
Proc. Natl. Acad. Sci. USA
Vol. 95, pp. 5942–5949. May 1998
Colloquium Paper
This paper was presented at the colloquium “Computational Biomolecular Science,” organized by Russell Doolittle, J.Andrew McCammon, and Peter G.Wolynes, held September 11–13, 1997, sponsored by the National Academy of Sciences at the Arnold find Mabel Beckman Center in Irvine, CA.
Electrostatic steering and ionic tethering in enzyme-ligand binding: Insights from simulations
REBECCA C.WADE*, RAZIF R.GABDOULLINE, SUSANNA K.LÜDEMANN, AND VALÈRE LOUNNAS
European Molecular Biology Laboratory, Meyerhofstrasse 1, 69117 Heidelberg, Germany
ABSTRACT To bind at an enzyme’s active site, a ligand must diffuse or be transported to the enzyme’s surface, and, if the binding site is buried, the ligand must diffuse through the protein to reach it. Although the driving force for ligand binding is often ascribed to the hydrophobic effect, electrostatic interactions also influence the binding process of both charged and nonpolar ligands. First, electrostatic steering of charged substrates into enzyme active sites is discussed. This is of particular relevance for diffusion-influenced enzymes. By comparing the results of Brownian dynamics simulations and electrostatic potential similarity analysis for triose-phosphate isomerases, superoxide dismutases, and β-lactamases from different species, we identify the conserved features responsible for the electrostatic substrate-steering fields. The conserved potentials are localized at the active sites and are the primary determinants of the bimolecular association rates. Then we focus on a more subtle effect, which we will refer to as “ionic tethering.” We explore, by means of molecular and Brownian dynamics simulations and electrostatic continuum calculations, how salt links can act as tethers between structural elements of an enzyme that undergo conformational change upon substrate binding, and thereby regulate or modulate substrate binding. This is illustrated for the lipase and cytochrome P450 enzymes. Ionic tethering can provide a control mechanism for substrate binding that is sensitive to the electrostatic properties of the enzyme’s surroundings even when the substrate is nonpolar.
Conceptually, the process of ligand-protein binding may be considered to consist of the following consecutive steps: 1, diffusion of the ligand to the entrance to the binding site on the protein surface; 2, diffusion of the ligand through the protein to the binding site; 3, rearrangement of the ligand in the binding site into its bound orientation (see Fig. 1). In some cases, the binding site is situated on the protein surface and diffusion through the protein is not necessary. Step 1 may involve diffusion in reduced dimensions or the ligand may be actively transported to the protein surface. However, in general the above three steps should be considered and electrostatic interactions can influence all three. Here, their role in the first two steps is considered.
The important influence of electrostatic interactions on the rates of diffusion of charged substrates toward the active sites of enzymes is now well established (1–3). Electrostatic steering is of greatest importance for diffusion-controlled enzymes because it is one of the main factors determining the catalytic rate. For these enzymes, the postdiffusional steps of the reaction have been so optimized that the diffusional association of substrate and protein has become the rate-limiting step.

FIG. 1. Schematic diagram showing how electrostatic interactions can influence the binding of a ligand (shaded) to a protein (outline). Step 1, electrostatic forces and torques can steer the ligand into its binding site on the protein. Step 2, electrostatic interactions such as salt links can affect the protein dynamics necessary for ligand access to binding sites shielded from solvent in “gated” binding. Step 3, electrostatic interactions, particularly salt links and hydrogen bonds, between ligand and protein can contribute to binding affinity and specificity and to the structural binding mode of the complex formed.
Enhancement of the diffusional association rates can be achieved by attractive electrostatic interactions between the substrate and the protein binding site. Here, we ask what enzyme features are necessary for electrostatic steering resulting in rapid ligand-protein association rates. By examining orthologs from different species, by means of Brownian dynamics (BD) simulations (4) and electrostatic potential similarity analysis, we identify common features important for their shared molecular function and find that these are confined to the close vicinity of the active site.
The role of electrostatic interactions in the next two steps of the ligand-protein binding process is more complex. Here, we focus on one type of interaction, which we shall term “ionic tethering.” This entails the formation of salt links between charged residues in the protein that affect conformational changes in the protein associated with or necessary for ligand binding. Salt-link formation between charged groups in the ligand and the protein can also contribute to ligand binding, but this will not be considered here. Instead, uncharged ligands, which cannot themselves engage in salt links, will be examined. Nevertheless, we show the importance of electrostatic interactions for the binding of nonpolar ligands and making binding sensitive to the surrounding environment. We
©
PNAS is available online at http://www.pnas.org.
| Abbreviations: BD, Brownian dynamics: BLAC. β-lactamase; SOD. superoxide dismutase; TIM, triose-phosphate isomerase. |
* | To whom reprint requests should be addressed. e-mail: wade@emblheidelberg.de. |
discuss possible mechanisms in the light of two examples that have been the subjects of recent calculations.
ELECTROSTATIC STEERING
One way to identify the features important for the electrostatic steering of a substrate toward its binding site on an enzyme is by site-directed mutagenesis. This approach has been employed to examine the fast association rates of superoxide dismutase (SOD) and acetylcholinesterase with their substrates, and barnase with the inhibitor barstar.
-
For human SOD, mutations were identified by using BD simulations to improve electrostatic steering, and, indeed, when the mutations were made, greater electrostatic enhancement of the rate was observed (5). resulting in a “superperfect” enzyme (6).
-
For acetylcholinesterase, a large number of mutations of charged residues was made, and these were shown to have little effect on the rate of substrate binding (7). This result was interpreted as evidence of lack of diffusion control and electrostatic steering. However, the rates for the mutants could be well reproduced in BD simulations, which demonstrated enhancement of rates because of electrostatic steering of substrate toward and inside the substrate-binding gorge (8, 9).
-
Barstar is the intracellular protein inhibitor of the extracellular ribonuclease barnase, and it binds very tightly with high on-rates. Even so, the rate of binding could be improved by mutation (10). The effects of mutations and ionic strength on the association rates could be well reproduced by BD simulations (11). The data show the dominance of certain residues on the protein binding faces in determining the electrostatic enhancement of the association rate.
Together, these results indicate that electrostatic enhancement of association rates arises mostly from the presence of a few charged residues close to the binding site.
Here, we take an alternative approach to site-directed mutagenesis, namely, comparison of diffusion-influenced enzymes from different species to find out what is required for electrostatically enhanced substrate binding rates. By relying on natural evolution, we are assured of examining fully functioning enzymes although they may not be fully optimized for electrostatic enhancement of substrate on-rates or fast reaction, as this may not be desirable in their in vivo environment. We examine three families of diffusion-influenced proteins, triose-phosphate isomerases (TIM), Cu,Zn-superoxide dismutases (SOD), and class A β-lactamases (BLAC), for which crystal structures are available from several organisms and whose kinetic properties have been measured (see Table 1).
Diffusional Control of Catalytic Rates. The primary indicator for diffusion control of an enzyme reaction is a fast catalytic rate that is dependent on the viscosity and ionic strength of the solvent. Both TIM and SOD are extremely fast, efficient enzymes with the rate-limiting step of their reactions under physiological conditions being the diffusion of substrate, glyceraldehyde 3-phosphate and superoxide, respectively, to the active site, Indeed, TIM has been described as a “perfect enzyme” (12, 13). The catalytic rates measured for TIMs from more than five species are all about 108 M–1·s–1 at 100 mM ionic strength (see ref. 14 and references in ref. 15), and viscosity dependence of the rates has been demonstrated (16). The catalytic rate has been measured for SODs from more than eight species, and all have rates of about 3×109 M–1·s–1 at 20 mM ionic strength (see references in ref. 17). The rates of SODs exhibit ionic strength dependence and decrease as the ionic strength increases (18). BLACs have been characterized as fully efficient enzymes with no single rate-determining step (19). They are partly diffusion-controlled for good substrates, such as benzylpenicillin with a single negative charge, and most have catalytic rates of 107 to 108 M–1·s–1 for such substrates at 100 mM ionic strength (20, 21).
BD Simulations. Experimental association rates were reproduced well for six variants of SOD (17) and four variants of TIM (15) by BD simulation. These results show that the main features influencing the catalytic rates are represented in the simulation model. The protein is represented by all atoms observed crystallographically plus modeled polar hydrogen atoms, with each atom assigned a partial charge and a van der Waals radius. The protein is immersed in a uniform solvent continuum. The electrostatic potential of the protein is computed from numerical solution of the finite-difference linearized Poisson-Boltzmann equation (22). The substrate is represented by a charged sphere (for SOD) or dumbbell (for TIM). The molecules are treated as rigid, and intermolecular hydrodynamic interactions are neglected. Comparison of simulations with and without a net charge on the substrate show that electrostatic interactions enhance the association rates for all the enzyme variants studied.
Electrostatic Potential Similarity Analysis. To quantify the common features in the electrostatic potentials of different variants of the enzymes, we carried out an electrostatic po-tential similarity analysis. The members of each family of enzymes were superimposed by matching α-carbons. Then
Table 1. Properties of the diffusion-influenced enzymes triose-phosphate isomerase (TIM), superoxide dismutase (SOD), and β-lacktamase (BLAC)
|
|
TIM |
SOD |
BLAC |
|
Substrate |
Glyceraldehyde 3-phosphate |
Superoxide |
Benzylpenicillin |
|
Net charge of substrate, c |
–1/–2 |
–2 |
–1 |
|
No. of protein variants compared |
4 |
6 |
4 |
|
Variants compared together with Protein Data Base identifier code (listed in order of increasing net charge) |
E.coli (ltre), yeast (lypi), chicken muscle.* T.brucei (5tim) |
Spinach (Isrd), frog (Ixso), yeast (Isdy), human (Ispd), bovine (2sod), P.leiognathi (lyai) |
E.coli (TEM-1) (Ixpb), B.licheniformis (4blm), S.albus G,† S.aureus (3blm) |
|
Net charges of proteins at neutral pH. e |
–12, –6, –2, +12 |
–8, –6, –4, –4, –2, +2 |
–6, –6, –4, +16 |
|
Range of sequence identity between proteins, % |
≈50 |
≈30–55 |
≈30–45 |
|
Measured kcatKm, M–1·s–1×10–8 |
1.0–8.4 |
25–39 |
0.03–0.8 |
|
Ionic strength for rate measurement, mM |
100 |
20 |
100 |
|
E.coli, Escherichia coli; T.brucei, Trypanosoma brucei; P.leiognathi, Photobacterium leiognathi; B.licheniformis, Bacillus licheniformis; S.albus, Streptomyces albus; S.aureus, Staphylococcus aureus. *Coordinates provided by P.Artymiuk. †Coordinates provided by O.Dideberg. |
|||
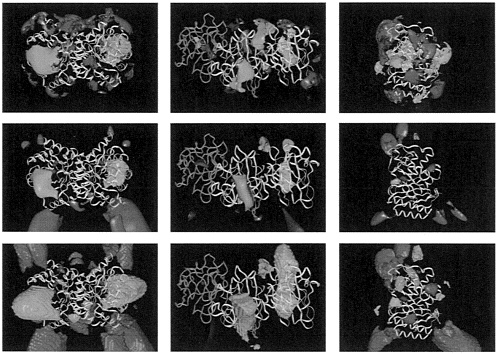
FIG. 2. Electrostatic potential comparison for variants of TIM (Left). SOD (Center), and BLAC (Right). (Top) Average potential contoured at ±0.4 kcal·mol– l·e–1 (1 kcal=4.184 kJ). (Middle) Similarity index with most conserved regions within contours at a level of 0.75 in all cases, except for the red contours in Center, which are at 0.85. (Bottom) Contours enclose regions where the sign of the electrostatic potential is conserved. In all cases, red represents regions of negative potential and blue represents regions of positive potential. Magenta solid spheres represent important active site-atoms: carboxylate oxygens of Glu-165 and amino nitrogen of Lys-13 in TIM. Cu and Zn ions in SOD, and the side chain of the catalytic Ser-70 in BLAC. The proteins are represented by ribbon plots of representative variants: chicken muscle TIM. bovine (yellow) and P.leiognathi (green) SOD, and TEM-1 BLAC. The dimers of all SODs studied except that from P.leiognathi superimpose well on the bovine SOD. Consequently, one monomer of P.leiognathi was superimposed on one monomer of bovine SOD instead of the complete dimer as done for the other SODs. Negative contours are not shown for the sign conservation plot in SOD (Center Bottom) for clarity. The similarity index, SI, is computed at points (i,j,k) around the proteins from the following formula, which is generalized to the comparison of N potentials. ϕ1, l=1, 2,…, N, from the Hodgkin formula for the comparison of two potentials (59):
SI=+1 when the potentials are all identical. SI=–1 when two potentials are opposite (N< 3). For small deviations, Δϕn, from the average potential, the decrease in the SI from its maximum (=1) is proportional to (Δϕn)2. This is because the SI can be rewritten as:
For example, when SI=0.85 for four potentials, ![]() The SI (and the average potential and sign conservation) are computed outside the molecules combined van der Waals volume as defined with atomic radii set at twice their normal values.
The SI (and the average potential and sign conservation) are computed outside the molecules combined van der Waals volume as defined with atomic radii set at twice their normal values.
average potentials, sign conservation, and similarity indices, as defined in the legend to Fig. 2. were computed as a function of position around the superimposed molecules. The results are shown in Fig. 2. The substrates considered are all negatively charged. The majority of the proteins are also net negatively charged. Thus, electrostatic enhancement of the association rates does not arise from nonspecific attraction between molecules due to monopole interactions. Instead, it arises from the nonuniform charge distribution of the proteins, which results in steering of the substrates toward the positively charged regions of the active sites.
The TIM and SOD enzymes considered here are homodimeric enzymes with two active sites, whereas BLAC is a monomer with one active site. The average potentials (Fig. 2 Top) all show attractive positive regions of potential over the active sites. On average, the electrostatic potential of the TIMs confine the substrate to a ring around the protein that includes both active sites. For BLAC, the positive active site potential is on average surrounded by a ring of negative potential and isolated from other positively charged regions of the protein surface.
The similarity index plots (Fig. 2 Middle) show the most conserved regions of the potentials for the variants of each
enzyme. The largest regions of positive potential are situated over the active sites for all enzymes. Regions of negative potential are also conserved away from the active sites but, as can be seen by comparison with the average potential maps, the potential in these regions is generally smaller in magnitude. Fig. 2 Bottom shows the regions where the sign of the potential is the same in all variants for each species. This gives an indication of the extent of the attractive potential acting on the substrate.
TIM In the TIMs, the positive potential at the active site is mostly because of the conserved active site Lys-13 and the conserved Lys-237 (chicken muscle TIM numbering). This conserved region of positive potential has a volume of about 6,000 Å3 and extends about 30 Å from the amino group of Lys-13 at 100 mM ionic strength. The large pincer-like regions of conserved, but small in magnitude, negative potential are mostly due to Glu-23, which is conserved in all four TIMs examined but not all known TIM sequences.
SOD. In the SODs, the positive potential over the active site channel is due primarily to the copper and zinc ions, the conserved Arg-141 (bovine SOD numbering), and a few other nearby positively charged residues that are not totally conserved. One of the SOD variants studied, that from the prokaryote Photobacterium leiognathi, displays a different dimerization mode from the other enzymes, which are eukaryotic (23). as shown in Fig. 2. It has a looser dimer interface than the other SODs, indicating that it may act partially as a monomer like the SOD from E.coli whose structure was solved very recently (24). Nevertheless, the structures of the monomers of all SODs are similar, and thus we compare the potentials around a superimposed monomer of each dimer. The region of attractive potential for the substrate is elongated, roughly following the shape of the active site cleft. It has a volume of ≈3,000 Å3 and extends up to 35 Å from the copper ion. While there is a common region of conserved positive potential as shown by the similarity index plot, the regions of positive potential in each of the proteins do not superimpose exactly. In particular, the P.leiognathi enzyme has a loop insertion known as the SSloop (green loop in the middle of Fig. 2 Top Center) containing two lysine residues, an aspartic acid, and a glutamic acid, that may compensate for the deletion of the 7,8 loop that is present in the other enzymes and contains one lysine and two glutamic acids. This increases the attractive potential near the SSloop (which is present to a lesser extent in the other SODs, as can be seen from the sign conservation map). It should also be noted that the magnitude of the positive potential in the active site is greater for the P.leiognathi enzyme than for the bovine enzyme, although its rate is identical (25). Although mutations have not yet been reported of charged residues in the SSloop. several studies of mutations of the charged residues in the 7,8 loop (Asp-130, Glu-131. Lys-134) and the nearby Lys-120 (not conserved in prokaryotic SODs) and Arg-141 have been made. Arg-141 has been shown to be particularly important electrostatically and mechanistically (26). The other residues are of lesser but significant importance for the catalytic rate, but the relative importance of each of the residues has been shown to differ in the different variants (5, 27).
BLAC. In the BLACs, the conserved positive potential runs along the active site cleft and is due primarily to Lys-234, which is conserved in all four enzymes, and Arg-244, which is present in all but the Streptomyces albus G enzyme (ABL numbering scheme). However the S.albus G BLAC has an arginine not present in the other three enzymes, Arg-220, whose guanidino group occupies a very similar position in the three-dimensional structure to that of Arg-244 in the other enzymes. While this appears to be a largely compensatory mutation, and is present in other BLAC sequences, it may be one of the reasons why the rate for the S.albus enzyme is lower than that of the other BLACs for benzylpenicillin. Lys-73, which is in the active site close to the catalytic Ser-70, is of less importance for the conserved attractive steering potential. The pKa of this residue is a subject of controversy (see ref. 28 and references therein), but here we note that neutralization of Lys-73 makes very little difference to the conserved positive potential region shown near the active site in Fig. 2 Middle Right. Lys-234, Arg-220, and Arg-244 have been the subject of mutational studies in several BLACs (20). These have shown that Lys-234 plays a role in both the initial recognition of the substrate and in the stabilization of the transition state, with the latter being dominant. Mutation to a nonpolar residue of Arg-244 or Arg-220 in the corresponding protein had a deleterious effect on the catalytic rate for charged substrates and indicated an important role for substrate binding. The conserved attractive potential region over the active site is much smaller for BLAC than for the other enzymes: it has a volume of about 200 Å3 and extends only 14 Å from the hydroxyl group of Ser-70.
Mechanistic Implications. Overall, these data show that the largest conserved regions of substrate-attracting electrostatic potential are near the active sites in all of the enzymes studied. This fact implies that the localized potentials at the binding site are sufficient for efficient electrostatic steering of substrate into the binding site. This is consistent with recent energetic analysis showing that the rate enhancement due to electrostatic interactions can be approximately estimated from the Boltzmann weighted average of the interaction energy in the binding site (29–31). However, the conserved local potentials vary in size and extent between the enzymes studied. While the volume of conserved attractive potential at the active site is twice as large for TIM as for SOD, it is much smaller for BLAC than for the other enzymes, indicating less electrostatic enhancement of rates, which is consistent with the lower measured rates for the BLACs. The local attractive potentials are largely provided by a few charged residues that are mostly highly conserved between orthologs. This permits enzymes with the same activity to have very different net charges (ranging from –12 to +12 e for the TIMs and –6 to +16 e for the BLACs examined). Thus, they can be highly efficient enzymes and fulfill different secondary functions or survive in different cellular environments.
IONIC TETHERING
Ionic Tethering Mechanisms. Ligand binding to proteins is frequently accompanied by conformational changes in the proteins such as loop motions, channel openings, or side-chain rotations. These may result in energy barriers to ligand binding and affect binding rates and their time dependence as described by gating theory (32, 33). We investigate the role of salt links as ionic tethers providing the protein with a means of controlling such conformational “gating.” Some of the possible mechanisms by which ionic tethers might act are as follows:
-
by thermodynamic stabilization of a conformation of the protein;
-
by kinetic stabilization of a conformation of the protein;
-
as devices to allow a certain degree of flexibility in the protein structure;
-
as devices dependent on pH. ionic strength, and dielectric of the environment; and
-
as devices to ensure specific interactions.
Protein folding is generally considered to be driven by hydrophobic interactions, and it is these interactions rather than interactions between charged groups (whose desolvation is unfavorable) that are thought to stabilize the folded states of proteins (34, 35). Nevertheless, comparison of the crystal structures of a number of proteins from mesophilic and thermophilic organisms shows that the latter often contain significantly more hydrogen bonds and salt links (36). More
over, calculations of the electrostatic free energy contribution to protein stability of salt links by using a classical continuum electrostatic model (34) show an overall tendency for salt links to be more stable in thermostable proteins than their mesostable counterparts (I. Shrivastava, V.L., and R.C.W., unpublished data). Stabilization due to the formation of ionic networks is indicated by mutational data (37) and calculations (38) showing that removal of salt links by mutation of their side chains tends to be more destabilizing when they participate in salt-link networks. These data suggest that, under certain conditions, salt links can thermodynamically stabilize a folded conformation of the protein relative to its fully or partially unfolded state. However, their increased presence in proteins from thermophilic organisms has also been attributed to “resilience” (39) or kinetic stabilization (40) which increases the kinetic barrier to unfolding. That is, if the protein is perturbed from its equilibrium structure, the long-range nature of charge-charge interactions in salt links will facilitate return to the original structure. Charge-charge interactions can be thought of as providing a smoother energy landscape funnel than short-range hydrophobic interactions. Thus, they can allow greater structural flexibility in proteins than hydrophobic interactions, although this is combined with greater specificity in the actual interactions formed. Moreover, the strength of salt links is dependent on the physical properties of the environment. Thus, protein conformational changes controlled by ionic tethers may be triggered because of a change in environment—e.g., pH, ionic strength, dielectric screen-ing—that alters the strength of a salt link.
Consider two situations: one in which a salt link tethers and stabilizes an enzyme in its active conformation under certain environmental conditions, and the other in which a salt link affects the opening of substrate-access and product-exit channels—i.e., gating perturbations from the equilibrium structure. In the former case, the ionic tether stabilizes one of several low-energy conformations. In the latter case, the ionic tether acts as a control of structural deviations from a low-energy conformation. We will illustrate the former by interfacially activated lipases and the latter by cytochrome P450s, which have buried active sites.
Lipase. Lipases catalyze the hydrolysis of uncharged ester substrates. Lipases undergo interfacial activation—i.e., their activity is greatly increased when they act on substrate at a lipid/water interface. Crystallographic studies (41–43) have shown that lipases possess a surface loop or “lid” over the active site that, upon activation, opens up to permit the binding of substrate. Upon opening, the lid’s hydrophobic face is exposed and its hydrophilic side is buried.
The properties of the active-site lid are probably best characterized in the lipase from Rhizomucor miehei. Crystal structures (41, 44) show that the main difference between the open and closed forms is the displacement of a helical lid of about 12 residues (see Fig. 3). The lid contains two charged residues, Arg-86 and Asp-91. In the open, inhibitor-bound form, Arg-86 is close to Asp-61. Evidence that these residues form an ionic tether that stabilizes the open form relative to the closed form in certain environments is provided by the following theoretical and experimental studies.
Molecular and Brownian dynamics simulations. The opening of the active-site lid has been simulated by molecular and Brownian dynamics (45–47). The time scale of the opening means that the loop must be artificially guided from closed to open states during the molecular dynamics simulations, which give information about the relative energies of the open and closed states. On the other hand, BD simulations, in which the lid is modeled as a simple chain of spherical residues rather than with an all-atom model, can be carried out on the relevant time scales and permit opening times to be estimated. The simulations show that opening of the lid is facilitated when the dielectric constant and polarity of the environment are re
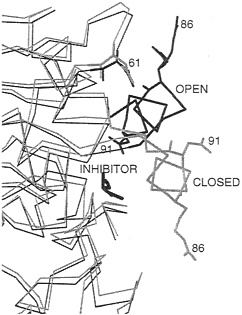
FIG. 3. Part of the α-carbon trace of two crystal structures of the lipase from R.miehei showing the positions of the helical lid in open (black) and closed (gray) forms of the enzyme. In the crystal structure of the open form of the enzyme, an inhibitor is bound in the active site. All non-hydrogen atoms are shown for selected titratable residues (numbered) involved in electrostatic interactions affecting the position of the helical lid. In the open form, Arg-86 in the lid is close to Asp-61.
duced, indicating that the open state is stabilized by electrostatic interactions. In BD simulations (46), the lid opens up in times on the order of 100 ns in a nonpolar low-dielectric medium, whereas the lid does not always open during simulations of 900 ns in a polar high-dielectric medium. BD simulations with Arg-86 and/or Asp-91 neutralized show their importance in activation, with the effect of Arg-86 being dominant. In the closed inactive conformation, Asp-91 experiences repulsive forces that tend to push the lid toward the active, open conformation. On opening, Arg-86 approaches Asp-61 to make a favorable ionic interaction stabilizing the open conformation. The crystal structure of the open form shows that the side chain of Arg-86 is disordered (41). Thus the interaction with Asp-61 does not result in the formation of a highly ordered hydrogen-bonded salt link but a less specific charge-charge interaction. The model in the BD simulations does not explicitly represent the side-chain atoms of Arg-86 and shows, therefore, that such less specific interactions can stabilize the open conformation of the lid.
Chemical modification and inhibition assays. Experimentally, the activity of the R.miehei lipase has been shown (48) to be reduced by chemical modification of arginines and the addition of guanidine before substrate. Chemical modification was shown to be greater for Arg-86 than for any other arginine in R.miehei. Inhibition by guanidine was not observed when the guanidine was added after addition of substrate, indicating that arginine residues are important only during activation. Inhibition experiments with guanidine also showed reduced activity for Hamicola lanuginosa and porcine pancreas lipases, although the reduction was smaller than for R.miehei lipase (70–88% vs. 26% residual activity) (48). Both these enzymes have arginine residues in the active-site lids, although at different positions from Arg-86 in the R.miehei lipase. No reduction in activity was observed for lipases without arginine
in the lid—e.g., Candida rugosa. The H.lanuginosa lipase has a glutamic acid (Glu-87) at the equivalent position to Arg-86 in the R.miehei lipase. Molecular dynamics simulations (47) indicate that this residue will make unfavorable electrostatic interactions in the open state that can be removed by neutralizing it. However, experiments show that the Glu-87→Ala mutant has decreased activity (35–70% residual activity) compared with wild type (49, 50). A possibility is that Arg-84 in the H.lanuginosa lipase approximately counters the effects of Glu-87 and makes favorable ionic interactions with the rest of the protein in the open state.
Role of ionic tethering in lipases. There are not strictly conserved charged residues in the lids that act as ionic tethers stabilizing the open form in the presence of a low-dielectric lipid and the closed form in high-dielectric aqueous solution. The lid residues, however, contribute to the different substrate specificities in different lipases. Thus, with the dual requirements of different substrate specificities and interfacial activation, lipases appear to have evolved alternative arrangements of charged residues (often arginines) in the lid to act as ionic tethers to control interfacial activation. Stabilization of the active lipase conformation is brought about, not by the formation of strong hydrogen bonds but by longer range precise charge-charge interactions, which may permit the enzyme more flexibility for efficient turnover. Such ionic tethers enable the binding of nonpolar substrates by the lipases to be sensitive to the electrostatic properties of its environment and contribute to the phenomenon of interfacial activation.
Cytochrome P450. Crystal structures show that the active site of cytochrome P450cam from Pseudomonas putida is buried in the protein, isolated from the solvent (51). Data for other cytochrome P450s (52) show that the active site is sometimes isolated from solvent and sometimes has an open channel to the active site lined on one side by a rather mobile F-G helix-loop-helix segment (see Fig. 4A). Clearly, in the case of cytochrome P450cam, protein motions are necessary for the substrate, camphor, to enter the active site. The binding of camphor to cytochrome P450cam can be considered as a two-step process: camphor first diffuses from the outside of the protein to the binding site, and then there is a low- to high-spin transition at the heme iron. Experiments show that the equilibrium constant for the diffusion step, and the accompanying enthalpy and entropy changes, are dependent on the dielectric constant and ionic strength of the surrounding solvent (53). The equilibrium constant is less sensitive to these properties when Asp-251 is mutated to Asn. Asp-251 participates in a tetrad of salt links in the crystal structure that connect the I helix, on which it sits, to the F helix (see Fig. 4A). This finding suggests that Asp-251 may influence substrate binding by participating in ionic tethers that regulate the opening and closing of the substrate access channel, which may involve motion of the F-G helix-loop-helix segment of the protein.
Electrostatic calculations of salt-link stability. To obtain an indicator of the energetic cost of perturbing the salt links to Asp-251, we computed their electrostatic contribution to protein folding stability by using a classical electrostatic continuum model (38). On average, the salt links in cytochrome P450cam [and in other proteins for which calculations have been done (34)], are neither stabilizing nor destabilizing. However, we found that the salt links to Asp-251 are exceptionally stable. The only salt links that were more stable were those to the propionate groups of the heme. This suggests that cytochrome P450cam has evolved particularly stable salt links to perform functional roles: keeping the heme group bound and regulating the opening and closing of the substrate/product access channel to the active site.
Thermal pathway analysis and molecular dynamics simulation. To probe the conformational changes for substrate access to and exit from the active site more explicitly, we performed two types of analysis: thermal pathway analysis and molecular
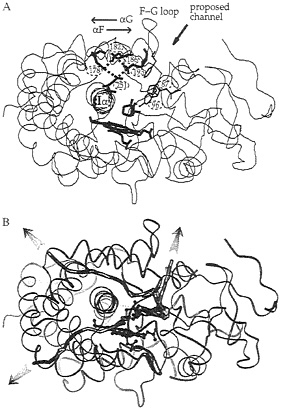
FIG. 4. Ribbon diagram of the crystal structure (51) of cytochrome P450cam with the buried heme and camphor substrate shown in bold. (A) The salt-link tetrad of residues involving Asp-251 is shown in bold. The region where a channel has been proposed, on the basis of crystallographic data (51, 60), to open up to allow ligand access to the active site is indicated. This channel is lined by aromatic residues whose side chains are shown (Tyr-96, Phe-87, and Phe-193). (B) Three representative camphor exit pathways derived by molecular dynamics simulation (54) are shown by thick lines that follow the position of the center of mass of the camphor as it escapes from the active site during the trajectories. The other trajectories simulated are clustered in the vicinity of each of these trajectories.
dynamics simulation (54). In thermal pathway analysis, the measured temperature factors in the crystal structures are analyzed to identify flexible regions where ligand channels may open (55). This analysis for cytochrome P450cam indicates three particularly mobile regions of the protein as candidates for ligand channels. Similar regions were located as exit channels for expulsion of camphor from the binding site during the molecular dynamics simulations. Representative trajectories for each of the three main channels are shown in Fig. 4B. These simulations were performed for times of approximately 100 ps. This is orders of magnitude less than the time it would actually take for camphor to escape from the active site. Therefore, simulations were performed with an additional artificial randomly oriented force applied to camphor to improve its sampling and enable it to find an exit channel in a short simulation time (54). The simulations show that perturbation of the salt links to Asp-251 is not necessary for expulsion of substrate from the active site, as their geometry is perturbed in only about half the trajectories generated. Surprisingly, relatively small and localized displacements of protein atoms, involving ≈0.5– to 2–Å shifts of backbone atoms and rotation
of a few side chains, are sufficient to permit camphor to escape from the protein.
Role of Asp-251 ionic tethers in substrate binding. What then is the reason for the dependence of camphor binding rates on salt links to Asp-251? Comparison of cytochrome P450cam with other cytochrome P450s indicates considerable flexibility in the F-G loop and the opening of a channel next to it as shown in Fig. 4A. This channel is the most often observed of the three classes of exit channel identified in the molecular dynamics simulations. Exit of camphor here during the simulations usually involves either perturbation of the F-G loop and Phe-193 or perturbation of the B' helix with rotation of Phe-87. Further evidence for this ligand channel is data from site-directed mutagenesis, which shows that mutation of Phe-87 to Trp reduces the camphor on-rate, mutation of Phe-193 to Cys increases the off-rate, and the introduction of Cys-Cys tethers to reduce the dynamics of the G helix affects on- and off-rates (56). A possible mechanism to explain the involvement of Asp-251 is that the salt links to Asp-251 regulate slower breathing motions of the protein by tethering the F-G helix-loop-helix to the I helix. These breathing motions allow for opening of the access channel, thus making the entrance and exit of substrate via the channel next to the F-G loop preferred over passage through the other channels identified in the calculations. Evidence for the modulation of the general dynamics of the protein by the salt links to Asp-251 also comes from photoacoustic calorimetry measurements of CO rebinding in cytochrome P450cam (57).
Role of ionic tethers in cytochrome P450s. Although residue 251 is conserved as Asp—or Glu—in cytochrome P450s, the salt links to Asp-251 are not conserved in the cytochrome P450s whose structures are available. However, in cytochrome P450cryf, Arg-185 in the G helix makes hydrogen bonds to the protein core that could affect the dynamics of the F-G flap in an analogous fashion to the salt-links to Asp-251 in cytochrome P450cam. This observation suggests that the control of protein dynamics permitting substrate access to the active site is tuned in each cytochrome P450 according to the particular substrates it acts upon and the efficiency and regio- and stereoselectivity required. In cytochrome P450s that bind large substrates, it may be possible to achieve sufficient desolvation of the catalytic site without the need to isolate the active site from the solvent. In cytochrome P450cam, a mechanism to bury the active site is likely to be crucial to its ability to catalyze a highly regio- and stereospecific reaction with remarkably little uncoupling side-reactions.
Insights into Ionic Tethering. The present examples demonstrate a role for ionic tethers in stabilizing an active conformation of an enzyme (lipase) under certain environmental conditions and regulating deviations from an enzyme’s (cytochrome P450’s) equilibrium structure affecting substrate binding. They make ligand binding sensitive to the electrostatic properties of the protein’s surroundings even when the ligand is nonpolar. The preceding example shows that the way in which ionic tethers affect ligand binding may be rather subtle. How widespread the phenomenon of ionic tethering is in proteins remains to be investigated. Ionic tethers are not involved in all major protein conformational transitions on ligand binding, but they can play a role in the binding of charged as well as uncharged ligands. For example, for sulfatebinding protein, it has been observed that two salt links affect kinetic dissociation rate constants by stabilizing the closed liganded form and modulating the rate of cleft opening (58). Further experimental and theoretical studies are necessary to fully understand the mechanisms of ionic tethering and its relation to ligand binding to proteins.
We thank Drs. P.Artymiuk and O.Dideberg for provision of coordinate sets. This work was partially supported by the European Union (Biotech CT94–2060). S.K.L. acknowledges an ErwinSchrödinger Fellowship granted by the Austrian Fonds zur Förderung der Wissenschaftlichen Forschung (JO1379-CHE).
1. Davis, M.E., Madura, J.D., Sines, J., Luty, B.A., Allison, S.A. & McCammon, J.A. (1991) Methods Enzymol. 202, 473–497.
2. Tan, R.C, Truong, T.N., McCammon, J.A. & Sussman, J.L. (1993) Biochemistry 32, 401–403.
3. Wade, R.C. (1996) Biochem. Soc. Trans. 24, 254–259.
4. Madura, J.D., Briggs, J.M., Wade, R.C. & Gabdoulline, R.R. (1998) in Encyclopedia of Computational Chemistry, eds. von Rague Schleyer, P., Allinger, N.L., Clark, T., Gasteiger, J., Kollman, P.A. & Schaefer, H.F. (Wiley, Chichester, U.K.), in press.
5. Getzoff, E.D., Cabelli, D.E., Fisher, C.L., Parge, H.E., Viezzoli, M.S., Banci, L. & Hallewell, R.A. (1992) Nature (London) 358, 347–351.
6. McCammon, J.A. (1992) Curr, Biol. 2, 585–586.
7. Shafferman, A., Ordentlich, A., Barak, D., Kronman, C., Ber, R., Bino, T., Ariel, N., Osman, R. & Velan, B. (1994) EMBO J. 13, 3448–3455.
8. Antosiewicz, J., McCammon, J.A., Wlodek, S.T. & Gilson, M.K. (1995) Biochemistry 34, 4211–4219.
9. Antosiewicz, J., Wlodek, S.T. & McCammon, J.A. (1996) Biopolymers 39, 85–94.
10. Schreiber, G. & Fersht, A.R. (1996) Nat. Struct. Biol. 3, 427–431.
11. Gabdoulline, R.R. & Wade, R.C. (1997) Biophys. J. 72, 1917–1929.
12. Albery, J.W. & Knowles, J.R. (1976) Biochemistry 15, 5631–5640.
13. Knowles, J.R. (1991) Nature (London) 350, 121–124.
14. Albery, W.J. & Knowles, J.R. (1976) Biochemistry 25, 5627–5631.
15. Wade, R.C., Gabdoulline, R.R. & Luty, B.A. (1998) Proteins, in press.
16. Blacklow, S.C., Raines, R.T., Lim, W.A., Zamore, P.D. & Knowles, J.R. (1988) Biochemistry 27, 1158–1167.
17. Sergi, A., Ferrario, M., Polticelli, F., O’Neill, P. & Desideri, A. (1994) J. Phys. Chem. 98, 10554–10557.
18. Argese, E., Viglino, P., Rotilio, G., Scarpa, M. & Rigo, A. (1987) Biochemistry 26, 3224–3228.
19. Christensen, H., Martin, M.T. & Waley, S.G. (1990) Biochem. J. 266, 853–861.
20. Matagne, A. & Frere, J. (1995) Biochim. Biophys. Acta 1246, 109–127.
21. Qi, X. & Virden, R. (1996) Biochem. J. 315, 527–541.
22. Davis, M.E. & McCammon, J.A. (1989) J. Comput. Chem. 10. 386–391.
23. Bourne, Y., Redford, S.M., Steinman, H.M., Lepock, J.R., Tainer, J.A. & Getzoff, E.D. (1996) Proc. Natl. Acad. Sci. USA 93, 12774–12779.
24. Pesce, A., Capasso, C., Battistoni, A., Folcarelli, S., Rotilio, G., Desideri, A. & Bolognesi, M. (1997) J. Mol. Biol. 274, 408–420.
25. Foti, D., Curto, B.L., Cuzzocrea, G., Stroppolo, M.E., Polizio, F., Venanzi, M. & Desideri, A. (1997) Biochemistry 36, 7109–7113.
26. Fisher, C.L., Cabelli, D.E., Tainer, J.A., Hallewell, R.A. & Getzoff, E.D. (1994) Proteins 19, 24–34.
27. Polticelli, F., Bottaro, G., Battistoni, A., Carri, M.T., DijnovicCarugo, K., Bolognesi, M., O’Neill, P., Rotilio, G. & Desideri, A. (1995) Biochemistry 34, 6043–6049.
28. Raquet, X., Lounnas, V., Lamotte-Brasseur, J., Frere, J.M. & Wade, R.C. (1997) Biophys. J. 73, 2416–2426.
29. Zhou, H.-X. (1996) J. Chem. Phys. 105, 7235–7237.
30. Zhou, H.-X., Briggs, J.M. & McCammon, J.A. (1996) J. Am. Chem. Soc. 118, 13069–13070.
31. Zhou, H.-X., Wong, K.-Y. & Vijayakumar, M. (1997) Proc. Natl. Acad. Sci. USA 94, 12373–12377.
32. McCammon, J.A. & Northrup, S.H. (1981) Nature (London) 293, 316–317.
33. Northrup, S.H., Zarin, F. & McCammon, J.A. (1982) J. Phys. Chem 86, 2314–2321.
34. Hendsch, Z.S. & Tidor, B. (1994) Protein Sci. 3, 211–226.
35. Wimley, W.C., Gawrisch, K., Creamer, T.P. & White, S.H. (1996) Proc. Natl. Acad. Sci. USA 93, 2985–2990.
36. Vogt, G., Woell, S. & Argos, P. (1997) J. MoL Biol. 269, 631–643.
37. Waldburger, C.D., Schilbach, J.E. & Sauer, R.T. (1995) Nat. Struct. Biol. 2, 122–128.
38. Lounnas, V. & Wade, R.C. (1997) Biochemistry 36, 5402–5417.
39. Aguilar, C.F., Sanderson, I., Moracci, M., Ciaramella, M., Nucci, R., Rossi, M. & Pearl, L.H. (1997) J. Mol. Biol. 271, 789–802.
40. Pappenberger, G., Schurig, H. & Jaenicke, R. (1997) J. Mol. Biol. 274, 676–683.
41. Brzozowski, A.M., Derewenda, U., Derewenda, Z.S., Dodson, G.G., Lawson, D.M., Turkenburg, J.P., Bjorkling, F., HugeJensen, B., Patkar, S.A. & Thim, L. (1991) Nature (London) 351, 491–494.
42. Derewenda, U., Brzozowski, A.M., Lawson, D.M. & Derewenda, Z.S. (1992) Biochemistry 31, 1532–1541.
43. van Tilbergh, H., Egloff, M.-P., Martinez, C., Rugani, N., Verger, R. & Cambaillau, C. (1993) Nature (London) 362, 814–820.
44. Derewenda, Z.S., Derewenda, U. & Dodson, G.G. (1992) J. Mol. Biol. 227, 818–839.
45. Norin, M., Olson, O.H., Svendsen, A., Edholm, O. & Hult, K. (1993) Protein Eng. 6, 855–863.
46. Peters, G.H., Olsen, O.H., Svendsen, A. & Wade, R.C. (1996) Biopliys. J. 71, 119–129.
47. Peters, G.H., Toxaerd, S., Olsen, O.H. & Svendsen, A. (1997) Protein Eng. 10, 137–147.
48. Holmquist, M., Norin, M. & Hult, K. (1993) Lipids 28, 721–726.
49. Holmquist, M., Martinelle, M., Berglund, P., dausen, I.G., Patkar, S., Svendsen, A. & Hult, K. (1993) J. Protein Chem. 12, 749–757.
50. Martinelle, M., Holmquist, M., Clausen, I.G., Patkar, S., Svendsen, A. & Hult, K. (1996) Protein Eng. 9, 519–524.
51. Poulos, T.L., Finzel, B.C. & Howard, A.J. (1987) J. Mol. Biol 195, 687–700.
52. Hasemann, C.A., Kurumbail, R.G., Boddupalli, S., Peterson. J.A. & Deisenhofer, J. (1995) Structure 3, 41–62.
53. Deprez, E., Gerber, N.C., Di Primo, C., Douzou, P., Sligar, S.G. & Hui Bon Hoa, G. (1994) Biochemistry 33, 14464–14468.
54. Luedemann, S.K., Carugo, O. & Wade, R.C. (1997) J. Mol. Model. 3, 369–374.
55. Carugo, O. & Argos, P. (1998) Proteins, in press.
56. Sligar, S.G. (1995) in Cytochrome P450: Structure, Mechanism and Biochemistry, ed. Ortiz de Montellano, P.R. (Plenum, New York), pp. 83–124.
57. Di Primo, C., Deprez, E., Sligar, S.G. & Hui Bon Hoa, G. (1997) Biochemistry 36, 112–118.
58. Jacobson, B.L., He, J.J., Lemon, D.D. & Quiocho, F.A. (1992) J. Mol. Biol. 223, 27–30.
59. Hodgkin, E.E. & Richards, W.G. (1987) Int. J. Quantum Chem.: Quant. Biol Symp. 14, 105–110.
60. Raag, R., Li, H., Jones, B.C. & Poulos, T.L. (1993) Biochemistry 32, 4571–4578.








