
Workshop Report
INTRODUCTION
The Topic
In the first meetings, Forum members concurred that antimicrobial resistance should be awarded high priority as a matter for concern—not, however. because the issue is a new one: penicillin-resistant strains of Staphylococcus aureus were isolated as early as 1945. By 1959, there was already enough evidence of resistance to stimulate a review article in the Journal of the American Medical Association.2 The sense of urgency among Forum members was provoked by the acceleration and accumulation of a number of variables, expressed as documented increases in:
-
the number of pathogens displaying resistance and, within these, a mounting number of multidrug-resistant strains;
-
the number of compromised hosts;
-
mortality attributable to antimicrobial resistance;
-
the speed with which resistant microbes can spread globally; and
-
the costs of health care deriving from resistant microbes.
These increases were, in turn, accompanied by decreases or limitations in:
-
the power of the antimicrobial armamentarium to deal with many resistant pathogens,
-
the amount of research and development dedicated to antimicrobials during a period when resistance was not generally seen as a major threat, and
-
funding for public health infrastructure.
The Workshop
The workshop agenda consisted of three major components: (1) resistance as a phenomenon and the major factors contributing to its increasing prevalence; (2) surveillance activities and needs; and (3) options for response. Its primary objectives were to extract those aspects of resistance that seemed most pressing, to inspect the potential of new scientific advances to spur needed diagnostic and therapeutic advances, and to examine areas of intervention most likely to return the biggest payback to investments of funding, research, advocacy, and education. A compendium of currently active systems that include surveillance of antimicrobial resistance is provided in Appendix A. Appendix B presents the outline of the global and national resistance surveillance system proposed in 1995 by the American Society for Microbiology (ASM) Task Force. Appendix C is a glossary. Appendix D contains the workshop agenda and participants list.
THE COSTS OF ANTIMICROBIAL RESISTANCE*
Background
Before proceeding to discuss the major categories of concern relative to antimicrobial resistance—that is, issues of surveillance, the potential offerings of new scientific developments, and the range of possible responses to the problem—workshop participants considered the costs of resistance. These costs can include such factors as the direct cost of time in a hospital, extra physicians' visits when antibiotics are ineffective, extra hospital days and hospitalizations due to community-acquired resistant infections, the costs of newer antibiotics to replace antibiotics to which bacteria have become resistant, and lost workdays and deaths.
The one study to date that has taken all of these factors into account used mathematical models to estimate the costs of resistance, including the effect of a resistant infectious agent that appears in one year on the cost to society in later years.3 Depending on whether or not death was a consequence, this study determined that in then-current dollars, the total societal costs of antibiotic resistance ranged from $150 million (without deaths) to $3 billion (with deaths) annually in the United States.
In 1990, the National Foundation for Infectious Disease estimated that the costs only of nosocomial (hospital-acquired) infections caused by antibiotic-resistant bacteria could be as high as $4 billion annually. The Centers for Disease Control and Prevention (CDC) estimated these costs at $4.5 billion when costs from both antibiotic-resistant and susceptible infections were included. In 1992,
the 19,000 deaths directly caused by nosocomial infections made them the eleventh leading cause of death in the U.S. population.4
In 1995, an analysis by the Office of Technology Assessment (OTA) of the United States Congress concluded that antibiotic-resistant bacteria generated costs of a minimum of $1.3 billion (1992 dollars) yearly in the United States. OTA emphasized that the estimate was a minimum, since it considered only in-hospital costs and the resistance of only six species of bacteria to just one antibiotic, and excluded the costs of multidrug resistance and all other costs.5
Recent Case Material
Because, as the OTA report also indicated, the passage of time, inflation, and an increase in the number of antibiotic-resistant infections would make any estimate of the costs of resistance significantly higher, there is interest in more current calculations. Workshop participants heard a recent analysis of the costs of one multiple drug-resistant pathogen in one major metropolitan area, which indicates that the costs of resistance to the U.S. economy now may be well above the 1989 estimates.
In New York City in 1995, methicillin-resistant Staphylococcus aureus (MRSA) infections cost almost a half-billion dollars and claimed 1,409 lives. Institutional infections represented 57 percent of those costs, as shown in Table 1; nosocomial infections accounted for 46 percent, of these institutional infections, 42 percent of total direct medical costs in dollars, and 62 percent of total mortality. Long-term care facility infections accounted for 11, 12, and 15 percent, respectively.
Table 1 Costs and Attribution of Staphylococcus aureus Infections, New York City, 1995
|
|
Total Direct Medical Costs |
||
|
Type of Infection |
Percent of Total Incidence |
No. of Infections |
Million Dollars |
|
Institutional infections |
|
|
|
|
Hospital-acquired (nosocomial) |
46.0 |
6,300 |
180.8 (42%) |
|
Long-term-care facility |
11.0 |
1,500 |
51.7 (12%) |
|
Community-acquired infections |
43.0 |
5,750 |
203.0 (46%) |
|
SOURCE: Robert Rubin, The Lewin Group, July 30, 1997. |
|||
Among hospital discharges in 1995, 13,550 had S. aureus infections. The cost to treat hospitalized patients with these infections was $435.5 million; the average cost per case was $32,110, almost double the average hospital charge for all New York City Primary Metropolitan Statistical Area (PMSA) discharges. Of the nosocomial infections, pneumonia, surgical site infections, and catheter-
associated bacteremia were the most expensive. Mortality averaged 10 percent compared to an in-hospital AIDS mortality rate of 14.1 percent. The average value of life lost was estimated at $105,000.
The limit of clinical ability to deal with S. aureus infections is best expressed in the percentages of these infections that were methicillin-resistant. Methicillin-resistant infections represented 20 percent of the incidence of all S. aureus infections, accounted for 21 percent of the costs of these infections, and were responsible for 41 percent of the mortality attributed to S. aureus infection. When the denominator is limited to nosocomial infections, these proportions rise to 29 percent of incidence, 32 percent of medical costs, and 48 percent of mortality. Not surprisingly, methicillin-resistant infections have higher per-case costs and attributable mortality than methicillin-susceptible S. aureus infections: $31,400 versus $27,700 per case, and 17 percent versus 8 percent, respectively.
More effective institutional infection control programs could decrease costs and mortality by reducing the incidence of S. aureus infections, especially if methicillin-resistant S. aureus were to be targeted. There would be other, very large benefits as well. Dealing with methicillin-resistance typically entails greater use of vancomycin, which in turn increases the prevalence of vancomycin-resistant enterococci (VRE) and, perhaps not too far in the future, vancomycin-resistant S. aureus (VRSA). Strains of S. aureus with diminished susceptibility to vancomycin have already been reported in Japan. These costs and the dynamics of antibiotic resistance suggest that the time has come for another comprehensive analysis, ideally including attention to the costs of resistance worldwide.
TRACKING THE PROBLEM: CURRENT APPROACHES TO SURVEILLANCE*
The purpose of surveillance is to ask and answer questions that will provide information for action. Its effectiveness is in large measure a function of who is posing the question and for what purpose. The surveillance of antimicrobial resistance has as its goal the gathering of information for several purposes at every level where health care is provided. Each level has different needs, and all are critical:
-
to help individual health care providers make rational clinical decisions;
-
to inform health facility managers about which antibiotics to include in their formularies for cost containment and, more importantly, for optimal patient care;
-
to assess the public health burden imposed by a resistant pathogen, and its importance relative to other resistant infections, for the national and regional policymakers who must decide budget allocations and program priorities accordingly;
-
to guide industry in new drug discovery, development, and marketing, and to provide the basis for drug licenser; and
-
to target and effectively implement prevention and control measures, and to design advocacy and public education accurately and productively.
These objectives dictate that much more needs to be known than simply which pathogens are becoming resistant to which drugs. For example, which patients have resistant infections? Are they randomly distributed across an entire population, or do they fall into certain risk groups, for instance, hospital patients, travelers returning from abroad, or individuals with high rates of past or current antibiotic use? Is the problem confined to a single group, or is it spreading into other groups and the population at large? Are there patterns to changes in the distribution of resistance, and how are these patterns instructive? What can be determined about trends in risk factors (e.g., drug use), and how do these differ by pathogen and location?
As a general matter, response to these questions requires close monitoring of treatment and illness outcomes. The inevitable variability in these responses further requires that data be gathered locally, not only for local use but for systematic aggregation to determine larger dynamics.
Historically, a number of problems have restricted efforts to monitor antimicrobial resistance. Since surveillance studies typically require the acquisition, shipment, and centralized testing of microorganisms, they are costly. Compromises are therefore made as to the number and types of institutions surveyed, demographics, the number and type of organisms studied, the geographic areas studied, and the frequency of assessments. There are other problems: the absence of standardized data to enable easy and rapid comparison of results; methodological differences between studies; delayed publishing and restricted availability of results owing either to proprietary sponsorship or to lack of interest among editorial boards; and poorly standardized methods for susceptibility testing and molecular epidemiology among nations.
Characteristics of an Ideal Resistance Surveillance System
The ideal system for surveillance of antimicrobial resistance would
-
be prospective, active, timely, and affordable;
-
be structured to permit the broadest possible access;
-
provide accurate incidence and prevalence rates, which would in turn require both numerator and denominator information (e.g., the number of isolates
-
tested and the number of resistant isolates), as well as a mechanism to permit exclusion of repeat isolates from the data pool;
-
include information that identifies organisms causing infection and those involved in colonization (i.e., the ability of a bacterium to remain at a particular site and multiply there);
-
gather data so as to permit categorization by region and locality, as well to discriminate between hospital or community and urban or rural sources;
-
gather information on antimicrobial use and treatment outcomes, especially treatment failure (the outcome of resistance);
-
be able to detect new resistance markers and therefore be dependent on standardized and reliable laboratory techniques, uniform criteria for determining resistance, appropriate specimens for culture, and adequate microbiologic validation;
-
be a national network representing all regions and levels of care, thus including both hospital and outpatient facilities;
-
computerize all participating laboratories, regularly collect electronic data, process and report in ongoing fashion, and integrate all databases at the national level; and
-
make surveillance data available to practitioners at the appropriate regional and local levels so that problems at these levels could be managed appropriately.
Local-Level Surveillance*
It is critical here to underscore the importance of data from the local level, not only as the foundation of national and international comprehension of antimicrobial resistance, how it develops, and what it means, but as the basis for local ability to deal with disease emergence. Case material from Minnesota on Campylobacter (this state's most frequently isolated bacterial enteric pathogen) illustrates the importance of understanding "microtrends" within larger patterns. In 1992, the proportion of all Campylobacter isolates in Minnesota that were resistant to fluoroquinolones was 1.5 percent; by 1996, it was 6.4 percent.
The point here is that this upward trend is actually a composite of two effects that have to be understood as independent phenomena, of comparable importance but with distinct dynamics. The first is an "indigenous" increase in the incidence of resistance—that is, an increase within the state—that is highest in summer months; the question of a possible relationship among the increase in the endemic rate of fluoroquinolone-resistant strains of Campylobacter, Food and Drug Administration (FDA) approval of fluoroquinolones for therapeutic use in poultry, and off-label use of fluoroquinolones remains unexamined. The second phenomenon is the increase in the first quarter of the year that comes from more
individuals' traveling outside the country, primarily to Latin America, who may acquire foodborne diseases due to widespread fluoroquinolone use in poultry in Latin America and then return with Campylobacter-resistant organisms—an ''exogenous" increase. Therapeutic use of fluoroquinolones in poultry has been prevalent in Latin America since the late 1980s; however, the FDA did not approve such use until 1994. Exogenous disease acquisition was also a factor in a complex dynamic involving the emergence in Minnesota of resistant Salmonella enteritidis, which proved to have been largely acquired through foreign travel in Europe, Africa, South America, and—in the great majority—Mexico.
National Systems
The United States Centers for Disease Control and Prevention*
The CDC has two major approaches for conducting surveillance of antimicrobial resistance. The first is the National Notifiable Disease Reporting System (NNDS). Because the legal authority to require disease reporting in the United States is vested in state governments rather than in the federal government, this system consists of information reported by state health departments on a weekly basis. The Council of State and Territorial Epidemiologists, with guidance from the CDC, recommends to states what they should require in the way of reporting, but states are under no obligation to comply with these recommendations. Although most states do in fact comply, the completeness of reporting is highly variable, depending as it does on state-level resources, priorities, and legal codes. This system provides essentially no information on antimicrobial resistance.
The second system, developed to compensate for some of the incompleteness and unevenness in the national system, consists of individual data collection efforts focused on individual diseases and involving direct reporting from different facilities. For example, data on gonococcal resistance are collected from a network of sexually transmitted disease (STD) clinics; on nosocomial pathogens, from a network of hospitals; on physician prescribing practices, from ambulatory care facilities; on foodborne pathogens, from a range of sources monitored by the CDC, FDA, and U.S. Department of Agriculture (USDA). There are defensible reasons for this variability: the differing epidemiology of each infection, the diversity of the prevention and control measures required, disparities in the research questions asked, and diversity in the partnerships needed to collect the data and address the particular problem at hand.
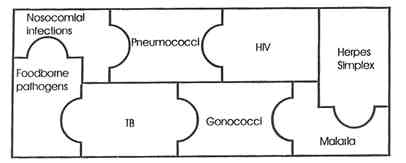
Figure 1. Centers for Disease Control and Prevention (CDC) "mosaic" of antimicrobial resistance surveillance systems. NOTE: Sources of nosocomial infections are primarily staphylococci, enterococci, and gram-negative bacilli. Foodborne pathogens are Salmonella, Escherichia coli,and Campylobacter. SOURCE: National Center for Infectious Diseases, CDC.
At the same time, although each CDC system may gather a fair amount of epidemiologic, microbiologic, and clinical information, none provides anywhere close to national coverage, and the linkages and coordination among them thus far have been quite limited (see Figure 1).
This raises several questions. One is how these different surveillance efforts might be integrated internally so as to provide a more solid and complete understanding of patterns and trends in resistance. Another is how these systems could be made to interact with the range of private-sector systems, including those implemented by universities, large managed care entities, or commercial enterprises, some of which may be funded by pharmaceutical companies and collect proprietary data. Other questions are how integration and expansion can be achieved and, very importantly, funded.
The Surveillance Network*
The Surveillance Network, or TSN, was developed by MRL Pharmaceutical Services, a private firm in Virginia specializing in diagnosis of infectious and immunological diseases. TSN is a U.S. national on-line network of 150 (by the end of 1997) hospital-based testing centers and independent laboratories chosen for their geographic, demographic, and methodological characteristics. MRL's philosophy was to incorporate these institutions in order to leverage existing testing capabilities and utilize data generated within the health care infrastructure. TSN has the ability to (1) assess and continuously improve testing; (2) detect the occurrence of antimicrobial resistance rapidly and analyze resistance
trends in real time; and (3) analyze data, also in real time, at a strain-specific level, using multivariate techniques.
TSN relies on the dynamic creation of two objective and interactive databases continuously expanded by the participating institutions. The first, the TSN database, contains more than 9.4 million strain-specific test results for 83 antibiotics tested against 649,000 bacterial isolates, representing more than 4,000 taxa and obtained from 426,000 patients; another 2.6 million records will have been added by the end of 1997. The size of this database relative to others is illustrated by Stenotrophomonas maltophilia: the largest study of this organism published to date contains information on approximately 170 strains; the TSN database currently has results for 4,331 strains. Data collected each day include selected patient information, microbial culture results, and quantitative and qualitative antimicrobial susceptibility test results.
The second database, TSN Archives, contains more than 13.7 million test results from the same participating laboratories, as well as less precise antibiogram-based historic data for 1992-1995, the years preceding the database period. These archived data are used mainly to track historical trends. MRL plans to expand the network globally and increase the database to include antifungal, antimycobacterial, and antiviral agents. Plans also call for establishing systems to include clinical and pharmacy information and collaborating with national and international public health organizations and researchers.
New software approaches were developed for collecting and analyzing TSN data and assessing their quality in an ongoing fashion. Data are automatically and electronically sent each day to MRL's data center in Reston, Virginia, where they pass through expert electronic systems that check for correctness, consistency, and epidemiologically significant events. They are then merged, at five-week intervals, into a national database. The databases can be queried from virtually anywhere via the Internet using proprietary software, but they are password-protected to ensure security and confidentiality for patients and participating hospitals, and all transmissions and Internet queries are encrypted.
TSN has already produced the following findings for the United States. The frequency of resistance to oxacillin in S. aureus is 27.5 percent and in nonaureus staphylococci, greater than 60 percent; among oxacillin-resistant S. aureus, 86.3 percent are resistant to ciprofloxacin, 89.2 percent to erythromycin, and 49.8 percent to gentamicin. TSN findings have also reinforced the urgency of identifying organisms at the species level. For instance, in the case of VRE, although laboratory results for unspeciated enterococci fail to demonstrate a significant resistance problem, results for Enterococcus faecium demonstrate that more than 50 percent of strains are, in fact, resistant to vancomycin.
The Canadian System*
Canada has undertaken several initiatives in the surveillance of antimicrobial resistance, including two more or less formal systems and several ad hoc programs.
-
The Canadian Hospital Epidemiology Committee (CHEC). The CHEC was initiated by the Canadian Infectious Disease Society and receives support from the Canadian counterpart of the U.S. CDC, the Laboratory Center for Disease Control (LCDC), and from industry. It consists of 23 hospitals in 9 of Canada's 10 provinces and will shortly include all 10. Detailed clinical data and information on organisms are collected, and all isolates are tested in a single dedicated center. The program has focused primarily on multidrug-resistant S. aureus (MRSA) infections, VRE, and Clostridium difficile.
-
The Canadian Bacterial Disease Network (CBDN). The CBDN, part of the federally funded Networks of Centers of Excellence program, is a Canada-wide consortium of researchers on bacterial disease. In 1993, Mount Sinai Hospital, University of Toronto, a node of the CBDN, established an ongoing cross-Canada surveillance program to monitor and study drug resistance in hospital and community pathogens. It has a current enrollment of more than 100 laboratories that service hospitals and community physicians. Isolates under study are processed centrally at Mount Sinai Hospital to ensure the accuracy of testing and to enable further investigation of the epidemiology and mechanisms of resistance. For example, the fact of 2,000 to 3,000 isolates yearly of Streptococcus pneumoniae and Haemophilus influenzae has allowed the rapid emergence of multidrug resistance in these organisms to be recognized.
-
Ontario Invasive Group A Streptococci Infections Surveillance Network. This program, which has been in place since 1992, monitors all invasive group A streptococcal infections in residents of the province of Ontario (population I I million). Each patient with an invasive infection of group A streptococci has the isolate, clinical information, and blood and tissue specimens, when possible and appropriate, forwarded to Mount Sinai Hospital as part of an ongoing study of the epidemiology and pathogenesis of this disease. This information is also forwarded to the Ontario Ministry of Health for the purpose of case follow-up and prophylaxis, where appropriate.
-
The Laboratory Proficiency Testing Program (LPTP). Through an agreement with the Ministry of Health of Ontario, the Ontario Medical Association (OMA) has been identified as an agent to examine and evaluate of the proficiency of performance of tests in clinical laboratories. LPTP is the unit within OMA that carries out this mandate. Established in 1974, LPTP has focused on ensuring that laboratories are aware of the importance and implications of new
-
and emerging multidrug-resistant pathogens, know how to detect them most accurately, and are able to identify them in blind surveys. LPTP has also conducted surveillance programs to monitor the emergence of such pathogens as VRE and methicillin-resistant S. aureus within the province.
-
Toronto Invasive Bacterial Diseases Network (TIBDN). This is a population-based, prospective surveillance program that monitors rates of invasive cases of group A and B streptococci, S. pneumoniae, Neisseria meningitidis, and Listeria monocytogenes in Toronto (population 3.5 million) and allows the study of the epidemiology of these pathogens. Also operated out of Mount Sinai Hospital, the program is funded in part by the LCDC, Physicians Services Incorporated (PSI), and CBDN.
-
Ad hoc surveillance programs. A number of provincial and national surveillance programs are carried out across Canada at a number of university affiliated hospitals to study the epidemiology of antimicrobial resistance. As in the United States, these are industry driven and funded with specific marketing goals. However, they also provide a valuable source of funding to allow point prevalence surveys to be carried out to determine prevalence and resistance rates of important hospital and community pathogens.
The Icelandic Surveillance System*
This small (population 270,000) homogeneous country is attempting to develop what might be considered a prototype of an ideal antimicrobial resistance surveillance system. It has been possible to develop uniform, standardized microbiological numerator and denominator data and information about antimicrobial use, as well as a national network. The system lacks merged databases, continuous processing and reporting, and the capacity for collecting outcome data, and is working on all three, independently and with other concerned entities.
The original goal of the program was to monitor all pneumococci with reduced susceptibility to penicillin and now has been expanded to include methicillin-resistant S. aureus, vancomycin-resistant enterococcus, and multiresistant Mycobacterium tuberculosis. The Department of Microbiology laboratory at the National University Hospital in Reykjavik, serves as a reference laboratory in addition to setting all of the standards and methods to be used in Iceland (according to National Committee for Clinical Laboratory Standards [NCCLS] Guidelines). This facility is also the only laboratory in the country that trains all technologists and physicians in microbiology.
Surveillance of resistance in Iceland is relatively easy because of its small homogeneous population and the relative isolation of the country. These factors also make it an ideal place to study the epidemiology of certain resistance traits. Close contact among the laboratories and a central laboratory that records all
strains along with basic information about the patient and infection facilitates the collection of data. In the future the system plans to merge health care, hospital, and pharmacy databases in an attempt to monitor how use and resistance trends affect the outcome of infections.
International Systems
CEM/NET*
International systems can both derive data from individual national systems and serve as a valuable supplement to these systems. An example of such a system is CEM/NET (Centro de Epidemiologia Molecular/Network for Epidemiologic Tracking of Antibiotic-Resistant Pathogens), an independent, international alliance between clinical microbiologists and molecular biologists. The former identify organisms, determine resistance rates, and provide characteristic strains for use by molecular biologists, who develop and streamline molecular fingerprinting methods and identify and track resistant genes. Scientific centers for this activity are, for molecular biology, the Institute of Biotechnology at the Universidade Nova de Lisboa in Portugal and the Laboratory of Microbiology at the Rockefeller University and, for clinical microbiology, the Microbiology Department at the National University Hospital in Iceland and comparable departments in other participating countries, currently 10 in number.6 Financial support is provided by the pharmaceutical industry, with in-kind support provided by participating institutions.
CEM/NET is prospective and problem oriented; its purpose is twofold. One is to serve as a base for collaborative projects between the core laboratories and individual scientists, providing molecular fingerprinting tools for the clinical microbiologist and access to antibiotic-resistant clinical isolates for the molecular biologist. The second is to analyze genetic and biochemical resistance mechanisms. New molecular fingerprinting technologies are transferred through working visits by scientists from different countries to the core laboratories in order to create an international network of independent, high-quality laboratories, which in the future will provide quality control and organization for the network as a whole. CEM/NET conducts training, organizes meetings and workshops, and conducts ongoing prospective studies. These have included a 20 center international study of MRSA, a study of respiratory tract pathogens and an interventional study in day care centers in Portugal and Iceland, a study of carriage of antibiotic-resistant genes in commensal staphylococci in healthy people, and development of DNA-based diagnostic assays for rapid speciation and detection of ß-lactam and glycopeptide resistance mechanisms in staphylococci, pneumococci, and enterococci.
World Health Organization (WHO) Initiatives*
At the global level, there is no system in place for the surveillance of antimicrobial resistance. WHO is establishing such a system as part of a program with the goal of reducing the rate of emergence and spread of antimicrobial resistance. Its premises are that to have any effect on the emergence of resistance, the use of antibiotics and antivirals has to be more rational, and that measuring the impact of any interventions to produce more rational use will require effective surveillance. The program has three main aims: (1) strengthening national laboratories in WHO member states, (2) helping to build the national infrastructures that make surveillance possible, and (3) promoting international coordination.
Program implementation will proceed in roughly the same sequence WHO has followed in Kenya in a pilot activity funded by the Pharmaceutical Research and Manufacturers of America (PhRMA) and the International Society of Infectious Diseases (ISID). The first step was a laboratory training course to improve susceptibility testing and resistance detection methods and to lay the foundation for coordinated surveillance activities among five hospitals, with a jointly developed national surveillance strategy as the ultimate goal. The second was a policy workshop for ultimate users of the data to be generated by the surveillance system, as the basis for changing the ways in which antibiotics are prescribed, used, and distributed. With the help of the WHO Collaborating Center for International Monitoring of Bacterial Resistance to Antimicrobial Agents, located in the CDC, laboratories in participating countries will have access to quality assurance measures that will contribute to the development of national quality assurance schemes. Given the great diversity among the countries in which resistance is a problem, a major challenge in establishing a global network for monitoring antimicrobial resistance is to determine exactly which organisms are to be monitored. With resistance to M. tuberculosis already covered by another WHO division, the initial emphasis will be on those organisms that are, in general, the greatest problems for the majority of member countries: S. aureus, S. pneumoniae, E. faecalis, E. faecium, Salmonella typhi, and Shigella dysenteriae. Neisseria gonorrhoeae data will be collected via the WHO Gonococcal Antimicrobial Surveillance Programme (GASP). Another area of effort will be a review of the antimicrobials on WHO's Essential Drugs List and the gathering of data on resistance to drugs in this category in key countries.
Finally, recognizing that there are a number of discrete surveillance activities scattered globally, the WHO strategy is to create a "network of networks" in order to make the data from these systems available more widely. The notion is to enroll these programs, provide them with assistance in quality assurance, determine definers and criteria for use of the data and then summarize them, and finally, organize the data by country for open access. The WHO philosophy is that despite the undeniable variance in data collection and analysis among these
different systems, the matter of resistance is too urgent to wait for harmonization. Its hope is that the energy dedicated to forming this network of networks will stimulate standardization of approaches.
SENTRY*
SENTRY is the first collaborative, worldwide, longitudinal antimicrobial surveillance program to provide timely data on both community- and hospital acquired infections with standard methodology. This project was launched in February 1997 in four regions; currently, there are 38 program sites in North America, 27 sites in 13 European countries, 10 sites in 7 South American countries, and 3 sites in Turkey. Japan, Australia, and countries in Asia and Africa are slated to join in 1998. Financial support for the activity has been provided by Bristol-Myers Squibb.
The program is the first to take up the recommendations of the 1995 ASM Task Force Report and study microbial epidemiology and antimicrobial resistance longitudinally and globally, utilizing reference quantitative methodology. The initial targets will be bacteria and antibacterial agents, with limited information on bloodstream isolates of fungi. The system will monitor both nosocomial and selected community-acquired infections through standardized quantitative methods, and will gather longitudinal data on resistance by both disease and pathogen site. Community-acquired respiratory tract pathogens; pneumonia in hospitalized patients; urinary tract infections; wound, skin and skin structure, and bloodstream infections will be monitored longitudinally for three to five years.
SENTRY aims to establish a worldwide, stable network of sentinel laboratories and to establish a very large library of well-characterized strains and data on drug usage as a basis for the analysis of use-related resistance or pathogen emergence. The program will collect data on the accuracy of locally used tests for susceptibility or identification, as well as outcome data on a specified therapeutic subset.
Molecular epidemiological tools are being used and information is being provided on a real-time basis to local institutions for possible epidemiological interventions. Currently, 55 antimicrobials are being monitored that incorporate a variety of drug-microorganism combinations. Laboratory-based education and training activity of research fellows is under way. Additionally, the principal investigators provide consultations for susceptibility testing methods, molecular techniques, epidemiology and infection control, and skills in medical writing for peer-reviewed publications.
The long-term durability and success of SENTRY and other resistance surveillance systems such as those described in Appendix A of this report will demand effective public- and private-sector partnerships, especially including the collabo-
rative formulation of constructive guidelines for a range of processes. There are many challenges facing the development of such partnerships: (1) defining the scope of the problem accurately; (2) designing systems that are geographically relevant; (3) identifying optimal partners to support surveillance, including members of the financial community and local, national, and international agencies; (4) implementing systems, monitoring output, and reevaluating needs on an ongoing basis; and (5) communicating timely data in the right forums.
Surveillance and the Laboratory*
Surveillance data come essentially from three sources: (1) active surveillance, (2) passive surveillance involving reference laboratories, and (3) outbreak investigations. Because antimicrobial resistance surveillance data are, and will continue to be, highly laboratory dependent, laboratory quality is crucial. Moreover, although the accuracy of data is always an issue in research, there are a number of reasons to be concerned about the accuracy of the surveillance data currently coming from laboratory sources. Concern is justified even when the data are from central laboratories; although the general presumption is that such facilities apply more rigorous quality control, proficiency testing and accrediting programs have found sizable proportions of test results from many central laboratories to be inaccurate.
Hospital laboratories, a major source of surveillance data, have numerous limitations. First, not all organisms are monitored and tested for resistance. For example, despite its intrinsic importance and the considerable media coverage of vancomycin resistance, many U.S. hospital-based laboratories continue to exclude from testing enterococcus isolates from urine and wound cultures, so that unknown numbers of vancomycin-resistant isolates are simply never recognized in these systems. In addition, hospitals are increasingly outsourcing testing to cut costs, often to out-of-state facilities, so that quality control is much reduced; whether these processes are subject to federal interstate commerce regulations is an open question.
Second, the testing methods employed may not be appropriate or correctly applied. In 1996, the American College of American Pathologists, a leading accrediting organization, sent out a strain of S. pneumoniae for proficiency testing. Thirty percent of the 2,100 participating laboratories were using inappropriate testing methods, and more than half of the laboratories that reported a minimum inhibitory concentration (MIC) result used a commercial product that the FDA had mandated be withdrawn from the market two years previously. Even laboratories perceived generally as producing accurate results may be testing and reporting results on antimicrobial agents that are not approved by the NCCLS for testing against specific organisms (i.e., testing staphylococci against third-
generation cephalosporin). In 1993, in a major proficiency-testing exercise, the CDC sent five enterococcal isolates of varying resistance to penicillin, ampicillin, and vancomycin to 92 laboratories in New Jersey; in 1995, the same survey was repeated in California (San Francisco), Minnesota, and Vermont. In all sites, there were significant problems in terms of the laboratory's ability to detect vancomycin resistance even in isolates with very high levels of resistance. Recent CDC testing of Staphylococcus isolates suggests that part of the problem may be poor performance of a widely used test for strains that have diminished susceptibility to vancomycin.
Third, many laboratories now use MIC panels that have only two or three dilutions of an antimicrobial agent, whereas in the past, seven or eight dilutions were tested. Since the quality control organisms used do not have values that are within the range of the test (i.e., scale values), it is difficult to know whether the test is working, which makes it even more difficult to assess trends or identify shifts in MICs.
Finally, many virulence-related factors in bacteria are expressed only in vivo. It is reasonable to ask to what extent this is true for resistance. Even the standardized methods for looking at phenotypic resistance are limited in the sense that they ignore the kinds of conditions that pathogens may actually be encountering and that may cause them to respond in ways undetectable with currently available tools. Lack of systematic data on treatment outcome further impairs understanding of in vitro-in vivo discrepancies.
There are few laboratories where testing cannot stand improvement and an alarming number where improvement is essential. One economical and straightforward source of improvement would be to revitalize and expand distribution of the updated NCCLS Guidelines, which no longer appear to be distributed regularly or as widely as needed. A related training issue has to do with the fact that many laboratories have replaced four-year degree medical technologists with less trained individuals, so that the gap between the bench and doctorate-level laboratory director has widened, a reality that has to be taken into explicit account in developing training programs.
Beyond their broader implications for surveillance, the accuracy, reliability, and consistency of laboratory data are qualities with immediate and vital clinical implications. In the absence of these data qualities, the eventual result is likely to be treatment failure, which—beyond its implications for mortality and morbidity in individuals—ultimately enhances antimicrobial resistance and reduces the therapeutic armamentarium for the population at large.
What Is Needed
There is presently no single global or national surveillance system for monitoring antibiotic resistance that answers to the ideal described at the beginning of this section. There are multiple surveillance activities scattered across the
globe that attempt in different ways and at different speeds to move toward the ideal. However, the cardinal features of these systems are that few have been longitudinal and as a group, they are almost totally uncoordinated and unstandardized (see Appendix A for a staff-compiled inventory of current surveillance activities that collect data on antimicrobial resistance). No country in the world today has a reliable, longitudinal, full-service antimicrobial resistance surveillance program with the comprehensive focus with the qualities outlined above. The result is that the magnitude and impact of the resistance problem are poorly understood. Redressing these deficits is crucial in global and national public health terms, and the most powerful case possible must be made for urgent and substantial response.
In 1995, an ASM Task Force recommended the immediate establishment of networks on a local, national, and global scale by the National Center for Infectious Diseases (NCID) at CDC and associated agencies, with the necessary fiscal support, for the surveillance of antimicrobial resistance in animals, humans, and food products. The system would monitor bacterial and fungal pathogens and representative populations concurrently; ensure the quality of participating laboratories; input, analyze, and make available data in simple, flexible, and timely fashion; and generally maximize potential for appropriate data-based interventions (ASM Task Force Recommendations for a national antimicrobial surveillance system are presented in Appendix B).
As straightforward as the ASM recommendations are, their elaboration and implementation, particularly when these have global dimensions, present formidable financial and political challenges that will require a level of coordination yet to be realized and a recognition that trade-offs and compromise are inevitable. No single system is likely to be able to perform the full range of necessary surveillance, so that harmonization of multiple systems and guidelines for the production of comparable data will be ongoing challenges. Establishment of universal breakpoints, standardization of quality control measures, open and timely access in friendly formats allowing specific queries, and development of widely relevant educational messages will all be part of the common task. Real partnerships will be essential as people and institutions with varying priorities try to achieve goals that may be similar in many ways but divergent in others.
System Design Issues
The design of surveillance systems raises many questions, including but not limited to the following:
-
Which pathogens and antimicrobial agents should be monitored? Where are the gaps in what is currently done?
-
What would be the most useful and cost-effective way to collect and analyze the critical body of data on antimicrobial usage?
-
Because the more data collected, the more expensive and complex the system becomes, what data are essential and what are the trade-offs?
-
What level of uncertainty is acceptable and what are the associated methodological issues?
-
What will be the further effects of the restructuring of health care delivery systems, downsizing, and cost containment on future surveillance capabilities, including the number of cultures that can be collected and the amount of susceptibility testing that can be performed?
-
How are multiple public and private systems to be coordinated? How can public health agencies interact with managed care companies? How much proprietary data on drug use and resistance will companies and health networks share with public health agencies?
-
How can the tensions among proprietary information, individual privacy, and community rights to protection from infection be resolved, especially when databases are to be merged, and what kinds of encryption systems have to be developed for shared use?
Funding for Surveillance
The ability to achieve these ideals, in the United States and globally, inevitably will be affected by fiscal considerations. In 1992, the most recent year for which aggregate figures are available, total investment in the United States, including federal, state, and local-level support, was $74.6 million, of which $42 million came from federal funds, $20.6 million from the states, and $12 million from the local level (see Table 2). However, when the $57.4 million in support dedicated to the surveillance of HIV/AIDS ($34.6 million), STDs ($13.2 million), and tuberculosis (TB) ($10 million) are subtracted from the total, only $16.8 million remained for the surveillance of all other infectious diseases. Of this amount, $55,455 from all sources was dedicated to dealing with antibacterial and antiviral drug resistance. These figures do not include private-sector investment in proprietary systems. 7
As for human resources, in 1992 there were 1,608 full-time public-sector professional positions involved in disease surveillance in the United States, of which 1,122, or 70 percent, were involved in the surveillance of HIV/AIDS, TB, and STDs. This left 486 positions to be apportioned throughout the 50 states and one territory for the surveillance of all other infectious diseases.
HOW CAN THE SCIENCES HELP?
Implications of Mapping the Genome*
The ability to sequence an entire genome to learn the arrangement of the nucleotide building blocks of the DNA that make up an organism, and the ability through bioinformatics8 to manage huge amounts of information, are the foundation for unlimited scientific advances in the development of new diagnostics and therapeutics for infectious disease. Gene sequencing has provided profound insights into the capacity of organisms to alter themselves, generated far greater comprehension of biochemical pathways and transport mechanisms, and permitted examination of areas possibly associated with virulence and new toxins.
It is clear that organisms have built-in mechanisms for evolution. During each replication, a type of coding slippage can occur that will cause certain genes to be expressed and others to be suppressed. This, in turn, can produce alterations in biosynthesis pathways and ultimately in cell surface antigens. Genomes also contain built-in splice mechanisms for changing adhesion molecules, the main mechanism for attachment to human cells, which can also produce alterations in antigens. As a consequence, each replication contains possibilities for change, a moment-to-moment capability to evolve so as to, for example, evade the human immune system or other potentially hostile forces. All this makes it surprising that antibiotic resistance is not actually an order of magnitude worse than it is.
Another benefit of these scientific breakthroughs is the ability to perform comparisons of specific genomes, which permits still more profound understanding of the evolution and transfer of genes. For instance, the sequencing of the present-day archeal genomes revealed that their ancestors may have been the source of many of our own (eukaryotic) genes. Their evolution occurred via processes that remain mysterious. Resolving such mysteries will eventually offer help in dealing with future problems in what may be very new ways.
Bioinformatics has been critical to managing the thousands of gene sequences now being identified by The Institute for Genomic Research (TIGR), whose intent is to sequence between 50 and 100 genomes over the next few years. The generally rapid growth in genetic information predicts that from 400,000 to 500,000 new genes will be sequenced within the decade, the majority of these coming from the microbial world. There is no dearth of potential targets, and the available database is already large, rich, and diverse. Knowing what is expressed in human disease and being able to detect and understand it will be extraordinarily useful but also very challenging, since even the 30,000 to 40,000 currently available new microbial genes will have to be narrowed down to the several dozen or so targets that are essential to the existence of a given pathogen and its ability to cause disease.
TIGR and other groups are also developing gene arrays that will allow for the simultaneous assessment of all genes within the genome, in turn permitting queries to be performed for different physiological conditions, as well as the assessment of variations in virulence and infectivity among different strains. Such arrays will, with almost immediate turnaround ( about 60 minutes), provide a wide range of information. This information would include identification of infectious organism(s) and the presence of genes associated with sensitivity or resistance to different antimicrobials; knowledge of gene content and function; and recognition of polymorphic shifts associated with changes in either resistance or infectivity, which will be of particular help to vaccine and drug manufacturers in selecting appropriate targets since the least appropriate genes are those that undergo high-frequency changes.
Table 2 Federal, State, and Local Support for Infectious Disease Surveillance by Disease Category, United States, 1992
|
|
Dollars |
|
|
|
|
|
Disease |
Federal |
State |
Local |
Total |
Percent |
|
AIDS/HIV |
25,794,280 |
7,478,557 |
1,317,359 |
34,590,196 |
46 |
|
TB |
4,085,098 |
2 ,987,606 |
2,884,901 |
9,957,605 |
13 |
|
STDs |
7,819,550 |
2,967,790 |
2,412,700 |
13,200,040 |
18 |
|
Vaccine preventable |
2,921,175 |
1,193,222 |
1,116,091 |
5,230,488 |
7 |
|
All other diseases |
1,535,059 |
5,830,516 |
4,193,480 |
11,559,055 |
15 |
|
Antibacterial/ antiviral drug resistance |
6,260 |
48,795 |
400 |
55,455 |
<1 |
|
Total |
42,161,422 |
20,506,486 |
11,924,931 |
74,592,839 |
100 |
|
NOTE: Includes data from 50 states and one territory. SOURCE: Osterholm MT, GS Birkhead, RA Meriwether. Impediments to Public Health Surveillance in the 1990s: The lack of resources and the need for priorities. Public Health Management Practice 2(4):11-15, 1996. |
|||||
Molecular Detection of Genes Associated with Antimicrobial Resistance *
Limitations of Phenotype-Based Detection Methods
Many of the standard methods for looking at antimicrobial resistance target a phenotype, which is essentially a behavioral characteristic. This is valuable as a means of measuring resistance per se, but considerable specificity and additional insights could be gained by looking at resistance from the genotypic level, since traditional phenotype-based detection methods are limited in several ways. Because resistance requires growth of an organism in pure culture and because some organisms are unculturable, fastidious, or slow or difficult to propagate at all, answers to clinically important questions about resistance may not be found in a timely fashion, that is, with enough speed to be of immediate clinical relevance. Furthermore, to understand phenotypic resistance in ways that are more broadly relevant, growth of the organism in question requires standardized conditions. Examining resistance from a behavioral perspective tells little or nothing about the mechanisms involved, nor does it provide information about just what conditions unique to the in vivo environment are essential for proper expression of a number of resistance determinants.
Knowledge About the Genetic Basis of Resistance
A fair amount is now known about the natural reservoir of resistant genes (i.e., about resistant gene pools) and about the fact that resistant genes are much more widely distributed than once thought, not only in human and related animal hosts but in the natural environment.
The reservoir of antimicrobial drug-resistant genes has been analyzed insufficiently. It is clear that genes have transferred not only between species within the same genus, but between genera and even across broader taxonomic boundaries. Furthermore, many of the antimicrobial-resistant genes in microbial pathogens are found in members of the generally beneficial commensal microflora, from which they may have been transferred. There remains, however, much to be learned about these transfer mechanisms, for example, about such complex phenomena as the transfer of genes between gram-positive and gram-negative bacteria, and from prokaryote to eukaryote; about self-transferable plasmids and conjugative transposons and resulting mosaic genes; and about the regulation of gene expression.
Advantages and Limitations of Genotype-Based Antimicrobial Resistance Detection
A major advantage of genotype-based resistance detection is that it does not generally require growth of the target organism. Direct detection of a gene in a clinical sample is now quite feasible, although most cases do involve prior DNA amplification. In many instances, either sequencing or the use of probes, in a filter-based method or solid-state high-density array, is available, and much can be automated. Because such methods are also rapid, in theory and often in practice their use can generate real cost savings to clinical management because the necessary information is available in a timely fashion so that chances for truly appropriate care would be substantially enhanced.
Finally, genotype-based detection targets the genetic basis of the fundamental mechanisms responsible for resistance, and disregards unrelated genes and nonspecific bacterial properties. Although simply finding a gene does not mean that it is necessarily expressed, in most cases, expression is either constitutive or induced under certain circumstances and selective pressures. An important corollary is that gene induction, and therefore expression of resistance, can be prevented by avoiding the stimulus for such induction (i.e., the antibiotic). Thus, by recognizing this genetic potential of an organism, one may be able to prevent the organism from realizing its potential.
Relying solely on genotype is not without problems. First, because there can be multiple and diverse genetic mechanisms for resistance to a single antimicrobial agent, scrutiny of a single mechanism does not mean that the screening has been comprehensive, as is the case with phenotype-based methods. Second, as discussed above, because genes are not always expressed, detection of a gene may or may not have clinical meaning in terms of expression potential and relevance. Third, present genotype-based methods are nonquantitative with respect to expression in enzymatic activity that can be measured as an informative endpoint. Finally, these methods are heavily technology intensive, which limits their accessibility and appropriateness for a number of settings, at least in their present stage of development. Thus, genotypic methods have to be accompanied by phenotypic analysis to provide a complete picture.
High-Density Oligonucleotide Arrays*
The emerging field of DNA diagnostics is being driven by a variety of new technologies for analysis of nucleic acids. Among these are mass spectrometry, surface (''chip") hybridization, and array-based technologies. Scientists at Affymetrix, a four-year-old biotechnology company in Santa Clara, California, have developed a DNA probe array that can trace mutations occurring in pathogens as
they develop resistance to antibiotics. The technology combines concepts familiar to both microbiologists and the computer industry, using photolithographic technology to synthesize allele-specific oligonucleotides, that is, short pieces of DNA, in a very high density on a dime-sized piece of glass. The arrays work on the principle of affinities of oligonucleotides for the complements they find (and hybridize or bond to) in the sequence specificities in whole genomes.
These arrays provide a platform for various experiments. They can be used to identify specific changes at the nucleotide level of a large segment of sequences (i.e., to genotype an organism) or to quantitatively measure changes in mRNA expression that may have significant biological implications. The arrays can also be used for genetic and physical mapping, covering whole genomes simultaneously.
The system has been used recently to investigate drug resistance and some of the phenotypic characteristics in M. tuberculosis and other Mycobacterium species. For example, the mutations that confer rifampicin resistance are confined to an 81-nucleotide segment that can easily be interrogated by this technology. Most of these mutations have been reported in the literature and can be identified by using DNA probe arrays. In addition, when the chip is exposed to a target genome that is not Mycobacterium so that hybridization is therefore incomplete, it will produce a highly informative, reproducible, species-specific pattern, or "fingerprint," that permits species identification. In other words, these arrays can provide species identification of the Mycobacterium at the same time they provide information on the nucleotide differences in specific sequences that confer drug resistance. This same strategy can be extended substantially to detect mutations in other clinically important genes simply by extending the number of genes on the array. Altogether, this reference comparison is a powerful tool for analyzing biologically important areas by measuring sequence differences.
These arrays can also be used to help understand the mRNA expression patterns that are exemplified by both the pathogen and the host. Experiments can be conducted at different hybridization and analysis stringencies and then categorized according to the amount of change in each gene—twofold, fivefold, tenfold, and so on. Categorizing data in this fashion provides a systematic analysis of expression and allows orderly interrogation as to what is happening along genetic pathways during the course of infection or transformation. For example, understanding which genes are increasing or decreasing their mRNA expression levels will allow for the identification of new functions and interrelationships among the 70,000 to 100,000 human genes. It will also identify which genes are essential for infection.
The challenges that lie ahead are to better comprehend the dynamic between the interactions of the host and human genome complement and how they interrelate to one another, which in turn will provide better insight into the nature and course of infections and, ultimately, more effective interventions.
Applications of Genomics and Bioinformatics to the Development of Anti-Infectives*
All the new technology that is increasingly becoming available does not change the fundamental steps required for target evaluation and screening. A medical need still must be identified, DNA sequences must be obtained, targets must be selected and a determination made as to their essentiality, and an assay must be developed. The significant difference is that target selection is no longer what it was even two to three years ago—a daunting, serial, one-by-one process that had to be based on known or newly established function. Now, all the potential available targets can be evaluated more or less simultaneously and target selection is determined by relationships among genomes. At least nine biopharmaceutical companies are currently using genomic techniques to develop new antibiotic targets.
Once the medical need is defined—for example, the ability to deal with antibiotic-resistant gram-positive (and, increasingly, gram-negative) bacteria—whole or partial sequences are obtained from a variety of public and private sources, including TIGR, the Internet, and companies selling proprietary data.
Bioinformatic techniques may then be used to select potential targets. These may be broad or narrow in spectrum, or they may be organism specific. In all cases, selection entails the application of a set of selection criteria and a process of comparison. For instance, S. aureus might be compared to mammalian, yeast, or other bacterial databases to identify which genes are shared and are therefore not specific to this organism, leaving genes that are specific or unique as the initial focus of interest. More specifically, S. aureus tRNA synthetases might be compared to different databases to find those synthetases that produce low or no homology and are therefore more likely to be good targets. Another approach would be to seek analogues of known proteins, for example, a family of gram-positive anchoring proteins with similar conserved motifs that might constitute potential targets.
The next step is to determine whether the targets selected are essential for the growth of the organism under various condition(s) of interest (e.g., minimal to rich media; different pH or temperature; or in vivo conditions). Determinations of essentiality can be made by using gene knockouts, by employing genomic footprinting methods, or by making temperature-sensitive mutants, the latter two methods being relatively rapid.
The penultimate step is the development of assays. These may be cell-free "genetic" assays based on phenotype, enzymatic assays, or binding assays. Because, even after selection and determination of essentiality, the number of potential targets will be quite large, companies are most interested in high throughput methods that will be able to simultaneously assess a number of targets, perhaps 10 to 50 in parallel. New technologies are still in development; of these, one of the most promising may be binding assays using mass spectrometry.
The final and perhaps most important step in target evaluation is screening for inhibitors of the gene targets. Because finding such inhibitors requires screening anywhere from hundreds of thousands to millions of compound samples, the necessary assays must be very robust so as to be compatible with today's high-throughput robotics and liquid handling systems. Fortunately, the very large natural product, compound, and combinatorial libraries required for this phase already exist and continue to grow. One of the interesting possibilities is looking for inhibitors of genomic sequences with no currently known function. Assay systems that provide better understanding of these targets will be truly revolutionary.
A Role for New Therapeutic Approaches in Combating Antimicrobial Resistance*
In addition to the pathways that lead microbes to change and develop resistance, there are pathways in the human host that are relevant to the development of resistance. One of the most important of these is the epithelial surface. As the primary site of disease entry, infection, and pathogen replication, this extensive system is a potentially key locus of selection for antibiotic resistance and, furthermore, may be responsible for maintaining resistant alleles and transmitting them in the general population. This raises the question of whether there is a role for targeting preventive and therapeutic interventions at epithelial surfaces so as to evade or forestall problems of antimicrobial resistance.
The principal large surface areas of epithelial tissue in the human body that are responsible for accepting and transmitting infectious disease agents are the gastrointestinal, oral-nasal-pulmonary, and genitourinary tracts and the conjunctiva of the eye. These body systems are lined with epithelial cells whose principal commonality is that they are protected by a robust immune system consisting of both a cellular immune response and an antibody-based (humoral) immune response. Adult human beings secrete about three grams of secretory antibodies (SIgA) daily across these tissues for protection from infection. In fact, prior to the advent of penicillin, there was a developing and fairly robust business in the application of antibody-based therapies to many of the infectious diseases for which antibiotics
came to be used as a general matter. However, no way has been found to stimulate the human immune system to produce secretory antibodies in quantities large enough for protection (e.g., mucosal vaccines), nor has it been possible to produce secretory antibodies for prophylactic or topical use.
There are, however, new possibilities for changing this picture. One is a process, developed at EPIcyte, by which secretory antibodies can be made in, literally, agricultural quantities by cloning the genes of the required proteins and expressing them in a single plant cell. The individual protein molecules produced in the plant are then directed to its endoplasmic reticulum, the organelle responsible for protein replication. Because plants have the ability to adapt their processes so as to secrete and store these proteins, they can replicate and produce the antibodies in bulk.
Another stratagem being developed by several biotechnology companies, including EPIcyte, is the production of second-generation molecules that would, in effect, coopt the polyimmunoglobulin receptor pathway from the "inside," or systemic subepithelial side of the mammalian pathway for SIgA production, to the "outside" in order to present molecules that are therapeutically targeted at the epithelial surface. This strategy may be of particular interest because it could provide a broader therapeutic window for molecules that now have a quite narrow one, such as the aminoglycosides and a number of other antibiotics whose efficacy may be waning. Another dimension of this line of research is the development of a class of molecules called "immunobiotics," modified SIgA's that will have an inherent capacity for transport across the epithelium for presentation on the epithelial surface, which is the site of the seed population for the distribution of antibiotic resistance.9
These molecules would then be used for different approaches to epithelial intervention, each of which will present developmental challenges: systemic, topical, and parenteral microbicides; vaccines to stimulate mucosal immunity, including attenuated live vaccines that would actually colonize the epithelia, as well as nonreplicating vaccines that would provide for vaccination directly on the epithelial surface; and "immunobiotics" targeted to epithelial tissues from the inside out.
Applications of Field Surveillance in the United States and Globally *
Surveillance for antibiotic resistance is dependent upon surveillance for infection, yet current global, national, and local systems are clearly inadequate for the detection of most infections and therefore woefully inadequate for tracking resistance in any coordinated way. Sexually transmitted disease is a useful example. A major study by WHO found that in 1995, among adults 15-49 worldwide, there were 333 million new cases of the four most common curable
STDs: Chlamydia, gonorrhea, syphilis, and trichomoniasis. North America accounted for 14 million of these cases, the overwhelming majority of them in the United States.
The greatest burden of sexually transmitted disease is borne by the women of the developing world, for whom these infections are the second largest contributor to death and disability, surpassed only by the rather large category of maternal causes. Among Western industrialized countries, the United States bears the greatest STD burden, and in 1994, the country spent almost $10 billion on treating the most common of these infections and their associated sequelae; this figure does not include the costs of HIV/AIDS. The $10 billion figure is 43 times higher than 1994 national expenditures on STD prevention ($231 million) and 94 times the amount spent on biomedical and clinical research ($105.4 million). Because of the fragmented character of health care in the United States and because STD surveillance is a somewhat disjointed and completely passive system, calculating STD surveillance expenditures is almost impossible, but the general perception is that funding allocations for STD surveillance are highly disproportionate to what has been described recently as an epidemic.10
One of the most significant problems in the surveillance of STDs is that it has depended on detection of individuals who are symptomatic, and such surveillance has typically required invasive diagnostic procedures, which many individuals try to avoid, even though a minimum of 50 percent of such infections are asymptomatic. However, the biotechnology advances of the last few years allow for new ways to track these diseases, and molecular techniques have been used locally, nationally, and globally, although their costs remain too high for universal application. There are now a number of nucleic acid amplification assays, importantly including polymerase chain reaction (PCR), ligase chain reaction (LCR), and transcription-medicated amplification (TMA) for detecting Chlamydia trachomatis, N. gonorrhoeae, and M. tuberculosis 11, as well as several multiplex assays, one of which detects herpes, syphilis, and chancroid from any genital ulcer and another that allows for detection of bacterial vaginosis, Chlamydia, gonorrhea, human papillomavirus, and trichomoniasis. These assays have much higher sensitivity and specificity than standard culture tests, can screen for both infection and resistant genes, and are cost-effective across a wide prevalence of infections and with different screening criteria. They are also much more versatile, noninvasive, and consumer sensitive; samples, which may be genital or ocular swabs or urine, are easy to collect; transport and storage requirements are simple; and results can be generated relatively rapidly (8-24 hours) since processing is semiautomated. Thus, these methods can be used in large populations to screen asymptomatic as well as symptomatic men and women, so that sampling bias can be avoided.
Such surveillance has been carried out in several local and national venues, in the United States and elsewhere. Beginning in 1994, PCR assays have been used, with treatment and intervention, in all STD and family planning clinics in Maryland's Region 3, which includes Baltimore, for routine screening of all
women presenting at these clinics. When the program began, chlamydia and gonorrhea prevalence rates were 13.6 and 25 percent, respectively; by mid-1997, they had fallen to 9 percent each. The program was later extended to high-school and middle-school students, the age cohorts at greatest risk of STD infection. Using PCR and LCR testing of urine samples collected from students presenting at the health clinic, the prevalence of Chlamydia infection was found to be 17.4 percent in females and 16.0 percent in males; gonorrhea rates were 4.7 and 5.7 percent, respectively. Those testing positive were given a full examination, confirmatory cultures were obtained, and treatment was provided.12
Another U.S. national project, funded by the U.S. Army, is screening all female military recruits for Chlamydia, gonorrhea, human papillomavirus, and Trichomonas, and mapping the distribution of these infections by region, state, and city. Of 8,262 new recruits, 9 percent were already infected with Chlamydia, and the highest percentage of those testing positive was under age 20. The highest infection rates were found in recruits coming from the southeast and northeast regions of the country. In the rural Rakhai District of Uganda, urine samples were collected from 10,000 individuals over an 18-month period and screened for Chlamydia and gonorrhea. Distributions of infection were found to be very similar to those in the United States: a chlamydia peak in very young teenage girls, a later peak in boys, and a falloff with increasing age. A subsequent intervention program dropped rates of both STDs by 80 percent.
The techniques used to test the samples gathered through such screening activities can also be used to assess resistance, an approach already in practice for HIV. The amplified DNA used to assess viral load in HIV-infected individuals in developed countries is probed for known resistant genes in the reverse transcriptase (RT) genome or the polymerase gene. These genes are well known since they were well mapped in the course of developing AZT (zidovudine), DDI (didanosine), D4T (stavudine), DDC (zalcitabine), and 3TC (lamivudine), the primary RT inhibitors, and later the polymerase inhibitors. The Line Immunoprobe Assay (LIPA), a new oligonucleotide probe assay, takes these mutations and wild-type genes and adheres them to a nitrocellulose strip similar to a Western blot assay. Following nested PCR of the RT and polymerase genes, the amplified products are hybridized to the oligonucleotide probes on the strip. Repeated assays over time then track the development of resistance to a given inhibitor (RT or polymerase) so that a switch can be made to an appropriate drug to which the resistant gene may not be present. The method is simple, easy, rapid (2 hours), and relatively inexpensive because it uses material already gathered and because the DNA has already been amplified for purposes of viral load testing. Such assays are critical in at least one respect. Although antivirals suppress replication, they also create selective pressure for resistant mutations; optimal HIV therapy uses multiple antiretrovirals (1) to reduce replication below the threshold for producing resistant mutants and (2) to impose the need for multiple mutations to achieve resistance.
What Is Needed
In sum, genomics sequencing and bioinformatics have opened up unparalleled opportunities in antimicrobial research, allowing the identification and selection of new gene targets based on specific medical needs. Because the functions of a very large proportion of targets remain unknown, the really new breakthroughs will come from developing much greater ability to find the inhibitors of such proteins. This will be an exact reversal of the history of antibiotic development to date: a purely empirical process of discovering new functions by finding out what a given antibiotic inhibits, a slow process that has thus far produced only about 15 or so classes of successful drugs.
Because resistance is so complex and dynamic at the genetic level, much more work is needed to understand the diversity and prevalence of resistant gene families, in nature and in the animal microflora that are the bridge to human contact, and to discern the origins of these genes and how they spread from one organism to another. Going beyond phenotype to tracking and attempting to comprehend gene flow can lay the foundation for anticipating problems that may arise with new agents and consequent interventions.
There are two major paths to enlarging these understandings. One would be development of a set of both broad-range and specific primers and probes for the known resistant gene families and, simultaneously, broad-range primers for targeting the unknown members of these families. Two potentially useful technologies are high-density arrays capable of assessing the presence of a wide variety of genes, known and unknown, and microarrays that display the entire genome of a particular organism to permit examination of the responses induced when the organism is grown in the presence of an antibiotic. It is important to remember that until more is known about how to correlate genetic sequence with behavior (i.e., with phenotype), phenotypic information will continue to be essential, both to basic knowledge and to tailoring response. For the foreseeable future, the two approaches must be seen as complementary. In this connection, one of the most compelling pieces of work will be to look at the interaction between pathogen and host in terms of the expression patterns and changes in gene levels that go on during the infection process, and the triggers and choke points at play in this dynamic.
In practical and immediate terms, some of the currently available molecular methods are clearly applicable only to research and reference laboratories; at present, their feasibility for most commercial or clinical laboratories is far more limited. One plausible strategy might be to select a set of sentinel hospitals that could serve as bases for global assessment of the prevalence and transmission of a few antibiotic-resistant genes, in other words, sites for the monitoring of gene flow and assessment of genetic diversity. This would surely require strengthening of the laboratories concerned and emphasis on effective data management to ensure that the proper responses occur in a timely manner. Also essential would
be the design of categories and pathways for reducing data sets into comprehensive packages for use by both clinicians and researchers.
SOURCES OF RESISTANCE AND ANTIBIOTIC USE*
Sources of Resistance
There are two primary factors in resistance emergence: the antibiotic as a selective agent in a particular environment and the resistant gene as the vehicle of resistance. These basic elements constitute what is, in effect, a "drug resistance equation." The ebb and flow of either component of the equation affects the magnitude of what may, or may not, become a resistance problem. If both elements of the equation are kept in check, the fact that drug-resistant organisms exist does not mean that they necessarily constitute a public health problem. If the two are not kept in check, drug-resistant traits will be both selected and propagated, so that a given environment will become rapidly populated with different kinds of resistant bacterial flora.
The variables on the antibiotic side of the equation are the amount of antibiotic used, the number of individuals in which it is used, and the geographic extent of its influence. The dynamics among these may translate into a level of density in which the natural microbial ecology is disrupted by selective pressure against bacterial strains that are susceptible to a given antibiotic and for strains that are resistant to it.
On the genetic side of the equation is the resistant gene and the factors that increase its transmission. Resistance traits will spread from cell to cell because bacteria have evolved the ability to exchange genes through a variety of mechanisms, not just to other members of their own genus and species but to other organisms in other genera. As these elements interact against the background of continual exchange of microbes among human, animal, and agricultural hosts, resistance is converted into a clinical and public health problem.
These dynamics are almost inevitably cumulative. The fact that an organism has become resistant to one antibiotic seems to help it become resistant to others. N. gonorrhoeae first became resistant to penicillin, then to tetracycline, then to the new fluoroquinolones, and treatment is now dependent on the cephalosporins. MRSA followed upon MSSA (methicillin-susceptible Staphylococcus aureus) as the acronym for the methicillin-resistant strain; this organism very rapidly accumulated resistance to other drugs as well, so that "MRSA" now denotes "multidrug-resistant S. aureus." The group A b-hemolytic streptococci are already resistant to the macrolides and tetracycline. Use of second- and third generation cephalosporins in hospital settings to treat resistant gram-negative bacteria selected for normally harmless enterococci, which were intrinsically
resistant to these antibiotics and so became prominent members of hospital acquired flora with their own multiplicity of resistances, most importantly to aminoglycosides and vancomycin. Now, vancomycin-resistant genes in enterococcus have appeared in Lactococcus lactis and Streptococcus bovis, and can be transmitted experimentally to S. aureus in the laboratory, a surprising and worrisome event.
Another critical aspect of resistance is that it begins essentially as a local matter. Although there are antibiotic-resistant bacteria everywhere, the problems they produce will differ, simply because the dimensions of the elements of the drug resistance equation differ. Vancomycin-resistant enterococci are a problem in the United States and Japan, but not in Europe. Cotrimoxazole-resistant pneumococci can be as high as 80 percent in Pakistan but just a few percent in Belfast, Ireland, or Memphis, Tennessee.
Resistance may manifest as an even more narrowly local phenomenon—in a community or hospital, or on a farm. The dynamics in each of these environments or ecosystems will vary and produce varying effects. In the United States, for example, resistance in pneumococci, gonococci, group A streptococci, and Escherichia coli arises primarily from antibiotic use in the community, whereas staphylococci and enterococci are developing resistance in hospital settings, and Salmonella and Campylobacter are becoming resistant on the farm. Yet even these patterns are not inevitable: In many parts of the developing world, drug-resistant Salmonella are a nosocomial rather than a farm problem.
At the same time, there is evidence that antibiotic resistance can be reversed or attenuated by reductions in antibiotic use. This possibility suggests the need for innovative ways of accomplishing such reversal, namely by restoring susceptible flora and thus extending the useful life of existing antibiotics.
Antibiotic Use*
The fundamental questions for addressing antimicrobial misuse and overuse are the following:
-
Does use impact resistance?
-
Is unnecessary use common?
-
Why does unnecessary use occur?
-
How can appropriate use be encouraged and inappropriate use discouraged?
Antibiotic use is patently widespread in the community, in the hospital, and on the farm. However, present knowledge of its magnitude depends largely on estimation and extrapolation; much better data gathering and analysis are clearly required. In the United States, an estimated 190 million daily defined doses of
antibiotics are prescribed in hospitals annually. Of the 145 million courses of therapy prescribed annually in community settings, 110 million courses are for outpatient use and 35 million for emergency department use. Of the approximately 50 million pounds of antibiotics produced in the United States annually, about half is used in animals for therapeutic purposes, disease prophylaxis, and growth promotion. Each year, an estimated 147 pounds of antibiotic are used per acre of farmed salmon and 40,000 to 50,000 pounds of antibiotic are sprayed on fruit trees for control of bacterial infections.13
Furthermore, not only are antibiotics used in all of these ecosystems, they have been overused. On-farm overuse seems to run from about 40 percent to as high as 80 percent, and physician use for community-acquired infections from 20 to 50 percent. Antibiotic overuse in hospital settings appears to range from 25 to 45 percent. Table 3 presents estimates of use in these different settings, the amount of use that is probably unnecessary, and the pathogens known to have developed antibiotic resistance in these environments.
The Hospital
The picture of the evolution of drug resistance in hospital settings clearly illustrates the fluidity of the resistance phenomenon as a whole. The dominant trend in hospital restructuring has been toward a decrease in overall size and total occupancy days, along with expansion in number of intensive care beds, so that many hospitals increasingly act as large intensive care units (ICUs). A significant correlate of this trend has been a shift in resistance patterns in the major categories of nosocomial pathogens, of which six are now multidrug-resistant: Acinetobacter, Enterobacter, Klebsiellae, Pseudomonas, MRSA, and VRE. Drug resistance of these pathogens is now more common in ICUs than in inpatient wards, and more common in inpatient wards than in the corresponding outpatient settings. It is hard to resist the conclusion that this correlates with the fact that in general, patients in ICUs receive an antibiotic on about 70 percent of ICU days; for inpatient wards, the figure is around 40 percent. These changes are not confined to the most vulnerable populations within hospital walls. As the resistant pathogens increase in number and the antibiotic armamentarium shrinks, transmission to the larger population will also increase and, in many cases, become a problem for the general public.
Table 3 Estimated Annual Antimicrobial Use in Humans and Animals in the United States, and Resistant Pathogens
|
Use |
Site of Use |
Uses |
Amount |
Use Estimated as Unnecessary |
Resistant Pathogens |
|
Humans |
Hospital |
Therapeutic, prophylactic |
~190 million annually defined daily doses |
25-45% |
Staphylococci, enterococci. gram-negative rods |
|
|
Community |
Therapeutic, prophylactic |
~145 million courses (110 million outpatient, 35 million emergency room) |
20-50% |
Pneumococci. gonococci. Group A streptococci. Escherichia coli. Mycobacterium tuberculosis |
|
Animals |
Farm |
Therapeutic |
~ 4 million pounds |
40-80% |
Salmonella Campylobacter |
|
|
|
Prophylactic/ growth promotion* |
~16 million pounds |
|
|
|
* ''Growth promotion" is a little-understood effect from incorporating low levels (200 grams or less per ton) of antibiotics in feed. How such levels affect growth is not clear: they may ward off undetectable, but consequential, minor infections, or they may have other effects. Growth promotion overlaps considerably with "disease prevention," that is, prophylactic actions taken to stave off spread of a disease. Both uses involve subtherapeutic doses, and decisions about these uses are almost always made by the producer acting alone (i.e., without the involvement of a veterinarian). The net effect is a great deal of what would be considered, in human medical practice, off-label use. SOURCE: U.S. Congress, Office of Technology Assessment. Impacts of Antibiotic-Resistant Bacteria, OTA-H-629. Washington, D.C., September 1995. Presented by Mitchell L. Cohen, Centers for Disease Control and Prevention, 1997. |
|||||
The Community
A major phenomenon in U.S. communities has been the dramatic rise over the past two decades in annual visit rates to office-based physicians for middle ear (otitis media) infections and in resistance to the antibiotics used for their treatment, leaving physicians a dwindling number of treatment options.14 In 1992, 23.6 million courses of outpatient antimicrobial therapy were prescribed in the United States for otitis media; another 17.9 million courses were prescribed for upper respiratory infections, 16.3 million for bronchitis, 13.1 million for pharyngitis, and 12.9 for sinusitis (see Figure 2). This is true even though the large majority of these infections are of viral etiology and therefore not responsive to antibiotics. The result has been a circular dynamic of tremendous selective pressure on the responsible organisms and the emergence of both intermediate- and high-level penicillin resistance, in an environment in which day care facilities have played a much increased role. Research in Iceland has found that there is a clear causal relationship between the rapid increase in penicillin- and multidrug-resistant pneumococci and antimicrobial use in community settings, a pattern that may well be typical in many other countries. 15
Figure 2. Outpatient antimicrobial therapy, United States, 1992. SOURCE: National Center for Infectious Diseases, Centers for Disease Control and Prevention. Adapted from McCaig LF and JM Hughes. Trends in antimicrobial drug prescribing among office based physicians in the United States. Journal of the American Medical Association 273(3):214-219, 1995.
The question was raised as to whether the general public might not also be contributing to imbalance in the antimicrobial resistance equation, unknowingly and with the best intentions. Industry has noted heightened public concern about infectious disease and has responded with cutting boards, mattress pads, socks, compounds that supermarkets can sell to spray on vegetables, and a proliferation of
lotions and detergents for hands, dishes, clothing, and babies—all of which incorporate, and advertise, antibacterial properties.16 The popular belief is that these products sterilize their environments, when it is known that some of them actually select for multiple antibiotic-resistant bacteria. What could be happening is that resistant microbial flora are being created in a different form so that one group of organisms is being replaced with another, with corresponding alterations in the home environment. Hospital patients, who are now being discharged from hospitals earlier than used to be the case, and are provided with drugs hitherto used primarily in hospital settings, are reentering home environments whose ecology may be in flux, with unknown consequences. Although lack of data makes these scenarios hypothetical at this point, there would seem to be little argument about the need for a more balanced public view of infection, the existence and benefits of susceptible microbial flora, and the simple virtues and large payoffs of good soap-and-water hygiene—most particularly and urgently—hand washing.
The Farm
The question of whether there is a causal relationship between antibiotic use in food production and development of antimicrobial resistance in human beings is complex, controversial, and hampered by many data limitations. It is also not new, having surfaced off and on for almost three decades. The issue has received fresh impetus, however, as a by-product of the generally heightened consumer concern about food safety and, in the scientific and policy communities, because of new, quite specific concerns. One such concern is the possibility of cross-resistance in Europe related to the use of avoparcin.17 The other has to do with DT104.*
Originally isolated in England in 1984 from a human specimen, Salmonella typhimurium DT (definitive type) 104 remained very rare until about 1990, when there was a rapid upsurge in isolation rates in humans and the first isolation in livestock. In 1993, the appearance of an epidemic strain (R-type ACSSuT) was confirmed with chromosomally encoded multiple drug resistance to ampicillin, chloramphenicol, streptomycin, sulfonamides, and tetracycline. Because these organisms were nevertheless susceptible to fluoroquinolone, veterinary use of fluoroquinolone for therapeutic purposes was approved in England in 1993, although subtherapeutic usage of human antimicrobial drugs has been prohibited there since 1970. Since then, however, DT104 has been isolated in a variety of domestic and feral animals, in England and the United States, and there has been a rapid rise in the proportion of Salmonella infections in the United States due to DT104, including two outbreaks. There also has been an apparent increase in the United Kingdom in the proportion of fluoroquinolone-resistant organisms, leaving only the extended-spectrum cephalosporins to treat this pathogen (Figure 3).
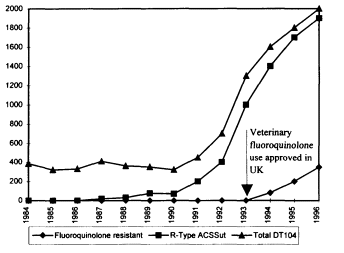
Figure 3. Antibiotic resistance among human Salmonella typhimurium DT104 isolates in the United Kingdom. NOTE: R-type ACSSut means that this strain of Salmonella is resistant to the antibiotics ampicillin, chloramphenicol, streptomycin, sulfonamides, and tetracycline at the concentrations commonly used for therapy of animal and human infections. SOURCE: Threlfall EJ Communicable Disease Report 6:155-159, 1996.
One hypothesis for the development of DT104 disease in human hosts is that the strongest risk factor is receiving an antibiotic for some other purpose prior to the onset of illness; this produces displacement of normal flora sensitive to the antibiotic, which in turn allows the overgrowth of the DT104 organism. Newly purchased farm animals have recently been found to constitute the principal point of entry of the pathogen into the farm ecosystem, where a plethora of other variables contribute to harboring and eventually transmitting it.
Factors in Inappropriate Use of Antibiotics
Critical factors in antimicrobial use and misuse are social, cultural, and economic influences that apply their own selective pressures on the development of resistance. Focus group research with pediatricians and parents of pediatric patients has cast light on some of these and revealed some paradoxes.18 Pediatricians all acknowledged that they overprescribed antibiotics and indicated that, all things being equal, they could reduce use from 20 to 50 percent. However, all things are not equal and there are substantial countervailing pressures on physicians, most significantly parental expectations and physicians' own uncertainties when faced with a sick patient or an anxious parent, without timely test results to undergird the process of diagnosis and without full clarity about best practice.
As for parents, although some voiced concerns about resistance and some were affected by sometimes contradictory restrictions placed by day care centers on childrens' attendance, most believed that decisions about the need for antibiotic use should be made by the physician. Thus, physicians feel pressured by parents to prescribe antibiotics, at the same time parents believe that such decisions ought to be made by physicians.
However, parents put a condition on their assignment of decision-making powers to the physician. They are willing to accept a physician's decision if it is accompanied by adequate communication: 86 percent of the parents in these focus groups would accede to not receiving an antibiotic if the physician took the time to explain why it was not necessary and to answer their questions.
At the same time, such perceptions would not protect a physician from liability if nonuse of an antibiotic proved to have been a poor clinical determination that resulted in excessive morbidity or in mortality. Still, establishment of a standard of care embracing the prudent use of antibiotics to prevent overprescription could serve as a defensive maneuver on the part of physicians. The matter of liability relative to antibiotic use and nonuse was noted as a nontrivial issue that remains to be explored.19
Table 4 summarizes these factors, adds to them, and includes two other sources of pressure: factors associated with the pharmaceutical industry and, of growing importance, factors deriving from the de facto restructuring of the U.S. health care system and the transcendent role of managed care in health care delivery and medical practice.
Strategies for Judicious Antibiotic Use
In many crucial respects, a major contributor to many of the factors listed in Table 4 is inadequate knowledge. This implies that there has been inadequate education—of physicians and patients, of veterinarians and farmers, and of decisionmakers in managed care organizations—about what antibiotics are, what they can do, when they are indicated, and the short- and long-term risks and costs of inappropriate antibiotic use. Some educational initiatives have been undertaken that offer examples for possible replication or expansion, for instance:
-
The Alliance for the Prudent Use of Antibiotics (APUA), established in 1981, has 15 national chapters and membership in more than 90 countries. It serves as a network for information exchange, provides support for country-based initiatives to track and curb antibiotic use and resistance at the local level, and educates providers and consumers about more prudent antibiotic use through a newsletter, gratis or low-cost distribution of key publications, and Internet links to other resources.20
Table 4 Factors Responsible for Inappropriate Antibiotic Use
|
Patient-Parent Factors |
Physician-Provider Factors |
Managed Care Factors |
Industry Factors |
|
Anxiety |
Real or perceived patient- parent pressure |
Cost-saving pressures to substitute therapy for diagnostic tests |
Misleading or erroneous advertising |
|
Misconceptions about: • what antimicrobials do • fever requiring antibiotics |
Economic concern for self (e.g., loss of clientele) and patients (missing work) |
Productivity incentives, increased patient load |
Promotion by retailers |
|
Belief in healing power of physician |
Litigation concerns |
Reduced appointment time per patient, less explanation time |
|
|
Return-to-work needs |
Physician fallibility: • inadequate knowledge • cognitive dissonance (i.e., knowledge but failure to act on it) |
Monitoring of rates of return visits to obtain prescription for antibiotic |
|
|
Day-care requirements |
|||
|
|
Physician self-esteem |
Responsiveness to patient complaints about "inadequate antibiotic use"; patient satisfaction surveys on which salaries are based |
|
|
|
Changing regulations in connection with off-label use |
|
-
The Fogarty International Center of the National Institutes of Health sponsored a study in 1987 that resulted in a report entitled "Antibiotic Use and Resistance Worldwide," which has been acknowledged as one of the initial attempts to bring recognition to these issues. 21
-
The Ontario Anti-Infective Review Panel in Toronto, Canada, an independent body composed of family physicians, specialists, and pharmacists, prepared Anti-Infective Guidelines for Community-Acquired Infections/1997 to help physicians make clinical decisions. Now in its second edition, the publication is funded by the Ontario Ministry of Health. Also in Canada, the Laboratory Center for Disease Control and the Canadian Infectious Disease Society have mobilized activity toward development of an integrated national plan for controlling antimicrobial resistance.
-
In 1994, a task force formed under the auspices of the Spanish Ministry of Health produced Antibiotic Resistance in Spain: What Can Be Done? which describes the problem and provides general recommendations for action.
-
The ASM, CDC, American Academy of Pediatrics, and American Academy of Family Practice have developed educational pamphlets and posters for patient education about antibiotic resistance and appropriate use.
-
Kaiser Permanente, Southern California Region, has a 24-hour pediatric and infectious disease support service to answer questions about the use of antimicrobials and provides feedback from its central laboratory to each medical center on local resistance patterns for E. coli, Klebsiella, Shigella spp., Salmonella, and other organisms.
-
Harvard Pilgrim Health Care in Boston has instituted audits of individual antibiotic-prescribing practices and outcomes, which are compared with outcomes of practices according to protocol; this information then flows into a collegial peer review and feedback process.
-
In Iceland, efforts at public and medical education have succeeded in changing parental attitudes, so that parents are less likely to ask a physician to prescribe an antibiotic and far more likely to ask if one is truly necessary. The effects of these shifts on outcomes are being studied.
-
Coverage of the resistance problem by the popular media has been expanding, perhaps most effectively in magazines aimed at the family market.
-
Increasing attention is being addressed to shortening courses of therapy or lowering dosages, partly motivated by the fact that in Japan and Latin America, antibiotic dosages in community practice are considerably lower than is normative in the United States, with apparently successful outcomes. A possibly informative model for purposes of reference is shorter-term application of antifungals in treating vaginal Candida infections. Nevertheless, since there is also evidence that subtherapeutic doses are more likely to select for resistance, research will obviously be required, as well as changes to labeling.22
See Table 5 for a summary of possible approaches toward remedying the factors that lead to inappropriate use of antibiotics.
Table 5 Potential Approaches to Modifying Behaviors and Policies in Connection with Antibiotic Resistance
|
Patient-Parent Behaviors |
Physician-Provider Behaviors |
Managed Care Policies |
Industry Policies |
|
Education • M.D. explanation of prescriptions to patients in terms of implications of unnecessary use • educational materials at point of care, pharmacies • information to popular media for public education (e.g., importance of hand washing) Economic incentives and disincentives (e.g., lower insurance fees for waiving drug coverage, medication copayments, formulary controls) Knowledge-based day-care policies (e.g., no requirement of antibiotic therapy for readmission) |
Alter patient expectations judicious antibiotic use Education • use of opinion leaders to educate providers • information support systems • feedback to clinicians on local resistance trends • journals, conferences, symposia • emphasis on critical role of hand washing Develop clinical practice protocols • literature-based guidelines • local review and consensus integrated into guideline development Analyze practice patterns • audits of antibiotic use vs. protocol and vs. peers • outcome analysis • peer feedback, discussion of outcomes |
Demonstrate economic advantages of judicious antibiotic abuse Provide guidance about proper antibiotic use via HEDIS and NCQA* Provide industry access to • local or regional infectious disease consultants • explanations of clinical practice protocols • feedback from patients, parents. providers regarding new practices and outcomes |
Demonstrate economic advantage (e.g., longer duration of efficacy) Promote responsible marketing and counter irresponsible marketing (opinion leaders) Facilitate antibiotic R&D • tax incentives • extended patent life for antibiotics |
RESISTANCE AND FOOD PRODUCTION: ISSUES AND NEEDS
The objectives of a single session of a Forum workshop on this large topic were necessarily modest.23 Its purpose was to ensure that the possibility of resistance associated with food production was explicitly incorporated into the workshop emphases on surveillance, diagnosis, and response. Greater attention has been focused recently on this issue, and deeper understanding is evolving.24 This session focused on S. typhrmurium DT104 as an example of a specific reason for concern and on the more general concerns of selected entities for whom some aspect of antibiotic resistance is a current or potential issue.
Concerns and Perspectives from Producers*
The National Pork Producers' Council (NPPC) represents the fourth largest agricultural sector in the United States, generating an estimated $66 billion in economic activity yearly and employing more than 764,000 people. Antibiotic resistance raises concerns for pork producers on two levels: as consumers concerned about their own health and as producers whose livelihood depends on the ability to use antimicrobials. Thus, the NPPC supports the recommendations of the ASM Task Force on Antimicrobial Resistance for (1) a national surveillance system, (2) strengthened professional and public education, and (3) increased research. Cooperative efforts will be required to achieve each of these objectives since veterinarians, physicians, and their clients share the need for the antimicrobial products that are critical to food safety and human and animal health.
A National Surveillance System
A newly formed CDC—USDA—FDA surveillance project focused on issues of susceptibility is a cooperative effort that will be extremely valuable in bringing stakeholders to the table, establishing regular lines of communication, and preventing duplication of effort. This undertaking raises several matters of practical and policy concern:
-
Despite the most carefully conceived, proactive, and effective educational effort, some level of resistance is inevitable; the challenge will be to achieve consensus beforehand about what level(s) of resistance will warrant intervention.
-
Assurance of appropriate and sustained funding of this monitoring process is another concern. Historically, in the agriculture sector, savings have been achieved by adding certain kinds of surveillance to the national animal health monitoring systems, which include sample selection. The question is whether
-
this will continue to be an alternative and, if not, what economies of scale can be realized in other ways.
-
Another concern is that all stakeholders be involved in data analysis and dissemination of results. Crucial entities are the American Veterinary Medical Association (AVMA) and the American Association of Swine Practitioners, as well as various affected producer groups.
Strengthened Professional and Public Education
The Council's Quality Assurance Program is an important educational vehicle. The program includes a Good Production Practices component, which provides a channel for professional veterinary input to the use of all drugs, including antibiotics. Each year the program convenes veterinarians and producers to review all aspects of pork production, including the prudent use of antimicrobials as well as the cost—benefit aspects of their use. Decisions to use antibiotics are a composite of the costs of a given treatment regime, transport, mode of administration, and timing, which are projected against the presumed benefits of the therapy.
Increased Research
Industry and government funding has supported research resulting in a number of innovative production techniques that not only have proved cost effective but have enabled producers to decrease the use of antibiotics except for direct treatment of disease: all-in-all out strategies, which move animals through production units as groups; segregated early weaning, in which pigs are weaned away from the sow according to the likelihood of pathogen transmission; and phased feeding, in which the nutritional needs of the animal are matched with the stage of production. Research has also shown that improved management has decreased the advantage of antibiotic use to improve growth efficiency, so that fewer antibiotics are used per animal even as herd size expands, a finding that will increasingly take hold throughout the industry as producers continue to be faced with the need for cost-effective production practices.
PCR, gene sequencing, and gel electrophoresis are powerful epidemiological tools that are presently expensive and time consuming. Veterinary and human medicine share a need for new diagnostics that are user friendly, clinically applicable, affordable, and of sufficient sensitivity and specificity to be useful; development of such tools would be a positive, proactive step in curtailing the development of resistance attributable to inappropriate antibiotic use.
The Ecology of Resistance on the Farm*
The case of DT104 offers an opportunity for seeing the farm environment as an ecosystem. This perspective differs, conceptually and in management terms, from the traditional, narrow focus on a clinically affected animal, a focus that excludes a number of significant variables, including the possibility that the rest of the herd might be subclinically infected. Field investigators have typically emphasized investigation of the disease agent and what antibiotic to employ, essentially ignoring evaluation of the host and its role in the ecosystem of transmission.
The risk factors for pathogen transmission on the farm are many and various. They include livestock housing, the vulnerability of feed to unsanitary factors, lack of isolation facilities, and high density of animals. Salmonella replicates in the intestinal tracts of cattle, rodents, flies, dogs, cats, birds, domestic pets, wild mammals, and humans, all inhabitants of the farm ecosystem. It also replicates or survives in feed, water (troughs, ponds, canals, wastewater lagoons, flush systems), dust, manure, and on contaminated surfaces not exposed to sunlight. The primary infection route in cows is oral, but it may also be ocular, nasal, via the streak canal,25 and possibly, rectal. Organisms are excreted in manure, oronasal secretions, urine, and milk. Furthermore, the animal's own biologic cycles can affect transmission; for example, changes in the rumen and immune status during the production cycle can increase susceptibility to infection.
Insufficient understanding of the systematic and complex relationships among these variables, ignorance of the risks of antibiotic resistance, and lack of acquaintance with cost-effective production alternatives are the greatest contributors to antibiotic misuse in the farm environment. Before resorting to regulation, more desirable correctives for this state of affairs would be improved communication, better education, and targeted research. Improved communication could help impede the spread of veterinary infections that, in epidemic form, demand extensive, high-volume uses of antibiotic therapy. Better education of producers and allied professions with respect to transmission of infectious diseases, basic prevention measures, and the nature of antibiotic resistance, particularly with respect to new findings, could reasonably be expected to affect farm management behavior in a positive direction. Finally, applied research targeted at better comprehension of the disease risks of the evolving farm ecosystem would provide empirical evidence on which to base critical and ultimately cost-effective sanitary interventions such as improvements in feed, water supplies, and housing for livestock.
U.S. Department of Agriculture*
Historically, the USDA has worked with a variety of government agencies to address issues of antimicrobial resistance related to agricultural concerns. In May 1997, the USDA, FDA, CDC, and EPA (Environmental Protection Agency) submitted to the President of the United States a proposed national food safety initiative; two of the five high-priority research areas focused on resistance. The first was antibiotic resistance: how it emerges, what factors influence it, and what alternatives exist in terms of agriculture production. The second area addressed the emergence in the last decade of foodborne pathogenic strains that are becoming increasingly resistant to traditional food safety practices and preservation technologies, a notable example being Escherichia coli 0157.
Another joint initiative in which the USDA is engaged is an attempt to understand foodborne antibiotic resistance, with emphasis on Salmonella. This activity involves parts of the USDA's Agricultural Research Service (ARS), Food Safety and Inspection Service (FSIS), and Animal Plant and Health Inspection Service (APHIS), the FDA's Center for Veterinary Medicine (CVM) and Center for Food Safety and Applied Nutrition, and the CDC. The activity is examining antibiotic resistance patterns in Salmonella, in cooperation with APHIS, USDA—ARS, FSIS, and animal diagnostic laboratories.
U.S. Environmental Protection Agency†
Each year for the past several years, an estimated 300,000 pounds of antibiotic pesticides have been applied to fruit trees and other crops for prophylactic or therapeutic use, by airplane or by ground spraying; both applications affect the entire orchard, so that the EPA considers environmental exposure to these pesticides to be high. Oxytetracycline has been used in this way for the past 20 years, streptomycin for the past 40; both have become less effective over time, and streptomycin is now ineffective in some areas. Of greatest concern, to growers and to the EPA's Office of Pesticide Programs, is the fact that Erwinia amylovora (fire blight), a highly destructive bacterial disease of pome fruits, has become resistant to both antibiotics. Furthermore, Erwinia is now known to have plasmids containing genes resistant to these same antibiotics for human use, so that there is a risk that marketed fruit might also transmit these resistant bacteria to consumers.
Thus, growers are attempting to have new antibiotics approved for application, and a Mexican company recently applied to the EPA to register gentamicin, an aminoglycoside presently approved only for human use. Comments from the CDC and the ASM raised concerns about potential gentamicin resistance and the
subsequent transfer of resistant strains to humans, as well as the more general risk of establishing precedent for similar uses of other antibiotics, notably fluoroquinolone. EPA's consequent refusal to act on the application has evoked considerable counterpressures that the agency feels inadequately equipped to handle for a number of reasons:
-
There are few hard data on the development and transfer of resistance caused by these agricultural uses, particularly solid quantitative data with well-described etiologic pathways.
-
More needs to be known about the extent of gentamicin resistance encountered in human pathogens and the etiology of any such resistance.
-
As a focus of research, the subject falls outside EPA's traditional risk assessment methodologies.
-
Fruit and vegetable producers, in the United States and other countries, have sizable and defensible economic concerns.
-
Finally, there is ambiguity about whether this particular request for registration is governed by the environmental clauses or the food safety requirements of the North American Free Trade Agreement (NAFTA) and/or the General Agreements on Tariffs and Trade (GATT).
Areas for Consideration and Action in Food Production
Resistance as a corollary or consequence of antibiotic use in food production poses quandaries that are especially controversial because their ramifications are economic and political, as well as global and national. So far, science has been of limited help in providing quantified evidence for their resolution. Workshop participants pointed to a number of areas in which research and action could be helpful.
Collaboration
-
The ecological character of antimicrobial resistance mandates an ecological approach to addressing what will be a persistent challenge. This will require that all stakeholders, in and outside government, be regularly and jointly engaged in identifying problems and laying down pathways toward their resolution. In the case of food production, the key stakeholders are the CDC, EPA, FDA, USDA, professional groups such as the AVMA and American Association of Swine Practitioners, and the commodity producer organizations.
-
Antibiotic resistance does not appear to be incorporated explicitly into discussions of food safety and the regulation and monitoring of imports, nor has the available scientific evidence inevitably been applied, suggesting the value of collaborative dialogue, perhaps led by WHO and including representation from
-
the World Trade Organization (WTO), European Union (EU), and U.S. Departments of State and of Commerce.
FDA's CVM is often asked for guidance regarding the use of antibiotics in veterinary therapeutics, but it has lacked access to the necessary data. Sharing data from veterinary record centers and diagnostic laboratories would make it possible for the FDA and other agencies to cooperate in applying molecular analysis to identify the persistence of susceptibility, the incidence and prevalence of resistance, and very importantly, any trends.
Research
The following topics surfaced as areas for research activity, ideally addressed sooner rather than later and as collaboratively as possible:
-
Analysis of the former practice in U.S. aviculture of the dipping of turkey eggs in gentamicin, which propagated gentamicin-resistant Enterobacteriaceae, which then entered animal and human populations;
-
A controlled, well-quantified study of the prevalence of drug-resistant organisms in agricultural areas currently treated with antibiotics vis-à-vis areas not using antibiotics, and any risks to human populations in the former;
-
Study of the role of wildlife and feral animals in the spread of E. coli 0157 among domestic farm animals;
-
Exploration of the potential for competitive exclusion therapies or ''probiotics," the constructive use of harmless or beneficial colonizing organisms in different areas of food production. Applied research in Europe might be instructive;
-
Exploration of off-label use in food production of antibiotics that retain critical roles in human health, most importantly, the quinolones, particularly given changes in the regulation of information on off-label use that are part of the FDA Modernization Act of 1997; 26
-
Exploration of whether management technologies can decrease subtherapeutic uses of antibiotics for growth promotion in animal husbandry as a consequence of more positive cost-benefit equations; and
-
Exploration of the prospects that new molecular diagnostics would be used in food production, particularly animal husbandry, were such tools to be developed.
Education
The following were also highlighted as important:
-
Systematic approaches to management practices by farmers and, therefore, systematic involvement of the USDA and producer organizations in developing the most effective strategies for doing so; and
-
Producer education regarding the potential negative effects of extralabel use of antibiotics without a valid veterinarian—client—patient relationship.
LEGAL AND REGULATORY CONCERNS
Matters of Law and Possible Responses*
The problem of antimicrobial resistance extends beyond science and public health into a domain of sizable legal and regulatory challenge. Globalization has permitted microbes to move freely around the world, yet attempts to globalize a coherent public health response are constrained by national borders and concepts of sovereignty. These constraints may in turn, and somewhat ironically, thwart the desires of individual nations to protect the health of their populations.
Law and public health historically have had an uneasy relationship because of the tension between the need to respect individual rights and the formal regulation required to simultaneously safeguard community rights, in this instance the right to health. The same is true among and within nations: a similarly delicate balance must be struck between the rights of localities and states to a certain autonomy and the rights of the larger community to protection from disease.
Efforts to develop and' implement a global public health strategy to address antimicrobial resistance must navigate among three different but interdependent levels of law: international, national, and local. The differences among these levels derive from systemic divisions in the authority to make and implement laws; this divided authority affects not only what can be sought from the law but where and how it can be sought. The interdependencies among the levels of law—as well as the interdependencies imposed by the increasing globalization of disease—are areas where coordinated legal strategies can be devised most effectively. The arguments for such strategies can be very pragmatic. The efficacy of legal reforms taken in individual countries to address resistance issues—for example, requirements for surveillance or the following of guidelines for antibiotic use—will ultimately be limited if other countries fail to do likewise. Similarly, the enforcement of new international legal duties for addressing resistance will be undermined if states do not translate such duties into national law. Because of these interdependencies, nothing less than a comprehensive legal vision that in-
tegrates international, national, and local laws will suffice in connection with the similarly interdependent, complex problem of antimicrobial resistance.
The three pillars of the public health strategy for confronting resistance are (1) surveillance, (2) rational use of antimicrobial drugs, and (3) research and development of new drugs. Legal issues arise in each domain.
Surveillance
Two legal issues stand out and permeate the application of law at every level. The first is the legal duty to report disease events. The second is concern for privacy protection and the management and uses of data generated in surveillance.
Reporting The first issue has to do with the substance and processes of reforming disease notification laws to incorporate specific duties for reporting resistance. The International Health Regulations (IHR) do not now include systematic notification of antimicrobial resistance, and WHO is drafting amendments to the IHR so that they will do so. The constraints here, however, have more to do with the fact that WHO has limited authority to enforce reporting under the IHR and has only rarely utilized the authority it does have. Additionally, WHO has limited resources to dedicate to reporting. To date, the WHO approach has been to depend on education and suasion. Compliance with the IHR is, therefore, essentially voluntary, so that historically it has been uneven at best. In addition, not all countries have the capacity for adequate identification and notification of disease, not to mention resistance. Often this occurs because there are so many other demands on essentially meager national health budgets; sometimes it is simply that priority has not been awarded to surveillance. Thus, the reportable disease data received by WHO are typically uneven in quality and in frequency.
There are also issues of enforcement in the United States. As noted earlier in this report, although the CDC coordinates the national public health notifiable disease reporting system, the legal authority to require disease reporting and to specify which diseases are "reportable" is vested in state governments; reporting to the CDC is essentially voluntary. Although all states do participate, reporting is far from uniform. For example, in 1995 the Council of State and Territorial Epidemiologists recommended that states add drug-resistant S. pneumoniae (DRSP) to their surveillance systems; some states still have not done so. Even among compliant states, the completeness of reporting is variable, quality is uneven, and almost no information on antimicrobial resistance is presently included. The CDC cannot mandate states to reform their laws regarding reporting generally or with specific regard to antimicrobial resistance. Like WHO, CDC depends on education and suasion; action is by invitation from the state level.
Privacy The second issue is privacy. Typically, surveillance systems must balance the privacy expectations of those infected with the scientific and medical need for epidemiologically useful information and the larger community's interest in protection from the spread of infectious diseases. This concern has proven especially acute in connection with sexually transmitted diseases, as the HIV/AIDS crisis amply demonstrated, but there are other sensitivities that have economic dimensions, for instance, the effects on tourism of published outbreak information.
In the United States, the dissemination of health information gathered by public health agencies is regulated largely by the Constitution and state statutes; however, a recent survey of these statutes indicates that although most states have nominal safeguards on public health privacy, they are often incomplete or inadequate. The problem is even more acute in connection with private entities, for instance, managed care organizations and companies whose business is collecting and selling health information; U.S. law currently offers weak protection of the informational privacy of such health records; in fact, legislation has been introduced in Congress calling for much stronger federal regulation in this regard. In contrast, the European Union has a law that places very strict conditions on the uses of health data, including a prohibition on the transfer of certain personal information to member states with insufficient data protection. Since this stricture would affect not just the United States but many other countries, it obviously has relevance to developing global surveillance of antimicrobial resistance. A pivotal question is whether governments can compel disclosure of privately gathered information in the interest of public health. All of these questions arise in connection with the potential use of cyberspace for resistance surveillance, which confronts the plethora of jurisdictional problems involved in regulation of Internet activity, not the least of which is privacy.
Rational Use of Antimicrobial Drugs
Despite consensus in the public health community on the need for more rational use of antimicrobial drugs, the strategy for achieving this has relied on encouraging voluntary behavioral changes through education and persuasion. The question has been raised as to whether this is sufficient, given the gravity of the situation, and whether public health officials and advocates should contemplate calling on the force of law to curb antimicrobial misuse. To do so would raise many legal questions.
International Issues A major issue here is the extent of WHO's authority. The organization has limited powers to adopt regulations, but these seem not to extend to creating rules regarding pharmaceutical use. WHO has not, for example, included any proposals dealing with rationalizing antimicrobial use in the revision of the IHR. Also, although the organization has the authority to adopt a
convention on the use of antimicrobials, it has in fact not done so. Some member states have routinely ignored the duty to report in a timely manner outbreaks of plague, cholera, and yellow fever, and may resist international legal obligations affecting the prescribing practices of their physicians. Were such duties to be accepted, requirements for monitoring and enforcement would be daunting and costs consequential.
International legal issues also arise in relation to the misuse of antibiotics in food production, alluded to in an earlier section of this report. Recent episodes, including the WTO decision against the European Union's ban on importation of U.S. beef raised with hormones and the near trade war between the United States and the EU over food inspection, suggest that legal issues in this area may proliferate. Since the U.S. Departments of State and of Commerce are customarily engaged in matters of this sort, guidance emerging from collaboration among CDC, EPA, and USDA could inform such debates.
One forum that might lend itself to discussion of a global approach to more rational antimicrobial drug use is the International Conference on Harmonization (ICH), the multilateral effort by the EU, Japan, and the United States to harmonize pharmaceutical regulatory systems. As currently structured, however, the ICH would not include other countries and regions, in many of which inappropriate use of antimicrobials is a serious problem.
U.S. National Issues Although state legislatures may have the power to regulate physician prescribing practices, attempts to do so might well evoke negative reactions by physicians and their medical associations. The alternative is self-regulation of the profession through guidelines and formalized peer reviews established by these associations or through the incentives and disincentives that managed care organizations have at their command.
At the federal level, Congress has access to the Commerce Clause for possible regulation of antibiotic use since pharmaceuticals are transported interstate; it would probably not be able to regulate prescription practices directly, since this authority rests with the states. The FDA can restrict post-approval marketing of new drugs designed for treating serious or life-threatening illnesses but has done so very rarely, a topic discussed later in this report. It would seem that the only regulatory strategy currently available to the FDA for dealing with resistance to existing antimicrobials is modifying labeling requirements, which is neither a simple process nor a clear-cut solution.
Research and Development
The key legal issues in stimulating more scientific research and the development of new antimicrobials include (1) intellectual property protection, (2) perceived antitrust law limitations on collaborative R&D efforts, and (3) regulatory approval procedures.
Intellectual Property Protection*
New Products
Of 10 new drugs developed by the biopharmaceutical industry, on average 3 will do well financially; the other 7 will not repay the initial investment, that is, the $450 million average cost of developing a drug through clinical trials and FDA approval. This very large investment in new drugs is reasonably well protected by the current system of U.S. patent law, and internationally, many countries also protect new intellectual property, including such very large countries as Brazil, China, and Mexico. The recent Agreement on Trade-Related Aspects of Intellectual Property (TRIPS) of the WTO also strengthened international rules on patent protection, although theft of patented agents remains a concern since some WTO member states still must amend their national laws to fulfill obligations under the agreement; for some countries, this will take years. In addition, the Hatch-Waxman Act of 1984 provides for patent term extensions under certain circumstances; similar protection is available in Europe and Japan, and other countries are being stimulated to enact comparable legislation.
Existing Products and Compounds†
A critical distinction in discussing patent protection is whether the subject product has been on the market (i.e., is market labeled), or whether it is a compound with possible development potential. One set of questions is raised when a new use is sought for an older product that has been on the market, whose patent protection may have expired, for which there may be a generic counterpart, and for which getting a new-use approval will require clinical trials, with associated costs in time and money. A somewhat different set of questions is raised with regard to compounds that companies have on their shelves, which may have antibiotic potential but were never commercially exploited and have lost patent protection.
Competing with shelved compounds is a new armamentarium derived from uniting combinatorial chemistry with efficient, high-throughput screening for efficacy, or perhaps using shelved compounds as platforms for new chemical diversity. Repeating the toxicity studies remains a costly endeavor, for which rapid-throughput procedures are only beginning in early stages of the development process.
A relevant example is nisin, a bacterially produced compound that shows promise for parenteral and oral use against multiple-drug-resistant infections. A composition-of-matter patent cannot be obtained because of the antiquity of the
molecule itself; thus, it cannot be protected as intellectual property in its basic form. One alternative protective strategy would be to construct a penumbra of protection through some combination of use patents, specific applications, and new formulations that are patentable; however, the pathway to a use patent is not clear since the basic molecule has been on the market as a food additive. Since the molecule is a gene-encoded peptide, the other strategy is to seek a genetic variant of the molecule. The fundamental message appears to be that although the science lends itself to revisiting existing compounds, protecting them as intellectual property is not straightforward. It is not clear that this lack of clarity deters pharmaceutical companies from this avenue of inquiry.
What seems more challenging is how to get maximum use from products already on the market that appear to have activity against resistant pathogens, but whose particular efficacy has not been documented so that these indications are not included in current labeling. Since there is no well-defined population in which to conduct a standard clinical trial in such cases and since safety is not at issue, what is needed is development of surrogate markers, in vitro technologies, or animal models that could support the relabeling and expedite the process. A related matter is whether some sort of patent extension might be afforded in such cases, since public health interests are being served at the same time that a pharmaceutical company is assuming the costs of the research required for relabeling. Neither the Hatch-Waxman nor the Orphan Drug Act protects antibiotics, but a possible model is the recently passed FDA reform legislation, which awards an additional six months of exclusivity when the Department of Health and Human Services asks a company to conduct pediatric studies of an existing product.
Targeted Drug Development
The subject of relabeling raises the prospects for what would be, in effect, tailored drug design, and the advantages and disadvantages of such an approach. One consequence of the explosion in genetic information is the possibility of more individualized therapies—in other words, drugs that would take human polymorphisms explicitly into account. Researchers will be able to probe research targets in human disease, in this case infectious disease, and aim at molecules designed for a specific subset of patients. It is already known—and the genotyping of Helicobacter pylori has been informative in this regard—that there will be both phenotypic and genotypic variations associated with specific microbes. A "customized" strategy would reduce the size of the user population, a factor in the evolution of resistance. In addition, the more restricted action of the molecule could be expected to reduce the chances of failure in drug discovery, clinical trials would be focused more economically on a specific user population, and development time overall should be shorter. Other benefits from this sort of discovery strategy would be better understanding of such phenomena as genetic susceptibility to infectious agents; individual variation in response to
drugs, especially including information about responders and nonresponders and potential for adverse events; and association of infectious agents with population subgroups having genetic susceptibilities to chronic inflammatory diseases. Finally, there is the reality factor: new technologies are already driving a trend toward collections of human polymorphisms to basically genotype whole populations.
At the same time, there are different kinds of disadvantages that are not trivial. The potential market for the resulting product would be much smaller because, in effect, licensing would be only for populations preselected as "good responders"; this implies significant pricing dilemmas, especially in terms of inclusion in managed care formularies and reimbursement, which are so price driven. Preselection might also risk narrowing the putative market unduly or defining an eligible target group so small as to not make investment in development practical. Targeting drugs to genotypes could also define individuals as better or worse responders and perhaps risk making them uninsurable or ineligible for managed care. For pharmaceutical companies and practitioners, prescribing algorithms for a large number of subpopulations could get dauntingly complicated and possibly press on the per-patient time limits already imposed in many managed care situations.
Another consideration is the implications of customized therapy for regulatory approval processes. As information accumulates on drug metabolism over the early and middle phases of drug development, the FDA is concerned with identifying vulnerable polymorphisms in trial populations; this concern, however, focuses on interactions with other commonly used drugs rather than on the degree of interaction between the drug and individual patient types. It is later in the development process that efforts are made to look at subgroups for safety information and different patterns of response as these occur. Still, the size of these subgroups is characteristically too small, even in clinical trials involving several thousand patients, to capture enough outliers to identify unusual patterns. Trials requiring analyses of many subgroups, large enough to be informative in this way, would entail greater size and cost, factors already much discussed in connection with new mandates for adequate inclusion of women and ethnic populations in clinical trials.
Collaborative Research and Development
It may be that in some circumstances, collaborative R&D efforts by several pharmaceutical companies might make scientific and economic sense, but companies point to difficulties posed by antitrust laws. In response to calls for collaborative R&D on a malaria vaccine, for instance, some companies have commented that such collaboration would be fraught with problems involving the sharing of intellectual property and the likelihood of falling afoul of legislation banning cartels. The reality may be less constraining than the perception, be-
cause both U.S. antitrust law and European Union competition law permit collaborative joint ventures within certain parameters.
Two examples may reward inspection in this connection, although their relevance is indirect. One is the permission recently granted by the Antitrust Division of the U.S. Department of Justice to a group of companies in the communications industry to unify the administration of 27 patents, on the grounds that doing so is not anticompetitive but, on the contrary, in the public interest. The other example is the Aviation Manufacturers Aircraft Association, formed in response to a virtual fiat by then-Assistant Secretary of the Navy Franklin Roosevelt, which required companies to unblock their patents against the background of a stagnating aircraft industry.
Whether patent blocking or patent pooling will be issues with respect to the development of new antimicrobials that depend on new genetic sequences is unclear, since the entire subject of the genome as intellectual property is also unclear, charged as it is with litigation and dissent around very fundamental notions of scientific discovery and who is to profit from it.
Regulatory FDA Responses to Antimicrobial Resistance
A call to streamline regulatory approval procedures is inevitably made when strategies for enhancing R&D investment are discussed. Given the likely passage of regulatory reform legislation, such a call at this time seemed extraneous and the workshop discussion therefore focused on a strategic option that had been addressed initially in the previous Forum workshop:27 the potential of restricted distribution to conserve the viability of new antimicrobials. A question also raised earlier but not pursued at this workshop was whether Congress could provide FDA with the authority to negotiate extended market exclusivity to manufacturers who would agree to restrictions on the marketing of antibiotics.
Restricted Distribution*
The issue here was whether the problem of inappropriate antibiotic use was not too serious to leave solely to provider and patient education and whether more formal, regulatory constraints on the distribution of new anti-infectives might have merit in terms of preserving the activity of such products and extending their usable life.
Although restricted distribution is available to the FDA as an option in the accelerated approval regulations for serious and life-threatening diseases, the
agency has very rarely used it. As noted above, prospective boundaries on market size could constitute a disincentive to industry R&D investment, disadvantaging companies whose product is narrowly restricted relative to companies whose products are not, especially at a time of increasing industrial effort to find new targets and develop new structural classes of drugs. Restriction could also compromise individual rights to therapy, partly because of limits on the user population and partly as a result of costs, because physicians might find themselves facing a choice between prescribing an inexpensive broad-spectrum drug and prescribing a costly drug of narrow spectrum. Thus, the sense of the workshop participants was that the concept of restriction was critical but would most effectively embrace a range of interventions aimed toward the objective of preserving the efficacy of products already in use:
-
using postmarketing surveillance as fully as possible to identify resistance;
-
exploring the potential of formularies used by managed care organizations to limit the use of drugs for which there already exist worrying levels of resistance;
-
quantifying the risks of inappropriate antibiotic use and developing descriptive models of the differences that could result from reduced use;
-
attempting to anticipate resistance by using enrichment procedures in the early stages of drug development to identify plasmids in the pertinent ecosphere that might become sources of resistance and their in vivo potential for "exchangeability" from natural sources;
-
more profoundly exploring broad ecological areas where resistance is known to be developing (e.g., fluoroquinolone and gentamicin use in food production);
-
raising existing levels of knowledge about actual drug usage in humans and animals;
-
exploring with WHO the use of its Essential Drugs List as a tool in dealing with the unrestricted availability of antimicrobials in some countries;
-
conducting research responsive to pressures for the approval of antimicrobials for over-the-counter use;
-
expanding outcome research on utilization (and nonutilization) of antibiotics, with a major objective incorporation of its findings into managed care policies.
SUMMARY OF AREAS FOR CONSIDERATION
Many areas, topics, issues, and options surfaced in the course of this workshop. Following is a summary of the key issues and options discussed.
Surveillance
Information Systems
ISSUE: No country, including the United States, has a reliable, longitudinal, full-service antimicrobial resistance surveillance program with comprehensive focus, nor is there a comprehensive database for monitoring trends in antimicrobial usage. Multiple surveillance activities around the globe are attempting in different ways and at different speeds to move toward the ideal depicted in this report, but these systems, as a group, are uncoordinated and unstandardized. Thus, the magnitude of the resistance problem and its impact are really unknown.
OPTION: The qualities needed are presented in the section of this report dealing with surveillance. Detailed recommendations for implementation of a comprehensive resistance surveillance program are outlined in the 1995 ASM Task Force Report (see Appendix B). These recommendations await funding, implementation, assumption or assignment of leadership, and formation of partnerships.
ISSUE: Research and information on the impact of rapidly increasing antimicrobial resistance in the community are lacking.
OPTION: Inclusion of information about the effects of resistance on the outcome of infections in systems of data collection.
Laboratory Systems
ISSUE: Some currently available molecular methods are clearly applicable only to research and reference laboratories; their feasibility for most commercial or clinical laboratories is at best limited.
OPTIONS:
-
Selection and strengthening of the laboratories in a set of sentinel hospitals to serve as bases for global assessment of the prevalence and transmission of the most critical antibiotic-resistant genes (i.e., sites for monitoring gene flow and assessing genetic diversity).
-
Design of categories and pathways for reducing data sets into comprehensive packages for use by clinicians and researchers.
ISSUE: The NCCLS Guidelines seem not to be as widely and regularly available as would be useful, and the processes and criteria for their development are not clear.
OPTIONS:
-
Expand distribution of NCCLS Guidelines and, if necessary, increase the frequency with which they are updated.
-
Train laboratory personnel in sentinel hospitals in standardized methodologies.
Law and Regulation
ISSUE: CDC cannot mandate states to reform laws regarding reporting, but must rely on education, persuasion, and invitation.
OPTIONS:
-
Exploration of whether increased resistance and rapid diminution of effectiveness of existing antibiotics might justify awarding greater authority to CDC to monitor and enforce legal duties regarding resistance, and consideration of the means by which this might be accomplished.
-
Consideration of ways to integrate issues of resistance into formulary development processes in pivotal managed care organizations, as well as the potential for inclusion of pharmaceutical industry representation on such committees for review and implementation of programs.
ISSUE: A global antimicrobial resistance network might require many countries to import equipment, software, and reagents.
OPTION: Drafting language for international agreement.
Response: Prolonging Effectiveness
Education
ISSUE: Many needs related to the modification of attitudes and behaviors among providers, patients, parents, managed care organizations, and the pharmaceutical industry may be most usefully considered as an integrated global strategy. Not the least of these is the need for ongoing education concerning infection control, hygiene, and sanitation in health facilities and the community in general.
OPTIONS: Table 5 of this report provides a listing of strategic areas for interventions meant to modify attitudes, behaviors, and, where applicable, policies among the major parties to the antimicrobial resistance problem.
Law and Regulation
ISSUE: Many groups are compiling practice guidelines for antibiotic use, possibly generating confusion and complicating their value to providers, for whom such guidelines might afford some protection from liability.
OPTIONS:
-
Speedy implementation of a joint project involving all pertinent professional societies in developing unitary guidelines (including checklists for providers to use in clinical settings) for antimicrobial use, perhaps analogous to the Report of the Committee on Infectious Diseases of the American Academy of Pediatrics (''Red Book"), implementing their extensive dissemination, and very importantly, updating them periodically based on annual data from longitudinal studies.
-
Greatly expanded research into outcomes of antibiotic misuse, nonuse, and prudent use in health care facilities and in the community, as the foundation for the articulation and revision of guidelines, the policies of the health professions and the full range of health care facilities, and the formularies of such facilities, including the WHO Essential Drugs List.
ISSUE: Some existing products seem to have activity against resistant pathogens but because this particular efficacy has not been documented, these indications do not appear in the labeling.
OPTIONS:
-
Development of alternative ways to define efficacy, for example, surrogate markers, in vitro technologies, and animal models to address lack of a well-defined population for clinical trials.
-
Exploration of the possibility of congressional authorization to extend patents for such products and the relevance of recent legislation adding six months of exclusivity when DHHS requests pediatric studies of an existing product, with the recognition that this topic is complex and difficult.
ISSUE: Shorter courses of full therapeutic levels of antibiotics may in some cases be feasible and perhaps encouraged, with a positive effect on volumes of selective pressure. The issue is complex since there is also evidence that subtherapeutic doses may select for resistance.
OPTION: Design and implementation of research on clinical outcomes from shorter courses of therapy, as the basis for subsequent updating of new practice guidelines and revisions to labeling.
ISSUE: The FDA is increasingly pressed to approve some prescription antibiotics and antifungals for over-the-counter use.
OPTION: Research and/or systematic marshaling of existing research to inform agency response.
ISSUE: Antibiotic use is widespread in hospital, community, and farm settings, yet knowledge of the magnitude of these uses depends largely on estimation and extrapolation.
OPTION: Improved data gathering and analysis, perhaps through national systems that would continuously monitor antimicrobial usage.
Response: Developing New Products
Incentives for Industry
ISSUE: There is said to be a perception in the pharmaceutical industry that collaborative development of new antimicrobials might be constrained by U.S. antitrust laws, although to what extent this is the case is unknown and the evidence is contradictory. On the one hand, both U.S. antitrust law and European Union competition law permit collaborative joint ventures within certain parameters; on the other, dispute about ownership of gene sequences is current and heated.
OPTION: Exploration of the extent to which these factors constitute disincentives explicitly for the development of new antimicrobial products and, if this should prove to be the case, further exploration focused on alternative solutions for the dilemmas identified.
Research
ISSUE: Resistance is so complex and dynamic at the genetic level that more work is needed to understand the diversity and prevalence of resistant gene families, both in nature and in the animal microflora that are the bridge to human contact, and to discern the origins of these genes and how they spread from one organism to another.
OPTIONS:
-
Studies of gene flow.
-
Research using enrichment procedures in the early stages of drug development to identify plasmids in the pertinent ecosphere that might become sources of resistance and their in vivo potential for "exchangeability" from natural sources.
Education
ISSUE: Limited data are available to describe the difference that prudent antibiotic use would make. Without such data, public education and advocacy are constrained.
OPTION: Quantification of the risks of injudicious antimicrobial use and development of descriptive and predictive models of the differences judicious use of antimicrobials would make, for purposes of policy development, advocacy, and action.
ISSUE: The community antimicrobial resistance equation is being affected to an unknown degree by proliferation of household and personal-use products with added antibacterial properties and by changes in hospital discharge patterns.
OPTIONS:
-
Intensified research into the effects of incorporating antimicrobials into items of daily personal use.
-
Intensified research into the effects of changes brought about by managed care on infection rates and antibiotic resistance patterns.
Agricultural Use
Research
ISSUE: Historically, there has been a paucity of hard data on the development and transfer of resistance produced by animal husbandry, aquaculture, and agricultural uses of antibiotics, particularly solid quantitative data with well-described etiologic pathways and data on trends in antimicrobial usage in veterinary settings. Lack of ready access to data from veterinary reference laboratories has been a limitation in this regard.
OPTIONS:
-
Collaboratively designed, implemented, and analyzed research on these dynamics, perhaps beginning with case studies (e.g., DT104, fluoroquinolones, gentamicin).
-
Collaborative access to data from veterinary reference laboratories.
ISSUE: An ecological understanding could help in a number of aspects of animal husbandry, including conditions that foster the enhancement of antimicrobial resistance.
OPTIONS:
-
Systematic, collaborative development, by the USDA, AVMA, FDA, and producer organizations, of strategies and educational materials to expand such understanding.
-
Development of cost-benefit and cost-effectiveness models of different on-farm antibiotic usages to enhance the public health community's understanding of farmer perspectives.
ISSUE: Applied research suggests that there is potential in using competitive exclusion therapies or "probiotics," that is, the constructive use of harmless or beneficial colonizing organisms in different areas of food production.
OPTION: Additional applied research.
ISSUE: There is ambiguity as to whether requests for registration of antibiotics for use on agricultural products that are exported are governed by the environmental clauses or the food safety requirements of NAFTA and/or GATT. Nor does antibiotic resistance appear to be incorporated explicitly into discussions of food safety and the regulation and monitoring of imports.
OPTION: Collaborative dialogue, perhaps led by WHO and including representation from the World Trade Organization, European Union, and U.S. Departments of State and of Commerce.
FINAL COMMENTS
Since the introduction of sulfonamides and penicillin more than 50 years ago, the world has become accustomed to the availability of antibiotics that are promptly and reliably effective, relatively free of side effects, and inexpensive compared to other medical and surgical interventions. The initial treasure trove is, however, all but exhausted. Yet, like cheap petroleum, the habit interferes conceptually and practically in market-incentive structures with the development of successors, and there is high risk that what remains of the treasure will be wasted by its imprudent use. The transition period as the market makes the necessary adjustments will be painful, and it is possible to imagine a scenario in which antibiotics with lower therapeutic indexes at thousands of dollars per course of treatment could instill a need for rationing and special development incentives, to great consumer distress, particularly in populations whose financial resources are constrained.
The evidence and opinions presented at this workshop suggest, nevertheless, that the transition from a historically generous armamentarium to one at least temporarily much less lavish could be mitigated by wiser policies, both to conserve what remains and to plan for what is to come; policies for the most cost-
effective use of antibiotics; evidence-based regulation, with transparent balancing of risks and benefits; and as already exemplified in genome projects, social investment in the underlying science needed to develop new antimicrobial agents. Also, because antimicrobial drug resistance is increasingly known to be a global problem, it can be addressed only with international cooperation, at a minimum in the acquisition and sharing of information. Whatever frictions might ensue from shaping and implementing such policies would be more than offset by the savings in medical and hospital costs and, most importantly, by the deaths and disability avoided.



































































