
This paper was presented at a colloquium entitled “Genetic Engineering of Viruses and Virus Vectors,” organized by Bernard Roizman and Peter Palese (Co-chairs), held June 9–11, 1996, at the National Academy of Sciences in Irvine, CA.
Immunization with DNA vaccines encoding glycoprotein D or glycoprotein B, alone or in combination, induces protective immunity in animal models of herpes simplex virus-2 disease
(herpes simplex virus/nucleic acid/glycoprotein)
WILLIAM L. MCCLEMENTS, MARCY E. ARMSTRONG, ROBERT D. KEYS, AND MARGARET A. LIU *
Department of Virus and Cell Biology, Merck Research Laboratories, West Point, PA 19486
ABSTRACT DNA vaccines expressing herpes simplex virus type 2 (HSV-2) full-length glycoprotein D (gD), or a truncated form of HSV-2 glycoprotein B (gB) were evaluated for protective efficacy in two experimental models of HSV-2 infection. Intramuscular (i.m.) injection of mice showed that each construction induced neutralizing serum antibodies and protected the mice from lethal HSV-2 infection. Dose-titration studies showed that low doses (≤1 μg) of either DNA construction induced protective immunity, and that a single immunization with the gD construction was effective. The two DNAs were then tested in a low-dosage combination in guinea pigs. Immune sera from DNA-injected animals had antibodies to both gD and gB, and virus neutralizing activity. When challenged by vaginal infection with HSV-2, the DNA-immunized animals were significantly protected from primary genital disease.
Genital infections caused by herpes simplex viruses (HSV) continue to present serious public health problems. In the United States, it is estimated that approximately 500,000 individuals become infected each year (1), adding to an infected population of between 40 and 60 million (2). A vaccine that prevented or ameliorated primary infection and thereby reduced transmission of HSV would be of great use in controlling this epidemic. Many different approaches have been used to develop such a vaccine. Early attempts using killed virus or viral extracts were largely unsuccessful (reviewed in refs. 2 and 3). In recent years, the major emphasis has been on subunit vaccines composed of recombinantly expressed viral proteins (3, 4). Used prophylactically, they are highly effective in experimental animal infection (5–7), but efficacy may depend on formulation with novel adjuvants not yet licensed for general human use. Live attenuated (8, 9) and replication-deficient (10) virus vaccines have shown promise in animals studies; however, concerns about the safety of infection with live HSV may limit broad acceptance for human use. An alternate approach has used recombinant adeno (11, 12), vaccinia (13–15), or varicella-zoster (16) viral vectors to deliver HSV antigens. Many of the safety concerns about HSV infection are avoided, whereas the advantage of in vivo expression of the immunogen is maintained. Unfortunately, other concerns about safety (17) and efficacy (18, 19) may limit the general use of these vaccines. The recent demonstrations in mice (20–23) and guinea pigs (24) that injection of DNA elicited protective immunity has suggested a new approach to developing vaccines for genital herpes.
The concept of DNA immunization grew from the observation in mice that i.m. injection of DNA encoding a reporter gene resulted in the in vivo expression of the reporter in myocytes near the injection site (25). Ulmer et al. (26) first demonstrated the efficacy of DNA immunization against viral infection using an influenza virus-infection model in mice. Injection of DNA encoding the influenza A virus nucleoprotein induced both humoral and cell-mediated immune responses, and protected the mice from lethal challenge. These findings were extended to other species for influenza (27, 28) and to other disease targets including bovine herpes (29), rabies (30), leishmaniasis (31), malaria (32), rabbit papilloma (33), and lymphocytic choriomeningitis virus (34).
The advantages of DNA immunization are simplicity, in vivo expression, and a common method for delivery and expression of diverse antigens. Cloned antigens can be manipulated by standard recombinant DNA methodology. In vivo expression is accomplished without the need for live infection or the construction of viral vectors, and the expressed protein or epitopes thereof have the potential to enter the major histocompatibility class I pathway and elicit CD8+ cytotoxic Tlymphocyte responses (26, 35). Immunization with a cloned gene offers the same specificity as a recombinantly expressed protein subunit vaccine without the complex manufacturing and formulation procedures sometimes required to ensure the immunogenicity of recombinantly expressed proteins. Antigens expressed in vivo may have a native conformation, undergo posttranslational modification, and therefore may contain epitopes for presentation to B cells, superior to those supplied exogenously. Additionally, because DNA immunization uses a common method for antigen delivery it is a potentially useful way to assemble and test multicomponent vaccines (28). In an effort to determine the feasibility of developing a multicomponent DNA vaccine for genital herpes, we have begun evaluating a two-component vaccine.
We cloned the HSV-2 full-length glycoprotein D (gD) and a truncated form of glycoprotein B (gB) into DNA vaccine vectors where high-level expression was under the control of the human cytomegalovirus immediate-early-protein promoter (36, 37). These DNA constructions were evaluated individually for induction of protective immunity in a mouse lethal-infection model. Unlike previous studies with HSV-1 gD or gB DNA (20, 23), or HSV-2 gD DNA (22), where multiple immunizations with high doses of DNA were used, we have titrated the DNAs to try to determine minimal effective doses,
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
Abbreviations: HSV, herpes simplex virus; FBS, fetal bovine serum; RD, rhabdomyosarcoma; gD, glycoprotein D; gB, glycoprotein B; GMT, geometric mean titers.
|
* |
To whom reprint requests should be addressed, e-mail: margaret_liu@merck.com. |
and we tested the effect of a single immunization. Having established that low doses of DNA were effective in the mouse model, we then tested the prophylactic effect of a low-dosage combination of these DNAs in the guinea pig vaginal-infection model. Genital disease in guinea pigs closely resembles that in humans (38, 39), and the model has been widely used to test potential vaccines (5, 6) and antiviral chemotherapies (40) for genital herpes.
MATERIALS AND METHODS
Viruses and Cells. Vero, baby hamster kidney (BHK)-21, and rhabdomyosarcoma (RD) cells, and HSV-2 strain MS were obtained from the American Type Culture Collection. HSV-2 strain Curtis was obtained from Andre Nahmias (Emory University, Atlanta). Virus was routinely prepared by infection of nearly confluent Vero or BHK cells with a multiplicity of infection of 0.1 at 37°C in a small volume of cell culture medium without serum. After 1 hr, virus inoculum was removed and cultures were re-fed with high glucose DMEM (BioWhittaker) supplemented with 2% heat inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 25 mM Hepes, 50 units/ml penicillin, and 50 μg/ml streptomycin (all GIBCO/ BRL). Incubation was continued until cytopathic effect was extensive; usually for 24–48 hr. Cell-associated virus was collected by centrifugation at 1800×g for 10 min at 4°C. Supernatant virus was clarified by centrifugation at 640×g for 10 min at 4°C, and stored at −70°C.
Mice and Guinea Pigs. Female BALB/c mice (Charles River Breeding Laboratories) and female Duncan Hartley guinea pigs (Harlan Sprague Dawley) were maintained and used in accordance with the Institutional Animal Care and Use Committee approved protocols.
Cloning and DNA Preparation. HSV-2 strain Curtis DNA, used as template for polymerase chain reactions (PCR), was prepared from nucleocapsids isolated from infected Vero cells (41). An 1182-bp fragment encoding the gD precursor gene was amplified by PCR (Perkin-Elmer/Cetus) using synthetic oligonucleotide primers (Midland Certified Reagent, Midland, TX), which corresponded to 5′ and 3′ end-flanking sequences for the HSV-2 gD gene and contained BglII restriction sites. A 2121-bp sequence encoding the amino terminal 707 aa of HSV-2 gB was amplified by PCR. Primers corresponding to the 5′ flanking sequence, and complementary to nucleotides 2110–2121 (42) were used to generate BglII restriction sites flanking the coding sequence, and to add the termination codon TAA immediately after nucleotide 2121. BglII-digested PCR-amplified fragments were ligated into vectors V1J or V1Jns (36, 37). Escherichia coli DH5α (GIBCO/ BRL) was transformed according to the manufacturer’s specifications. Candidate plasmids were characterized by restriction mapping, and the vector-insert junctions were sequenced using the Sequenase DNA sequencing kit, version 2.0 (United States Biochemical). The gD-coding sequence, originally cloned in V1J, was subcloned into V1Jns. For simplicity, the final gD and truncated gB plasmid constructions were designated gD-2 and ΔgB-2, respectively. Large-scale DNA preparations were essentially as described (36).
Expression of Recombinant Proteins. Plasmid DNA was precipitated onto RD cells (ATCC CCL136) by the calcium phosphate method using Pharmacia CellPhect Kit reagents according to the manufacturer’s instructions, except that 5 or 15 μg of DNA per well were used. After 48 hr, cell lysates were resolved by electrophoresis and then transferred to nitrocellulose membranes. Immunoblots were processed with an anti-HSV gD monoclonal antibody (Advanced Biotechnologies, Columbia, MD) or sheep anti-HSV-2 antiserum (ViroStat, Portland, ME) and developed with an enhanced chemiluminescence detection kit (Amersham).
Immunization. In all cases, DNA dose refers to the total amount of DNA injected per animal per round of immunization; one-half of the total was delivered to each injection site. Mice were anesthetized by i.p. injection of a mixture of 2 mg ketamine HCl (Aveco, Fort Dodge, IA) and 0.2 mg xylazine (Mobley, Shawnee, KS) in saline. The hind legs were shaved with electric clippers and washed with 70% ethanol. Each quadriceps muscle was injected with 50 μl of DNA diluted into sterile saline just prior to use. Control animals were shamimmunized with saline or vector DNA. Mice were 5–6 weeks old at the time of the first immunization. Guinea pigs, weighing 400–550 g at the time of the first immunization, were anesthetized by subcutaneous injection of 22 mg ketamine plus 5 mg xylazine/kg. The hind legs were washed with 70% ethanol and each quadriceps muscle was injected i.m. with 100 μl of DNA or saline.
Serology. Sera were assayed for HSV-specific responses in ELISAs using either HSV glycoproteins partially purified from HSV-2 Curtis-infected BHK cell lysates (mouse sera) or recombinantly expressed gD and ΔgB purified from recombinant baculovirus-gD-and baculovirus-gB-infected SF21 cultures (guinea pig sera). Recombinant viruses were constructed using the BacPAK Baculovirus Expression System (CLONTECH) pBacPAK8 transfer vector and Bsu361-digested BacPAK6 virus and gD and ΔgB coding sequences from gD-2 and ΔgB-2, respectively. Glycoproteins from HSV-2 or baculovirus-gD-infected cultures were purified by Lentil Lectin Sepharose chromatography (Pharmacia) essentially as described (43). Truncated gB was purified from clarified culture medium adjusted to 0.1 mM MnCl2, 0.5% Nonidet P-40, batch adsorbed at room temperature to Lentil Lectin Sepharose 4B, and eluted as described. For the ELISA, glycoproteins were diluted to 5 μg/ml total protein in 50 mM carbonate buffer (pH 9.5), 100 μl per well was applied to Maxi-sorb 96-well plates (Nunc) and allowed to absorb at 4°C overnight. All subsequent incubations were carried out in 100 μl volumes for 1 hr at room temperature and plates were washed four times with phosphate buffered saline (PBS, pH 7.2), with or without one distilled water wash between steps. Dilution buffer (920 mM Tris·HCl, pH 7.5/137 mM NaCl/2.7 mM KCl/0.5% gelatin/0.05% Tween 20) was used as a blocking agent, as well as for the serial dilution of immune sera and the dilution of alkaline phosphatase-labeled goat anti-mouse (Boehringer Mannheim) or goat anti-guinea pig (Accurate Chemicals) IgG. The ELISA was developed with 1 mg/ml p-nitrophenylphosphate in 10% diethanolamine (pH 9.8), 0.5 mM MgCl·6 H20 at 37°C, and optical absorbance was read at 405 nm. Serum dilutions were scored as positive if the OD405 signal exceeded by more than 3 SD the mean OD405 signal (six replicates) of sera from sham-immunized mice at the same dilution, or if the OD405 signal exceeded by >0.1 OD unit, the signal of the guinea pig’s preimmune serum at the same dilution. The reciprocal of the last sample dilution scored positive was taken as the endpoint titer. Individual endpoint titers were used to calculate geometric mean titers (GMT). For purposes of calculation, sera negative at the lowest dilution tested were assigned endpoint titers equal to the reciprocal of the next lower dilution if the dilution series had been extended. ELISA titers are HSV-specific as originally shown by lack of measurable ELISA titer in sera from naive or saline-immunized animals, and by the lack of reaction of immune sera with antigen prepared from mock-infected BHK cell lysates.
Neutralization Assays. Sera from DNA- or saline-immunized animals were heat inactivated at 56°C for 30 min prior to serial dilution in DMEM/2% heat-inactivated FBS. Fifty microliters of each dilution were delivered to duplicate wells in a sterile polypropylene, 0.5 ml 96-well plate (Marsh Biomedical Products, Rochester, NY). HSV-2 stocks were diluted to 4000 plaque forming units (pfu)/ml and 50 μl of virus was added to sample wells and the plate incubated
overnight at 4°C. Guinea pig complement (Cappel) was diluted 1:4 in DMEM/2% heat-inactivated FBS and 50 μl was added to each sample well. After a 1-hr incubation at 37°C, 100 μl of serum-free medium was added and then each reaction mixture was used to infect confluent Vero cells in 12-well cluster plates (Costar) incubated for 1 hr at 37°C. Inocula were aspirated, monolayers were overlaid with 1 ml of 1×MEM containing 5% heat-inactivated FBS, 1×basal medium Eagle vitamins, 10 mM L-glutamine, 25 units/ml penicillin, 25 μg/ml streptomycin, 12.5 mM Hepes, 0.5% carboxymethylcellulose and plates were incubated at 37°C for 48 hr. Monolayers were stained with 1% basic fuchsin in 50% methanol/10% phenol and the number of plaques determined. A serum dilution was considered neutralization-positive if plaque numbers were ≤50% of those obtained in parallel control assays using pooled sera from saline-immunized control mice or preimmune serum from the same guinea pig at the same dilution.
Statistical Analysis. Mouse survival data were analyzed using the log-rank test in the SAS procedure LIFETEST (44). Guinea pig daily lesion scores were analyzed by the two-tailed Student’s t test. For comparison of overall disease among groups of guinea pigs, mean lesion scores were analyzed by the Kruskal-Wallis test followed by a multiple comparison test at the P<0.05 significance level.
RESULTS
Cloning and in Vitro Expression of Cloned Proteins. The coding sequences for full-length gD and the amino-terminal 707 aa of gB were cloned from HSV-2 strain Curtis viral DNA by PCR methods into the eukaryotic expression vectors V1J or V1Jns (36, 37) and were designated gD-2 and ΔgB-2, respectively. The plasmids were characterized by restriction mapping and sequence analysis of the vector-insert junctions. Over the regions sequenced, the gD clone was identical with that published for HSV-2 strain G (45) and the gB clone sequence was identical with that published for HSV-2 strain 333 (42). The ability of gD-2 or ΔgB-2 plasmids to express the encoded protein was demonstrated by transient transfection of RD cells (not shown). Immunoblot analysis of gD-2 DNA-transfected RD cell lysates with an anti-HSV-2 gD monoclonal antibody detected a protein with a molecular weight of approximately 60 K not present in mock-transfected RD cell lysates. Immunoblot analysis of conditioned medium from ΔgB-2 DNA-transfected RD cells and cell lysates with sheep anti-HSV-2 antiserum detected a protein with an apparent molecular weight of 106 K not present in controls, and found that a majority of the protein was in the medium. The observed size was consistent with a 707 aa truncated form of gB and, because this truncation deleted the transmembrane and cytoplamic domains from HSV-2 gB (46), the expressed protein was not expected to be cell associated.
Serology of Mice Immunized with gD- or gB-Expressing DNA. The biological effects of immunization with gD-2 or ΔgB-2 DNA were first investigated in separate dose titration experiments in mice. Animals were immunized by i.m. injection of DNA or were sham-immunized with saline at weeks 0 and 7. Sera obtained at week 10 were assayed in an HSV-specific ELISA. Table 1 shows the seroconversion results and reports the GMT±SEM attained for each dose group. In these assays, pooled sera from the saline-injected control mice were used as the negative controls. The results indicated that injection of each DNA construction resulted in gene expression in vivo and the induction of substantial antibody responses, even at low doses. At the lowest dose tested, 0.8 μg of gD-2 DNA, eight of nine immunized mice developed detectable antibody responses.
Representative sera from both gD-2 DNA- and ΔgB-2 DNA-immunized animals were surveyed for HSV-2 (strain Curtis) neutralizing activity at dilutions of 1:10, 1:100, and
Table 1. Effect of DNA immunization on antibody development in mice
|
DNA dose, μg* |
No. seropositive |
No. sera tested |
GMT(log10) |
±SEM |
|
gD-2 |
||||
|
200 |
9 |
9 |
4.44 |
0.22 |
|
100 |
10 |
10 |
4.69 |
0.31 |
|
50 |
9 |
10 |
4.38 |
0.33 |
|
25 |
8 |
8 |
4.26 |
0.23 |
|
12.5 |
10 |
10 |
4.15 |
0.30 |
|
6.3 |
10 |
10 |
4.21 |
0.33 |
|
3.1 |
10 |
10 |
3.89 |
0.18 |
|
1.6 |
7 |
10 |
2.56 |
0.35 |
|
0.8 |
8 |
9 |
3.75 |
0.43 |
|
Saline |
|
|
(0.48)† |
|
|
ΔgB-2 |
||||
|
30 |
10 |
10 |
4.88 |
0.16 |
|
10 |
10 |
10 |
4.58 |
0.18 |
|
3 |
10 |
10 |
4.47 |
0.15 |
|
1 |
10 |
10 |
4.18 |
0.15 |
|
Saline |
|
|
(0.48)† |
|
|
*Dose given at weeks 0 and 7; sera obtained at week 10. †By convention, pooled sera from saline-injected mice were defined as negative at all dilutions tested and assigned endpoint titers equal to the reciprocal of the next lower dilution had the series been extended; in these cases 1:3. |
||||
1:1000. Fifteen sera from mice injected with doses of gD-2 DNA, ranging from 3.1 to 100 μg, were tested; 13 were neutralization-positive at 1:10; of those, 11 were also positive at 1:100, and of those, 5 were positive at the 1:1000 dilution. The two negative sera (from the 50 μg dose group) also had low ELISA endpoint titers (log10≤2.00 and 2.52, respectively). A more limited survey of sera from the animals immunized with 30 μg of ΔgB-2 DNA found that of three sera tested, all were positive at 1:10, two at 1:100, and none at 1:1000. These results indicated that immunization with either DNA construction was capable of inducing HSV-2 neutralizing antibodies in mice.
Effect of DNA Immunization on Lethal Infection of Mice. Immunized and control (saline-injected) mice were challenged by i.p. injection of HSV-2 and observed daily for survival. Fig. 1A shows the effect of gD-2 DNA immunization on survival; significant protection from death (P<0.001) was achieved for each dose. Eighty-two of eighty-six gD-2 DNA-immunized mice survived challenge. The survival results for the ΔgB-2 DNA-immunized mice are in Fig. 1B. Although ΔgB-2 DNA did not appear to be as effective as gD-2 DNA, significant protection from death was found for each dose tested (P<0.01 for the 30, 10, and 3 μg groups, and P=0.027 for the 1 μg group). Thus, low dose immunization with either gD-2 or ΔgB-2 DNA induced antibody responses in mice which protected them from lethal HSV-2 infection. However, while the protection from death was significant, infection was not completely prevented. During the observation period, some animals exhibited transient morbidity: failure to groom, failure to thrive, or a hunched posture. Infection was confirmed in some animals by the detection of antibodies to nonstructural HSV proteins in convalescent sera (not shown).
Sham immunization with saline was used as the control in dose titrations. To confirm that protection was dependent upon the HSV coding sequence rather than injection of DNA per se, groups of 10 mice were immunized with either 12.5 or 1.6 μg of gD-2 DNA, or 12.5 μg of vector V1J DNA. Ten weeks after a single immunization, sera were analyzed by ELISA. The logic GMT (±SEM) for the group injected with 12.5 μg of gD-2 DNA was 3.89 (±0.97) and that for the 1.6 μg group was 2.49 (±1.20). None of the sera from mice immunized with vector DNA were seropositive; the log10 GMT of 0.48 was
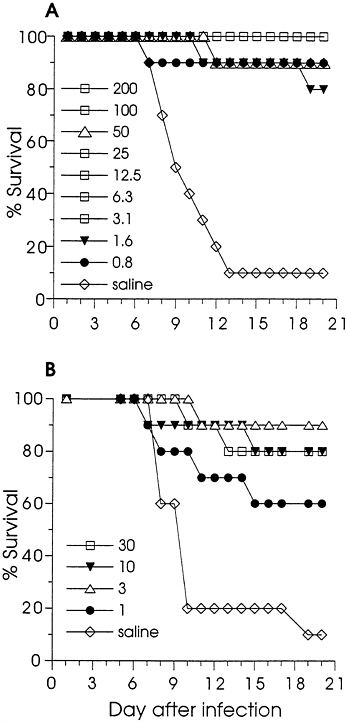
FIG. 1. The effect of DNA immunization on the survival of mice infected by i.p. injection with HSV-2. Mice were immunized twice with gD-2 (A) in a 2-fold dilution series, with ΔgB-2 (B) in a half-log dilution series, or with saline. The doses (in μg) are indicated on the figure. The numbers of mice in each group are noted in Table 1. Mice were challenged by i.p. injection with 0.25 ml (105.7 pfu) of a clarified stock of HSV-2 strain Curtis, and were observed for 3 weeks for signs of disease and survival.
background. Fig. 2 reports the survival data for these animals following i.p. challenge at 11 weeks. Both groups immunized
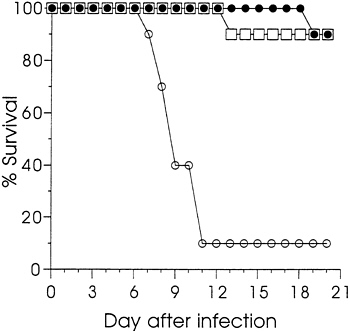
FIG. 2. The effect of immunization with gD-2 DNA or vector DNA on the survival of mice infected by i.p. injection with HSV-2. Mice were immunized with 12.5 μg (□) or 1.6 μg of gD-2 (●), or with 12.5 μg (○) of vector V1J. Viral challenge was as described in Fig. 1.
with gD-2 DNA were significantly protected from death compared with those immunized with the vector (P<0.001). Survival of the vector-injected animals was similar to that found for the saline-injected animals in the experiments summarized in Fig. 1. These results confirmed that protection was dependent upon the gD coding sequence. Furthermore, they showed that protective immunity could be established with a single injection of gD-2 DNA. Additional studies in mice (not shown) and guinea pigs (not shown) comparing plasmids ΔgB-2 or gD-2 with vector, or with control plasmids that expressed influenza viral proteins, also found that protection was dependent on the presence HSV protein-coding sequences.
Serology of Guinea Pigs Immunized with a Mixture of gD-and gB-Expressing DNA. The lethal-infection model was useful for confirming the in vivo activity of the gD-2 and ΔgB-2 DNA, and for establishing that low DNA doses were effective; however, this infection model may not be relevant to human disease. Therefore, the guinea pig vaginal-infection model was used to assay the effects of immunization with a combination of low doses of gD-2 and ΔgB-2 DNA. Seven guinea pigs were immunized with a DNA mixture containing 3 μg of gD-2 DNA and 10 μg of ΔgB-2 DNA at weeks 0 and 6; 14 control guinea pigs were sham-immunized with saline. Sera, obtained at 9 weeks, were analyzed for anti-gD and anti-gB antibodies using antigen-specific ELISAs. Results are shown in Table 2. All of the DNA-immunized animals developed ELISA titers to both gD and gB; individual endpoint titers were ≥300. None of the sham-immunized animals were positive at the lowest dilution
Table 2. Effect of immunization with a combination of gD-2 and ΔgB-2 DNA on antibody development in guinea pigs
|
|
|
No. positive sera |
log10 ELISA GMT (±SEM) |
||
|
Immunization |
No. of animals |
Anti-gD |
Anti-gB |
Anti-gD |
Anti-gB |
|
gD-2+ ΔgB-2 DNA |
7 |
7 |
7 |
2.62 (0.14) |
3.05 (0.20) |
|
Saline |
14 |
0 |
0 |
0.48 (0)* |
0.48 (0)* |
|
*For purposes of GMT calculation, sera negative at all dilutions tested were assigned an endpoint titer equal to the reciprocal of what would have been the next lower dilution had the dilution series been extended; in this case 1:3. |
|||||
(1:30) tested. The results indicated that both DNAs in the mixture were expressed. These sera were also assayed for HSV-2 neutralizing antibodies. Immune sera from all seven DNA-immunized animals were neutralization-positive at a 1:10 dilution, six of seven at 1:100, two at 1:1,000, and none at 1:10,000. None of four randomly-selected representative sera from the sham-immunized control animals were positive at dilutions of 1:10 or 1:100.
Effect of DNA Immunization on Vaginal Infection of Guinea Pigs. At 10 weeks, all of the DNA-immunized and 8 of the sham-immunized guinea pigs were challenged by the introduction of HSV-2 strain MS into the vagina. As a control for any effect the manipulations used in the infection procedure might have on the scoring of external disease, the remaining six sham-immunized animals were mock-infected using an inoculum prepared from mock-infected Vero cells. The severity of the primary disease was assessed by the lesion scoring system described in the legend to Fig. 3. The course of primary disease is summarized in Fig. 3 by reporting the mean daily lesion scores on days 3–14 following infection. All animals in the infected control group developed severe external disease. From day five onward, this group’s scores were significantly higher than those of the DNA-immunized or the mock-infected control groups (P<0.01). In contrast, none of the DNA-immunized animals developed severe disease, and the scores for this group were statistically indistinguishable from those of the mock-infected group. The overall primary disease, as measured by the means of all lesion scores, was significantly lower for the DNA-immunized group compared with the infected control group (P<0.001), but was not significantly different from the mock-infected control group (P=0.92). The scores for the mock-infected group were taken as the experimental background (see Discussion).
The DNA-immunized animals were further distinguished from the infected controls in that none of them developed signs of systemic disease. In contrast, six of the eight sham-immunized-infected guinea pigs showed signs of severe systemic infection: five retained urine on 2 or more days, one developed partial paralysis of the hind limbs, and five animals became moribund during the observation period and required euthanization (Fig. 3). None of the mock-infected animals showed signs of systemic disease. This challenge study indicated that immunization with low doses of DNAs that encode HSV-2 full-length gD and a truncated form of gB protected guinea pigs from HSV-2-induced primary genital disease.
DISCUSSION
Our data show that immunization with DNA encoding full-length HSV-2 gD or a truncated form of HSV-2 gB induced immune responses in mice and protected them from lethal challenge with HSV-2, and that a combination of these two DNAs protected guinea pigs from primary genital disease. It had been shown previously that multiple immunizations with much higher doses of gD DNA or gB DNA could induce protective immunity in mouse- (20–22) and guinea pig (24)-HSV-infection models. In contrast, our study has found that protective immunity could be induced with low doses of DNA, and in the mouse model with only a single immunization.
When gD-2 DNA was titrated over a 250-fold concentration range in mice, all doses tested induced serum antibodies and protected mice from lethal infection. The level of protection induced by the 0.8-μg dose could not be distinguished statistically from that induced by the highest dose tested. In subsequent titration studies, we found that a single injection of as little as 50 ng of gD DNA could induce detectable antibody responses, although doses of 500 ng were required to obtain consistent seroconversion (unpublished observations), and that a single immunization with 1.6 μg of gD-2 DNA protected mice from lethal i.p. challenge (Fig. 2). Titration of ΔgB-2 DNA in mice showed that two immunizations of as little as 1 μg resulted in significant protection.
Recently, Bourne et al. (24) reported that guinea pigs could be successfully immunized with high doses of DNA. Three 250-, 100-, or 50-μg injections of an HSV-2 gD expression vector (similar to our gD-2 construction) resulted in significant protection from vaginal challenge with HSV-2. We had found previously that guinea pigs immunized twice with 100 μg of ΔgB-2 DNA either alone or in combination with 100 μg gD-2 DNA were significantly protected from primary genital disease and subsequent recurrence (unpublished data). The success of low-dosage immunizations in the mouse model suggested testing a low-dosage combination in guinea pigs. For this study, 10 μg of ΔgB-2 DNA was chosen because we had found previously that this amount gave only partial protection from vaginal challenge (unpublished data). The 3 μg gD-2
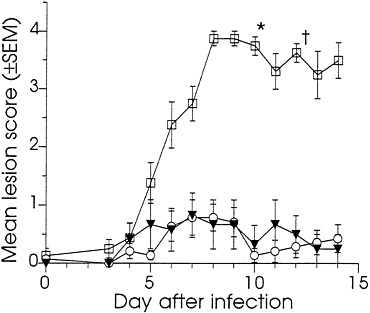
FIG. 3. The effect of immunization with gD-2 and ΔgB-2 DNA in combination on preventing primary HSV-2-induced genital disease in guinea pigs. Immunized and sham-immunized guinea pigs were infected by application of HSV-2 strain MS to the vagina and external genital skin. One hour prior to infection, the vaginal closure membrane was ruptured with a saline moistened cotton swab. The vagina and external skin were then swabbed with 0.1 N NaOH. Virus was introduced using a cotton swab dipped into a clarified HSV-2 MS-infected Vero cell lysate diluted in tissue culture medium to 106.7 pfu/ml. The swab was inserted into the vagina, twisted back and forth five times, then removed and wiped over the external genitalia. To ensure infection, virus application was repeated 1 hr later. For the mock-infected group, the inoculum was prepared from mock-infected Vero cells. Animals were caged randomly and evaluated daily by observers blinded to the study groups. On day 3, the vagina was swabbed with a moistened calcium alginate swab, which was eluted into 2 ml of virus transport medium (Carr-Scarborough Microbiological, Stone Mountain, GA). Infection was confirmed by reisolation of virus, a positive response in the HERPCHEK kit (DuPont), or appearance of symptomatic disease and the development of antibodies to nonstructural HSV proteins. The severity of external disease was quantified using a visual scoring system adapted from that described by Stanberry et al. (38). Numerical scores were assigned to specific disease signs using the following scale: 0, no disease; 1, redness or swelling; 2, several (≤3) small vesicles; 3, several (≤3) large vesicles; 4, large ulcers with maceration. Scores of 0.5, 1.5, 2.5, and 3.5 were assigned to disease of intermediate severity. Daily mean lesion scores were calculated by dividing the sum of a group’s lesion scores by the number of observations. In the case of death during the observation period, the final score assigned to that animal was carried through to the end of the observation period. Animals were immunized with 3 μg of gD-2 +10 μg of ΔgB-2 and challenged with HSV-2 (○) n=7, sham-immunized with saline and challenged with HSV-2 (□) n=8, or sham-immunized and mock-infected (▾) n=6.*, Three animals euthanized; †, two animals euthanized
DNA dose was chosen to be lower than any dose previously tested by us in guinea pigs. The detection of neutralizing serum antibodies, and responses to both gD and gB by ELISA, indicated that these levels of DNA were effective. Furthermore, the combination at this dose was highly effective in preventing primary genital disease. Following challenge, control animals developed severe disease characterized by high lesion scores and the systemic involvement; in contrast, the DNA-immunized guinea pigs were nearly free of disease. Mean lesion scores for the DNA-immunized group were statistically indistinguishable from background. This background was established by inclusion of a mock-infected control group. The assignment of non-zero scores to some control animals on some observation days (Fig. 3) was likely the result of the slight irritation caused by the infection procedure. There was no evidence that these scores were due to HSV infection transmitted from infected cage mates; all mock-infected control animals were HSV seronegative 4 weeks after the challenge (data not shown). The extent of disease in those DNA-immunized animals which did develop lesions was lower than that seen in the infected control animal. The highest score attained by any DNA-immunized animal was 2.0 on 2 successive days. In contrast, seven of the eight infected controls were scored 4.0 on 2 or more successive days.
It has been postulated that due to competition for DNA uptake or expression, or antigen competition, immunization with DNAs in combination might result in reduced responses to the individual components. The combination of gD-2 and ΔgB-2 DNA did not appear to compromise the response to either component. Moreover, the protection achieved with this low-dosage combination was as good as, or better than, that seen in similar challenge studies using 100-μg doses of gD-2 DNA or ΔgB-2 DNA alone (unpublished observations). Because the combination of gD-2 and ΔgB-2 DNA could induce responses to the broader spectrum of epitopes contained in two separate antigens, it had the potential to be more effective than either component alone. The results are consistent with the combination being more effective than the individual components; however, that cannot be concluded from this study because the comparison was not made directly. Further titrations of gD-2 DNA and ΔgB-2 DNA, both individually and in combination, are in progress to address the question directly, and to establish minimally-effective doses.
Because of the small number of surviving control guinea pigs, latent infection and recurrent disease could not be evaluated. It has been shown with protein subunit vaccines (5, 6) and recently with high-dose DNA immunization (24) that significant reduction in primary genital disease also resulted in reduced latent infection and decreased recurrence. The extent of protection against primary disease found in the study presented here suggests that prophylactic immunization with a low-dose combination of gD and ΔgB-2 DNA would be effective against recurrence; experiments to evaluate this are ongoing.
We have not yet identified which DNA-induced immune responses are protective in our challenge models. Injection of gD-2 or ΔgB-2 DNA, individually or in combination, induced substantial neutralizing serum antibody titers, which may fully account for the protection observed. Others have shown by passive transfer that antibody alone can be protective in some mouse infection models (47, 48). However, DNA immunization has the capacity to elicit cell-mediated, as well as humoral immune responses (26, 35). Recently, Manickan et al. (20) showed that in mice immunized with HSV-1 gB DNA, a CD4+ cytotoxic T-lymphocyte response protected the animals from zosteriform infection with HSV-1. We have detected antigen-specific lymphoproliferative responses in mice and guinea pigs immunized with the gD-2 or ΔgB-2 DNA, but have not yet shown the induction of cytotoxic T lymphocytes (unpublished observations) and cannot rule out a contribution of cell-mediated immunity to the protection observed in these studies. The relative protective roles of humoral and cell-mediated immunity induced by immunization with gD-2 and ΔgB-2 DNA are yet to be defined and may depend on the infection model used.
We have demonstrated that immunization with low doses of DNA was highly effective in generating protective immunity in two animal models of HSV infection and we found, in mice, that a single immunization was protective. (Single immunizations have not been evaluated in the guinea pig model.) These results suggest that simple i.m. injection has the potential to be an efficient form of DNA delivery, and support the feasibility of developing DNA vaccines for human use where low dose and limited numbers of injections are desirable characteristics. We also found that the combination of gD-2 and ΔgB-2 DNA induced immune responses to both proteins and was effective in preventing HSV-2-induced mucosal disease. This result supports the concept that multivalent vaccines can be made by simply combining DNAs, and provides a starting point for the development of such a vaccine for genital herpes.
To date, gD and gB have been the focus of DNA vaccine development (20, 22–24) just as they have been for protein subunit vaccines (reviewed in ref. 3). However, the inherent simplicity of DNA immunization should allow the rapid identification of additional immunogens for inclusion in multivalent DNA vaccines for HSV-induced disease. It is now possible to scan the genomes of complex pathogens for novel immunogens (49) and readily test their capacity to elicit protective immunity. As potentially useful immunogens are identified, they can be easily evaluated in the context of an existing DNA vaccine. Using DNA immunization as both a discovery tool and as a method of delivering combinations of antigens should expedite the development of vaccines with greater potency and breadth of protection. Clearly this approach needs extensive evaluation before clinical efficacy and safety are demonstrated, but these early results with the combination of gD-2 and ΔgB-2 DNA are encouraging.
We wish to thank Mr. Timothy Schofield (Merck Research Laboratories Biometrics Department) for statistical analyses of the data.
1. Roizman, B. (1991) Rev. Infect. Dis. 13, Suppl. 11, S892–S894.
2. Whitley, R.J. & Meignier, B. (1992) in Vaccines: New Approaches to Immunological Problems, ed. Davies, J.E. (Butterworth-Heinemann, Boston), pp. 223–254.
3. Burke, R.-L. (1993) Semin. Virol. 4, 187–197.
4. Burke, R.L. (1991) Rev. Infect. Dis. 13 (Suppl. 11), S906–S911.
5. Stanberry, L.R., Bernstein, D.I., Burke, R.L., Pachl, C. & Myers, M.G. (1987) J. Infect. Dis. 155, 914–920.
6. Stanberry, L.R., Myers, M.G., Stephanopoulos, D.E. & Burke, R.L. (1989) J. Gen. Virol. 70, 3177–3185.
7. Sanchez-Pescador, L., Burke, R.L., Ott, G. & Van Nest, G. (1988) J. Immunol. 141, 1720–1727.
8. Whitley, R.J., Kern, E.R., Chatterjee, S., Chou, J. & Roizman, B. (1993) J. Clin. Invest. 91, 2837–2843.
9. Meignier, B., Longnecker, R. & Roizman, B. (1988) J. Infect. Dis. 158, 602–614.
10. Farrell, H.E., McLean, C.S., Harley, C., Efstathiou, S., Inglis, S. & Minson, A.C. (1994) J. Virol. 68, 927–932.
11. McDermott, M.R., Graham, F.L., Hanke, T. & Johnson, D.C. (1989) Virology 169, 244–247.
12. Gallichan, W.S., Johnson, D.C., Graham, F.L. & Rosenthal, K.L. (1993) J. Infect. Dis. 168, 622–629.
13. Wachsman, M., Aurelian, L., Smith, C.C., Lipinskas, B.R., Perkus, M.E. & Paoletti, E. (1987) J. Infect. Dis. 155, 1188–1197.
14. Aurelian, L., Smith, C.C., Wachsman, M. & Paoletti, E. (1991) Rev. Infect. Dis. 13 (Suppl. 11), S924–S930.
15. Cantin, E.M., Eberle, R., Baldick, J.L., Moss, B., Willey, D.E., Notkins, A.L. & Openshaw, H. (1987) Proc. Natl. Acad. Sci. USA 84, 5908–5912.
16. Heineman, T.C., Connelly, B.L., Bourne, N., Stanberry, L.R. & Cohen, J. (1995) J. Virol. 69, 8109–8113.
17. Lane, J.M., Ruben, F.L., Neff, J.M. & Millar, J.D. (1969) N. Engl. J. Med. 281, 1201–1208.
18. McDermott, M.R., Smiley, J.R., Brais, P.L.J., Rutazroga, H.E. & Brenenstock, J. (1984) J. Virol. 51, 747–753.
19. Cooney, E.L., Collier, A.C., Greenberg, P.D., Coombs, R.W., Zarling, J., Arditti, D.E., Hoffman, M., Hu, C.S.L. & Corey, L. (1991) Lancet 337, 567–572.
20. Manickan, E., Rouse, R.J.D., Yu, Z., Wire, W.S. & Rouse, B.T. (1995) J. Immunol. 155, 259–265.
21. Manickan, E., Yu, Z., Rouse, R.J.D., Wire, W.S. & Rouse, B.T. (1995) Viral Immunol. 8, 53–61.
22. Kriesel, J.D., Spruarce, S.L., Daynes, R.A. & Araneo, B.A. (1996) J. Infect. Dis. 173, 536–541.
23. Ghiasi, H., Cai, S., Slanina, S., Nesburn, A.B. & Wechsler, S.L. (1995) Antiviral Res. 28, 147–157.
24. Bourne, N., Stanberry, L.R., Bernstein, D.I. & Lew, D. (1996) J. Infect. Dis. 173, 800–807.
25. Wolff, J.A., Malone, R.W., Williams, P., Chong, W., Acsadi, G., Jani, A. & Felgner, P.L. (1990) Science 247, 1465–1468.
26. Ulmer, J.B., Donnelly, J.J., Parker, S.E., Rhodes, G.H., Felgner, P.L., Dwarki, V.J., Gromkowski, S.H., Deck, R.R., DeWitt, C.M., Friedman, A., Hawe, L.A., Leander, K.R., Martinez, D., Perry, H.C., Shiver, J.W., Montgomery, D.L. & Liu, M.A. (1993) Science 259, 1745–1749.
27. Fynan, E.F., Webster, R.G., Fuller, D.H., Haynes, J.R., Santoro, J.C. & Robinson, H.L. (1993) Proc. Natl. Acad. Sci. USA 90, 11478–11482.
28. Donnelly, J.J., Friedman, A., Martinez, D., Montgomery, D.L., Shiver, J.W., Motzel, S.L., Ulmer, J.B. & Liu, M.A. (1995) Nat. Med. 1, 583–587.
29. Cox, G.J.M., Zamb, T.J. & Babiuk, L.A. (1993) J. Virol. 67, 5664–5667.
30. Xiang, Z.Q., Spitalnik, S., Tran, M., Wunner, W.H., Cheng, J. & Ertl, C.J. (1994) Virology 199, 132–140.
31. Xu, D. & Liew, F.Y. (1994) Vaccine 12, 1534–1536.
32. Sedegah, M., Hedstrom, R.C., Hobart, P. & Hoffman, S.L. (1994) Proc. Natl. Acad. Sci. USA 91, 9866–9870.
33. Donnelly, J.J., Martinez, D., Jansen, K.V., Ellis, R.W., Montgomery, D.L. & Liu, M.A. (1996) J. Infect. Dis. 713, 314–320.
34. Yokoyama, M., Zhang, J. & Whitton, J.L. (1995) J. Virol. 69, 2684–2688.
35. Wang, B., Ugen, K.I., Srikantan, V., Agadjanyan, M.G., Dang, K., Refaeli, Y., Sato, A.I., Boyer, J., Williams, W.V. & Weiner, D.B. (1993) Proc. Natl. Acad. Sci. USA 90, 4156–4160.
36. Montgomery, D.L., Shiver, J.W., Leander, K.R., Perry, H.C., Friedman, A., Martinez, D., Ulmer, J.B., Donnelly, J.J. & Liu, M.A. (1993) DNA Cell Biol. 12, 777–783.
37. Shiver, J.W., Perry, H.C., Davies, M.-E., Freed, D.C. & Liu, M.A. (1996) in DNA Vaccines, eds. Liu, M.A., Hilleman, M.R. & Kurth, R. (N.Y. Acad. Sci., New York), Vol. 772, pp. 198–208.
38. Stanberry, L.R., Kern, E.R., Richards, J.T., Abbott, T.M. & Overall, J.C., Jr. (1982) J. Infect. Dis. 146, 397–404.
39. Stanberry, L.R. (1994) Semin. Virol. 5, 213–219.
40. Bravo, F.J., Stanberry, L.R., Kien, A.B., Vogt, P.E. & Kern, E.R. (1993) Antiviral Res. 21, 59–72.
41. Denniston, K.J., Madden, M.J., Enquist, L.W. & Vande Woude, G. (1981) Gene 15, 365–378.
42. Stuve, L.L., Brown-Shimer, S., Pachl, C., Najarian, R., Dina, D. & Burke, R.L. (1987) J. Virol. 61, 326–335.
43. Pachl, C., Burke, R.L., Stuve, L.L., Sanchez-Pescador, L., Van Nest, G., Masiarz, F. & Dina, D. (1987) J. Virol. 61, 315–325.
44. Lee, E.T. (1980) Statistical Methods for Survival Data Analysis (Lifetime Learning Publications, Belmont, CA), pp. 122–156.
45. Lasky, L.A. & Dowbenko, D.J. (1984) DNA (N.Y.) 3, 23–29.
46. Bzik, D.J., Debroy, C., Pederson, N.E. & Person, S. (1986) Virology 155, 322–333.
47. Dix, R.D., Pereira, L. & Baringer, J.R. (1981) Infect. Immun. 34, 192–199.
48. Balachandran, N., Bacchetti, S. & Rawls, W.E. (1982) Infect. Immun. 37, 1132–1137.
49. Barry, M.A., Lai, W.C. & Johnston, S.A. (1995) Nature (London) 377, 632–635.







