
6
Molecular and Cellular Mechanisms of Radon-Induced Carcinogenesis
The exposure of cells to densely ionizing radiation, such as radon alpha particles, can initiate a series of molecular and cellular events that culminates in the development of lung and other cancers (Hall 1994). That flow can now be described in outline, starting with the deposition of clusters of ionizations and ending in the development of cancer (Cox 1994). Ionization leads to cellular damage, DNA breakage, accurate or inaccurate repair, apoptosis, gene mutations, chromosomal change, and genetic instability (Kronenberg 1994; Ward and others 1990; Ward 1988). Radiation-induced molecular changes result in the gain and loss of functions in critical regulatory genes, which permit cells to escape from normal controls and become invasive unregulated malignancies. The process of malignant transformation involves a series of changes that follow, at least roughly, a functional and temporal sequence by which cells gradually and progressively escape from normal tissue control and acquire independence, diversity, and invasive properties (figure 6.1). Molecular changes associated with radiation carcinogenesis have mainly been investigated after higher doses and dose rates than those experienced from background levels of radon exposure. Therefore these changes are described qualitatively and the extent to which any or all occur in tissues in which a small proportion of cells have experienced a single alpha particle track remains to be determined.
Cells at Risk
Inhalation of radon results in exposure of lung cells to alpha irradiation from radon progeny, which are deposited in the mucus layer and can result in exposure
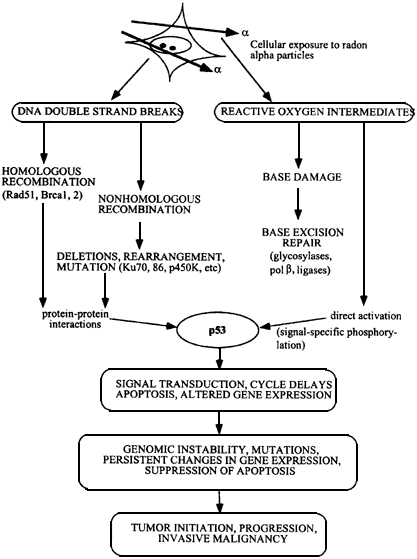
Figure 6.1
Flow chart showing development of malignant cells from initial α-particle damage to cells. DNA strand breaks are repaired by homologous or nonhomologous (illegitimate) double strand break rejoining, and damaged bases by base excision repair. Activation of p53 protein, initiates pathways leading to cell cycle delays and apoptosis, and surviving cells may contain gene deletions, rearrangements, amplifications, and persistant genomic instability. Mutations in oncogenes, loss of function in tumor suppressors, and loss of heterozygosity produces a heterogeneous population of cells which escapes from normal cell and tissue homeostasis to become malignant.
of epithelial cells from unilateral sources on the surface. Ingestion of waterborne radon might, on first impression, similarly expose the cells of the stomach lining. After ingestion, however, radon travels as gas molecules with high mobility through cell membranes, and cells may receive a more uniform exposure. Stem cells and other proliferating cells of the stomach are found in bands at the bases of the necks of narrow invaginations of the stomach wall that constitute the secretory glands of the stomach wall (Nomura 1996). Stem cells and other proliferative cells of the stomach are major targets of radon alpha particles, but cells of the small intestine are also potential targets. After ingestion of water, radon passes into the small intestine with a half-time of about 15–20 minutes. Radon can therefore be absorbed into the bloodstream from both the stomach wall and the small intestine. The resulting exposures to most cells of the body will then be through bloodborne radon. From that point of view, the stomach might be at greatest risk of exposure from ingested, aqueous radon. The transfer of dissolved radon from water to air and its later inhalation constitute another route by which the lungs can be at risk.
Implicit in these scenarios is the idea that the cells most likely to become malignant are the stem cells and proliferative cells that retain the capacity for continued division and can fix and express permanent genetic change. Malignant cells often retain characteristic enzymatic and cellular features of their tissue of origin, so the differentiation and specialization programs of cells might be altered but not completely abrogated by the malignant-transformation process. Alpha-particle damage to genetic material becomes fixed as permanent alterations to gene structure and expression as a result of processes that involve DNA repair, replication, and cell division. The stem cells of epithelial tissues are embedded in crypts; this renders them relatively inaccessible to direct contact with ingested or inhaled radon. Stem cells will, however, still be exposed to alpha irradiation from the lumen or blood stream, from intercellular and intracellular water, and after inhalation from decay products that plate out and act as additional sources of radiation damage. An additional factor to be considered is the potential role of chronic stomach infections. A large fraction of the normal human population carry Helicobacterpylori infections in the stomach that can cause gastritis and, in severe cases, ulcers. The inflammation and proliferation associated with these infections can be a factor in the induction and progression of stomach cancer and have been regarded as risk factors (McFarlane and Munro 1997).
Cellular Damage Induced by Radon Alpha Particles
Alpha particles create dense ionization that leaves tracks of ion-pair clusters across cells and tissues. Cells that suffer an alpha-particle track through the nucleus are severely injured. At the low exposure conditions under consideration from waterborne and airborne radon in the home, however, less than 1% of the cells in the bronchial epithelium would experience an alpha-particle track per
year. For comparison, it requires an exposure of 100 WLM to reach the level at which the average exposure to stem cells reaches one alpha particle per nucleus (Harley and others 1996). Therefore, complex considerations of dose rates and total doses that are important for miners or other people with high occupational exposure are unimportant in consideration of domestic exposure (Brenner and others 1995; Brenner 1992) (see also BEIR VI report National Research Council 1999).
Alpha particles traverse a cell in less than 10-12 seconds and deposit energy corresponding to about 10–50 cGy (Jostes 1996). As the particles slow down, they deposit increasing amounts of energy (linear energy transfer, or LET) per unit length of track, reaching a maximum at the end of their track at what is known as the Bragg peak. The relative biologic effectiveness (RBE) of an alpha particle is therefore variable along its track according to whether the LET reaches a maximum at the Bragg peak (Brenner and others 1995). The average track through a spherical cell nucleus can cross many individual strands of DNA, depositing energy in the form of clusters of ionizations, and produce corresponding numbers of double-strand breaks. These breaks have a complex chemistry and have been described as multiply locally damaged sites (MLDSs) (Ward 1990). Because of the track structure and the tightly coiled nature of DNA in the nucleus, there is likely to be a nonuniform distribution of DNA breaks with an excess of small fragments which might get lost or incorrectly positioned in the process of rejoining (Ritter and others 1977).
Ion clusters can also produce reactive oxygen intermediates which can damage individual DNA bases, and at high doses, alter intracellular signal transduction, reduce macromolecular synthesis, and trigger processes that resemble those from inflammatory cytokines involved in other kinds of tissue injury. A series of early experiments in the 1950 and 1960s used collimated beams of alpha particles and other kinds of radiation and demonstrated the relative importance of nuclear, cytoplasmic, and extracellular irradiation (Munro 1970b; 1970a; Smith 1964). Those experiments showed that nuclear damage was potentially lethal; nonnuclear damage could also produce detectable effects, such as reduced DNA synthesis, but it was not lethal. Extracellular damage involved reactive oxygen intermediates that could be prevented by catalase (which degrades hydrogen peroxide) (Dendy and others 1967). More recent and technically sophisticated experiments in which the effects of single alpha particles can be estimated or observed have resulted in essentially similar conclusions (Hei and others 1997; Hickman and others 1994).
Lethality of Alpha-Particle Tracks Through Cells and Tissues
The dose required to produce an average of one lethal hit to a cell (the D37) corresponds to about 1.2–1.5 alpha particles per spherical nucleus (Jostes 1996). Flattened cells can withstand more tracks (up to 15 or even more), each of
which crosses shorter distances through the nucleus. Lethality can be related to the net absorption of a particular amount of total energy per cell, measured along a total path length through the nucleus—either a single track through a spherical nucleus or several shorter tracks through a flat nucleus. Calculations indicate a constant probability of 0.03–0.08 for a lethal event per micrometer of track (Jostes 1996). All radon alpha-particle effects at the low doses associated with environmental exposure from water occur from the passage of single particles through a small proportion of the cells in a tissue, so the dose-effect relationship will be a linear function of dose, with no dose-rate effects. This is true because variations in exposure change the number of cells hit by an alpha particle, rather than the amount of damage per cell. To calculate cancer risk it is then necessary to know the probability that a hit cell will undergo transformation, and the latent period and its age distribution before transformation to malignancy is complete. The latent period for single cells exposed to single tracks of alpha particles is unknown, but if it were long compared to the lifespan of the individual, the cancer risk would be correspondingly reduced, as suggested by Raabe (1987).
The important cellular subpopulation for carcinogenesis is not that of the rare cells killed by alpha-particle damage, but that of the cells that survive either with direct damage to their genetic material or with altered genomic stability. Because the calculated D37 is more than one alpha particle per cell in very low exposures, such as to ambient air or water, most exposed cells should survive, because it is extremely rare for any cell to be hit more than once. That might also account for the strong synergism displayed between radon exposure and cigarette-smoking: initial radon exposure leaves a viable, damaged cell, which is then stimulated further by the carcinogens found in cigarette smoke (Moolgavkar and others 1993; Brenner and Ward 1992).
Low-dose exposure also raises the question of whether radon alpha particles can give rise to radiation hormesis—the phenomenon whereby very low radiation doses are stimulatory and beneficial (Ueno and others 1996). If hormesis occurs through a stimulation of some kind of repair, the low stimulating dose must induce an excess repair capacity that can mend not only the damage caused by the initial dose, but also pre-existing endogenous cellular damage. That has been observed for repair of mitochondrial oxidative damage (Driggers and others 1996) but, evidence generally is indirect and difficult to obtain. Evidence of radiation hormesis is consequently controversial and will not be further considered here. Although extranuclear damage and extracellular ionization might play a role in some biologic effects (known as bystander effects), they are unlikely to play an important role in cell-killing (Hickman and others 1994; Dendy and others 1967). The flow of events that follow the production of DNA damage and other forms of cellular damage is therefore critical in understanding the development of malignancies.
Transformation of Cells by Alpha Particles in Vitro
Low doses of alpha particles which simulate radon exposure have been used to achieve malignant transformation of cultured cells in studies aimed at measuring their biological effectiveness and estimating carcinogenic hazards. In general, normal diploid cells, with the exception of some hamster embryo cells, have extremely low transformation rates after irradiation. Studies of transformation therefore often use cells such as mouse 3T3 in which genetic changes have already occurred that increase their overall genetic instability and hence their transformability. Although many of these studies generated linear dose-response curves over the dose ranges used (Miller and others 1996; 1995; Brenner and others 1995; Ling and others 1994), some indicated a nonlinear response with greater effectiveness at the lowest doses (Martin and others 1995; Bettega and others 1992). Considerable uncertainty, therefore, still exists about the precise shape of the dose-response relationship for transformation of cells in culture, and by implication, also for carcinogenesis. The results in general do not permit a definitive answer to be obtained for the shape of the dose-response curve at the lowest doses and dose rates, but at the same time there is no compelling evidence to adopt any one particular nonlinear dose-effect relationship. The many and varied biological changes over long time periods that are involved in carcinogenesis, which are discussed in the following sections, indicate that many factors can be expected to influence the shape of the dose-response relationship.
DNA Damage and its Repair—The Caretaker Genes
The gene products responsible for sensing damaged DNA and carrying out repair, euphemistically called the cellular caretakers (Kinzler and Vogelstein 1997), involve a number of enzymatic systems capable of mending single-and double-strand breaks in DNA and excising damaged and mismatched bases. Double-strand breaks are the most important kinds of damage resulting from radon alpha particles. They can be repaired through at least two pathways: homologous recombination (figure 6.2) or nonhomologous recombination (figure 6.3) (Sargent and others 1997). Repair through homologous or nonhomologous recombination involves complex sets of enzymes, which share components with enzymes and gene products associated with the generation of immunoglobulin diversity, such as RAG1 and RAG2 (Melek and others 1998) and with mitotic and meiotic recombination (Jeggo and others 1995; Jeggo 1990).
Most mammalian somatic cells are in the prereplicative, G1, phase of the cell cycle and double-strand break repair appears to involve the nonhomologous, or illegitimate, end-joining reactions (Jeggo and others 1995). In large part, that is because the homologous chromosomes in a diploid G1 nucleus are widely separated, so nonhomologous recombination can occur at about 104 times the efficiency of homologous recombination (Godwin and others 1994; Benjamin and
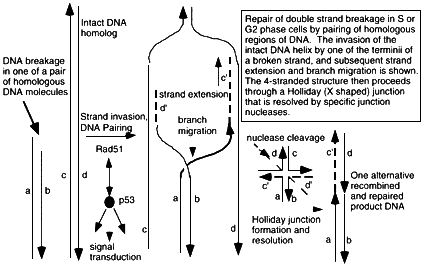
Figure 6.2
Mechanism of double strand break repair by homologous recombination through hybridization of the broken DNA strands sequences on the undamaged homolog. A DNA terminus is paired with the intact DNA by the action of pairing proteins including Rad51 and many other associated proteins that modulate its functions and carry out the numerous steps of pairing, elongation of DNA termini, and migration of hybridizing junction regions. Conformation changes produce a Holliday junction (a + form, 4-stranded junction) which is a strong binding site for p53, and which is resolved into separate DNA double helices containing regions of exchange, by junction-specific nucleases. The extent of sequence overlap can be very long, up to kilobases in length, and requires exact matching of DNA along most of the length of the hybrid molecules. Rad51-dependent DNA pairing is suppressed by p53-rad51 interaction, which is also a route for initiating intracellular p53-dependent signal transduction pathways. Broken double stranded DNA indicated by a,b; recipient intact strands by c,d; strands created by strand extension c', d'. De novo synthesis indicated by ——. Repair of a double strand break will require two of these homologous exchange events, one for each terminus. Some resolved DNA products may be visualized at the chromosomal level in mitotic cells as a sister chromatid exchange.
Little 1992). The relative importance of those pathways can vary with cell-cycle stage, tissue type, developmental stage and species. Direct measurement of DNA breakage and repair indicates that double-strand breaks can be rejoined rapidly—within a few hours. There is, however, a residuum of unrepaired damage that is greater for densely ionizing radiation, such as alpha particles, than for x rays (Ager and others 1991; Iliakis 1991; Iliakis and others 1990; Ward 1990). Although it is unknown if high levels of alpha-particle damage saturate DNA-repair systems, such potential saturation would not be relevant at low-dose ambient
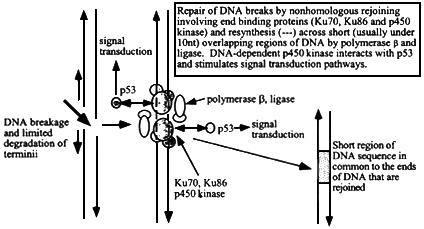
Figure 6.3
Mechanism of nonhomologous (illegitimate) recombination at sites of double strand breakage in DNA. The ends are sites of association of end-binding proteins, Ku70, Ku86, and p450 DNA-dependent kinase. After limited exonucleolytic degradation, short single stranded DNA termini (that may not necessarily be from either side of the original break) with a few nucleotides that can form base-pairs will hybridize and local regions can then be patched by DNA polymerase b and ligase. The extent of sequence overlap is very short, usually less than 10 nucleotides (more often 1 to about 5). The p450 kinase interacts with p53 and initiates intracellular signal transduction pathways.
radon exposure in which all damaged cells receive single alpha-particle tracks and the maximal track length through the nucleus is still below the D37 dose required for cell-killing. More important, even at low doses, is whether any kinds of damage are completely irreparable and whether repair is always accurate. Persistent genetic changes caused by radiation must then be caused by repair that misassembles broken termini from distant regions of the genome and triggers a lasting genetic instability.
Homologous rejoining involves matching of a broken fragment with the corresponding region on the undamaged homologous chromosome followed by strand invasion and reconstruction of the damaged region by replication of the sequence information in the intact homologue (figure 6.2). It requires that the two homologues are within range of each other; consequently, it might be more important for replicating cells in late S and G2 phases of the cell cycle when sister chromatids are in close apposition (Takata and others in press; Sonoda and others 1998; Thompson 1996) and contributes to increased radio-resistance in these phases of the cell cycle (Cheong and others 1994). This form of double-strand repair is likely to be highly accurate because of the use of sequence information from the intact chromosomal homologue (chromatid) in reconstructing the broken DNA. The Rad51 protein
plays a major role in carrying out the initial pairing reaction during homologous recombination, and dense complexes can be detected in the nuclei of irradiated cells (Haaf and others 1995), which are thought to be part of the homologous rejoining complexes (Scully and others 1997b; Scully and others 1997a). Rad51 is inhibited by association with the tumor suppressor p53 (Buchhop and others 1997) and interacts with the breast-cancer-specific gene products Brcal and Brca2 (Scully and others 1997b; Scully 1997a). Knockout of the Rad51 and the Brcal and Brca2 genes result in early embryo death (Lim and Hasty 1996; Tsuzuki and others 1996); this suggests a complex regulatory scheme for homologous recombination during development and carcinogenesis.
The nonhomologous recombination pathway for repair of radiation-induced DNA breakage in somatic cells involves an end-to-end rejoining reaction in which broken ends of DNA are braced by a set of supporting proteins. The gap between DNA ends is bridged by overlapping single-strand termini that are usually less than 10 nucleotides long (more commonly one to five long) and a set of proteins, including Ku70, and Ku86, p450 kinase, and DNA ligase IV (Kirchgessner and others 1995; Lees-Miller and others 1995; Getts and Stamato 1994; Rathmell and Chu 1994; Smider and others 1994; Taccioli and others 1994; Anderson 1993) (figure 6.3). The p450 kinase interacts with p53, the major signaling protein that regulates cell-cycle control, apoptosis, and the transcription of many downstream genes (Elledge and Lee 1995; Kastan and others 1995; Lane 1993). Defects in p450 have been associated with the systemic combined immunodeficiency (scid) phenotype in mice (Kirchgessner and others 1995). Knockout of the Ku70 and Ku86 genes renders cells more sensitive to ionizing radiation but, unlike the genes involved in homologous recombination, does not result in embryo death.
The rejoining reaction results in a junction made by an overlap of a few bases at each terminus with additional possibilities of single-base or larger insertions, deletions, or mismatches. No consistent DNA-sequence motifs have been found in these short regions of sequence overlaps, despite direct investigation of micro-satellite repeats and telomere and triplet repeat sequences. Insertions can be many kilobases long and can come from locally produced fragments or from single-strand invasion into proximal regions of DNA. The ends involved in rejoining reactions are not necessarily those from either side of the initial break but can be from other breaks made by the same alpha track. The intervening stretch of DNA can then be lost, with consequent chromosomal rearrangement. These losses and rearrangements can involve many kilobases of DNA, producing the losses, deletions, and rearrangements of genetic material which are hallmarks of genetic effects caused by densely ionizing radiation (Zhu and others 1996; Kronenberg and others 1995; Nelson and others 1994; Phillips and Morgan 1994). The process of DNA breakage and rejoining therefore initiates a major change in signal transduction and cellular regulation that can persist over many cell generations (see discussion of genetic instability below).
The ion pairs that do not directly damage DNA can produce reactive oxygen intermediates. These intermediates influence the stability of p53 with downstream effects on cell regulation and can activate many cellular systems that are sensitive to the redox state of the cell, such as the fos/jun transcriptional regulators (Xanthoudakis and others 1996). Reactive oxygen intermediates can produce oxidative damage, of which 8-oxy-guanine is a major product. Oxidations are produced in DNA and in both deoxyribose and ribose triphosphates. Oxidized nucleotides can be incorporated into DNA and RNA, and lead to either DNA mutations or transcription and translation errors. Oxidized nucleotides can be eliminated from the nucleotide pool by MutT, which hydrolyzes 8-oxo-dGTP and 8-oxo-rGTP to monophosphates, thereby removing the oxidized bases from the pool of DNA and RNA precursors (Taddei and others 1997). MutT activity reduces mutations from naturally occurring oxidative reactions by a factor of about 104.
Oxidative damage involves production of damaged individual bases, such as 8-oxy-G, and many other products in DNA that cause point mutations by mispairing during DNA replication (Singer 1996) and that are repaired by the base-excision repair system. Base excision involves a set of glycosylases with limited ranges of substrate specificity (uracil, 3-methyladenine, formamidopyrimidine, glycosylases and others). The glycosylases remove damaged bases (Cunningham 1997; Singer and Hang 1997), leaving apurinic sites that are later cleaved by apurinic endonuclease (Hang and others 1996), and the gaps are replaced by polymerase β and completed by ligase I or III (Sancar and Sancar 1988). Several base-excision repair enzymes have multiple additional functions: the AP endonuclease is also known as Ref-1 and reduces the oxidized transcriptional regulators fos/jun (Xanthoudakis and others 1996); and pol β and ligase III are linked by structural protein XRCC1, which interacts with poly (ADP-ribose) polymerase (PARP) (Caldecott and others 1994). PARP is a major chromatin protein that is activated by DNA strand breaks and can exhaust the cellular NAD content by polymerization and hydrolysis (Cleaver and Morgan 1991).
DNA breaks and other base damage therefore are the assembly points for complex, multifunctional, multipurpose structures that signal their presence to many other cellular processes and within which repair and genetic changes occur. The combined actions of these cellular caretakers produces surviving cells that bear the permanent marks of alpha particle exposure, including deletions, insertions, amplifications, point mutations, and altered cellular regulation (Kronenberg and others 1995; Kronenberg 1994).
Deletion Mutagenesis and Chromosomal Changes Caused by Densely Ionizing Radiation
The end result of DNA breakage and rejoining is the deletion, insertion or rearrangement of various amounts of genetic material, from a few base pairs
through many kilobases to cytogenetically visible chromosomal changes (Sankaranarayanan 1991). Chromosomal fragments that are not rejoined can be excluded from interphase nuclei and can form micronuclei. These micronuclei, which encapsulate p53 (Unger and others 1994) can be scored as a quantitative measure of chromosomal damage in somatic and cultured cells. The size of deletions that persist in surviving cells is determined by the initial spacing of DNA double-strand breaks and by the presence of vital genes in the intervening sequences. Deletion sizes associated with loss of function of the adenine phosphoribosyl transferase (APRT) gene, for example, are generally smaller than those associated with loss of function of the hypoxanthine phosphoribosyl transferase (HPRT) gene because of the presence of vital genes closer to APRT than HPRT (Park and others 1995; Nelson and others 1994; Fuscoe and others 1992; Morgan and others 1990; Thompson and Fong 1980). Deletion sizes and junction positions are markedly nonrandom in both the chromosomal HPRT gene and in episomal vectors that carry reporter genes. The positions of DNA breaks and the efficiency and precision of their repair are therefore strongly influenced by chromatin structure and attachment of DNA to nucleosomal and matrix proteins and the functions of flanking genes. In an experimental cell-culture system in which a single human chromosome bearing a marker gene is carried in a hamster cell line (the AL cell line), very few of the human genes are required for cell survival, and alpha-particle damage can produce very large deletions that involve most of the chromosome (Hei and others 1997; Ueno and others 1996). This situation cannot apply to most chromosomes in a normal cell, in which deletion sizes consistent with survival will be limited by the presence of important genes distributed throughout the genome.
Control of Cellular Responses to Damage—The Arbitrator Gene
One gene product, the p53 protein (figure 6.1), plays a critical role in regulating the multitude of responses that are elicited in damaged cells, especially those involving cell-cycle arrest and apoptosis, and interacts with numerous other regulatory and repair proteins (Elledge and Lee 1995; Kastan and others 1995; Lane 1993; 1992). The p53 protein is a rapidly synthesized, but short-lived, multifunctional protein which interacts with a wide array of other cellular and viral proteins and binds to DNA in both sequence-specific and sequence-independent fashions. In the presence of damage (either DNA breaks or reactive oxygen intermediates) the lifetime of p53 increases, it is phosphorylated at specific sites that depend on the particular signal, and it acts as a transcriptional activator with downstream effects on many other genes, especially stimulating transcription of p21, which then inhibits cell-cycle progression. Alpha-particle irradiation at low exposures has been shown to result in p53 stabilization in more cells than could have experienced alpha-particle tracks: this suggests that reactive oxygen intermedi-
ates generated outside the nucleus could result in substantial changes of cell regulation (Hickman and others 1994).
The level of damage and of consequent p53 function plays a major role either in causing cell-cycle delays (through activation of the p21 gene, which blocks cells in the G1 phase) or in initiating apoptosis. Several of the protein complexes involved in DNA breakage and repair interact with p53, including the homologous and nonhomologous recombination complexes, and the transcription-factor component of nucleotide-excision repair, so the action of repair systems leads into the signal-transduction pathways regulated by p53. Mutations in p53 are important events that occur frequently at some stage in tumor progression and are found in over 50% of all human tumors (Greenblatt and others 1994; Hollstein and others 1991). The consequent functional changes alter many facets of cellular and gene regulation. These mutational changes in p53 do not necessarily constitute the first genetic event in carcinogenesis; for example, they can occur early in sunlight-induced skin tumors (Brash and others 1991) but late in colon cancers (Kinzler and Vogelstein 1996). The p53 protein might, in fact, play a multitude of roles in cancer, from the initial response to DNA damage, through tumor initiation and progression, to final malignancy.
Apoptosis—The Undertaker Genes
Cells die by several routes depending on cell and tissue type and on the particular endogenous and exogenous signals experienced. Unrepaired chromosomal damage can cause ''mitotic death'' when cells attempt to divide; massive damage can cause necrosis, with rapid collapse of the nucleus and permeabilization of the membranes; and a regulated cell-suicide process, apoptosis, that involves activation of proteases (caspases) and nucleases that degrade the cell components in a controlled fashion can occur (figure 6.4) (Cohen 1997). Apoptosis is activated by a wide variety of complex interrelated regulatory and signal-transduction pathways initiated by specific cellular signals, by external irradiation, and by endogenous generation of oxidative products. Some of these processes may be markedly nonlinear functions of dose, since apoptosis is a tissue response which eliminates cells that have suffered more than a critical amount of damage.
Apoptosis is an important feature of normal cell and tissue function, especially when tissue remodeling is involved during embryo development, during wound healing, and after exposure to radiation or chemicals. Apoptosis involves a family of specific proteolytic enzymes (caspases) and a specific nuclease that cleaves DNA at internucleosomal sites and produces characteristic DNA fragmentation (Enari and others 1998). Apoptosis is a complex, regulated process that involves both activators and inhibitors. These molecules fine-tune a cell's response to endogenous damage, modify its redox state, and respond to its immediate environment (Enoch and Norbury 1995; Guillouf and others 1995; Kastan
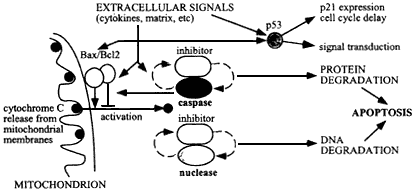
Figure 6.4
Mechanism of apoptosis, initiated by damage to cellular molecules or cellular signals during tissue remodeling or wound healing, resulting in cytochrome C release from mitochondria. This release is regulated by Bax/Bc12 on the outer mitochondrial membranes, and results in activation of caspases and an apoptotic-specific nuclease. These degradative enzymes normally are associated with specific caspase-sensitive inhibitors, so that once apoptosis is set in train an autocatalytic process results with positive feedback to produce irreversible cellular degradation.
and others 1995; Kumar 1995; Leonard and others 1995; Caelles and others 1994; Canman and others 1994; Jacobson and Evan 1994; Meyn and others 1994; Lowe and others 1993; Waddick and others 1993; Uckun and others 1992). Fluctuating oxygen levels leading to oxidative bursts and the production of reactive oxygen intermediates can trigger apoptosis through their activation of p53. The mitochondria play an important role in the initial events leading to apoptosis, and one of the first signals is the release of cytochrome C into the cytoplasm (Reed 1997), which, with dATP activates a caspase cascade involving especially caspase-3 (Li and others 1997). The gene product Bc1-2 is on the outer mitochondrial membrane, where it regulates ion flow and, under conditions of normal expression, suppresses apoptosis. Its expression is induced by p53 (Pourzand and others 1997; Chen and others 1995). Two other proteins, Bax and Ced-4, bind to Bcl-2 and are inactive in bound form, but on release they further stimulate the release of apoptosis-initiating factors, which eventually activate the caspase class of proteases (Kumar 1995). One pathway to apoptosis is thus determined by the ratio of Bax/Bcl-2 expression. Many other proteins are involved in apoptosis, including many members of the caspase family of proteases and caspase inhibitors which regulate the process in different tissues and under various stresses. Other pathways by which apoptosis is activated involve the cytokines, such as TNF1-alpha, and the signal transducer and activator protein STAT1 (Kumar and
others 1997) and c-myc, which interacts with Bc1-2 (Bissonnette and others 1992; Evan and others 1992; Fanidi and others 1992). The level of ATP also influences apoptosis (Eguchi and others 1997), and one of the early targets for caspases, PARP, can drain the cell of NAD and ATP in the presence of excess DNA breakage (Shah and others 1996).
Apoptosis is a normal process of cell elimination that can clear abnormal cells from the population. If apoptosis is no longer functional, abnormal cells can persist and expand in the population. The loss of apoptosis can therefore play an important role in clonal expansion during carcinogenesis. Additional genetic changes can then occur, moving a cell population through multiple stages required for the emergence of a fully malignant phenotype. Along the way, irradiated cell populations and tumor cells develop instabilities and mutator phenotypes that favor further diversity.
Initial Genetic Changes in Carcinogenesis—The Gatekeeper Genes
Investigation of rare cancers with strong hereditary factors—such as subsets of colon, breast, retina, and skin cancers—has suggested that genetic alterations are involved in carcinogenesis (Fearon 1997). In general, tissue-specific alterations in a small number of genes act as critical rate-limiting steps that allow cells to escape from normal controls on growth and function and to develop into autonomous populations. The genes that normally exercise these tissue-specific controls have been figuratively called gatekeepers (Kinzler and Vogelstein 1997). The autonomous cell populations acquire functions that include the suppression of apoptosis, independence from extracellular matrix signals, invasive behavior, genomic instability, the activation of oncogenes, and the inactivation of tumor suppressors. Oncogenes, such as ras, are genes that are activated by mutation or translocation and act as dominantly acting genes that induce malignant properties. Tumor suppressors, such as Rb, are genes that maintain normal cell and tissue homeostasis and whose loss permits unregulated cell proliferation to begin.
The ordered progression of genetic changes involved in carcinogenesis is most clearly understood in colon carcinogenesis (Kinzler and Vogelstein 1996). One of the earliest genes to be affected is the APC gene; it is followed by changes in DCC, p53, ras, and others. Error generation in this process is enhanced by mutations in mismatch-repair genes (Arnheim and Shibata 1997). In colon carcinogenesis, APC mutation seems to be required early, before ras mutation; when ras mutations are observed first in colon polyps, the growths are usually benign and regress; when APC mutations occur before ras mutations the tissue usually progresses to other changes that result in malignancy (Jen and others 1994). It is unlikely that such an ordered sequence of mutations or gene loss will be as clear and precise in most tissues, but the principle of an approximate sequential order to the earlier genetic changes involved in carcinogenesis seems reasonable.
The APC gene can therefore be regarded as an example of a rate-limiting gatekeeper gene that presents an initial barrier to be overcome in the initiation and progression of colon cancer (Kinzler and Vogelstein 1997). In the retina, the Rb cell-cycle regulatory gene appears to play the gatekeeper role (Newsham and others 1998); in sunlight-induced nonmelanoma squamous carcinoma of the skin, p53 plays this role (Brash and others 1991); in breast cancer it might be the Brcal and 2 genes (Couch and Weber 1998); in basal cell cancers it might be the signal-transduction pathway involving genes called "hedgehog" and "smoothened" (Xie and others 1998; Epstein Jr 1996; Johnson 1996). In the progression of stomach cancer, p53 mutations occur earlier than in the small intestine, and tumors with mutations in EGFR-1 are more aggressive than those with p21 (WAF1) mutations. Amplification of c-erbB-2 and of some specific chromosomal regions and loss of heterozygosity in a region containing thymine glycosylase have been reported in stomach cancers.
When a gatekeeper gene can be clearly identified, it should contain mutations or rearrangements that are characteristic of the initiating damage, in that these changes represent some of the earliest genetic events in carcinogenesis. Tissue-specific genes might be similarly involved in the initiation of cancers of lung, stomach, and other tissues by exposure to radon and radon progeny alpha particles. Some of the initial genetic changes resulting from alpha-particle irradiation, such as deletions and rearrangements, are distinctive and might leave characteristic genetic changes, or "fingerprints," on gatekeeper genes and on others activated early by exposures, thus aiding in their identification. But deletions and rearrangements are also common events during tumor progression because of their inherent genomic instability, so alpha-particle fingerprints might be obscured in advanced tumors.
Tumor Growth and Nutrition—The Caterers
Tumors rapidly outgrow the capacity of diffusion from pre-existing blood supplies to provide the oxygen, nutrients, and growth factors required to sustain their growth and expansion. Anoxic regions of tumors that develop far from blood vessels have been shown to contain elevated amounts of p53 indicative of their abnormal state (Graeber and others 1994). Consequently, a critical factor in tumor growth is the capacity to stimulate new blood vessel growth—angiogenesis. Angiogenesis is achieved by a combination of mechanisms. Tumors secrete stimulators of new blood vessel formation (vascular endothelial growth factors) and reduce the presence of inhibitors (Boehm and others 1997; Folkman 1996). Because the growth of new blood vessels involves the proliferation of essentially diploid, normal endothelial cells, these do not exhibit the genomic plasticity of tumor cells and are subject to normal cell regulation. Proliferative endothelial cells exhibit characteristics and gene-expression profiles different from those of
mature established blood vessels and so might even constitute a unique target for cancer therapy (Boehm and others 1997).
Genetic Instability in Irradiated Cell Populations—The Diversifiers
Damage caused by high-and low-LET radiation exposure appears to create a genetically unstable state in which further chromosomal and genetic changes can be observed many generations after the exposure. That was first observed for alpha particles by Kadhim and others (1994; 1992) who detected chromatid and chromosomal type aberrations in clonal descendants and nonclonal cultures of both mouse and human hematopoietic stem cells. The instability is not confined to high-LET radiation, and it can even be induced by ionization produced outside the nucleus. Abnormal karyotypes were observed several passages after irradiation; this indicated that heritable changes were transmitted to progeny cells and resulted in new chromosomal rearrangements during later cell cycles. There is evidence that those changes can involve a wide variety of genetic events, including rearrangements, gene amplification, and mutation. DNA sequence rearrangements can lead to mutations, the production of new fusion genes, or changes in gene regulation by position effects that are known to be involved in chromosomal activation of oncogenes in several human and rodent malignancies (Rabbitts 1994). The mechanism of instability might involve rearrangements that result in inappropriate gene expression that then triggers later genetic events. Alternatively, it could involve persistent changes in gene expression through p53 and other gene products that act as altered transcriptional regulators.
The high frequency of chromosomal abnormalities and mutations in human cancers indicates that a "mutator" phenotype is often involved in multistep carcinogenesis (Loeb 1994; 1991). The spontaneous-mutation rate in normal diploid cells is insufficient to account for the high frequency of mutations in cancer cells. Rather, the genomes of cancer cells are unstable, and this results in a cascade of mutations that cumulatively enable cancer cells to bypass the host regulatory processes (Loeb 1994). The development of genetic instability, especially the capacity for gene amplification, is acquired in stages through preneoplastic to fully neoplastic cells, and this capacity appears to depend on the progressive loss of p53 function (Tlsty 1996; Tlsty and others 1995).
DNA damage of various kinds is particularly effective in inducing genomic instability, whether produced by α-particles or x rays or endogenously. For example, an anoxia-inducible endonuclease activity has been reported that cleaves DNA without specificity for sequence (Stoler and others 1992). That activity could account for the induction of gene amplification in anoxic cells and could be associated with break-related genomic instability. Repeat sequences, such as interstitial telomere-like repeats might also be hot spots for recombination, break-
age, and chromosome fusion (Alvarez and others 1993; Ashley and Ward 1993; Bouffler and others 1993; Day and others 1993; Hastie and others 1990). Even chromosomal rearrangements that appear stable, such as balanced translocations, are not as secure as normal chromosomes and show declines in frequency with time after radiation exposure in vivo (Tucker and others 1997). A frequent result of chromosomal instability in tumor progression is the loss of a chromosome and the reduplication of the homologue; the chromosome number is maintained with the loss of heterozygosity (LOH). That can result in the loss of a normal gene and the duplication of mutant genes. Recent analysis of a large number of tumors indicates that LOH can involve an exceedingly large variety of genome-wide alterations even for a single tumor type (Kerangueven and others 1997).
The processes of mutation, insertion, deletion, rearrangement, loss of heterozygosity, reduced apoptosis, radiation-induced genomic instability, and the continued replication and proliferation of stem cells lead to a number of critical changes in genes along the paths that result in malignancies. Each tissue might require changes in specific genes, possibly in a particular sequential progression, for complete malignancy to emerge. The need for an ordered set of changes leads to the concept of fingerprints: characteristic mutations in tissue-specific rate-limiting genes that need to be altered early to allow tumor progression (Dogliotti 1996).
Mutations In α-Particle-Induced Tumors—The Fingerprints
It would be expected on the basis of in vitro work, that radon alpha-particle-induced cancers of the lung and other tissues would contain characteristic mutations, fingerprints, in critical gatekeeper genes that initiate carcinogenesis (Dogliotti 1996). Genetic changes that occur during tumor progression are likely to involve many genes but would lack characteristic fingerprints. The strongest example of a carcinogenic fingerprint is the detection of C to T mutations at the 3' C in dipyrimidine sites in nonmelanoma skin cancers, representing mispairing at sites of sunlight-induced photoproducts in DNA (Brash and others 1991). Carcinogenesis, however, is a highly selective process in which many genetic changes are pruned by selective constraints before the fully malignant cell types with genetic variability, unregulated growth, and invasive properties emerge. Many large α-particle-induced deletions might therefore be inconsistent with emergence of these properties and be lost from such a population. Deletions with a range of sizes up to complete gene loss, however, have been observed in the Rb and p53 genes in murine tumors induced by ionizing radiation (Zhang and Woloschak 1997). The mutations observed in α-particle-induced tumors will therefore be a subset of the full spectrum of genetic changes that are produced initially. But with the prevalence of genomic instability in tumors, specific deletions and rearrangements might easily become obscured; this would make a search for α-particle-induced fingerprints difficult.
A potential fingerprint of α-particle damage at the whole-chromosome level has been suggested. Because of the physical distribution of α-particle tracks, there might be a much lower ratio of interchromosomal exchange aberrations to intrachromosomal exchanges compared to the ratio of these exchanges induced by either low-LET radiation or chemically induced damage (Brenner and Sachs 1994). Again, after the scrambling of the genotype associated with tumor progression, this ratio would be extremely difficult to assess in advanced tumors.
There have been only a few analyses of tumors known to be induced by radon or other α-particle exposure. One set of results is from miners who experienced high radiation doses and dose rates—doses that might not correspond to the exposures expected from domestic situations. The evidence of signature mutations is not strong. A report described point mutations in codon-249 and 250 of the p53 gene, but these could have been spontaneous events or induced by the molds or cigarette-smoking associated with miners' working conditions (Vahakangas and others 1992). The presence of signature mutations in the p53 gene therefore remains to be established. Alternatively, if p53 is not the critical, rate-limiting gatekeeper gene for lung carcinogenesis, signature mutations might yet be identified when the appropriate genes are known and investigated.
Epidemiologic, Biophysical, And Cell-Based Models Of Radon-Induced Carcinogenesis
To obtain estimates of risk posed by exposure to radon in air or drinking water, it would be ideal to trace the complete process from α-particle exposure to cancer, on a quantitative, biologic, and molecular basis and to incorporate such difficult issues as individual and subpopulation variations in susceptibilities (see BEIR VI, National Research Council 1999). Unfortunately that is not yet feasible. Instead, the problem of risk estimation has been approached from a variety of avenues. One is through strict epidemiologic relationships between numbers of cancers and exposure and the use of the linear no-threshold dose-response curves used commonly in radiation risk estimates. Another approach introduces biophysical models of radiation action based on radiation tracks, total doses and dose rates, damaged sites in DNA, and breaks and their rejoining and, from these considerations, reaches interpretations of risk versus dose. Most often, this approach has been used to explain such phenomena as the inverse dose-rate effects relevant at doses higher than those that would arise from domestic exposures (Brenner 1994; Elkind 1994). The approach still does not pay specific attention to the particular genetic changes involved in cancers, and a more detailed attempt to interpret carcinogenesis on a quantitative basis has incorporated changes in cell cycles, proliferation kinetics, cell-killing (Luebeck and others 1994), and other biological processes alluded to in this chapter (Luebeck and others 1994). Because of the large numbers of variables involved, these approaches are still too difficult, computationally, to incorporate into a completely predictive model.
Instead, the biologic approach gives a mechanistic underpinning to the epidemiology and biophysical interpretations of risk. Together, they lead to a more comprehensive understanding of cancer risks posed by low ambient radiation exposures and provide a rational basis for quantitative exposure risk assessment and mitigation by multimedia approaches to risk reduction.



















