
Proc. Natl. Acad. Sci. USA
Vol. 95, pp. 5884–5890, May 1998
Colloquium Paper
This paper was presented at the colloquium “Computational Biomolecular Science,” organized by Russell Doolittle, J.Andrew McCammon, and Peter G.Wolynes, held September 11–13, 1997, sponsored by the National Academy of Sciences at the Arnold and Mabel Beckman Center in Irvine, CA.
Photoactive yellow protein: A structural prototype for the three-dimensional fold of the PAS domain superfamily
JEAN-LUC PELLEQUER*, KAREN A.WAGER-SMITH†, STEVE A.KAY†, AND ELIZABETH D.GETZOFF*‡
*Department of Molecular Biology and The Skaggs Institute for Chemical Biology, †National Science Foundation Center for Biological Timing, Department of Cell Biology, The Scripps Research Institute, 10550 N.Torrey Pines Rd., La Jolla. CA 92037.
ABSTRACT PAS domains are found in diverse proteins throughout all three kingdoms of life, where they apparently function in sensing and signal transduction. Although a wealth of useful sequence and functional information has become recently available, these data have not been integrated into a three-dimensional (3D) framework. The very early evolutionary development and diverse functions of PAS domains have made sequence analysis and modeling of this protein superfamily challenging. Limited sequence similarities between the ~50-residue PAS repeats and one region of the bacterial blue-light photosensor photoactive yellow protein (PYP), for which ground-state and light-activated crystallographic structures have been determined to high resolution, originally were identified in sequence searches using consensus sequence probes from PAS-containing proteins. Here, we found that by changing a few residues particular to PYP function, the modified PYP sequence probe also could select PAS protein sequences. By mapping a typical ~150-residue PAS domain sequence onto the entire crystallographic structure of PYP, we show that the PAS sequence similarities and differences are consistent with a shared 3D fold (the PAS/PYP module) with obvious potential for a ligand-binding cavity. Thus, PYP appears to prototypically exhibit all the major structural and functional features characteristic of the PAS domain superfamily: the shared PAS/PYP modular domain fold of ~125–150 residues, a sensor function often linked to ligand or cofactor (chromophore) binding, and signal transduction capability governed by heterodimeric assembly (to the downstream partner of PYP). This 3D PAS/PYP module provides a structural model to guide experimental testing of hypotheses regarding ligand-binding, dimerization, and signal transduction.
A large and growing set of multidomain protein sensors and transcription factors involved in signal transduction include PAS domain sequences (http://www.whoi.edu/biology/hahnm.html). The PAS acronym was coined originally (1) to describe the ~270-residue region encompassing two direct sequence repeats (PAS-A and PAS-B) of ~50 residues each, that had been identified in the Drosophila Period clock protein (PER) (2), the vertebrate Aryl hydrocarbon receptor nuclear translocator (ARNT) (3). and the Drosophila Single-minded (SIM) (2). These three proteins are involved in regulation of circadian rhythms, activation of the xenobiotic response, and cell fate determination, respectively. More recently, PAS domains have been found in many other proteins, including histidine-kinases (4), light receptor and regulator proteins (5), clock proteins (6, 7), sensor proteins (oxygen/redox sensors), ion channels (5), and a Ser/Thr kinase with a putative redox sensing or flavin-binding domain, in which PAS regions are named “LOV” (light, oxygen, or voltage) (8). These PAS-containing proteins occur in a wide range of living organisms including: eubacteria, archaca, cyanobacteria, fungi, plants, insects, and mammals (5). PAS-containing proteins have been categorized (5) into three functional subgroups: (i) transcription activators [DNA-binding proteins with both basic helix-loop-helix (bHLH) and PAS sequence motifs], (ii) sensor modules of two-component regulatory systems (oxygen sensor, nitrogen fixation, sensor kinase, etc.), and (iii) ion channels (in eucarya).
One function of the PAS domain is to mediate protein-protein interactions (9, 7). Dimerization has been demonstrated for many transcriptional activators such as the aryl hydrocarbon receptor (AHR), ARNT, SIM. hypoxiainducible factor 1 (HIF-1), Member Of the PAS superfamily (MOPs), and the trachealess (TRH) protein. Dimerization is known to be mediated by both the bHLH region of these transcription activators and by their PAS repeats (9–14). Some PAS-containing proteins lack a bHLH region (PER) but can still either homodimerize (9) or heterodimerize with other bHLH-PAS-containing proteins (9–11) through their PAS domains in vitro. A second function for PAS domains is ligand and/or cofactor binding, as is the case for AHR (15) and for the heme-binding bacterial O2-sensing protein FixL (16).
Sequence similarities between the PAS repeats and photo-active yellow protein (PYP), a self-contained bacterial blue-light photoreceptor (17–19) implicated in negative phototaxis (20), were identified by Lagarias (21), who probed a sequence database with a 43-residue consensus sequence constructed from the PAS-A and PAS-B domains of phytochromes, which are the red and far-red photoreceptors in plants. Recently, many more PAS domains have been shown to exhibit sequence similarities with PYP (22, 7, 23, 5, 4), and sequence alignments have been extended into the “S2” region that is located immediately C-terminal to the original PAS repeats (5) and into a second more C-terminal region of the PAS B sequences, termed “PAC” (4). PYP also exhibits the functions characteristic of PAS domains: sensing (of light), binding of ligand/ cofactor (chromophore), and signal transduction through protein-protein interaction (with the downstream partner of PYP).
©
PNAS is available online at http://www.pnas.org.
‡ | To whom reprint requests should be addressed. E-mail: edg@scripps.edu. |
Here, we present the hypothesis that the entire PYP protein fold is the structural prototype for the modular, three dimensional (3D), PAS A and PAS B domain folds in PAS-containing proteins. We define the PAS/PYP module with a length of ~20–150 residues that matches the length of the PYP sequence and encompasses the entire PYP protein fold. PYP therefore appears to be both structurally and functionally a prototypical PAS domain, with a modular single-domain fold that links sensing and ligand binding to signal transduction via dimerization with another protein. A 3D molecular model of the PAS B domain of the human protein ARNT, based on the PYP structure, provides a detailed structural framework for integrating and differentiating sequence and functional data and suggests specific regions to test experimentally for involvement in ligand binding, dimerization, and signal transduction.
METHODS
Sequence Alignment. Representative members of the ARNT family were aligned to each other using the program PILEUP (24) from the Genetics Computer Group (GCG) suite (25). Then several PAS-containing sequences, identified from previous publications (4, 5, 26) and obtained from the National Center for Biotechnology Information Entrez Web service (http://www.ncbi.nlm.nih.gov/Entrez/) were then successively added to the alignment with the PILEUP program. Finally, the sequence of PYP was added manually to complete the sequence alignment (see Fig. 2). Automatic alignment of the PYP sequence with the PAS domain sequences is hindered by the presence of a few apparent mismatched residues that inflate the alignment score (mainly Gly→hydrophobic). Such residues are discussed in detail later.
Molecular Modeling. Using the alignment in Fig. 2, we built a 3D model of the PAS B domain of the human ARNT protein, based on the coordinates of dark-state PYP (2phy.pdb) (27). Nonidentical side chains were replaced using the program XFIT (28). Side-chain conformations were taken from a rotamer dictionary based on the library of Tuffery et al. (29). In each case, the most common rotamer was used unless it displayed strong steric clashes with any backbone atoms. Alternative rotamers were used for residues 33, 34, 62, 63, and 112 (PYP numbering is used throughout this paper), and a significant deviation from the standard rotamers was necessary to fit residue 29. The resulting ARNT model was energy minimized with the XPLOR program (30). using the conjugate gradient method (31) and the CHARMM22 all-atom parameter set (32). The dielectric constant was set to one. The shifted electrostatic and the switched van der Waals functions were selected using a cut-on value of 6.5 Å and a cut-off value of Å. Nonbonded interactions were cutoff beyond 9 Å. Hydrogen atoms were added with the HBUILD program (33), and their positions were energy-minimized until the norm of the gradient was <0.1. Then, while the backbone was kept fixed, all side-chain positions in the model were energy-minimized with the electrostatic energy term turned off, until the norm of the gradient was <0.5. This was followed by a short minimization of all atomic positions (norm of the gradient <2.0) in order to remove any remaining clashes between side chain and main chain atoms. Two-residue insertions at the positions 87 and 98, and a single-residue deletion at position 69 were introduced with the program TURBO
RESULTS
The PAS/PYP Module. PYP provides a structural and functional prototype for the 3D fold of the PAS domain superfamily (Fig. 1), which we name the PAS/PYP module. PYP is a self-contained, bacterial blue-light photoreceptor, with an unusual fold characterized by a central six-stranded β-sheet with N- and C-terminal β-strands in the center (27). The overall PYP fold breaks down into four segments: (i) the N-terminal helical lariat (residues 1–28), including helices al and α2, (ii) the first three-stranded half of the central β-sheet (residues 29–69), including the β1, β2 hairpin, two short intervening α-helices (α3 and α4), β3, and an overlapping turn of π-helix, (iii) the helical connector (residues 70–86), composed predominantly of the long α5-helix that diagonally crosses the β-sheet, to connect the two edge β-strands, and (iv) the last three-stranded half of the central β-sheet, including β4, a connecting loop, and the β3, β6 hairpin. PYP has a hydrophobic core on each side of the central β-sheet (27). The N-terminal helical lariat caps one side of the β-sheet to form the smaller hydrophobic core. The remaining helices and loops, together with the central β-sheet, surround the 4-hydroxycinnamoyl chromophore, to form the larger hydrophobic core. Helix α3 and flanking residues contribute the hydrogenbonding network for the phenolic hydroxyl at the tip of the chromophore.
The PAS core (Fig. 1), the second segment of the PAS/PYP module, provides the photosensing active site of PYP and roughly corresponds to the traditional repeating PAS sequence motif of ~50 amino acids (1–3). This key portion of the PYP structure, ending at the Cys69 attachment site for the chromophore, forms the majority of the immediate environment of the chromophore, provides all residues that hydrogen bond the chromophore, and supplies the Arg52 gateway (27). The Arg gateway likely participates in PYP heterodimerization with a downstream signal transduction protein, by moving and pro

FIG. 1. A proposed PAS/PYP 3D fold illustrated on the PYP structure. The N-terminal cap, colored in purple, contains residues from 1 to 28. The PAS-core, colored in gold, is the domain where higher sequence homology is found among various members of the PAS-containing molecules. It spans from residue 29 to 69. The helical connector, colored in green, includes a short loop followed by the helix α5 and spans residues from 70 to 87. The β-scaffold, colored in blue, contains the last three strands of PYP, spanning residues from 88 to 125.
viding solvent access to the chromophore during the long-lived, bleached, signaling intermediate of the PYP light cycle (36).
The β-scaffold, the fourth segment of the PAS/PYP module, provides a long platform with a characteristic β-sheet twist that supports the PAS core and completes the central six-stranded PYP β-sheet. This β-scaffold (Fig. 1) approximately matches the PAC sequence motif described by Ponting and Aravind (4). Within this β-scaffold, the end of the fourth β-strand plus the ω-loop (37) connecting β4 and β5 wrap around the PAS core to complete the chromophore environment. In the PAS ‘‘S-box” nomenclature created by Zhulin and coworkers (5), the S1 box corresponds to the last three-fourths of the PAS core, and the S2 box covers most of the β-scaffold.
The central β-sheet of the PAS/PYP module is protected from solvent by the two remaining segments: the N-terminal cap protects one side, whereas the helical connector combines with parts of the PAS core to protect the other. The PAS-related LOV (light, oxygen, or voltage) sequence motif encompasses the PAS core, the helical connector, and the β-scaffold. This sequence region was identified by Briggs and coworkers (8) in the plant protein NPH1 (which participates in the signal-transduction pathway for phototropism) and in a family of proteins regulated by environmental factors that could change their redox status.
Sequence conservation between PYP and PAS domains. PAS domain sequences occur in all three kingdoms of life, and act in a multitude of regulatory, sensing, and signal transduction pathways. Thus, sequence similarities are limited and may be further obscured by the functional variation of buried active-site residues that would otherwise exhibit the relative conservation expected by inward-facing residues of the hydrophobic core. To evaluate the limited sequence similarity between PYP and PAS domains, we compiled a full-length PYP sequence alignment with a set of PAS domain sequences (Fig. 2). We used automated protein sequence alignment to align the full-length sequences of closely related PAS proteins (Fig. 2), including the original PAS trio of PER, ARNT, and SIM, as well as the more recently discovered mammalian CLOCK proteins and their homologues. PYP sequences from Ectothiorhodospira halophila (38), corresponding to the crystallographic structure, and three other bacteria were added manually, starting from the sequence registration within the PAS core identified by Lagarias et al. (21) using a phytochrome PAS repeat consensus sequence as the search probe. ARNT was chosen as the major PAS protein family to include and model-build because of the many known sequences from

FIG. 2. Similarities revealed by multiple sequence alignment of several members of the PAS-containing proteins and members of the PYP family. The alignment was performed using the program PILEUP in the GCG suite (25) starting from the ARNT molecules, then adding each PAS-containing molecule in the list. PYP molecules were manually aligned on the top (see Methods). White letter amino acids are conserved in both PYP and PAS-containing proteins. Red letter amino acids highlight significant differences between PYP and PAS-containing proteins. The secondary structure of PYP is displayed on the top using the color coding from Fig. 1. Helices are represented by “noodles”, strands by arrows and loops by lines. Accession numbers from the SwissProt database, as extracted using the Entrez web service (http://www.ncbi.nlm.nih.gov/Entrez/protein.html) are P16113 (pyp.ecto), X98888 (pyp_rhodosp). X98889 (pyp_rhodoba), M19029 (sim_fly), U33427 (trh_fly), U22431 (hifa_human), U51627 (mop3_human), X03636 (per_fly), U10325 (arnt_mouse), M69238 (arnt_human), D45239 (arnt_rabbit), and AF020426 (arnt_fly). Other sequences were obtained through the Entrez service by searching full names of proteins (pyp_chroma, clock_mouse, and arnt_trout).
diverse species, their relatively unambiguous alignment with PYP sequences, and the importance of ARNT as a common regulatory partner in many PAS protein heterodimers (12, 13, 39, 40). In trial alignments, we discovered that the PAS-B sequences align better with PYP than do their more N-terminal PAS-A counterparts. As shown in Fig. 2, we found that the sequence alignment of PYP with PER and with these bHLH-PAS-containing transcriptional activators can be extended both N- and C-terminally to encompass the entire PYP single-domain fold.
The diversity of PAS domain sequences and the low sequence similarity among the more distant members made sequence alignment challenging but also provoked an interesting discovery. Automated alignment with standard programs failed to properly align PYP sequences with the PAS proteins. Similarly, when searching the nonredundant protein sequence database with the BLAST server (http://www.ncbi.nlm.nih.gov/BLAST) the PYP sequence alone failed to select any PAS-domain proteins (although PAS protein sequences can successfully select PYP; ref. 21). However, we discovered that by simply changing a few functionally key PYP residues (G29 into F, G47 into V, and R52 into Y). the modified sequence was now able to pick a member of the ARNT family in a BLAST search. These residues were chosen because their specific roles in PYP would not likely be conserved in any other PAS domain proteins. As shown in Fig. 2, PYP-conserved G29 and G47 are replaced by large hydrophobic residues in the PAS domain sequences. In sequence evolution, the conservation of glycine residues often results from their unrestricted backbone dihedral angles, freed by the absence of a Cβ atom. However, in the PYP structures, neither G29 nor G47 occupies a region of ϕ, ψ dihedral space forbidden to other larger amino acids. Instead, substitution of these glycines in PYP appears to be spacially prohibited by the proximity of adjacent side chains that make key interactions with the chromophore of PYP which is unlikely to be present in PAS domain proteins. In PYP, substitution at G29 would likely interfere with buried E46, which forms an important salt bridge with the phenolic oxygen of the deprotonated chromophore of dark-state PYP (27). Likewise, substitution at PYP G47 would likely interfere with buried Y42, which hydrogen bonds with the same phenolic oxygen of the PYP chromophore. During the PYP photocycle, R52 actively participates in signal transduction by undergoing conformational changes that allow the photoisomerized chromophore access to solvent (36). Although these three PYP function-specific residues would not require conservation in PAS proteins, standard substitution matrices used for automated sequence alignment cannot reasonably accommodate the resulting sequence substitutions and thus fail to perform appropriate sequence alignments. Such alignment peculiarities may often stymie sequence alignment programs and preclude sequence alignment where no structural information is available.
At the sequence level, the alignment presented in Fig. 2 identifies several interesting features. First, only two short insertions into PYP are required: between residues 87 and 88 (PYP numbering is used throughout) and between residues 98 and 99. Second, a single short gap in SIM, TRH. and HIF-1α occurs near the N terminus. Third, PYP C69, which carries the chromophore, is deleted. Fourth, the following residues mostly are conserved among all sequences: V4, D34, G37,139, N43, G51, P54, V57, I58, G59, K60, N61, F63, P68, D71, F79, F92, Y94, V120, and F121. Fifth, the following residues represent the major differences between PYP and the PAS domains: G29, Y42, E46, G47, R52, D65, A67, T/A70, E/D93, and D/A97. These sequence conservations and differences are clarified at the 3D level, based upon the PYP structure.
Structural Consequences of Sequence Conservation and Differences. As PAS domains function to mediate macromolecular interactions along signalling pathways, the development and analysis of 3D structural models for specific PAS/ PYP modules is immediately useful for designing experiments to probe function. We explored the potential structural and functional consequences of significant sequence similarities and differences by examining the structural roles of the conserved residues in PYP (Fig. 3) and by mapping residues exhibiting sequence conservation and major sequence differences onto the molecular model of the ARNT PAS B domain (Fig. 4).
In general, both the greatest sequence conservation (Figs. 2 and 3) and the most striking sequence differences (Fig. 2) occur within the PAS core (Fig. 4), which represents the “active site” and chromophore ligand-binding region of PYP. Each of the 20 natural amino acids does not have the same probability of being structurally conserved. Pro residues, because of their backbone conformational restriction, and Gly residues, especially those with left-handed backbone conformations, often are evolutionarily conserved within a protein family. Both Gly and Pro residues frequently contribute to kinks and turns in the polypeptide chain. Pro 54 in PYP is
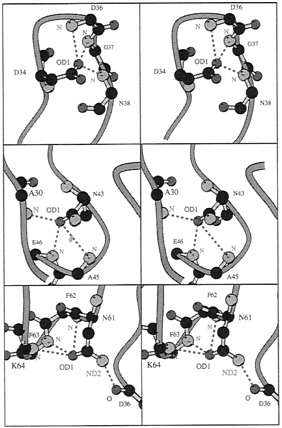
FIG. 3. Detailed residue interactions for conserved residues that form specific side-chain to main-chain hydrogen bonds in PYP and appear to be retained in PAS-containing molecules. First, Asp 34 OD1 hydrogen bonds to three backbone nitrogens from residues D36, G37, and N38 (2.87 Å, 3.08 Å and 2.89 Å, respectively). Most residues at position 34 have an atom OD1 (Asp, Asn) or similar (OG1 in Ser, Thr). Second, Asn 43 OD1 hydrogen bonds to three backbone nitrogen atoms: A30, A45, and E46 (2.96 Å, 3.55 Å, and 3.06 Å). All residues 43 in Fig. 2 have an atom OD1 or OE1. Third, Asn61 OD1 and ND2 hydrogen bond to three backbone nitrogens and one backbone oxygen: F62. F63, K64, and D36 (3.39 Å, 3.09 Å, 3.00 Å, and 2.96 Å). Almost all residues 61 in Fig. 2 have an atom OD1 or OG1. Drawing made with the program MOLSCRIPT (46).
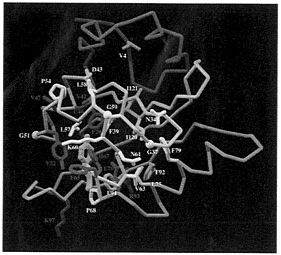
FIG. 4. The PAS-domain of the human ARNT protein modeled from the PYP crystal structure (2phy.pdb). The Cα trace is represented by a tube. The N-terminal cap is colored in magenta, the PAS-core in gold, the helical connector in green, and the β-scaffold in blue. Conserved side chains between PYP and PAS-containing proteins are displayed in white. Amino acids that significantly vary between PYP and PAS-containing molecules are drawn in red. PYP’s chromophore is displayed in yellow in the same orientation as in the PYP molecule. Most conserved residues are located in the hydrophobic core of the PAS-core domain (in gold). Most of significantly variant amino acids (in red) occur in the vicinity of the chromophore pocket as expected from molecules that carry different biological functions. In the ARNT model, H67 and F29 occupy the chromophore pocket. Figure displayed using the Application Visualization System (AVS) (AVS, Waltham, MA).
conserved in all PAS domains shown in Fig. 2. PYP has five left-handed Gly residues: G7 and G59 participate in type II β-turns, G37 mediates a β-bulge, G51 ends α3, and G86 ends the helical connector α5. In the PAS domain proteins of Fig. 2, P54 is conserved completely; G51 and G59 of PYP are conserved predominantly or substituted with residues (Asp, Asn, and Glu) that are fairly tolerant of left-handed backbone conformations; G7 and G37 of PYP also are mostly conserved but show more unusual substitutions; and the remaining PYP residues with left-handed backbone conformations (G86 and Q99) occur near regions tolerant of insertions, making analysis of their conformations difficult.
Other residues likely to be conserved are those that make critical hydrogen bonds required either for proper folding or for stabilizing a particular fold. In particular, residues that form key side-chain hydrogen bonds to main-chain atoms of other residues are expected to be conserved. Residues D34, N43, K60, and N61 all serve this function in PYP, and their counterparts in the PAS domains of Fig. 2 should be able to maintain this hydrogen-bonding function. Asp 34 in PYP can make up to three hydrogen bonds to peptide nitrogen atoms from residues 36–38 (Fig. 3, Top), This requires an appropriately positioned hydrogen-bond acceptor atom like OD1 of Asp or OD1 from Asn (ARNT, TRH, Fig. 3, Top) or OG1 from Ser/Thr (PER, CLOCK, HIF-1α). Conversely, the Asn at position 43 also can make up to three hydrogen bonds to nitrogen backbone atoms of residues 30, 45, and 46. This particular interaction also can be mediated by the atom OD1 of the Asp in ARNT, PER, Clock, MOP3, SIM and HIF-1α or an OE1 from a Glu in TRH (Fig. 3, Middle). In these two examples, hydrogen bonds stabilize a turn structure. The last example involves hydrogen bonds between a conserved Asn in PYP and ARNT at position 61 where both their hydrogen bond donor and acceptor atoms can make contacts with other backbone atoms (Fig. 3, Bottom). Although, this Asn is not fully conserved in every PAS domain (Fig. 2), the hydrogen bonds to OD1 that mediate π helix formation are conserved by most residues at position 61 (Asn/Ser/Thr residues, Fig. 3, Bottom).
The segments of the PYP protein outside of the PAS core exhibit lesser sequence conservation. Here, those residues that are conserved across all or most of the PAS domain protein families (Fig. 2) generally have identifiable roles in defining the secondary and tertiary structure of PYP. Both the helical connector and the β-scaffold show lesser sequence conservation (Fig. 2), consistent with their apparent role in maintaining appropriate secondary structural elements for the PAS/PYP module. The largest length variations in the sequence alignment are located at PYP positions 87–88 in the helical connector, which was observed previously to differ among PAS domains (5). The only other insertion in PYP maps to the turn joining the first two strands (β4 and β5 of the β-scaffold, Figs. 2 and 4). This insertion, found in every PAS domain sequence in Fig. 2, may highlight a significant structural difference distinguishing PYP from other PAS-containing proteins. The N-terminal cap, which is located on the opposing side of the module from the PYP active site, appears the least conserved among PAS domain proteins and in some cases may be substituted with other structures capable of protecting the central β-sheet from solvent.
Hydrophobic residues are usually buried in the core of a protein and therefore should show some conservation of their hydrophobic character. However, unlike the specificity of particular side-chain to main-chain hydrogen bonds, the non-specific nature of hydrophobic packing allows more liberty in sequence variation. In particular, complementary substitutions of hydrophobic residues to maintain a well packed hydrophobic core is frequently observed (41). Hydrophobic core residues conserved between PYP and PAS domains include: V4, 139, V57, I58, F63, F79, F92, Y94, V120, and F121, which are respectively conserved as or replaced by V4, V/F39, L/V57, L/V58, Y/M/V/L63, F/Y/H79, F/Y92, L/M/F/A94, I/F120, and I/V121 in PAS domains (Figs. 2 and 4).
PAS Domain Differences in the PYP Chromophore Environment. Although PYP and PAS-containing proteins share sensor and signal transduction functions, PYP incorporates both functions within a single, small, globular domain of 125 residues, whereas the much larger multidomain PAS-containing proteins like the phytochromes segregate these functions to different domains. The covalently bound 4-hydroxycinnamoyl chromophore of PYP that is necessary for light-mediated negative phototaxis is not found in any of the well characterized PAS domain proteins. Hence, it is expected that residues of the chromophore environment will differ between PYP and PAS domains. Indeed, most of the PYP residues that are not shared with the other PAS domain proteins interact with the chromophore. In the PAS domain proteins in Fig. 2, chromophore-bound PYP Cys69 is deleted, the inward-facing hydrophilic side chains (Y42, E46, and T50) that form the hydrogen-bonding network stabilizing the phenolic hydroxyl at the buried tip of the 4-hydroxycinnamoyl chromophore (27, 42) are replaced with hydrophobic residues, and the R52 side chain that forms the gateway of the chromophore to solvent (27, 36, 42) is converted to Tyr.
In the ARNT model (Fig. 4), the cavity created by the absence of the PYP chromophore is partly filled by very conserved H67, which can make a buried salt bridge with the conserved E/D70 in the PAS domains (Fig. 4). However, a predominantly hydrophobic cavity about one-half the size of a heme remains, caused by the reduction in size of residues Y42, F62, and F96 in PYP becoming V42, I62, and S96 in ARNT. This cavity might provide insights into the ligand-binding properties of the PAS domains including possible specific
hydrophilic interactions with conserved residues H67 and E70. Interestingly, PAS-conserved ARNT F29, replacing PYP G29, also can be directed into the central cavity because PYP E46 has been replaced with a smaller hydrophobic residue (Cys in ARNT). This explains why G29 was identified as PYP-specific during a BLAST search, as described above. A large hydrophobic residue at this location can help to fill the void left by the absence PYP’s chromophore.
The last major sequence differences between PYP and PAS-containing proteins in the chromophore environment are located at positions 93 and 97, where negative charges are replaced by positive charges. These residues are part of a surface array of positive charges that includes R93, R95, K97, W99, and W101 in ARNT and in most PAS domains. These residues are very close to the insertion located at position 98 in PYP (Fig. 4), which was modeled to be a turn (consistent with predictions by the program DSSP; ref. 43). In PYP, this turn is positioned at the entrance of the chromophore pocket and may be important for a putative interaction with a partner to accomplish the role of PYP in signal transduction.
Protein-Protein Dimerization Interface. During PYP’s light cycle, the chromophore undergoes a trans-to-cis isomerization and the protein rearranges slightly to accommodate the new chromophore configuration (36). The largest movements, taking place in the chromophore and the side chain of R52, presumably send the signal to the unknown downstream partner of PYP. leading eventually to negative photoaxis. Therefore, the surface of PYP surrounding these moving residues (Fig. 4) likely provides the interaction face for heterodimerization with the signaling partner of PYP. In other PAS-containing proteins, the PAS domains mediate homo- or hetero-dimer formation, in most cases with other PAS domains. We suggest that the interaction face of PYP is the prototype for the dimerization interface of the PAS domain protein super family.
This putative dimerization interface includes residues from three regions: (i) a central region (51–68), that includes residues located within 10 Å of Y52, (ii) the loop 95–103, that includes the insertion in the β scaffold, and (iii) two residues from α3 in the PAS core domain that are H44 and R45 in ARNT. Residues that are exposed to solvent are displayed in Fig. 5A. The molecular surface area of these regions are shown in Fig. 5B. Exposed side chains are H44, R45, Y52, Q53, Q55, E56, K60, F65, R95, K97, N98, Q98A, E98B, W99, W101, and R103. Almost all of the residue types found in this interface can form specific contacts to other amino acids and are characteristic of other protein-protein interfaces. The loop composed of residues 95–103, which differs between PYP and the PAS-containing proteins in Fig. 2. might be involved in the recognition specificity for PAS dimerization because of the low sequence homology among PAS domains in this area (Fig. 2).
DISCUSSION
Although sequence comparison alone is insufficient to demonstrate the proposed structural similarities between PYP and the PAS domains, low sequence homology was expected because of the evolutionary diversity among PAS-containing proteins, which populate all three kingdoms of life. Instead, the similarity and potential homology identified between PYP and the PAS domain superfamily is corroborated by finding that a modified PYP sequence, replacing only three residues specific to PYP function, allows automated selection of several PAS domain sequences from a nonredundant protein sequence database, albeit with resulting low scores, as expected. The potential homology between PYP and PAS domains is further supported by our ability to generate from the PYP crystallographic structure, a well behaved molecular model for the PAS domain from ARNT, a member of the PAS domain superfamily. Indeed, only two insertions and a single deletion were
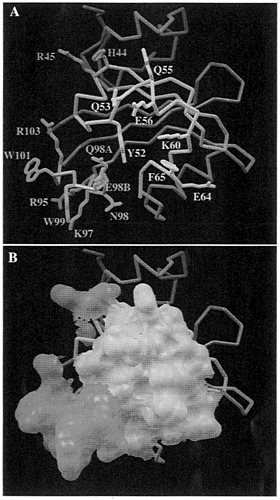
FIG. 5. Predicted PAS functional interactions. (A) Amino acid side chains that might participate in a protein-protein interaction are highlighted. The central segment, formed by residues 51–68, is located within 10 Å of the residue Y52 (in yellow). The second area, from residue 95–103 (in cyan), is made of a loop in which an insertion occurs at position 98 in each PAS-containing molecule. The third area is made by two residues (orange) adjacent to the central segment (yellow). H44 and R45, for which their side chains point toward the solvent. (B) same as A but the molecular surface for these residues is displayed.
needed for building a 3D model of the PAS-B domain of human ARNT, which exhibits a root-mean-square deviation of 0.76 Å from PYP’s 3D structure, and comparable quality and stability. Moreover, from the resulting molecular model of the ARNT PAS domain, it is possible visually to identify and understand similarities and differences between PYP and the PAS domains, supporting the proposition for a PAS/PYP prototypical fold. Given that the PAS/PYP module hypothesis is valid, the 3D model of the ARNT PAS-B domain provides useful insights concerning the two major known functions of PAS domains: protein-protein interaction and ligand binding.
Protein-protein interactions between the ubiquitous partner ARNT and PAS-containing proteins AHR, SIM, MOPs, TRH, and HIF-1α (9–11, 13, 14, 44, 45) have been identified in vitro and in vivo. These interactions are mediated by both the bHLH region and the PAS domains. Because the bHLH region is a self-dimerizing structural motif (3), the PAS domains evidently
supply the recognition specificity needed for these interactions, rather than driving the interactions per se. This hypothesis is supported by recent work from Zelzer and coworkers (14) revealing that the swapping of PAS domains between SIM and TRH confers the functional specificity of the PAS domain, rather than that of the parent protein. Based on the experimentally determined structures of PYP light-cycle intermediates (36), we propose that the region of the PAS/PYP fold that is involved in protein-protein interaction is centered around residue 52, as shown in Fig. 5. This hypothesis can be tested by site-directed mutagenesis of residues highlighted in this interface (Fig. 5).
Several PAS-containing proteins bind ligands and/or cofactors. To date, however, mapping the ligand binding to the PAS domain itself has been demonstrated only for the FixL (16, 21, 4) and AHR (15) proteins. The PAS domain of FixL binds heme, whereas the PAS-B domain of AHR binds dioxin and other poly-cyclic aromatic hydrocarbons. Interestingly, for both molecules, the minimum size of the PAS domain that is able to bind the ligand is ~130 residues, the size of the entire PAS/PYP module. Because of a reduction in size of several side chains in ARNT compared to PYP, the 3D model of the ARNT PAS domain displays an internal cavity large enough to accommodate a medium-sized ligand (one-half a heme). Thus, the region previously occupied by the PYP’s chromophore is a logical choice for a ligand pocket.
In summary. PYP appears to exhibit all of the major structural and functional features characteristic of the PAS domain superfamily: a modular domain of ~125–150 residues, a sensor function linked to ligand or cofactor binding, and signal transduction capability governed by heterodimeric assembly. Thus, we propose the testable hypothesis that the entire PYP protein fold is the structural prototype for the modular, 3D, PAS-A and PAS-B domain folds in PAS-containing proteins. This PAS/PYP module provides a structural model to guide experimental testing of hypotheses regarding ligand-binding, dimerization, and signal transduction in PAS proteins.
We thank Christopher Bruns and Christopher D.Putnam for preliminary sequence searches, alignments, and analyses; C.Bruns for help with the illustrations; and C.Bruns, C.D.Putnam, Ulrich K. Genick, and John Tainer for useful criticism and discussion. Research on PYP is funded by the National Institutes of Health Grant GM37684 (to E.D.G.).
1. Nambu, J.R., Lewis, J.O., Jr., Wharton, K.A. & Crews, S.T. (1991) Cell 67, 1157–1167.
2. Crews, S.T., Thomas, J.B. & Goodman, C.S. (1988) Cell 52, 143–151.
3. Hoffman, E.C., Reyes, H., Chu, F.-F., Sander, F., Conley, L. R., Brooks, B.A. & Hankinson, O. (1991) Science 252, 954–958.
4. Ponting, C.P. & Aravind, L. (1997) Curr. Biol. 7, 674–677.
5. Zhulin, I.B., Taylor, B.L. & Dixon, R. (1997) Trends Biochem. Sci. 22, 331–333.
6. King, D.P., Zhao, Y., Sangoram, A.M., Wilsbacher, L.D., Tanaka, M., Antoch, M.P., Sleeves, T.D.L., Vitaterna, M.H., Kornhauser, J.M., Lowrey, P.L., et al. (1997) Cell 89, 641–653.
7. Kay, S.A. (1997) Science 276, 753–754.
8. Huala, E., Oeller, P.W., Liscum, E., Man, I.-S., Larsen, E. & Briggs, W.R. (1997) Science 278, 2120–2123.
9. Huang, Z.J., Edery, I. & Rosbash, M. (1993) Nature (London) 364, 259–262.
10. Lindebro, M.C., Poellinger, L. & Whitelaw, M.L. (1995) EMBO J. 14, 3528–3539.
11. McGuire, J., Coumailleau, P., Whitelaw, M.L., Gustafsson, J.-A. & Poellinger, L. (1996) J. Biol. Chem. 270, 31353–31357.
12. Jiang, B.-H., Rue, E., Wang, G.L., Roe, R. & Semenza, G.L. (1996) J. Biol Chem. 271, 17771–17778.
13. Hogenesch, J.B., Chan, W.K., Jackiw, V.H., Brown, R.C., Gu, Y.-Z., Pray-Grant, M., Perdew, G.H. & Bradfield, C.A. (1997) J. Biol. Chem. 272, 8581–8593.
14. Zelzer, E., Wappner, P. & Shilo, B.Z. (1997) Gene Dev. 11, 2079–2089.
15. Fukunaga, B.N., Probst, M.R., Reisz-Porszasz, S. & Hankinson, O. (1995) J. Biol. Chem. 270, 29270–29278.
16. Monson, E.K., Weinstein, M., Ditta, G.S. & Helinski, D.R. (1992) Proc. Natl. Acad. Sci. USA 89, 4280–4284.
17. Meyer, T.E. (1985) Biochim. Biophys. Acta 806, 175–183.
18. Meyer, T.E., Yakali, E., Cusanovich, M.A. & Tolin, G. (1987) Biochemistry 26, 418–423.
19. Meyer, T.E., Tollin, G., Hazzard, J.H. & Cusanovich, M.A. (1989) Biophys. J. 56, 559–564.
20. Sprenger, W.W., Hoff, W.D., Armitage, J.P. & Hellingwerf, K.J. (1993) J. Bacteriol. 175, 3096–3104.
21. Lagarias, D.M., Wu, S.-H. & Lagarias, J.C. (1995) Plant Mol. Biol. 29, 1127–1142.
22. Linden, H. & Macino, G. (1997) EMBO J. 16, 98–109.
23. Crosthwaite, S., Dunlap, J.C. & Loros, J.J. (1997) Science 276, 753–754.
24. Feng, D.F. & Doolittle, R.F. (1987) J. Mol Evol. 25, 351–360.
25. Devereux, J., Haeberli, P. & Smithies, O. (1984) Nucleic Acids Res. 12, 387–395.
26. Hahn, M.E., Karchner, S.I., Shapiro, M.A. & Perera, S.A. (1997) Proc. Natl. Acad. Sci. USA 94, 13743–13748.
27. Borgstahl, G.E.O., Williams, D.R. & Getzoff, E.D. (1995) Biochemistry 34, 6278–6287.
28. McRee, D.E. (1992) J. Mol. Graphics 10, 44–47.
29. Tuffery, P., Etchebest, C., Hazout, S. & Lavery, R. (1991) J. Biomol Struct. Dyn. 8, 1267–1289.
30. Brünger, A.T. (1992) X-PLOR, A system for X-ray crystallography and NMR (Yale University, New Haven, CT), Version 3.1.
31. Powell, M.J.D. (1977) Math. Program. 12, 241–254.
32. Brooks, B., Bruccoleri, R., Olafson. B., States, D., Swaminathan, S. & Karplus, M. (1983) J. Comp. Chem. 4, 187–217.
33. Brünger, A.T. & Karplus, M. (1988) Proteins 4, 148–156.
34. Roussel, A. & Cambillau, C. (1989) TURBO-FRODO in Silicon Graphics Geometry Partners Directory (Silicon Graphics, Mountain View, CA), Version 5.2.
35. Laskowski, R.A., MacArthur, M.W., Moss, D.S. & Thornton, J.M. (1993) J. Appl. Crystallogr. 26, 283–291.
36. Genick, U.K., Borgstahl, G.E.O., Ng, K., Ren, Z., Pradervand, C., Burke, P.M., Srajer, V., Teng, T.-Y., Schildkamp, W., McRee, D.E., et al. (1997) Science 275, 1471–1475.
37. Leszczynski, J.F. & Rose, G.D. (1986) Science 234, 849–855.
38. Baca, M., Borgstahl, G.E.O., Boissinot, M., Burke, P.M., Williams, D, R., Slater, K.A. & Getzoff, E.D. (1994) Biochemistry 33, 14369–14377.
39. Wang, G.L., Jiang, B.-H., Rue, E.A. & Semenza, G.L. (1995) Proc. Natl. Acad. Sci. USA 92, 5510–5514.
40. Kallio, P.J., Pongratz. I., Gradin, K., McGuire, J. & Poellinger, L. (1997) Proc. Natl. Acad. Sci. USA 94, 5667–5672.
41. Getzoff, E.D., Tainer, J.A., Stempien, M.M., Bell, G.I. & Hallewell, R.A. (1989) Proteins Struct. Funct. Genet, 5, 322–336.
42. Genick, U.K., Devanathan, S., Meyer, T.E., Canestrelli, I.L., Williams, E., Cusanovich, M.A., Tollin. G. & Getzoff, E.D. (1997) Biochemistry 36, 8–14.
43. Kabsch, W. & Sander, C. (1983) Biopolymers 22, 2577–2637.
44. Ohshiro, T. & Saigo, K. (1997) Development (Cambridge, U.K.) 124, 3975–3986.
45. Sonnenfeld, M., Ward, M., Nystrom, G., Mosher, J., Stahl, S. & Crews, S. (1997) Development (Cambridge, U.K.) 124, 4571–4582.
46. Kraulis, P.J. (1991) J. Appl. Crystallogr. 24, 946–950.






