller, D.G., Garcia, J.V. & Lynch, C.M. (1993) Methods Enzymol. 217, 581–599.
47. von Kalle, C., Kiem, H.P., Goehle, S., Darovsky, B., Heimfeld, S., Torok-Storb, B., Storb, R. & Schuening, F.G. (1994) Blood 84, 2890–2897.
48. Bauer, T.R., Miller, A.D. & Hicksein, D.D. (1995) Blood 86, 2379–2387.
49. Chesebro, B. & Wehrly, K. (1985) Virology 141, 119–129.
50. Burns, J.C., Friedmann, T., Driever, W., Burrascano, M. & Yee, J.K. (1993) Proc. Natl. Acad. Sci. USA 90, 8033–8037.
51. Kasahara, N., Dozy, A.M. & Kan, Y.W. (1994) Science 266, 1373–1376.
52. Somia, N.V., Zoppe, M. & Verma, I.M. (1995) Proc. Natl. Acad. Sci. USA 92, 7570–7574.







