
3
Atmospheric Chemistry and Evaluation of Environmental Effects of Fire Suppressants
Laboratory studies, atmospheric measurements, and numerical models of the atmosphere have provided important evidence for the significant effects of chlorine and bromine on stratospheric ozone in the last few decades and have confirmed the role of bromine-containing halons in ozone depletion. This chapter examines compounds being considered as replacements for halon and the approaches being used to evaluate their potential effects on Earth's stratospheric ozone and climate, including their contribution to radiative forcing—the greenhouse effect—and other possible environmental impacts. The chapter's opening section briefly describes how researchers came to learn of the link between release of man-made chlorine- and bromine-containing compounds and depletion of ozone.
Reduction of Ozone—Mechanisms and Effects
Earth's atmosphere is made up of regions, classified by the vertical trends in temperature (Figure 3.1). The stratosphere, where most of the atmospheric ozone resides (see Box 3.1), extends from ~10 to 50 km above the surface. Stratospheric ozone is important because it absorbs much of the intense ultraviolet (UV) radiation from the sun, dissociating into O2 and O. The O2 and O recombine to form ozone, which can then absorb more radiation. When it is absorbed by cells, UV radiation can lead to cellular damage, which can be manifested directly as cancer, for example, or as DNA damage that is transferred to later generations.
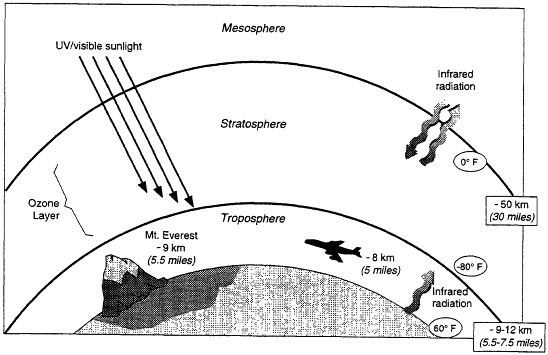
Figure 3.1
Regions of Earth's atmosphere. Roughly 90% of the ozone is in the stratosphere, with most of the rest in the troposphere.
|
Box 3.1 How Much Ozone Is There in the Stratosphere? If all the ozone in the atmosphere were brought down to sea level, it would be merely 2.5 to 4.5 mm thick. However, this amount of ozone can absorb most of the harmful UV radiation entering Earth's atmosphere. Atmospheric scientists use the Dobson Unit (DU) to measure the amount of ozone overhead, i.e., from the ground to outside the atmosphere. 1 DU is equal to 1 millicentimeter of ozone at sea-level pressure (0°C). So, in general, the amount of column ozone is between 250 and 450 DU. In Antarctica, the ozone column has been measured to be as low as 125 DU, i.e., a reduction of ozone by a factor of two or three. |
The potential for ozone depletion in the stratosphere became an important topic when the first commercial supersonic transport aircraft were proposed in the late 1960s.1 At that time, scientists noted that nitrogen oxide (NOx) emissions from engine exhaust associated with aircraft operating in the stratosphere could be involved in a catalytic cycle leading to ozone loss.2 This realization helped lead to the U.S. withdrawal from this potential market. In 1974, Rowland and Molina3 proposed that chlorofluorocarbons (CFCs) emitted by human activities at Earth's surface could pass though the troposphere to the stratosphere. In the stratosphere, UV radiation from the sun is absorbed by the C-Cl bonds in CFCs, leading to rupture of the bonds and liberation of Cl atoms. The Cl atoms can participate in a catalytic cycle that leads to conversion of ozone to oxygen molecules. Later laboratory studies showed that bromine and iodine atoms also participate in similar reactions and can also affect ozone.4 The recognition that anthropogenic emissions of gas could lead to destruction of ozone in the stratosphere led to the Nobel Prize in chemistry being awarded in 1995 to P. Crutzen, M. Molina, and F.S. Rowland.
Although the amounts of CFCs and halons released into the atmosphere are small in terms of the total amount of gas there, they have a great impact on the global ozone balance for three reasons: (1) Ozone is in a constant state of "flux"—it is made and destroyed by natural processes that define a delicate balance (Box 3.2). (2) The production of ozone is controlled by solar input that does not undergo dramatic fluctuations. Removal of ozone from the atmosphere is controlled by catalytic processes, set in motion by small concentrations of natural and synthetic chemicals that can destroy a large number of ozone molecules without being destroyed in the process. Depletion is accelerated when the halogen atoms chlorine, bromine, and iodine are present; thus changes in the "balance" lead to a lower level of ozone. (3) A large fraction of the anthropogenic (and natural) reagents released at Earth's surface can be transported to the stratosphere if they are chemically stable in the troposphere. Because halons and CFCs are very stable in the troposphere, a large fraction of the released amounts reach the stratosphere, where they are quickly broken apart to release halogens that are active in destroying ozone. The parameter that defines how much of the released amounts reach the stratosphere is the atmospheric lifetime (Box 3.3).
Initially, ozone depletion was just a hypothesis based on laboratory data. It was unclear whether other trace gases in the atmosphere would interfere with the ozone removal cycles. Starting in the early 1970s, numerical models were developed to simulate the interactions of trace gases under atmospheric conditions, and the models showed that ozone depletion should occur. The trace gas concentrations simulated by these models compare favorably with measured concentrations, which lends credence to the models. However, direct comparison of model-calculated change in ozone with observed change is more difficult. Because the expected globally averaged ozone decrease is small, on the order of a few percent over a decade, detecting such change in the atmosphere requires extracting this long-term trend from large seasonal cycles (~30%) and interannual variations (~10%). It was not until the early 1980s that ground-based and satellite data were sufficient to determine a clear trend.
Direct measurements now show that stratospheric ozone depletion has occurred during the past two decades.5 In fact, the extent of ozone depletion is larger than predicted by the models based on gasphase chemistry. It has been shown in laboratory experiments that heterogeneous (gas-particle) chemistry on cold particles can occur such that a Cl atom can destroy even more ozone molecules than
|
Box 3.2 How Is Ozone, O3, Made and Destroyed in the Stratosphere? What Determines the Ozone Abundance? Ozone is produced via the photolysis of molecular oxygen by solar radiation: In addition to O atoms, other reactants, many of which are naturally occurring, can also destroy ozone. For example, OH and HO2 radicals, which are present in the stratosphere, can catalytically destroy ozone: As with OH and HO2, chlorine and bromine can also destroy ozone. Examples include: The balance between production, via photolysis of oxygen, and loss via catalytic ozone destruction cycles, described above, determines the mean level of ozone in the atmosphere. Since the production rate of ozone is essentially constant, any enhancements in the loss processes, such as introduction of bromine compounds into the stratosphere, will lead to a lower level of ozone. |
thought previously.6 The change in the vertical distribution of ozone has been measured and the trend deduced.
The ozone depletion observed in the upper stratosphere (Figure 3.2) is consistent with the Rowland and Molina hypothesis. For the lower stratosphere, where the majority of the ozone loss has occurred, it is now clear that such reactions also take place in/on sulfuric acid aerosols, which are always present at low levels in the stratosphere. Volcanic eruptions can greatly enhance the number of these droplets and increase the effectiveness of bromine and chlorine in destroying ozone. Thus, following the eruption of Mt. Pinatubo in 1991, there was a measurable decrease in stratospheric ozone abundance. The large loss of ozone during the 1990s in the lower stratosphere can be attributed, in part, to the eruption of Mt. Pinatubo, which increased the number of particles on which heterogeneous chemistry can occur and helped make the connection between the role of halogen chemistry and ozone changes.7 A semi-quantitative understanding of the entire ozone loss has emerged. The release of man-made chlorine and bromine compounds is the primary cause of ozone depletion.
The Antarctic ozone hole, formed in the Antarctic stratosphere during the springtime, caught the attention of the atmospheric sciences community in the mid-1980s.8 During August and September 1987—the end of winter and beginning of spring in the Southern Hemisphere—aircraft equipped with many different instruments for measuring a large number of chemical species were flown repeatedly over Antarctica.9 Among the chemicals measured were ozone and chlorine oxide, the reactive chemical identified in the laboratory as one of the participants in ozone-destroying chain reactions. On the first flights southward from the southern tip of South America, relatively high concentrations of ozone were measured everywhere over Antarctica. By mid-September, however, the instruments recorded low
|
Box 3.3 Concentrations and Lifetimes The tropospheric concentration of a halon or a replacement compound is dependent on the rate of its emission into the atmosphere and its atmospheric lifetime. Measurements of halons 1211 and 1301 show that their current global mixing ratios (the ratio of volume density or concentration to the volume density of air) are about 2.5 and 2.0 parts per trillion by volume (pptv), respectively, and are currently increasing at about 2 and 6% per year, respectively.1,2,3 These rates of increase have slowed appreciably in recent years, consistent with the reduction in production and emission of these compounds. Despite such small concentrations, production of these compounds has been halted because of the capability of the bromine they contain to destroy ozone. Atmospheric models indicate that halons 1211 and 1301 are essentially nonreactive in the troposphere and are destroyed through photolysis in the stratosphere, resulting in atmospheric lifetimes of about 20 and 65 years, respectively.4,5 Because of their long atmospheric lifetimes, the destruction of these halons generally releases their bromine into the stratosphere, where the bromine has the greatest impact on ozone. The long atmospheric lifetimes also imply that halons already emitted will be releasing bromine into the stratosphere for several decades to come.
|
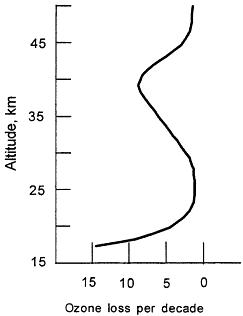
Figure 3.2
Observed trends in vertical distribution of ozone for mid-latitudes (30-50°N) during the 1980s, based on satellite, balloon, and groundbased observations.
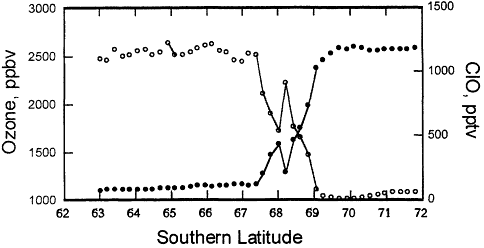
Figure 3.3
NASA aircraft-based measurements of ozone and reactive chlorine (ClO) made in September 1987 over Antarctica. These data demonstrate the relationship between high reactive chlorine and low ozone inside the Antarctic ozone "hole."
Source: National Aeronautics and Space Administration.
concentrations of ozone in regions where there were high concentrations of chlorine oxide, and vice versa, as shown in Figure 3.3. Flights later in September showed even less ozone over Antarctica, as the chlorine continued to react with the stratospheric ozone (see Figure 3.3).
Independent measurements made from airplanes, balloons, satellites, and the ground have provided a detailed understanding of the chemical reactions in the Antarctic stratosphere. In the winter, the lower polar stratosphere can reach temperatures sufficiently cold (less than approximately -80°C, or -112°F at 20 m) that stratospheric clouds can form. These clouds facilitate heterogeneous chemical reactions that allow the release of active chlorine in sunlight. The chemical reactions related to the clouds are now well understood through study under laboratory conditions mimicking those found naturally.
The now well-documented phenomenon of the Antarctic ozone hole has demonstrated the cause-and-effect relationship between release of anthropogenic chlorine and bromine and depletion of ozone. Ozone loss has also been observed in the Arctic and, at least in part, is related to processes similar to those occurring in the Antarctic. One of the primary reasons for concern about depletion of stratospheric ozone is the increase in UV radiation that results from the decrease in ozone. Simple physics clearly shows that such UV increases must occur. It is difficult to detect UV changes at the ground caused by small changes in ozone in the stratosphere. In contrast, it is relatively easy to detect the increased UV radiation reaching the surface as a result of the large loss of stratospheric ozone over the Antarctic in springtime. The observations shown in Figure 3.4 agree with the predictions and clearly make the connection between ozone-depleting substances and changes in UV radiation at Earth's surface.
Atmospheric Lifetime, Ozone Depletion Potential, and Global Warming Potential as Indicators of Environmental Impact
Compounds being considered as replacements for halon include perfluorocarbons, hydrofluorocarbons, hydrochlorofluorocarbons, and several other compounds, such as CF3I and SF6. These compounds are listed in Table 3.1, along with their atmospheric lifetimes and calculated ozone depletion potentials. Only individual compounds and their effects are examined here, although the effects of a mixture can be evaluated in terms of the proper ratios of the individual effects. The atmospheric lifetime of such compounds is important in determining their potential effects on ozone and climate, as is their calculated ozone depletion potential (ODP) and global warming potential (GWP).10
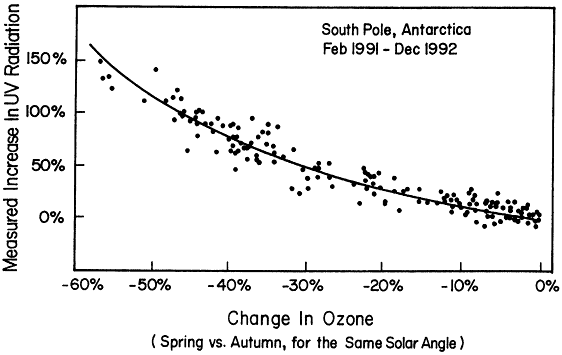
Figure 3.4
Measured increase in erythermal ultraviolet (UV) radiation at the South Pole. These data demonstrate the effects of reduced ozone in the Antarctic ozone "hole" in allowing more UV radiation to reach the surface.
Source: C.R. Booth and S. Mandronich, "Radiation Amplification Factors: Improved Formulation Accounts for Large Increases in Ultraviolet Radiation Associated with Antarctic Ozone Depletion," pp. 39-42 in Ultraviolet Radiation in Antarctica: Measurements and Biological Research, C.S. Weiler and P.A. Penhale, Eds., AGU Antarctic Research Series, Vol. 62, American Geophysical Union, Washington, D.C. (1994).
Atmospheric Lifetime
After emission into the atmosphere, the time scale for removal of a gas—its atmospheric lifetime—is generally defined as the ratio of total atmospheric burden to integrated global loss rate. The lifetime is the time it takes for the global amount of the gas to decay to l/e, or 36.8% of its original concentration after initial emission into the atmosphere. The lifetime must take into account all of the processes determining the removal of a gas from the atmosphere, including photochemical losses within the atmosphere (typically due to photodissociation or reaction with OH), heterogeneous removal processes (e.g., loss into clouds or into raindrops), and permanent removal uptake by the land or ocean. Atmospheric lifetimes of a number of gases have been determined based on current knowledge of these loss processes.
As shown in Table 3.1, atmospheric lifetimes of greenhouse gases of interest as replacements for halon range from a few days (e.g., for CF3I, owing to its photolysis at near-ultraviolet wavelengths) to thousands of years (e.g., for SF6 and the perfluorocarbons). Lifetimes of HCFCs and HFCs range from very short periods for gases such as HCFC-123 that react rapidly with hydroxyl (OH) to lifetimes comparable in length to those of the halons.
Table 3.1 Atmospheric Lifetime and Calculated Ozone Depletion Potential (ODP) for Halons and for Potential Replacements
Potential Effects on Ozone
Understanding of the depletion of stratospheric ozone has led to the need for simple measures for comparing the impact on ozone of different compounds as a scientific guide to public policy. The concept of ozone depletion potential has proven to be a useful index for gauging the effects on ozone from CFCs, halons, and their replacements.
Effectiveness of Chlorine and Bromine in Ozone Destruction
The chlorine and bromine catalytic mechanisms are particularly efficient at destroying ozone. The chlorine and bromine catalytic cycles can occur thousands of times before the catalyst is converted to a less reactive form such as HCl or HBr. Because of this cycling, relatively small concentrations of reactive chlorine or bromine can have a significant impact on the amount and distribution of ozone in the stratosphere. In the lower stratosphere, atmospheric and laboratory measurements indicate that heterogeneous chemistry on particles leads to enhanced effects on ozone from chlorine and bromine by helping to convert less reactive species to the forms of bromine and chlorine that can react catalytically.
Bromine is much more effective at destroying ozone in the lower stratosphere than is chlorine, as much as 100 times more efficient below 20 km. 11,12,13 The emissions and corresponding amount of brominated compounds or halons in the atmosphere are much smaller, however, than those of the chlorinated compounds. As a result, while not negligible, bromine's impact on the current atmosphere is smaller than the effects from increasing chlorine.
CF3I and other compounds containing iodine have been suggested as replacements for halons. Iodine reaching the stratosphere is even more effective than bromine and over 1,000 times more effective than chlorine at destroying ozone in the lower stratosphere.14,15
Other suggested replacements are primarily composed of carbon, fluorine, and hydrogen. None of these compounds affects ozone. It has been suggested that CF3 radicals produced by dissociation of some of these compounds, such as from the dissociation of HFC-134a, could affect ozone; recent studies show, however, that these radicals do not have any significant effects on ozone.16,17
How Much Have Halons Affected Stratospheric Ozone?
The concept of chlorine/bromine loading18 provides a useful means of examining the relative effects of different chlorine and bromine compounds on stratospheric ozone. By combining atmospheric observations with analyses from atmospheric models, chlorine/bromine loading provides a measure of the amounts of chlorine and bromine available to react with ozone. The changes in chlorine/bromine loading with time, based on past and projected emissions of halocarbons, are assumed to be proportional to changes in ozone using the effects of chlorine and bromine in determining trends in ozone depletion over recent decades. Of particular interest is the chlorine/bromine loading in the lower stratosphere, where observations indicate that most of the ozone loss has occurred.
Figure 3.5 shows the chlorine/bromine loading in the lower stratosphere for known past emissions of CFCs, halons, and other halocarbons and for projected emissions corresponding to those expected following the Copenhagen Agreement modifications to the Montreal Protocol (note that bromine is assumed to be 60 times as effective as chlorine in affecting ozone in this analysis). Taking into account uncertainties associated with the effects of bromine on ozone relative to the effects of chlorine, these analyses suggest that halons account for about 7 to 12% of the ozone decrease in the lower stratosphere through 1995, while total bromine, including effects from human-related emissions of methyl bromine, accounts for 15 to 25% of the ozone decrease in this region. Bromine has almost no effect on determining the ozone decrease in the upper stratosphere, thus implying smaller effects from halons (3 to 5%) and total bromine (9 to 12%) on globally integrated ozone distribution.
Ozone Depletion Potential
The concept of ozone depletion potential (ODP) provides a cumulative measure of the expected effects on ozone from the emissions of a gas relative to one of the gases of most concern with respect to ozone change, namely CFC-11 (CFCl3).19,20,21,22 This concept is an integral part of national and international policy considerations to protect ozone, including the Montreal Protocol and its amendments, and the U.S. Clean Air Act. ODPs provide an important index for analyzing a new
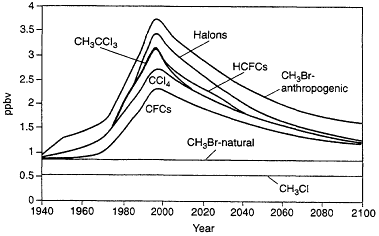
Figure 3.5
Chlorine/bromine loading in the lower stratosphere for known past emissions of CFCs, halons, and other halocarbons and for projected emissions corresponding to those expected following the Copenhagen Agreement modifications to the Montreal Protocol.
chemical's potential to affect ozone relative to CFCs, halons, or other replacement compounds being considered.
The ODP of a gas is defined as the change in total ozone per unit mass emission of the gas relative to the change in total ozone per unit mass emission of CFC-11. Time-dependent ODPs can also be defined that provide information on a compound's effects on ozone on a shorter time scale. However, the steady-state values have been preferred and have been used in regulatory considerations. ODPs are currently determined by two different means: by calculations with two-dimensional models of the global atmosphere23,24 and by the semiempirical approach developed by Solomon et al.25 The two approaches give similar results.
By definition, the ODP for CFC-11 (CFCl3) is 1.0, and the calculated ODPs for other banned CFCs are all greater than 0.4. The Clean Air Act calls for policy actions on compounds whose ODPs are equal to or greater than 0.2. As listed in Table 3.1, the steady-state ODPs for halons are all extremely large, much greater than 1.0, reflecting the reactivity of bromine with ozone, whereas the ODPs for all of the hydrofluorocarbons (HFCs), the perfluorocarbons (PFCs), and sulfur hexafluoride are all near zero, reflecting the inability of their degradation products to participate in catalytic ozone destruction cycles.26 Although the HCFCs do contain chlorine and can affect ozone, the ODPs of those being considered as halon replacements (Table 3.1) are all small, with values of 0.02 to 0.05. The effects on ozone per unit emission of one of these HCFCs would be less than one hundredth of the effect on ozone caused by the halon it would replace. The short lifetimes of these HCFCs and their lack of bromine result in the reduced effect on ozone.
Although iodine is extremely reactive with ozone, the ODP for surface emissions of CF3I is less than 0.01 (a recent analysis by Connell et al.27 suggests that the ODP is less than 0.006); because of its reactivity in the troposphere, very little iodine would be expected to reach the stratosphere. However, this ODP value is subject to significant uncertainty because of uncertainty in the understanding of iodine chemistry (e.g., lack of data on such reactions as IO with O3, ClO, or BrO) and the physical processes (e.g., the effects of fast vertical transport from convective processes in transferring iodine from the lower to the upper troposphere) affecting the iodine distribution in the troposphere and stratosphere.
In evaluating alternatives to halon, time-dependent ODPs are also useful to examine because they provide insight into a compound's short-term effects on stratospheric ozone following its emission (while steady-state ODPs indicate integrated effects over longer time scales). As discussed in recent international assessments,28 the ODPs for the HCFCs are much larger at short time scales of a few years
than they are at steady state. The short atmospheric lifetimes of these compounds imply that, compared to CFC-11, they release chlorine in the lower stratosphere more quickly, and can result in a more immediate (but small) effect on ozone. However, the time-dependent ODPs for these HCFCs are still much smaller than the ODPs for the halons they would replace.
Potential Effects on Climate
Much of the concern about effects of human activities on climate has centered on carbon dioxide (CO2) because of its importance as a greenhouse gas and also because of the rapid rate at which its atmospheric concentration has been increasing. However, other greenhouse gases account for about half of the overall increase in the effect of radiative forcing on climate.29 Although halons, because of their small concentrations, are currently only a minor contributor to increased radiative forcing, it is important to consider the possible role of any replacements in affecting future climate.
Several different indices have been used as measures of the strength of the radiative forcing on climate from different greenhouse gases. The indexing approach for greenhouse gases that has gained the widest acceptance is the concept of global warming potential (GWP) originally developed by the Intergovernmental Panel on Climate Change (IPCC). 30
Radiative Forcing
The radiative forcing of the surface-troposphere system (owing, for example, to a change in greenhouse gas concentration) is defined as the change in net irradiance (in watts per meters squared, Wm-2 ) at the tropopause after allowing for readjustment of stratospheric temperatures to stratospheric equilibrium. The tropopause is the reference point because it is considered, in a global and annual mean sense, that the surface and the troposphere are closely coupled.
A key factor affecting the radiative forcing associated with a gas is the location of the wavelengths at which it absorbs infrared radiation. The spectral region from about 8 to 12 µm is referred to as the ''window'' because of the relative transparency of the atmosphere to radiation over these wavelengths. Most of the non-CO2 greenhouse gases with the potential to affect climate, including halons and most of their replacements, all have strong absorption bands in the atmospheric window region. Relatively small changes in the concentrations of these gases can produce a significant increase in radiative forcing.
As the concentration of a greenhouse gas becomes high, it can absorb most of the radiation in its energy bands; once any of its absorption wavelengths become saturated, it is unable to absorb more energy at those wavelengths, and a further increase in its concentration has a diminishing effect on climate. This is called the band saturation effect. For example, the radiative forcing attributable to further increases in carbon dioxide concentrations in the current atmosphere will increase as the natural logarithm of its concentration because of this effect. Also, at the wavelengths where water vapor and carbon dioxide strongly absorb infrared radiation, the greenhouse effect of other gases will be minimal. However, absorption by other gases, such as the halons or other halocarbons, at wavelengths that are not saturated varies linearly with concentration. Another important consideration in radiative absorption is the band overlap effect. If a gas absorbs at wavelengths that are also absorbed by other gases, then the effect on radiative forcing of increasing its concentration can be diminished.
In addition to the direct forcing effect from emission of a gas into the atmosphere, the net radiative forcing can also be modified through indirect effects relating to chemical interactions on other radiatively important constituents. For example, emissions of halons result in stratospheric bromine that can destroy stratospheric ozone, which is also a greenhouse gas.31 Such indirect effects need to be considered when candidate replacement compounds are evaluated for potential effects on climate.
Table 3.2 Radiative Forcing Due to Halon 1301 and Potential Replacements
Relative Radiative Forcing per Molecule or Mass
Relative radiative forcing represents a comparison of radiative forcing on a molecule-per-molecule or kilogram-per-kilogram basis for the different greenhouse gases. It is generally given relative to CO2 or CFC-11. A radiative transfer model of the atmosphere is used to determine the radiative forcing attributable to small perturbations of these gases relative to present-day conditions. Small perturbations are used in the calculations in order to prevent the marked nonlinear absorption of carbon dioxide, methane, and nitrous oxide from affecting the radiative forcing for these gases. Table 3.2 shows radiative forcing on a per-molecule basis relative to CFC-11 for halons and their possible replacements.
The values for radiative forcing for halon 1301 and possible replacements listed in Table 3.2 are all within a factor of four of each other and are roughly of the same order of magnitude as the values attributable to CFC-11. On the other hand, the radiative forcing for CFC-11 is 3,970 times greater than that for CO2 on a per-unit-mass basis and is 12,400 times greater than that for CO2 on a per-molecule basis. These results suggest that all of the replacement compounds could be important greenhouse gases if their atmospheric concentrations became large enough.
What Effects Could Replacements for Halons Have on Climatic Change?
Current concentrations of halon 1301 in the atmosphere (about 0.003 ppbv) produce a radiative forcing on climate that is 0.0005 that owing to increases in carbon dioxide concentrations over the last two centuries. Even over recent decades the effect on climate of the rapid increases in halon concentrations would still be small relative to the effect of the increase in CO2 concentration. Increased halon concentrations due to use by the U.S. Navy account for an even smaller fraction of the radiative forcing effects on climate.
Global Warming Potential
Akin to the concept of ODP, the concept of global warming potential has been developed to provide a simple representation of the relative effects on climate resulting from a unit-mass emission of a greenhouse gas. Values for GWP are expressed as the time-integrated radiative forcing from the instantaneous release of a kilogram (i.e., a small mass emission) of a trace gas expressed relative to that of a kilogram of the reference gas, CO2:
where n is the time horizon over which the calculation is considered; F([x(t)] is the radiative forcing in response to the changing concentration of species x after the pulse emission at time t = 0; [x(t)] is the time-decaying concentration of that gas; and the corresponding quantities for the reference gas are in the denominator. The radiative forcing responses are derived from radiative transfer models. The trace gas concentrations, [x(t)], remaining after time t are based on the atmospheric lifetimes of the gas in question. The reference gas has been taken generally to be CO2, since this allows a comparison of the radiative forcing role of the emission of the gas in question to that of the dominant greenhouse gas that is emitted as a result of human activities.
The best choice of integration time horizon in evaluating GWP has been the subject of much discussion and controversy. Unlike the case with ODP, the complexities of treating CO2 and the carbon cycle prevent integration of GWP to steady state. There is, however, no given value of integration time for determining GWP that is ideal over the range of uses of this concept. Values for GWP are generally calculated over three time horizons, for 20, 100, and 500 years. It is believed that these three time horizons provide a practical range for policy applications.32 GWPs determined for the longest integration period provide a measure of the cumulative chronic effects on climate, while integration over the shortest period is representative of near-term effects. GWPs evaluated over the intermediate 100-year period appear generally to provide a balanced representation of the various time scales for climatic response. The best choice of time horizon will depend on the specific analysis being considered. It is necessary to balance the effects of near-term responses in comparing greenhouse gases with consideration of the long-term persistence of any environmental effects from long-lived gases.
Table 3.3 Direct Global Warming Potential of Halon 1301 and of Candidate Replacements
Direct GWPs Table 3.3 summarizes the GWPs for halons and their replacements included in the new IPCC assessments. Because their radiative forcing is of similar magnitude, the replacements with the longer atmospheric lifetimes have the largest GWPs. In fact, the GWPs for many of the replacements are as large as or larger than the GWPs for halon 1301 (other halons have not been evaluated), particularly for the 20-year integration time. The shorter-lived compounds, such as HCFC-123, HCFC-124, and HFC-32, have appreciably smaller GWPs, particularly at the 100- and 500-year integration periods. The GWPs for CF3I are extremely small (<3 even for the 20-year integration) owing to its atmospheric lifetime of only a few days.
The GWPs for the perfluorocarbons and for SF6 are all much larger than any of the GWP values for CFCs or halons. The very long atmospheric lifetimes of these gases lead to extremely large GWPs. These large GWPs imply potentially large effects on climate over long time scales, with the actual effect on climatic radiative forcing dependent on the magnitude of emissions into the atmosphere.
Indirect GWPs Possible effects on ozone from emissions of halons or their replacements also need to be considered. Daniel et al.33 have estimated the indirect GWPs for effects on ozone from a variety of halocarbons, including CFCs, halons, and HCFCs, in an attempt to clarify the relative radiative forcing roles of different classes of ozone-depleting compounds. Decreased ozone from CFCs and halons should decrease the radiative forcing on climate. Daniel et al. found that the net GWP of halocarbons depends strongly on the each compound's effectiveness for ozone destruction. Halons are likely to have negative net GWPs, while those of CFCs are likely to be positive over both 20- and 100-year time horizons. These analyses, however, are still subject to remaining uncertainties about the causes of ozone decreases in the lower stratosphere. These indirect effects are not included in the values given in Table 3.3.
Summary
The concepts of ozone depletion potential and global warming potential provide important guides to the potential of halon replacements to destroy stratospheric ozone and to affect radiative forcing on climate. In general, unless the amounts of replacement compounds produced and emitted into the atmosphere are much larger than the amounts of the compounds they replace, the compounds being considered as halon replacements are not expected to have any appreciable effect on stratospheric ozone.
However, the perfluorocarbons and SF6 have extremely long atmospheric lifetimes (> 1,000 years) and their large GWPs suggest that emissions of these compounds could lead to significant concerns about radiative forcing on climate.
Other Environmental Effects
A chemical compound introduced into the atmosphere at the ground will be dispersed throughout the troposphere. It takes a few months for the compound to be dispersed within the hemisphere in which it is released. The time constant for cross-equatorial transport is about 1 year. On the same time scale, a portion of the chemical will also be transported to the stratosphere. Material is continuously recycled between the troposphere and the stratosphere. An inert molecule spends 90% of its time in the troposphere. Once transported to the stratosphere, a compound has an average residence time of about 3 years before it is transported back to the troposphere.
The extent to which a compound can be dispersed globally depends on the competition between transport and removal processes. Removal from the atmosphere is achieved by chemical or photochemical reactions that alter the identity of the compound, or by physical means such as rainout. Figure 3.6 provides a schematic representation of the life cycle of a chemical, A, released to the atmosphere. It is dispersed between the ground, tropospheric, and stratospheric reservoirs. Possibilities exist for the compound to be transformed into an intermediate product, B, and/or final products C and D in each reservoir.
If the compound is insoluble and chemically inert, the material will retain its identity as A, and all of it will stay in the atmosphere. Eventually, the material will be dispersed evenly throughout the atmosphere with a constant volume mixing ratio.* One concern in this case is that the concentration of A in the atmosphere would reach a sufficient level that it could block infrared radiation emitted from the surface, leading to greenhouse warming. Given the radiative properties of most molecules, the greenhouse warming effect starts to become significant when a few teragrams (1 teragram = 1012 gm) of the compound are present in the atmosphere so that its mixing ratio approaches the parts per billion by volume level. The GWP of a compound provides a relative measure of its greenhouse warming impact on a unit mass emitted basis (see discussion in preceding section).
A more typical situation is presented by a chemical whose reactivity in the atmosphere is sufficiently slow (removal rate slower than 1% per day) that it can be transported globally before it is chemically
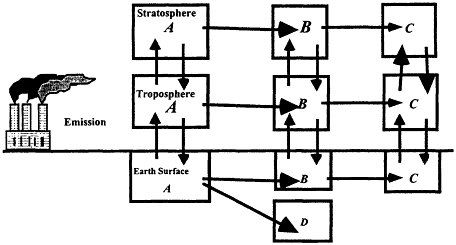
Figure 3.6
Schematic representation of a chemical's life cycle. The horizontal arrows represent chemical and/or physical processes. Vertical arrows represent transport between reservoirs.
removed. The final degradation products (C) for the halocarbons are CO2 from the carbon atoms, and hydrogen halides (i.e., HF, HCl, HBr, and HI) from the halogen atoms. The amount of CO2 produced by the oxidation of halocarbons is negligible when compared to the magnitude of the other CO2 sources (e.g., fossil fuel combustion). The hydrogen halides are removed from the atmosphere by rainout. The resulting acid deposition is negligible compared to that from other sources present in the atmosphere.34 With the exception of fluorine, the halogen atoms (i.e., Cl, Br, and I ) coexist with the hydrogen halides in the atmosphere. Cl, Br, and I atoms participate in catalytic chemical cycles that can deplete stratospheric ozone; F atoms do not. F atoms react rapidly with H2O vapor and with CH4 to give HF. The H-F bond is extremely strong. HF is an efficient and permanent sink for F atoms in the atmosphere, and as a result F atoms do not participate in catalytic chemical cycles.35 The value of the ozone depletion potential assigned to a compound is a combined measure of the amount of free radicals that it delivers to the stratosphere and the catalytic efficiency of the free radicals in ozone removal. ODPs measure the relative effects of different compounds on ozone on a per unit mass emitted basis, not on the basis of absolute concentrations oft he halogen radicals delivered to the stratosphere. For a detailed discussion of ODPs and their derivation, see the discussion in the preceding section.
Intermediate products may also be formed during the atmospheric degradation of halon replacements. The question addressed here is whether any of these products will have any significant environmental impact. There are three issues to be considered:
- If the atmospheric concentrations of the intermediate products (B) and final products (C) are sufficiently high, their greenhouse warming effects should be added to the global warming potential of the parent compound.
- The intermediate products may react with other trace gases in the atmosphere and perturb their natural balance in the atmosphere. The effect on ozone is of particular interest.
- The ecological effects of the deposition of intermediate and final products on the ground should be assessed. A necessary step in assessing these effects is the estimation of the concentrations of each chemical and their by-products in each reservoir.
Estimating Atmospheric Concentrations of Intermediate Degradation Products
The atmospheric concentration of a substance depends on the rate at which it is emitted to, or produced in situ, in the atmosphere and the rate at which it is removed from the atmosphere. The local
chemical removal rate can be characterized by a time constant that is related to the inverse of the reaction rate. For two species with the same emission or production rates, the one with the longer lifetime will have a larger concentration in the atmosphere. For all of the CFCs, HCFCs, and HFCs that have been examined, the lifetimes of the intermediate atmospheric degradation products are always much shorter than those of the parent compounds. As a result, the atmospheric concentrations of the intermediate products are always much smaller than those of the parent compounds.
Chlorofluorocarbons (CFCs) and halons are essentially inert in the troposphere but are reactive in the stratosphere. Intermediate degradation products from CFCs and halons usually have small concentrations in the stratosphere because there are abundant high-energy UV photons in the stratosphere to dissociate them. The initial sequence of reactions leads rapidly (within a few minutes) to the formation of intermediate oxidation products. One example of an intermediate product is CF2O from CFC-12 (CF2Cl2). CF2O is further degraded in the atmosphere to form HF and CO2. The atmospheric concentration of CF2O is expected to be about a factor of 1,000 less than that of CFC-12. Concentrations of CF2O observed in the atmosphere are in agreement with expected concentrations calculated using known photochemical processes.36
Hydrochlorofluorocarbons (HCFCs), hydrofluorocarbons (HFCs), and hydrogenated halons react with the hydroxyl radical in the troposphere. Although their primary loss is in the stratosphere, some halons (1211, 1202, and 2402) do have a small fraction of their photodissociation in the troposphere. Intermediate products produced in the troposphere from the degradation of HCFCs and HFCs are subject to removal by rainout within a few day37 As a result, the concentrations of the intermediate products in the atmosphere are very small.
Estimating the Concentrations of the Intermediate Products on the Ground
Some of the intermediate degradation products produced in the troposphere may be removed and deposited back to the ground by rainout and dry deposition before they can be photochemically converted into CO2 and the respective halogen halides. A simple estimate of the upper-limit rainwater concentration of the intermediate product can be obtained as follows. If one assumes that 1 kiloton of compound in the atmosphere would produce 1 kiloton of the intermediate degradation product, the annual total rainfall of 5 × 1017 liter would imply an average concentration of the intermediate product at 2 ng/liter for a 1 kiloton/yr emission. More sophisticated estimates can be obtained by taking into account the yield of the intermediate product, the spatial distribution of the gas-phase precursors, and the distribution of precipitation. This can be achieved using a three-dimensional chemistry-transport model. An example of such a study of the global-scale deposition of CF3COOH (TFA) produced during the atmospheric oxidation of HFC-134a can be found in Kanakidou et al.38 In addition, regional-scale acid deposition models can be used to examine the variations on local and regional scales.
Once the rainwater reaches the ground, one needs to assess if the intermediate product can be degraded in the natural environment, e.g., by microbial action in the ground water. Tromp et al.39 pointed out that in certain water bodies, the normal evaporation cycle will remove the water, leaving behind the solute. In the absence of a degradation mechanism, the solute could accumulate to concentrations that are much higher than those found in rainwater.
Degradation Pathways for Candidate Alternative Fire Extinguishing Agents
The halon alternatives that have been identified consist of a number of HCFCs, HFCs, and PFCs and CF3I. They can be separated into the following groups:
|
• Halogenated methanes: |
HCFC-22 (CHClF2), HFC-23 (CF3H), IFC-12I1 (CF3I) |
|
• Halogenated ethanes: |
HCFC-123 (CF3CHCl2), HCFC-124 (CF3CHClF), HFC-125 (CF3CHF2), HFC-134a (CF3CH2F) |
|
• Halogenated propanes: |
HFC-227ea (CF3CHFCF3), HFC-236fa (CF3CH2CF3) |
|
• Perfluorinated halocarbons: |
PFC-218 (CF3CF2CF3), PFC-31-10 (CF3CF2CF2CF3). |
Degradation Pathways for Halomethanes and Haloethanes
The atmospheric degradation mechanisms of halomethanes and haloethanes have been the subject of several workshops. The results are available in a 1990 WMO report.40
Atmospheric degradation of halomethanes and haloethanes is initiated principally by the reaction with the OH radicals. The products are given in Table 3.4. The degradation pathways for the typical haloethane are illustrated in Figure 3.7, after Cox and Lesclaux41 and Wallington et al.42 The detailed discussion that follows focuses on haloethane of the form CX3CYZH, where X = Cl or F, and Y and Z can be H, Cl, or F. The HFC/HCFC molecule reacts with OH or O(1D), giving rise to haloalkyl (R) radicals, labeled A in Figure 3.7. Haloalkyl radicals add molecular oxygen rapidly (within 1 microsecond) to give haloalkyl peroxy radicals, labeled B2. Haloalkyl peroxy radicals undergo a variety of reactions and within a few minutes are converted into hydroperoxides (ROOH), labeled B1; alkyl peroxynitrates (ROONO2), labeled B3; and alkoxy (RO) radicals, labeled C. As shown in Figure 3.7, a variety of processes (reaction with OH radicals, thermal decomposition, and photolysis) return the hydroperoxides and peroxynitrates to the peroxy and alkoxy radical pool. While the atmospheric lifetimes of hydroperoxides and peroxynitrates vary depending on their chemical identity and the region of the atmosphere in which they are formed, these compounds typically have lifetimes in the range of several hours to several days. Haloalkoxy radicals are reactive species and have lifetimes that are less than 1 second. There are several possible degradation pathways for the haloalkoxy radicals. Alkoxy radicals can decompose by two different bond cleavage routes, C-C bond cleavage (path P1) to form fragments labeled D1, or C-Cl cleavage (path P3) to form the acetyl halide (CX3CYO), labeled D2. For alkoxy radicals containing an α-H atom, reaction with O2 (path P2) to form an acetyl halide is a possible atmospheric fate. The CX3CF2O radicals undergo C-C bond cleavage via path P1 exclusively. CF3CCl2O and CF3CFClO radicals decompose by C-Cl bond cleavage (P3) exclusively. For CF 3CHFO radicals there are two important pathways. The first is C-C bond scission and the second is reaction with
Table 3.4 Atmospheric Degradation Products of Selected Fluorocarbons
|
Fluorocarbon |
Structural Formula |
Intermediate Degradation Products |
Final Degradation Products |
|
HCFC-22 |
CHClF2 |
CF2O |
HF, HCl, CO2 |
|
HFC-23 |
CF3H |
CF2O, CF3OH |
HF, CO2 |
|
IFC-13I1 |
CF3I |
CF2O, CF3OH |
HF, HI, CO2 |
|
HCFC-123 |
CF3CHCl2 |
CF3C(O)Cl |
HCl, CF3COOH |
|
HCFC-124 |
CF3CHClF |
CF3C(O)F |
HCl, CF3COOH |
|
HFC-125 |
CF3CHF2 |
CF2O, CF3OH |
HF, CO2 |
|
HFC-134a |
CF3CH2F |
HC(O)F, CF3OH, CF3C(O)F, CF2O |
HF, CF3COOH, CO2 |
|
HFC-227ea |
CF3CHF CF3 |
CF3OH, CF3C(O)F, CF2O |
HE, CF3COOH, CO2 |
|
HFC-236fa |
CF3CH2 CF3 |
CF3CO CF3, CF3OH, CF2O, CO |
HF, CO2 |
|
PFC-218 |
CF3CF2 CF3 |
|
HF, CO2 |
|
PFC-31-10 |
CF3CF2CF2 CF3 |
|
HF, CO2 |
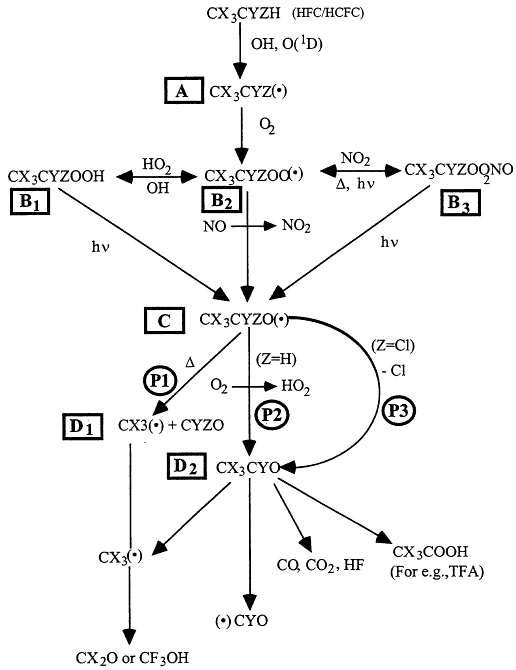
Figure 3.7
Degradation pathways for haloethane of the form CX3CYZH, where X = Cl or F, and Y and Z can take the form of H, Cl, or F. The symbol "(•)" denotes a radical species, and D denotes thermal decomposition. Source: Adapted from R.A. Cox and R. Lesclaux, "Degradation Mechanisms of Selected Hydrochlorofluorocarbons in the Atmosphere: An Assessment of Current Knowledge" in Scientific Assessment of Stratospheric Ozone: 1989, Vol. II, World Meteorological Organization, Global Ozone Research and Monitoring Project—Report No. 20, Geneva (1990); and T.J. Wallington, W.F. Schneider, D.R. Worsnop, O.J. Nielsen, J. Sehested, W.J. Debruyn, and J.A. Shorter, "Environmental Impact of CFC Replacements—HFCs and HCFCs," Environ. Sci. Technol. 28, 320A-326A (1994).
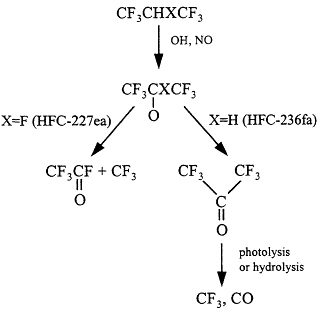
Figure 3.8
Degradation scheme for halopropanes.
O2. The relative importance of these two processes is dependent on temperature, O2 partial pressure, and total pressure. Because of this dependence, the branching will vary with altitude. For example, recent laboratory measurements43,44 for the CF3CHFO radical decomposition indicate that approximately 30% of these radicals will degrade to CF3C(O)F. CX3CH2O radicals react with O2 to form the aldehyde, CX3 CHO, and HO2 radicals. The aldehydes and acid halides are soluble in water and will then react further to form halogenated acids or hydrogen halides and carbon dioxide. The probable degradation products for the haloethanes are summarized in Table 3.4.
In the atmosphere CF3 radicals form CF3O radicals, which react with hydrocarbons and NO to give CF3OH and COF2, respectively.45 The atmospheric fate of COF2, CF3OH, and the acetyl halides is dominated by incorporation into cloud-rain-seawater followed by hydrolysis. Hydrolysis of CF 3COF (produced by HCFC-123, HCFC-124, and HFC-134a) gives trifluoroacetic acid, CF3COOH. Hydrolysis of COF2 and CF3OH gives HF and CO2, which, at the concentrations expected from HFC degradation, are of no environmental concern.
Degradation Schemes for Halopropanes
Two partially fluorinated propanes are under consideration as halon replacements: HFC-227ea (CF3CHFCF3) and HFC-236fa (CF3CH2CF3).
Atmospheric oxidation of HFC-227ea gives rise to CF3C(O)F molecules and CF3 radicals (Figure 3.8).46,47 The fate of the CF3 fragments is discussed in the section immediately above. The atmospheric fate of CF3C(O)F is incorporation into rain-cloud-seawater, followed by hydrolysis to give trifluoroacetic acid, CF3COOH.
Atmospheric oxidation of HFC-236fa gives hexafluoroacetone (CF3COCF 3).48 The fate of CF3COCF3 is either photolysis or interaction with water surfaces. While the relative importance of these two processes has not been measured, it seems likely that they are of comparable importance. The lifetime of CF3COCF3 with respect to photolysis has been estimated to be 3 days.49 Photolysis will generate CF3 radicals and CO. CF3 radicals are converted into CF3O radicals that are converted into
COF2, which is then hydrolyzed to give HF and CO2. Hydrolysis of CF3COCF3 produces the sesquihydrate CF3C(OH)2CF3, the fate of which is unknown.
Atmospheric Degradation of Perfluorocarbons
The atmospheric degradation of perfluorocarbons (PFCs; CF4, C2F6, C3F8, C4F10, etc.) has been investigated by Ravishankara et al.50 The usual mechanisms by which organic compounds are removed from the atmosphere—such as reaction with OH radicals and with O(3P) atoms, and photolysis in the lower atmosphere—are of no importance for perfluorocarbons. As a result, perfluorocarbons have extremely long atmospheric lifetimes (2,000 to 50,000 years). Ravishankara et al.51 reported that the most important removal mechanism for perfluorocarbons is photolysis by absorption of Lyman-α vacuum UV radiation at 121.6 nm at high altitudes (> 60 km) in the mesosphere and thermosphere. Two perfluorocarbons are currently under consideration as halon replacements: PFC-218 (C3F8), and PFC-31-10 (n-C4F10). By analogy to the existing database for other perfluorocarbons, it is anticipated that the atmospheric lifetime of these compounds is 2,000 to 3,000 years.52 As discussed above, the long atmospheric lifetimes of PFC-218 and PFC-31-10 lead to high GWP values for these species. Photolysis of PFC-218 and PFC-31-10 will give CF3, C2F5, and C3F7 radicals. These radicals are the same as those produced in the atmospheric degradation of HFC-23 (CF3H), HFC-125 (C2F5H), and HFC-227ca (C2F5CF2H), and their atmospheric transformation into the final products shown in Table 3.4 is well established. There are no known closed-shell (i.e., non-radical) intermediate degradation products of PFCs.
Assessment of the Environmental Impacts—ODP and GWP—of the Intermediate Products
Since the ODP and GWP are relative measures on a per unit mass basis, it is important to look at the atmospheric concentration of a degradation product relative to that of its respective parent compound. There appears to be no long-lived intermediate that will lead to high concentration in the atmosphere. While there has been one report that in the presence of O2 microbial degradation of trifluoroacetic acid (CF3COOH) produces CHF3,53 which has a lifetime of several hundred years, other workers have been unable to reproduce this result.54 The available evidence shows that the contributions to indirect GWP of the atmospheric degradation products of HFCs, HCFCs, and PFCs are negligible compared to the direct GWP of the parent compounds.
There have been suggestions that CF3Ox radicals formed by CF3 fragments may react with ozone in catalytic cycles, leading to depletion of stratospheric ozone. However, recent modeling and laboratory studies 55,56 ,57 and work by Wallington et al.58 have shown that there is no impact of CF3Ox, or any other HFC degradation intermediate, on stratospheric ozone. As with HFCs, perfluoroalkanes have no impact on stratospheric ozone.59
Questions have also been raised as to whether intermediate products (if they contain chlorine and bromine atoms) can act as carriers of additional chlorine/bromine atoms from the troposphere to the stratosphere. These questions have been answered by recent experimental studies that have shown that the lifetimes of the intermediate products are much shorter than the transport time to the stratosphere, and hence the intermediate oxidation products do not act as carriers of chlorine/bromine atoms to the stratosphere.
Accumulation of Degradation Products in the Biosphere
Given the expected emission rates and the kinetic data available, it is unlikely that the concentrations of the intermediate products in the global atmosphere will approach the parts per trillion level. Hydrolysis of COF2 and CF3OH gives HF and CO2, which, at the concentrations expected from HFC degradation, are of no environmental concern. Trifluoroacetic acid (CF3COOH) has been identified as a
degradation product in several of the candidate agents for fire extinguishment Thus far, no biotic or abiotic destruction mechanism has been clearly established for CF3COOH, and consequently it is thought to be quite stable in water.60 CF3COOH is not toxic toward animals but does have a mild herbicidal effect.61 The global average concentration of CF 3COOH expected in rainfall as a result of the atmospheric degradation of HFCs is many orders of magnitude below that observed to have an impact on plant systems.62 However, uncertainties concerning the persistence of CF3COOH in ground water allow for the possibility that CF3COOH could accumulate in seasonal wetlands over long time periods (![]() years).63 Very recently, Frank et al.64 have reported the detection of CF3COOH in lake, river, spring, and ocean water samples at levels which are orders of magnitude greater than can be accounted for by man-made emissions. The work of Frank et al. implies that there are large unknown sources of CF3COOH. It seems unlikely that the concentration of CF3COOH formed during the atmospheric degradation of the halon replacement compounds will have any significant adverse global environmental impact. Research is needed to better establish the environmental inventory and fate of CF3COOH.
years).63 Very recently, Frank et al.64 have reported the detection of CF3COOH in lake, river, spring, and ocean water samples at levels which are orders of magnitude greater than can be accounted for by man-made emissions. The work of Frank et al. implies that there are large unknown sources of CF3COOH. It seems unlikely that the concentration of CF3COOH formed during the atmospheric degradation of the halon replacement compounds will have any significant adverse global environmental impact. Research is needed to better establish the environmental inventory and fate of CF3COOH.
References
1. CIAP Monograph 3, The Stratosphere Perturbed by Propulsion Effluents, U.S. Department of Transportation, Washington, D.C. (1975).
2. P.J. Crutzen. ''The Influence of Nitrogen Oxides on the Atmospheric Ozone Content,'' Q. J. R. Meteorol. Soc. 96, 320-325 (1970).
3. F.S. Rowland and M.J. Molina, "Chlorofluoromethanes in the Environment," Rev. Geophys. Space Phys. 13, 1-36 (1975).
4. World Meteorological Organization, Scientific Assessment of Ozone Depletion (1994).
5. R. Stolarski, R. Bojkov, L. Bishop, J. Staehelin, and J. Zawodny, "Measured Trends in Stratospheric Ozone," Science 256, 342-349 (1992).
6. World Meteorological Organization, Scientific Assessment of Ozone Depletion (1994); World Meteorological Organization, Global Ozone Research and Monitoring Project, Report No. 37D, Geneva (1995).
7. World Meteorological Organization, Scientific Assessment of Ozone Depletion (1994); World Meteorological Organization, Global Ozone Research and Monitoring Project, Report No. 37D, Geneva (1995).
8. J.C. Farman, B.G. Gardiner, and J.D. Shanklin, "Large Losses of Total Ozone in Antarctica Reveal Seasonal ClOx/NOx Interaction," Nature 315, 207-210 (1985).
9. J.G. Anderson, D.W. Toohey, et al. "Free Radicals Within the Antarctic Vortex: The Role of CFCs in Antarctic Ozone Loss," Science 251, 39-46 (1991).
10. D.J. Wuebbles, "Weighing Functions for Ozone Depletion and Greenhouse Gas Effects on Climate," Ann. Rev. Energy Environ. 20, 45-70 (1995).
11. World Meteorological Organization, Scientific Assessment of Ozone Depletion (1994); World Meteorological Organization, Global Ozone Research and Monitoring Project, Report No. 37D, Geneva (1995).
12. S. Solomon, J.B. Burkholder, A.R. Ravishankara, and R.R. Garcia, J. Geophys. Res. 99, 20929-20935 (1994).
13. D.J. Wuebbles, P.S. Connell, and K.O. Patten, "Evaluating the Potential Effects of Halon Replacements on the Global Environment" in Halon Replacements: Technology and Science, A.W. Miziolek and W. Tsang, Eds., American Chemical Society, Washington, D.C. (1995).
14. S. Solomon, R.R. Garcia, and A.R. Ravishankara, J. Geophys. Res. 99, 20491-20499 (1994).
15. D.J. Wuebbles, P.S. Connell, and K.O. Patten, "Evaluating the Potential Effects of Halon Replacements on the Global Environment" in Halon Replacements: Technology and Science, A.W. Miziolek and W. Tsang, Eds., American Chemical Society, Washington, D.C. (1995).
16. World Meteorological Organization, Scientific Assessment of Ozone Depletion (1994); World Meteorological Organization, Global Ozone Research and Monitoring Project, Report No. 37D, Geneva (1995).
17. T.J. Wallington, W.F. Schneider, J. Sehested, and O.J. Nielsen, "Hydrofluorocarbons and Stratospheric Ozone," Faraday Discussions 100, 55 (1995).
18. World Meteorological Organization, Scientific Assessment of Ozone Depletion (1994); World Meteorological Organization, Global Ozone Research and Monitoring Project, Report No. 37D, Geneva (1995).
19. D.J. Wuebbles, J. Geophys. Res. 88, 1433-1443 (1983).
20. World Meteorological Organization, Scientific Assessment of Ozone Depletion, 1991, World Meteorological Organization, Global Ozone Research and Monitoring Project—Report No. 25, Geneva (1991).
21. World Meteorological Organization, Scientific Assessment of Ozone Depletion (1994); World Meteorological Organization, Global Ozone Research and Monitoring Project, Report No. 37D, Geneva (1995).
22. D.J. Wuebbles, "Weighing Functions for Ozone Depletion and Greenhouse Gas Effects on Climate," Ann. Rev. Energy Environ. 20, 45-70 (1995).
23. World Meteorological Organization, Scientific Assessment of Ozone Depletion, 1991, World Meteorological Organization, Global Ozone Research and Monitoring Project—Report, No. 25, Geneva (1991).
24. World Meteorological Organization, Scientific Assessment of Ozone Depletion (1994), World Meteorological Organization, Global Ozone Research and Monitoring Project, Report No. 37D, Geneva (1995).
25. S. Solomon, M.J. Mills, L.E. Heidt, and A.F. Tuck, J. Geophys. Res. 97, 825-842 (1992).
26. T.J. Wallington, W.F. Schneider, J. Sehested, and O.J. Nielsen, "Hydrofluorocarbons and Stratospheric Ozone," Faraday Discussions 100, 55 (1995).
27. P.S. Connell, D.E. Kinnison, D.J. Bergmann, K.O. Patten, D.J. Wuebbles, R.G. Caniel, C.K. Williamson, A.W. Miziolek, and R.E. Huie, "Environmental Effects of Halon Replacements: Considerations for Advanced Agents and the Ozone Depletion Potential of CF31," Proceedings of the Halon Options Technical Working Conference, Albuquerque, N. Mex., May 7-9 (1996).
28. World Meteorological Organization, Scientific Assessment of Ozone Depletion (1994); World Meteorological Organization, Global Ozone Research and Monitoring Project, Report No. 37D, Geneva (1995).
29. Intergovernmental Panel on Climate Change (IPCC), Radiative Forcing of Climate Change, Cambridge University Press, Cambridge, Great Britain (1995).
30. D.J. Wuebbles, "Weighing Functions for Ozone Depletion and Greenhouse Gas Effects on Climate," Ann. Rev. Energy Environ. 20, 45-70 (1995).
31. A.A. Laths, D.J. Wuebbles, and J.A. Logan, A. Geophys. Res. 95, 9971-9951 (1990).
32. Intergovernmental Panel on Climate Change (IPCC), Radiative Forcing of Climate Change, Cambridge University Press, Cambridge, Great Britain (1995).
33. J.S. Darnel, S. Solomon, and D.L. Albritton, J. Geophys. Res. 99 (1994).
34. See, for example, J.P. Friend, in Scientific Assessment of Stratospheric Ozone, Vol. II, Chapter X (1990).
35. T.J. Wallington, W.F. Schneider, J. Sehested, and O.J. Nielsen, "Hydrofluorocarbons and Stratospheric Ozone," Faraday Discussions 100, 55 (1995).
36. J.A. Kaye, A.R. Douglass, C.H. Jackman, R.S. Stolarski, R. Sander, and G. Roland. "Two-Dimensional Calculation of Fluorine-Containing Reservoir Species," J. Geophys. Res. 96, 12865-12881 (1991).
37. See Wallington et al., Chapter 3 in Halon Replacements: Technology and Science, ACS Symposium Series 611, American Chemical Society, Washington, D.C. (1995).
38. M.F. Kanakidou, F.J. Dentener, and P.J. Crutzen, "A Global Three-Dimensional Study of the Fate of HCFCs and HFC-134a in the Troposphere," J. Geophys. Res. 100, 18781-18801 (1995).
39. T.K. Tromp, M.K.W. Ko, J.M. Rodriguez, and N.D. Sze, "Potential Accumulation of a CFC Replacement Degradation Product in Seasonal Wetlands," Nature 376, 327-330 (1995).
40. World Meteorological Organization, Scientific Assessment of Stratospheric Ozone: 1989, Vol. II, WMO Global Ozone Research and Monitoring Project Report No. 20, Geneva (1990).
41. R.A. Cox and R. Lesclaux, "Degradation Mechanisms of Selected Hydrochlorofluorocarbons in the Atmosphere: An Assessment of Current Knowledge," in Scientific Assessment of Stratospheric Ozone: 1989, Vol. II, World Meteorological Organization, Global Ozone Research and Monitoring Project, Report No. 20, Geneva, (1990).
42. T.J. Wallington, W.F. Schneider, D.R. Worsnop, O.J. Nielsen, J. Sehested, W.J. Debruyn, and J.A. Shorter, "Environmental Impact of CFC Replacements—HFCs and HCFCs," Environ. Sci. Technol. 28, 320A-326A (1994).
43. T.J. Wallington, M.D. Hurley, J.C. Ball, and E.W. Kaiser, "Atmospheric Chemistry of Hydrofluorocarbon 134a: Fate of Alkoxy Radical CF3FHO, Environ. Sci. Technol. 26, 1318-1324 (1992).
44. E.C. Tuazon and R. Atkinson, "Tropospheric Degradation Products of CH2FCF3 (HFC-134a), J. Atmos. Chem. 16, 301-312 (1993).
45. T.J. Wallington, W.F. Schneider, D.R. Worsnop, O.J. Nielsen, J. Sehested, W.J. Debruyn, and J.A. Shorter, "Environmental Impact of CFC Replacements—HFCs and HCFCs," Environ. Sci. Technol. 28, 320A-326A (1994).
46. R. Zellner, G. Bednarek, A. Hoffmann, J.P. Kohlmann, V. Meyers, and H. Saathoff, "Rate and Mechanism of the Atmospheric Degradation of 2H-heptafluoropropane (HFC-227)," Ber. Bunsenges. Phys. Chem. 98, 141-146 (1994).
47. T.E. Mogelberg, J. Sehested, M. Bilde, T.J. Wallington, and O.J. Nielsen, "Atmospheric Chemistry of HFC-236fa: Spectrokinetic Investigation of the CF3CHO2CF3 Radical, Its Reactions with NO, and the Fate of the CF3CHOCF3 Radical," J. Phys. Chem. 100, 8882-8889 (1996).
48. T.E. Mogelberg, J. Platz, O.J. Nielsen, J. Sehested, and T.J. Wallington, "Atmospheric Chemistry of HFC-236fa: Spectrokinetic Investigation of the Radical CR3CHO2CF3 Radical, Its Reactions with NO, and the Fate of the CF3CHOCF3 Radical," J. Phys. Chem. 99, 5373-5378 (1995).
49. T.E. Mogelberg, J. Platz, O.J. Nielsen, J. Sehested, and T.J. Wallington, "Atmospheric Chemistry of HFC-236fa: Spectrokinetic Investigation of the Radical CR3CHO2CF3 Radical, Its Reactions with NO, and the Fate of the CF3CHOCF3 Radical," J. Phys. Chem. 99, 5373-5378 (1995).
50. A.R. Ravishankara, S. Solomon, A.A. Turnipseed, and R.F. Warren, "Atmospheric Lifetimes of Long-lived Halogenated Species," Science 259, 194-199 (1993).
51. A.R. Ravishankara, S. Solomon, A.A. Turnipseed, and R.F. Warren, "Atmospheric Lifetimes of Long-lived Halogenated Species," Science 259, 194-199 (1993).
52. Intergovernmental Panel on Climate Change (IPCC), Radiative Forcing of Climate Change, Cambridge University Press, Cambridge, Great Britain (1996).
53. Visscher et al., Nature 369, 729 (1994).
54. AFEAS Workshop on the Environmental Fate of Trifluoroacetic Acid (1994).
55. A.R. Ravishankara, S. Solomon, A.A. Turnipseed, and R.F. Warren, "Atmospheric Lifetimes of Long-lived Halogenated Species," Science 259, 194-199 (1993).
56. M.K. Ko, N.D. Sze, J.M. Rodriguez, D.K. Weisenstein, C.W. Heisy, R.P. Wayne, P. Biggs, C.E. Canosa-Mas, H.W. Sidebottom, and J. Treacy, "CF3 Chemistry: Potential Implications for Stratospheric Ozone," Geophys. Res. Lett. 21, 101-104 (1994).
57. Review in Chapter 12 of World Meteorological Organization, Scientific Assessment of Ozone Depletion (1994).
58. T.J. Wallington, W.F. Schneider, J. Sehested, and O.J. Nielsen, "Hydrofluorocarbons and Stratospheric Ozone, J. Chem. Soc., Faraday Discuss. 100, 55-64 (1995).
59. T.J. Wallington, W.F. Schneider, J. Sehested, and O.J. Nielsen, "Hydrofluorocarbons and Stratospheric Ozone, J. Chem. Soc., Faraday Discuss. 100, 55-64 (1995).
60. AFEAS Workshop on the Environmental Fate of Trifluoroacetic Acid (1994).
61. T.J. Wallington, W.F. Schneider, D.R. Worsnop, O.J. Nielsen, J. Sehested, W.J. Debruyn, and J.A. Shorter, "Environmental Impact of CFC Replacements—HFCs and HCFCs," Environ. Sci. Technol. 28, 320A-326A (1994).
62. T.J. Wallington, W.F. Schneider, D.R. Worsnop, O.J. Nielsen, J. Sehested, W.J. Debruyn, and J.A. Shorter, "Environmental Impact of CFC Replacements—HFCs and HCFCs," Environ. Sci. Technol. 28, 320A-326A (1994).
63. T.K. Tromp, M.K.W. Ko, J.M. Rodriguez, and N.D. Sze, "Potential Accumulation of a CFC Replacement Degradation Product in Seasonal Wetlands," Nature 376, 327-330 (1995).
64. Frank et al., Nature 382, 34 (1996).























