
2
Scientific Background for HSCT Concerns
Atmospheric Structure and Dynamics
The simplest model for qualitatively understanding the observed distribution of trace constituents in the stratosphere consists of advection by a single meridional cell in each hemisphere with uniform rising in the tropics, poleward drift, and, by continuity of mass, a subsiding flow in the extratropics. Such a circulation was proposed by Brewer (1949) who pointed out that the observed aridity of the stratosphere (water-vapor mixing ratios of a few ppmv) could be understood only if water vapor were transported into the stratosphere by an upwelling circulation confined to the tropics, where air parcels would be "freeze-dried" as they went through the cold tropical tropopause. Somewhat later Dobson (1956) pointed out that poleward and downward advection by this type of mean circulation was consistent with the observed maximum concentration of ozone in the lower polar stratosphere, far from the region of maximum photochemical production. The "Brewer-Dobson" model, as it is now called, clearly demonstrated the importance of the global-scale circulation in determining the observed distribution of ozone. It also provided a partial, but by no means complete, model for transport in the stratosphere.
Stratospheric-Tropospheric Exchange
Stratospheric-tropospheric exchange is better understood now than it was at the time of the last NASA assessment. The work of Holton et al. (1995) described below provides a simple way to view this exchange, as far as the HSCT is concerned.
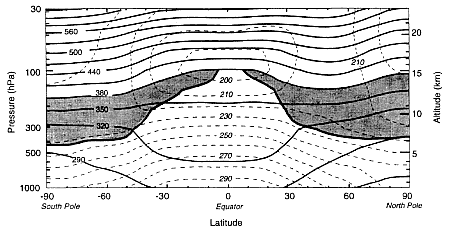
FIGURE 1
Latitude-altitude cross-section for January 1993 showing zonally averaged potential temperature (solid contours) and absolute temperature (dashed contours). The heavy solid contour (cut off at the 380K isentrope approximates the tropopause outside the equatorial zone, while the 380K isentrope approximates the equatorial tropopause. The shaded areas denote the "lowermost stratosphere", where isentropic surfaces span the tropopause. (From Holton et al., 1995; reprinted with permission of the American Geophysical Union.)
Earth's atmosphere is classified in terms of a number of concentric layers. The two lowest layers, the troposphere and the stratosphere, are of primary concern for aircraft-related issues. Figure 1 shows in solid contours the meridional distribution of zonally averaged potential temperature (a measure of entropy) and in dashed contours the absolute temperature in January 1993 from the surface to 25 km altitude. The heavy dark line is the tropopause, which is the separation between the troposphere and stratosphere; the stratosphere extends up to 50 km altitude, and only the lower portion is shown. The shaded areas include the portion of the stratosphere in which lines of constant potential temperature (isentropes) connect the stratosphere and troposphere; in the field of atmospheric dynamics this region is called the "lowermost stratosphere". Above the shaded areas is the portion of the stratosphere in which the isentropic surfaces do not cross the tropopause; this region is called the "overworld" (see Holton et al. 1995), a specific technical term used to avoid confusion with qualitative statements such as "lower" or "middle'' stratosphere. The temperature inversion throughout the stratosphere confers stability on the air against vertical mixing and vertical motion, but it does not inhibit horizontal motions. In contrast, tropospheric air undergoes strong vertical mixing, mainly because of moist convection.
As shown schematically in Figure 2, stratosphere-troposphere exchange oc-
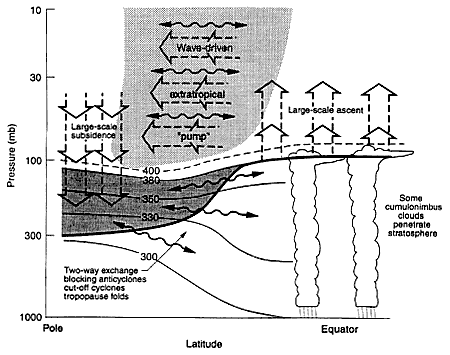
FIGURE 2
Dynamical aspects of stratospheric transport and stratosphere-troposphere exchange. The tropopause is shown by the thick line. Thin lines are isentropic (constant potential temperature) surfaces labeled in kelvins. The heavily shaded region is the "lowermost stratosphere" where isentropic exchange occurs by tropopause folding and small-scale mixing. The region above 380K is the "overworld", in which isentropes lie entirely in the stratosphere. Light shading denotes wave-induced forcing of the global-scale vertical circulation (shown as broad arrows). The wavy double-headed arrows denote quasi-adiabatic meridional transport by eddy motions. (From Holton et al., 1995; reprinted with permission of the American Geophysical Union.)
curs through both isentropic transport (i.e., transport along surfaces of constant potential temperature) and mixing in the lowermost stratosphere. Exchange of air between the overworld and the troposphere, however, occurs only through a global-scale cross-isentropic circulation driven by the pumping action of wave-induced forces in the extratropical stratosphere. This wave-induced pumping pulls air upward and poleward from the tropical troposphere into the stratosphere and pushes it poleward and downward into the extratropical lowermost stratosphere (Holton et al., 1995). Note that vertical transport from the overworld into the lowermost stratosphere, not quasi-horizontal exchange across the tropopause,
is the rate-limiting process for exchange between the chemically important region of the stratosphere and the troposphere.
These considerations are important to AESA because they show that aircraft flying in the overworld represent a qualitatively different situation with respect to atmospheric effects from aircraft flying in the lowermost stratosphere, because of the persistence of exhaust components. The proposed fleet of HSCTs would spend most of its flight time in the overworld, while the current subsonic fleet flies in the upper troposphere and the lowermost stratosphere. Thus, transport and exchange processes for a supersonic fleet's exhaust components, and the time scales on which they operate, are expected to be quite different from those for the subsonic fleet emissions.
Differences in transport between the overworld and the lowermost stratosphere were also evident in the time evolution of stratospheric radioactive debris following the nuclear bomb tests of 1961–1962. For years after the conclusion of the bomb tests, all forms of radioactive fallout occurred with especially high intensity at mid-latitudes in the spring. Carbon-14 (written 14C), in the form of gaseous carbon dioxide, moved rapidly with longitude around the globe in both regions of the stratosphere. The residence time of 14C deposited in the overworld was about 2 years if deposited between 17 and 20 km and about 5 years if deposited between 25 and 30 km (Telegadas, 1967, 1971; Telegadas et al., 1972; Kinnison et al., 1994a). Both gaseous 14C and the heavy-metal radioactive debris incorporated in stratospheric aerosols had stratospheric residence times of about six months if they were deposited in the lowermost stratosphere. The longer residence times of 14C in the overworld, by comparison with 14C and other radioactive debris in the lowermost stratosphere, demonstrate that for transfer of HSCT exhaust from the stratosphere to the troposphere, the rate-limiting step is the relatively easily evaluated vertical transport from the overworld into the lowermost stratosphere, not the intermittent, very complicated, quasi-horizontal exchange across the tropopause.
A simple mental picture of stratosphere-troposphere transport for both subsonic-aircraft and HSCT exhaust is to visualize the lowermost stratosphere as a large reservoir for chemical constituents that is replenished from above by diabatic descent across the boundary of the overworld, and undergoes horizontal leakage on its equator-facing side, especially in the springtime, as well as some down-directed leakage. Exhaust gases from subsonic aircraft accumulate continuously in this reservoir. The HSCT exhaust gases emitted in the overworld leak slowly (years) by diabatic descent into the lowermost stratospheric reservoir, where they also accumulate. In the spring a large "gate valve" opens for several weeks, and a large amount of air with its contained aircraft exhaust is flushed horizontally into the troposphere along the isentropes. The average residence time of subsonic exhaust gases in the stratosphere is about 6 months, but average residence time of HSCT exhaust gases in the stratosphere is more than 2 years.
Natural Ozone Distribution and Transport
The impacts of HSCT emissions must be assessed in the context of ozone's natural climatology. The global distribution of ozone by latitude and altitude is illustrated in Figure 3. The upper panel presents observed ozone concentrations. Relatively high ozone concentrations are found in an arching band from pole to pole, with maximum concentrations at ~19 km at the poles and ~23 km in the mid-latitudes. The lower panel of the figure shows the distribution of ozone mixing ratios by latitude and altitude. (A concentration is an absolute quantity, whereas a mixing ratio is the number of a given type of molecule relative to the sum of all molecules present. The decrease in atmospheric density with altitude makes both quantities useful.) The two heavy lines added to Figure 3 show two local ozone photochemical-replacement times, which are computed by dividing the local ozone concentration (from the upper panel) by twice the local rate of oxygen photolysis; the upper curve shows where the replacement time is 1 month, and the lower curve where it is 4 months. The band between these two curves can be viewed as separating the regions under chemical and dynamical control. The "chemically controlled region" is the ozone-formation region, where the ozone steady state is controlled primarily by local chemistry, but also by the past transport of ozone-destroying species in this region. The "dynamically controlled region" is the ozone-reservoir region, where the ozone steady state is controlled primarily by atmospheric dynamics, but also by the history of ozone destruction in this region.
During late fall, the tilting of Earth's axis in orbit shades the parcels of air above that hemisphere's polar region from sunlight. This "polar night" quenches both ozone production and loss, thereby "freezing" the existing high local ozone mixing ratio. This high mixing ratio is then further increased as ozone in the middle stratosphere is transported into the polar-night region by quasi-horizontal eddy motions. Within a few months, the ozone in the polar vortex is transported downward into the lower overworld by diabatic circulation (Rosenfield et al., 1994). Horizontal eddy diffusion, with net transport toward lower latitudes as shown in Figure 2, then replenishes ozone in the pole-to-pole arching band of maximum concentration shown in the upper panel of Figure 3.
Ozone concentrations in this maximum-concentration band thus depend on the chemistry in the mid-stratospheric region where the air entered the polar night, as well as on the dilution that occurred as the air descended into the lower overworld and spread into lower latitudes. Ozone production is quenched sooner than ozone destruction in the air entering polar night, so the ozone mixing ratio decreases. The changing photochemical steady state in that region also affects ozone concentration. The rate at which the air is transferred into the polar night thus can have an impact on the mixing ratios in both the polar-night region and the band of maximum concentration. Atmospheric-model results need to be compared with observations to determine how well the models currently repre-
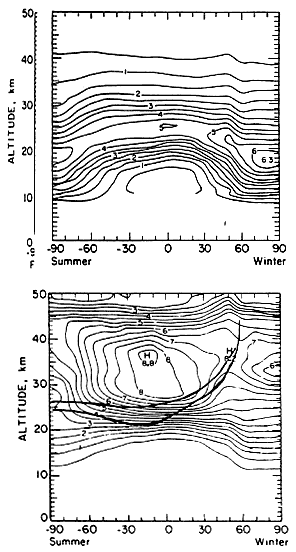
FIGURE 3
Contour plots showing examples of two-dimensional distributions of ozone. Upper panel: Concentration (number density) in units of 1012 molecules cm -3. Lower panel: Mixing ratios in parts per million by volume. The superimposed heavy lines indicate where the local ozone photochemical replacement time ([O3 ]/2j[O2]) is 4 months (lower line) and where it is 1 month (upper line). The area above and to the left of the upper heavy line may be interpreted as the ozone-formation region; the area below and to the right of the lower heavy line may be interpreted as the ozone-reservoir region. (Upper panel is from Solomon et al., 1980; lower panel is modified from that article. Both reprinted with permission of Birkenhaeuser Publishers.)
sent this feature. Long model runs are needed to fully simulate these evolving processes.
Our quantitative understanding of the chemical and dynamical processes that control ozone distribution has been greatly improved by AESA-sponsored field campaigns. In 1990 (see Prather et al., 1992), AESA worked out plans and priorities for extensive simultaneous measurements of multiple species in the stratosphere using the NASA ER-2 aircraft up to the top of its flight range (20 km altitude), and later plans were made to use a proposed new unmanned aircraft in the 20 to 27 km altitude range. (Ultimately, development of the unmanned aircraft did not proceed fast enough for it to be used, and balloon measurements had to be substituted.) Stolarski and Wesoky (1995) describe the multiple-flight Stratospheric Photochemistry, Aerosols, and Dynamics Expedition (SPADE), made in 1992–1993. In this campaign, almost all reactive species were measured simultaneously to provide definitive answers to some important chemical questions (discussed later in this chapter). The simultaneous observations of long-lived gases, in conjunction with recently developed understanding of atmospheric dynamics, provided investigators with quantitative new information about transport. Tracer data obtained by SPADE, and by more recent campaigns discussed under Atmospheric Measurements, have yielded extremely useful information about atmospheric motions that provides new structure and details to the Brewer-Dobson general-circulation model. These ideas are now being incorporated into models (see, for example, Plumb, 1996), and underlie the discussions of the formation and distribution of natural ozone in this report.
Transport Barriers Within the Overworld
Given the importance of transport history in understanding the distribution of ozone, it is desirable that the spatial and temporal variability of transport be well understood in order to account properly for transport in models. A key issue in this regard is the existence of so-called "barriers" to transport.
It has long been recognized that the tropopause acts to some extent like a quasi-permeable barrier to constituent transport and stratosphere-troposphere exchange. In more recent years it has become clear that there are also quasi-permeable transport barriers within the stratosphere itself. Such barriers separate the tropics from the mid-latitudes and the mid-latitudes from the winter polar vortex. These barriers are typically characterized by strong gradients in the zonal wind and steep gradients in various chemical species. Erosion at the edges of such barriers produces filamentary structure in chemical constituents determining the degree of "leakiness" across such barriers is key to understanding the fate of aircraft emissions and of other long-lived chemical constituents; the importance of such "transport barriers" for the HSCT assessment is clearly recognized in the 1995 NASA assessment.
The increase in global coverage of trace constituents in the stratosphere provided by the Upper Atmosphere Research Satellite (UARS) has made possible many advances in understanding the development and maintenance of the polar and subtropical barriers in the region above about 20 km, where the dynamics is dominated by planetary Rossby wave breaking (Plumb et al., 1994).
The theoretical behavior of the transport barriers in the bottom layer of the overworld (between about 16 and 22 km, or the 380–480K isentropes) is less well understood than their behavior at higher altitudes. In the 16–22 km region the upward extensions of tropospheric disturbances, including synoptic-scale extratropical storms and continental-scale monsoon circulations, can play a considerable role in transport. Evidence from a number of sources suggests a higher degree of leakiness in the 16–22 km altitude range across both the polar and subtropical transport barriers than at greater altitudes (see, e.g., Minschwaner et al., 1996). Since 18–21 km is the altitude range at which HSCT emissions will occur, it is important that accurate quantitative estimates (including temporal and spatial variability) be developed for the rates at which mass and trace constituents are transported between the mid-latitude northern hemisphere air corridors and the polar vortex as well as those corridors and the tropical-upwelling region. This is one of the objectives of the Stratospheric Tracers of Atmospheric Transport (STRAT) mission that is supported by the AESA project. It should be noted, however, that the 20 km level is the ceiling for ER-2 operations, and much of the new information obtained by AESA concerning atmospheric composition and dynamics is limited to altitudes below 20 km. Thus, evidence suggesting a change in transport regime at about this altitude must be treated with caution, because above this level nearly all observations are based on satellite remote sensing.
Meridional wave transport and mixing are inhibited at the edge of the polar winter vortex, so the vortex region is relatively isolated chemically. There is also evidence that an analogous inhibition of wave transport occurs in the subtropics. The possible existence of such a subtropical mixing barrier has led to suggestions that, to a first approximation, the transfer of trace constituents from the equatorial troposphere upward through the lower equatorial stratosphere can be viewed as occurring in a "tropical pipe" that is isolated from contact with the extratropical lower stratosphere (Plumb, 1996). (In other words, near the equator there is limited meridional mixing in the 16–24 km altitude band.) However, the tropical pipe is not airtight. Analysis of recent in situ observations of molecules with a wide range of lifetimes (Minschwaner et al., 1996; Volk et al., 1996) suggests that about 50 percent of air in the tropical ascent region at 21 km originates in the mid-latitudes. Substances emitted in the mid-latitudes thus have a significant probability of reaching the altitudes (about 25 to 38 km) at which NOx chemistry is dominant in ozone destruction, as do emissions occurring on the significant fraction (38 percent) of flight paths that are above the tropics (see Chapter 3).
Gas-Phase Ozone Chemistry
Ozone is produced from oxygen by far-ultraviolet (UV) solar radiation (Figure 3, middle panel) in a two-step process: O2 + UV ⇒ 2 O followed by O + O2 ⇒ O3 (twice). Ozone is destroyed in the chemically controlled region almost as fast as it is made, setting up a "photochemical steady state". Throughout the stratosphere, ozone is continually destroyed by several homogeneous gas-phase chemical reactions, including direct ozone destruction by the reaction O + O3 ⇒ 2O2, and by reactions with active atoms and radicals, illustrated here for the case of nitrogen oxides (NOx), nitric oxide (NO), and nitrogen dioxide (NO 2). At first sight it might appear that the nitrogen oxides present at a few times 108 molecules cm-3 could not destroy much ozone, which is present at a few times 1012 molecules cm-3. Paul Crutzen showed (Crutzen, 1970), however, that ozone destruction does occur by way of the homogeneous catalytic cycle:

Many cycles can occur. Both NO and NO2 art regenerated every cycle, whereas two molecules of ozone are lost for each cycle, since the oxygen atom in the second step is formed from sunlight and ozone (O3 + UV ⇒ O2 + O).
There are other catalytic cycles perpetuated by trace concentrations of atoms and free radicals that destroy ozone in the stratosphere and troposphere. These processes are classified by the chemical family that contributes the catalytic chain carriers: (i) free radicals HOx derived from water, H, OH, HOO; (ii) atoms and radicals ClOx derived from chlorine (Cl2), Cl, ClO; and (iii) atoms and radicals BrOx derived from bromine (Br2), Br, BrO. The great majority of ClO x and BrOx radicals come from photolysis of organo-halogen compounds. Bromine reactions parallel chlorine reactions in most respects, but for the radical—radical reactions that constitute the rate-limiting steps in catalytic ozone-destruction cycles, bromine reactions are faster than the corresponding chlorine reactions. For aircraft problems, the most important cycle in the HOx family (out of five in all) and the most important cycle in the ClOx family are written below:
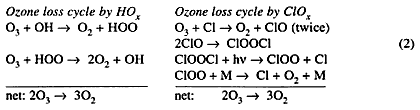
Each of these ozone-destroying cycles is diverted into a null cycle by NO:
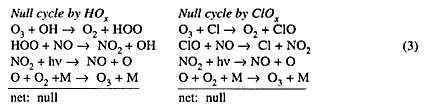
As directly demonstrated by atmospheric measurements, in the lower stratosphere NOx catalysis is a minor mechanism, and ozone loss is dominated by HOx catalysis with significant contributions from ClO x and BrOx (Wennberg et al. 1994; Stolarski et al., 1995). Atmospheric measurements also show that catalytic destruction due to NOx is one of the significant background atmospheric ozone-loss mechanisms in the middle and upper stratosphere (Stolarski et al., 1995; Jucks et al., 1996). According to model calculations (see Figure 5 in Chapter 3), ozone destruction by NOx is about 60 percent of the total in the tropics, 70 percent at mid-latitudes, and 80 percent in sunlit polar regions in the ozone-formation region between 28 and 38 km.
Chemically active species NO2, OH, Cl, ClO, and BrO are reversibly converted to inactive ''reservoir" species HNO3, ClONO2, BrONO2, and HCl through the reactions:
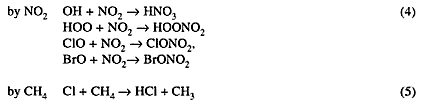
Solar radiation or hydroxyl radicals OH slowly re-convert the reservoir species to active radicals.
The two null reaction cycles in Equation (3) and the reactions in (4) are of importance in understanding the role of NOx in the natural atmosphere and in an atmosphere with aircraft-added NOx. In the natural atmosphere NOx reduces ozone in the ozone-formation region in the middle stratosphere, and thereby determines the magnitude of ozone in its region of maximum concentration around 23 km. NOx mitigates ozone destruction by HOx and ClOx systems (Equations 3 and 4), however, and thus protects lower-stratospheric ozone from almost total destruction by ClOx and HOx. The proper interpretation to be given to the null cycles in Equation (3) is that nitrogen oxides reduce the ozone reduction by HOx and ClOx systems, not that NOx "produces" ozone.
In the lower atmosphere, UV photolysis of oxygen is not the only mechanism by which ozone is formed. Natural "smog" reaction mechanisms were first explained by Crutzen (1973). The essence of these mechanisms is the slow
photochemical combustion of carbon monoxide or methane, or volatile organic compounds (VOCs) such as acetone. The reactions are cyclic in HOx and NOx but consumptive of VOCs. The number of ozone molecules produced by these reactions is generally a small multiple of the number of carbon atoms in the molecule that is consumed. However, the net effect of these reactions on ozone depends on the ambient [NO]/[O3] ratio. Low NO permits O3 destruction by HOO, while high NO permits the production of O3.
Heterogeneous Chemistry in the Stratosphere
Stratospheric aerosols arising from natural sources play a key role in the chemistry of the ozone layer, because of reactions on their surfaces. Under some circumstances, such as large volcanic eruptions, they even influence stratospheric dynamics. Since the emissions from a fleet of HSCTs might become a significant additional source of stratospheric aerosols, it is important that reactions on aerosols be better understood.
Discovery of the Antarctic ozone hole in 1985 sparked coordinated campaigns of field measurement, laboratory studies, and modeling, including reconsideration of the potential role of heterogeneous processes in the chemistry of ozone at mid-latitudes. The special perturbed chemistry of the polar stratosphere requires the presence of at least one of the two types of polar stratospheric cloud (PSC) particles. The particles that compose PSCs are responsible for the catalytic heterogeneous chemistry that leads to the formation of the ozone hole (Molina et al., 1987; Tolbert et al., 1987; Solomon, 1990). These particles "activate" the reservoir species of chlorine (e.g., HCl and ClONO2) and turn them into labile species (e.g., Cl 2), and "deactivate" the labile species of NOx and turn them into inactive HNO3.
Heterogeneous reactions that have been incorporated into chemical models of the Antarctic ozone hole include:
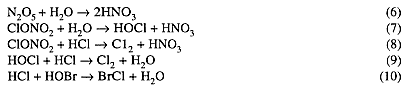
At mid-latitudes, stratospheric temperatures are far too high to permit formation of PSCs. (Although ice particles can form at the tops of strong thunderstorms near the tropical tropopause, the air upwelling from the troposphere there contains mostly CFCs and N2O, not HCl and ClONO2.)
Figure 4 represents some interesting data obtained during the SPADE campaign. Instruments aboard the NASA ER-2 aircraft simultaneously measured many reactive species in the stratosphere up to the ER-2's 20 km flight ceiling.
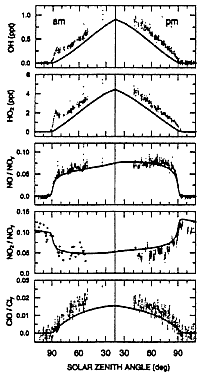
FIGURE 4
Diurnal variations of free radicals (OH, HO2, NO, NO2, and ClO) measured at 19 km, 37°N during SPADE. The lines show calculations made by a photochemical model. (Adapted from Salawich et al., 1994b; reprinted with permission of the American Geophysical Union.)
The measured data are plotted as points on each panel of the figure; for comparison, a line shows values calculated by a local atmospheric model specifically suited to the air mass in which the measurements were made. On Figure 4, time starts a few hours before sunrise, continues throughout the day, and ends a few hours after sunset. There is good agreement between model and measurement for the ratio NO/NOy and for the ratio NO2/NOy, where NOy is the sum of NO, NO2, HNO3, and all other molecules containing the NO group. However, there is a pulse of OH and HOO radicals shortly after sunrise; some substance had been accumulating all night and was dissociated by the early morning sunlight, and the observed OH and HOO were higher than the modeled values throughout the sunlit day. Also, the observed ratio ClO/Cl was higher than the model values, which is consistent with high OH, since the reaction OH + HCl = H2O + Cl increases active chlorine, including ClO.
It has been proposed that the heterogeneous hydrolysis of bromine nitrate
plays an important role in the bromine and HOx chemistry at mid-latitudes. Hanson and Ravishankara (1995) have suggested that reaction (11) may be the
source of the early-morning peak of OH and HOO and the higher-than-predicted OH and HOO (see also Salawitch et al., 1994b). Stolarski et al. (1995) reported unpublished work that tested this hypothesis: Inclusion of bromine nitrate hydrolysis in the LLNL and CSIRO models improves the agreement between models and measurements as far as the early peak and the excess OH and HOO are concerned, but it causes predicted NOx to fall below observed values. There remains some suspicion of missing chemistry involving OH at altitudes below 20 km (see also Lary et al., 1996).
Hofmann and Solomon (1989) argued that volcanic eruptions could lead to enhanced depletion of ozone at mid-latitudes, according to laboratory studies by Mozurkewich and Calvert (1988), Tolbert et al. (1988), and Worsnop et al. (1988) of the kinetics of reaction (6) on liquid sulfuric acid and water surfaces. Depletion occurs through a reduction in the abundance of NOx, and a consequent increase in the importance of ClOx-and HOx-catalyzed destruction of ozone (Equations 3 and 4). Changes in ozone observed by means of Dobson spectrophotometers, ozonesondes, and satellite-based instruments subsequent to the 1982 eruption of El Chichón, summarized by Hofmann and Solomon (1989), were consistent with the results of a photochemical model incorporating reactions on aerosol, but the uncertainties were large. Additional laboratory studies confirmed the potential importance of reaction (6), so modeling studies incorporated the occurrence of (6) on sulfate aerosol into reaction schemes for describing mid-latitude ozone chemistry (Rodriguez et al., 1991). Considerable improvement in agreement between predicted and observed vertical patterns of ozone depletion caused by chlorofluorocarbons and high ClO concentrations was obtained as a result, and the significance of the region below 20 km became apparent.
The eruption of Mt. Pinatubo during the second Airborne Arctic Stratospheric Expedition (AASE II, September 1991-March 1992) provided an early opportunity to test the aerosol hypothesis. Measurements were made of NOx/NOy and ClO/Cly at mid-latitudes before and after the onset of the effect of the Pinatubo eruption. The measurements showed a change in these ratios that could not be explained with gas-phase chemistry alone (Fahey et al., 1993). Models that included heterogeneous reactions improved prediction of the ratios of radical species involved in ozone chemistry for a large range of aerosol abundances (Kinnison et al., 1994b). Similarly, models that included aerosol reactions agreed well with ratios measured in situ during SPADE (November 1992–May 1993), whereas gas-phase-only models did not (Salawitch et al., 1994b). In a more recent modeling study, Solomon et al. (1996) confirmed the importance of heterogeneous chemistry in driving ozone depletion in the northern mid-latitudes.
Of course, the composition, size, and phase (solid or liquid) of aerosols vary greatly with latitude, altitude, and season. Aerosols composed primarily of sulfuric acid and water, or aerosols of mixed composition (including nitric acid), may dominate at any one time and place. The significance of aerosols for stratospheric chemistry at mid-latitudes, in addition to their role in polar ozone loss,
has become clear over the last decade. Many of the major uncertainties remaining in the chemistry of the lower stratosphere involve heterogeneous chemistry occurring on atmospheric aerosols at all latitudes, and on polar stratospheric clouds. Heterogeneous chemistry at middle and polar latitudes exhibits a range of behavior determined by characteristics of aerosols that evolve with ambient temperature (Molina et al., 1993; MacKenzie et al., 1995; Peter, 1997).
At mid-latitudes, where aerosols are composed largely of sulfuric acid and water, only the hydrolysis of N2O5 and BrONO2 may prove important. At the lower temperatures characteristic of polar regions, a supercooled ternary solution (called STS) of nitric acid, sulfuric acid, and water forms PSCs under conditions at which nitric acid trihydrate (NAT) would be expected to form in equilibrium (Hamill et al., 1996). The kinetics of some heterogeneous reactions have been studied on a variety of surfaces, including liquid and solid sulfuric-acid solutions, ice, and NAT, but PAEAN has found no reports of kinetic studies on STS. It should be noted, however, that the physical and chemical changes in PSCs caused by HSCT emissions, and the magnitude of the response of polar ozone chemistry to those PSC changes, will depend on both microphysical properties such as the aerosol surface-area-to-volume ratio and the specific composition of the aerosol (e.g., the proportions of NAT, STS, and sulfuric acid tetrahydrate). For example, STS and NAT absorb nitric acid with different efficiencies (Mackenzie et al., 1995), and the rates of several of the key heterogeneous reactions depend on the aerosol composition. Several of these factors are not yet understood quantitatively.
Atmospheric Measurements
Observations show that the mixing ratios of long-lived species (e.g., N2O and CH4) show a simple, compact relation in the lower stratosphere (Ehhalt et al., 1983; Fahey et al., 1989a,b). This relationship results from the fact that the surfaces of constant mixing ratio (isopleths) are similar for all species whose quasi-horizontal mixing times are shorter than the chemical-loss times (Holton, 1986; Plumb and Ko, 1992). Successful field measurements of multiple reactive components by programs such as AASE, ASHOE/MAESA*, and SPADE were carried out using the NASA ER-2 aircraft (see Wennberg et al., 1994; Salawitch et al., 1994a,b; Cohen et al., 1994; Stimpfle et al., 1994; Jaeglé et al., 1994), and the data from more recent campaigns such as STRAT. The use of the mixing-ratio relationships in a novel graphical approach has yielded useful information about atmospheric dynamics, leading to the development of the ''tropical pipe" model and revealing that 2-D models cannot predict the NOy/O3 ratio well (Murphy et al., 1993).
These recent expeditions have shown that HOx radicals (ultimately derived from H2O and O3) are the principal catalysts at mid-latitudes for removing O3 at altitudes below 20 km (Wennberg et al., 1994). These results represent a major shift from the previously held notion that NOx radicals were the principal catalysts for removing O3 in this region of the atmosphere (Logan et al., 1978; McElroy and Salawitch, 1989). If the rates of ozone destruction caused by HOx, ClOx and BrOx, and NOx (according to SPADE data) are plotted against altitude between 15 and 20 km, HOx is clearly the most important, the halogens are a close second, and NOx the slowest (Wennberg et al., 1994).
Remote sensing methods measure several reactive species at altitudes above 20 km. The UARS limb-sounder measurements are less dense in time and space than what is really needed for evaluating the effects of aviation, and the UARS data set does not include all of the radical species whose concentrations are needed to "close" the chemistry. Nonetheless, it provides useful information on several of the crucial chemically important species in the chlorine, nitrogen, and hydrogen families, and it is the best source of data on the behavior of long-lived trace constituents at altitudes above 20 km. Observations of the spatial and temporal variability of such trace constituents, together with high-resolution Doppler imager (HRDI) wind data and meteorological analyses from the U.K. Meteorological Office and the National Centers for Environmental Prediction, can be used in diagnostic studies to assess transport and mixing rates in the 16–30 km altitude range.
From measurements of O3, HO2, OH, NO, NO2, and ClO, Jucks et al. (1995) have inferred ozone-destruction rates between 24 and 38 km for HOx, ClOx, BrOx, and NOx. They find that while HOx reactions are the dominant ozone destroyer below 20 km, NOx reactions are the dominant ozone destroyer above 24 km. Figure 5, which appears in the next chapter, illustrates the altitudinal and latitudinal dependence of the various ozone-destroying processes.















