
Biotechnology and the Human Genome Project
Charles R. Cantor
Director, Center for Advanced Biotechnology
College of Engineering
Boston University
Boston, Massachusetts
The Department of Energy (DOE) has played and is poised to continue to play a unique role in biology. This role is well symbolized by the Office of Health and Environmental Research (OHER). OHER is the only entity I know that covers the dimensions from the whole world, such as global climate change, through all kinds of environmental issues down through properties of individual DNA molecules. OHER constitutes a unique vertical integration, and I strongly encourage that this be continued.
My introduction to the Human Genome Project began at a meeting in Alta, Utah, in December 1984 that was cochaired by Mort Mendelson and Ray White. It was a very exciting meeting. It was called to deal with the issue of mutation detection in the Hiroshima-affected cohort, but it rapidly became an intense brainstorming session in which those present reached the consensus that if one wanted to detect mutations de novo in humans at the DNA level, one would have to have the analytic power to sequence the human genome.
Shortly after Alta, a few additional meetings were held, and a famous memorandum was written by Charles DeLisi to A1 Trivelpiece, suggesting the possibility of a DOE-funded human-genome project. This year is the 10th anniversary of the Coldspring Harbor genome meetings, and it is appropriate that the cover of the booklet for that meeting showed a photocopy of the memo from DeLisi to Trivelpiece.
The most-remarkable thing about this project, looking back 10 or 15 years, is that, although all of us had a clear picture of the cost that would be involved and the number of base pairs that would be involved, I, for one, never thought about how many people would be involved. Thousands of people are now working in a field, genomics, that was not named 12 years ago.
There is a flow of biologic information (table 1) from DNA to RNA to proteins to 3-dimensional structures to assemblies of structures and pathways to interactions with small molecules. This flow is described by words such as genome, gene expression, proteome (the world of proteins), phenome (the word of function), and, most important to the private sector, drugs and chemistry.
The biologic information starts as 1-dimensional, but it becomes 3-dimensional and even 4-dimensional. It is more difficult to deal with things in higher dimensions, so we are going to be more challenged as we move over the next decade from relatively easy 1-dimensional molecules, DNA and RNA, to difficult multidimensional objects, like functional assemblies of proteins.
Our ability to handle biologic information, with today' s technology, varies from very poor to very good. If we have small-molecule-binding data to characterize or biologic function to observe (someone has blue eyes or blond hair, and so on), we can interpret readily. For most people, trying to look directly at the information in DNA is like trying to translate hieroglyphics; even at the RNA level, for most of us, trying to make sense out of sequence data
TABLE 1. The Flow of Biologic Information
|
|
DNA |
RNA |
Proteins |
Folded Structures |
Function: Assemblies Interactions Pathways |
Small-Molecule Binding |
|
Catchword |
Genome |
Gene expression |
Proteome |
Phenome |
Proteome |
Drugs, chemistry |
|
Dimension |
1-D information |
1-D information |
1-D information |
3-D structure |
3-D structure |
Time-dependent events |
|
Interpret ability of raw data |
''Hieroglyphics'' |
"Greek" |
Greek" |
Picture |
Readable |
Readable |
|
Complexity |
Human 3 × 109 base pairs |
100,000 genes |
100,000 proteins |
2,000 motifs |
>1012 combinations |
>1040 small molecules |
|
Species dependence |
All different |
Most different |
Most different |
Most same |
Most same |
Most same |
|
Relative abundance |
All genes 1:1 |
1:10,000 dynamic range |
1:100,000 dynamic range |
— |
— |
— |
is like trying to read Greek without knowing the language. Many of the DNA data that are most easily obtained—sequence data—are the most difficult to interpret.
A few key numbers characterize the field of genomics. The human genome is the haploid genome; 1 copy is 3 × 109 base pairs of DNA. Current estimates of the number of genes cluster around 100,000. Usually, 1 gene codes for only 1 protein, so there are roughly 100,000 proteins. The motifs mentioned in the previous presentation are the fundamental building blocks of 3-dimensional structures. No one knows how many there are—possibly only a few thousand. When proteins are combined into assemblies and functional pathways are created, the number of such entities can be astronomical. The number of known small molecules is far in excess of 106, and the number of potential small molecules is close to unlimited.
As one goes from person to person or from species to species, we are frighteningly different at the DNA-sequence level. However, as one moves toward higher order structures and into the world of function, similarities among various species are much greater. Thus, depending on which viewpoint one takes, the environment is either a difficult problem viewed at the level of DNA sequence or a simpler problem viewed at the level of common biologic function.
Different dynamic ranges are encountered as we progress from the world of DNA to the world of function. We do not know enough to be rigorously quantitative, but DNA, roughly speaking, is monotonous. Most genes are present in a 1-to-1 ratio. However, at the level of gene expression or protein synthesis, dynamic ranges that are fairly formidable appear.
STATUS OF THE GENOME PROJECT
The genome project is doing well. Maps have been made, and sequences have been completed.* The number of completed genome sequences is changing almost dally (table 2). These are all very small genomes. The biology emerging from this knowledge is astounding, but our ability to interpret the sequences is so poor that the number of genes that code for an RNA product, rather than a protein product, is uncertain. As Jurgen Brosius and others have shown, the numbers listed for such genes in table 2 are probably wrong. As a result of the first microbial-genome projects, we are already drowning in important and fascinating information. As more genomes, especially those of microorganisms, are sequenced rapidly over the next few years, the surfeit will get greater and greater.
The results in table 3 are less optimistic. The table shows the progress in completing the sequence of the 6
TABLE 2. Genomes with Known Nucleic Acid Sequence
|
Species |
No. DNA molecules |
No. kb DNA |
Largest DNA |
No. ORFsa |
No. genes for RNA |
|
M. genitalium |
1 |
580 |
580 |
470 |
38 |
|
M. pneumonia |
1 |
816 |
816 |
677 |
39 |
|
M. jannaschii |
3 |
1,740 |
1,665 |
1,738 |
~45 |
|
H. influenza |
1 |
1,830 |
1,830 |
1,743 |
76 |
|
Synechoncystis sp. |
1 |
3,573 |
3,573 |
3,168 |
? |
|
E. coli |
1 |
4,639 |
4,639 |
4,200 |
? |
|
S. cerevisiae |
16 |
12,068 |
1,532 |
5,885 |
455 |
|
a DNA sequences that appear capable of being translated into proteins long enough to be functional. |
|||||
canonical organisms that were picked by the National Research Council Committee on the Human Genome in the late 1980s. The 2 smallest have been completed, but even 5 years ago there was a considerable amount of sequence information available on these organisms.
Caenorhabditis elegans sequencing is going extremely well, and the estimates are that it will be completed in about another year, but Drosophila-, mouse-, and human-sequencing projects have just begun, and the numbers shown in table 3 are hard to come by and hard to stand by. It is difficult to know what is redundant in the sequence databases.
PRIVATE-SECTOR ACTIVITIES
One remarkable thing about the genome project was how aggressively, forcefully, and, I think, effectively the private sector entered the project and in many ways attempted to skim the cream. I would like to say it was predictable. I would like to say we anticipated it, but I think hardly anyone did.
Table 4 shows, as of April 1, 1997, a list of some of the public companies whose sole declared mission is genomics. The aggregate market value of these companies is $2.5 billion. Other companies are not public, so they do not have value that we can easily calculate. At least 1 major additional public company, Affymetrix, should be added to the list; its valuation is surely in the $0.5 billion range. It is easy to do simple-minded calculations on these numbers and conclude that the private-sector effort in genomics is already greater than the government-funded effort worldwide. And it is likely to continue—many of the large pharmaceutical companies are beginning to mount substantial genome efforts of their own.
TABLE 3. Progress in DNA Sequencing of Chosen Targets
|
|
|
Finished Sequence, Mb |
|
|
|
Organism |
Complete genome, Mb |
June 1992 |
February 1997 |
Comment |
|
E. coli |
4.6 |
3.4 |
4.6 |
Complete |
|
S. cerevisiae |
12.1 |
4.0 |
12.1 |
Complete |
|
C. elegans |
100 |
1.1 |
63.0 |
Cosmids |
|
D. melanogaster |
165 |
3.0 |
4.3 |
Large contigsa only |
|
M. musculus |
3,000 |
8.2 |
24.0 |
Total, assuming 2.5× redundant |
|
H. sapiens |
3,000 |
18.0 |
31.0 |
In contigs >10 kb |
|
|
|
|
116.0 |
Total, assuming 2.5× redundant |
|
a Sets of overlapping clones or DNA fragments. |
||||
TABLE 4. Values of Some Genome Companies (April 1, 1997)
|
Name |
Market Valuation (millions of dollars) |
|
Human genome sciences |
673 |
|
Incyte pharmaceuticals |
559 |
|
Millennium pharmaceuticals |
332 |
|
Genset |
322 |
|
Myriad genetics |
275 |
|
Sequana therapeutics |
131 |
|
Microcide |
128 |
|
Genome therapeutics |
123 |
|
Total |
2,543 |
I think it is important, as we go forward, to ask how we should optimize the roles, separate as they are, of the private and public sectors. Who is going to do what? I do not have a simple answer, but I will try to point out places where I think it is critical for the public sector to keep focused on issues that the private sector is not going to focus on, because the scale on which the private sector operates and the speed at which it operates are impressive. That all these companies exist and have raised so much capital means that DNA-sequencing information is viewed as valuable. That is no surprise. Once one assembles a large DNA-sequencing effort, as exists now in both the public and the private sectors, it is a valuable resource. The challenge is to use that resource effectively, and this challenge is sometimes confusing.
Selecting Sequencing Targets
An open reading frame (ORF) is a gene. Some examples of the density of open reading frames are shown in table 5. In Methanococcus jannaschii , the archibacterium that is part of the DOE microbial-genome project, there is an extremely high density of genes—1 gene for every 1,000 base pairs. In sequencing this organism, one gets useful information very efficiently. In E. coli, the ORF density is more or less the same. In yeast, the density is about 1 gene per 2,000 base pairs. But in the human (we can only estimate, because we do not know the complete sequence), it is 1 gene per 30,000-50,000 base pairs. If one blindly decides to sequence human DNA today, functional units of information will be discovered at only 1/50 to 1/30 the rate for bacteria, if one does simple genomic sequencing.
The challenge is to figure out whether direct human-genome sequencing is worthwhile. Alternatively, given, say, the ability to sequence 100 million base pairs a year, how does one obtain the largest amount of information? I will argue, as devil's advocate, that we should sequence the genes first. We did not know how to do that 10 yr ago when the genome project was started, so it was not part of the plan. But we know how to do it today. If we
TABLE 5. Density of Open Reading Frames
|
Species |
Kb/ORF |
|
M. jannaschii |
1.001 |
|
E. coli |
1.105 |
|
S. cerevisiae |
2.051 |
|
Human |
30-40 (estimate) |
focus on sequencing the genes in the human and many other organisms, we are going to gain important biologic information much faster than if we just sequence the human chromosomes from end to end.
There is another reason to recommend this approach. When the human-genome project was founded, the lie was told that we are going to complete the DNA sequences from telomere to telomere on each chromosome. That is not true. It cannot be done; not with any technology that we have today. Centromeres, as have been elegantly studied by Bob Moyzis and others, are monotonous. There are millions of base pairs of simple repeated sequences with occasional variations. Even if one could figure out how to sequence such regions, most of them are probably unclonable. Doing such monstrous stuff—the last 10% of the genome—might require almost 90% of the total effort, and probably very little could be learned for it.
We should sequence all the genes and then "declare victory" once it is done. As technology gets better and costs go down, we can fill in all the other garbage, which today is not very enlightening.
GENOMICS IN THE FUTURE
I want to turn to the future of genomics. The first issue to address is the design of optimal strategies. The genome project has been tremendously successful thus far. As has been said many times, the project is ahead of schedule and it is under cost. I wish other large science projects were doing as well.
Economies Of Scale
A significant reason that the genome project is so successful is that some of its features, such as genomewide mapping versus individual-gene mapping, give an obvious economy of scale. One can argue about the numbers, but huge economies of scale have been gained in some genome-related manipulations. Leroy Hood' s presentation described transcript display as an alternative to looking at 1 gene expression at a time. It is obvious that there is an advantage. However, it is not clear that anyone has figured out how to do an equivalently effective display of proteins.
Mutation detection, if one has to do it by direct DNA sequencing, is very inefficient because one has to sequence millions of base pairs to find 1 mutation. If someone can come up with a method of comparative sequencing that projects only the differences, we will gain an enormous economy of scale.
Wherever one can move human-biology problems into simpler organisms—such as yeast or worms, which have short life spans—it affords a large economy of scale. It is not obvious to me that there is any economy of scale in making all possible transgenic mice, a task I am sure people think about. Similarly, in structure determination: At this point, although we have gained speed, it is not obvious yet what the economies of scale are.
One reason DOE deserves enormous praise is that it has focused a considerable fraction of its resources on the development of new and enabling technologies that promise in the future, or even right now, to gain economies of scale. Array technology is something that I knew would be covered in Leroy Hood's presentation, so, in spite of its importance, I will not dwell on how arrays gain an economy of scale because of parallelism.
Another potential new method that could provide an economy of scale is mass spectrometry (MS). It allows both high-speed work and multiplexing; many things can be done at once through multiple labels. Other new technology that seems promising is the microfluidic circuit, which is essentially a laboratory on a chip. This promises both parallelism and speed. The last set of potentially new techniques incorporates the power of intelligently used biology, whether through genetic selections or genetic manipulation. This is one of the ways that we might be able to get some economy of scale in studies of function.
The Challenge Of Genetic Testing
Thus far I have presented a very rosy picture, but not everything is rosy. At the beginning of the human-genome project, we promised that by the end of the project we would be able to do "genetic fingerprinting" and, depending on how good the technology became, look at the 100-1,000 most-common genetic diseases, predispositions, or environmental risk factors in a single relatively inexpensive test—everyone could be genetically pro
filed at birth. That promise looks more remote today. The reason is not that our technology has not improved; in fact, it has improved remarkably. The reason is that the problem of genetic testing has turned out to be much harder than we thought it would be.
The picture of a genetic disease that I think most of us had 10-15 yr ago was hemoglobin S. One base change, 1 allele, caused the same disease in everyone affected. That picture is not representative. Figure 1 shows the result of the last time my co-worker, Joel Graber, peeked into the European Bioinformatics Institute mutation database for p53, a gene important in breast cancer, colon cancer, and probably many other cancers. The figure shows the spectrum of known mutations; the vertical axis is the number of times that a particular mutation has been seen, and the horizontal axis is the position in the gene. The complexity of the mutation spectrum is extraordinarily complicated, and it is typical. If one wanted to do a diagnostic test to guarantee that someone was free of any of these known disease-causing alleles, one would have to sequence the whole gene or come up with a totally new technology that would allow inference that the gene was normal without sequencing it. To make things worse, if we look at the existing data on p53, about 5,000 mutations are known. Almost all of them are single-base substitutions. We can think of fancy tricks to find larger changes easily, but these are not larger changes; they are all single-base substitutions, so methods need to be developed to attack this problem. Today, many of the methods are not user friendly.
Today, mutation detection usually requires accurate DNA sequencing. Now that we know what the mutation
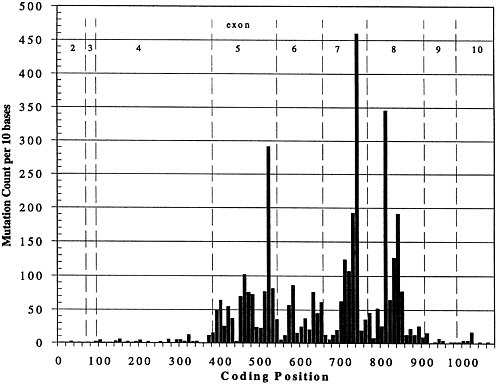
FIGURE 1. Spectrum of mutations seen in p53 expressed sequence.
spectrum is like, the challenge for genetic fingerprinting is the following: Suppose that we are willing to try to score 6 × 106 people at birth; this is 0.1% of the total diploid human genome. Such a goal is ambitious, but it is not ridiculous. From just a few big genes like BRCA1 and BRCA2, one might get up to this scale fairly quickly.
A number that always depresses me is that them are 150 × 106 births a year, so to do worldwide genetic screening the annual mutation-detection sequencing requirement is 150 × 106 × 6 × 106 = 9 × 1014 bases, which is a very big number. Dividing by 300 yields the sequencing capacity needed per day, 3 × 1012. If we had some magical instrument that could do, not 105 as today's instruments can do, but 109 bases per day, we would need about 3,000 instruments worldwide to satisfy the demand. That is probably equivalent to the number of magnetic resonance imagers worldwide, so it is a reasonable number, but the number that is not feasible with today's technology is this 109 bases per day per instrument. Optimized current technology will probably get us another order of magnitude beyond the current 105, but for that we need new technology. Array-chip hybridization in principle could go to 107 or 108 bases per day with proper design.
Those numbers are not preposterous. As E. coli synthesizes its DNA, when it replicates it is actually doing so at the rate of 2 × 108 bases per day, and it is reading the sequences accurately. But it is not telling us what the answer is.
The Potential Of DNA Mass Spectrometry
We need to develop new methods if we are going to attack problems like human variability and human pharmacogenetics. The current methods will not do it, and I cannot resist using this occasion to mention my favorite among the new methods. DOE has invested a great deal in MS programs, and the effect of this and other investment worldwide is beginning to pay off: In the last year, from being a fantasy, DNA-sequence analysis by MS has become a reality. The method used is called MALDI-MS, matrix-assisted laser desorption ionization MS.
The appeals of this particular form of MS are several. The spectra are simple, the instruments are simple, and the measurements are rapid—they take less than 1 s/spectrum. In fact, they could be pushed to 1 ms or maybe even 1 µs if one wanted to.
In MALDI-MS, the sample is combined with an organic crystal, put on a surface, and hit with a laser pulse. The crystal is vaporized, and, it is hoped, the DNA molecule that one is interested in stays intact. It is accelerated by an electric field, hits the target, and is detected.
The impressive thing about the MALDI approach, as opposed to most technologies used by biologists, is that it yields a very-high-resolution measurement. If one measures the time of flight—how long it takes the molecule to get to the target—the resolution for short pieces of DNA is 1 part per thousand. If one puts the ion into a stable orbit, essentially as a tiny synchrotron, and measures what is called ion cyclotron resonance with Fourier-transform MS the resolution is 100 times higher.
The challenge is to figure out how to use these techniques. Although they are powerful, they are limited to relatively short pieces of DNA, and several physical problems, fairly severe, are posed in achieving high resolution as one pushes to longer pieces. Nature has been kind in designing nucleic acids, almost as though she had the mass spectrometer at heart when she chose the bases, in that they differ from one another substantially in mass. The worst case is A versus T, which is 9 daltons (D), but in general different bases are easily distinguishable. A piece of DNA of length L can have L3 different base compositions, or L3 different masses, so the possibility of looking at many things at once is very rich.
A 30-base unit of DNA has a mass of 9,000 D. The resolution limit of 1/1,000 of that is 9 D. Therefore, with the possible exception of distinguishing an A from a T, relatively simple MS can distinguish almost everything in 30-base DNA fragments.
When confronted with a potentially novel sequencing method, people always ask, Has anything actually been sequenced with it? I would like to describe a recent pilot project to demonstrate the feasibility of DNA sequencing with MS. The work has been done by a consortium of researchers in Germany and the United States. Robert Cotter at Johns Hopkins; Hubert Koester at the University of Hamburg; and a company named Sequenom, with laboratories both in Germany and in the United States; are involved.
What is done is just ordinary, solid-state sequencing chemistry, but instead of examination of DNA-fragment
lengths with electrophoresis, the sample is mixed with a crystalline organic matrix that is blasted into the vapor phase by laser excitation, and the sequence is read from the masses of the fragments detected. The technique is called MALDI time-of-flight mass spectrometry. The data are getting better and better in our hands and in other people's hands week by week. For example, a run of Gs, which is very difficult to work with in ordinary electrophoretic sequencing, is well resolved with MS.
To demonstrate feasibility, we have sequenced 4 of the exons of p53 with mass spectrometry. We used a walking primary technique, because our read length is short and because we wanted to read through each primer in turn to make sure that the primers were not covering up mutations. The results were sharp peaks and accurate sequencing data.
Those results are encouraging, but they do not mean that MS is ready to replace the thousands of gel-based sequencers in current use. There is a problem in that MALDI-MS is hard to automate. MALDI yields excellent data, but in most conventional MS one has to search around the sample to find what is called a "sweet spot." If one simply hits the sample with a laser at random, no useful data are obtained. A manual search has to be performed, usually under the trained eye of an experienced person looking for the one little place in the sample that gives good MS results.
Recently, our group of collaborators finally solved that problem. We put our samples into wells etched in single-crystal silicon. This resulted in quite reproducible data without the need to hunt. The sample holder is 1 cm2 piece of silicon with 100 little holes in it. The time is coming when we will be able to analyze DNA very rapidly with MS. However, I chose those words very carefully. I said "analyze," not "sequence," because I do not think that MS is a smart way to sequence DNA. It can do something better than sequence: It can check the sequence by measuring the mass. The peaks seen for DNA fragments with MS are so sharp and so well defined that any variation from the normal shows up as a detectable change in mass.
For most applications where the sequence is nominally known, we can check it; if the sequence is normal, we do not have to determine the sequence. Consider the problem as finding a single mutant base in p53, which contains 1,200 bases. If one has to ask whether each base is normal as one reads through the sequence, it is a waste of time because the answer is yes, the base is normal in perhaps all cases or all but one. With MS, it will be possible to look much more quickly at fragments of a gene and make sure that they are normal; sequencing will not be needed except in the very rare cases in which they are not normal.
I would like to describe 1 final example of DNA analysis with MS. In current human-gene mapping, in forensics, in many other applications of DNA technology, we like to work with the lengths of alternating simple repeating sequences, like ACACACAC. Because these sequences are rather unstable, their lengths vary among different people. Short tandem repeats are 1 of the 2 powerful sets of markers that we have to look at in the human genome and other genomes. These are conventionally analyzed with gel electrophoresis. To make a long story short, gel-electrophoretic patterns are unexpectedly complicated; they are difficult to read, and we now know, because of the MS results with identical samples, that some of the gel-electrophoretic results are artifacts of electrophoresis.
We have seen a number of cases in which the mass spectrum clearly indicates only a single allele (repeat length) and gel electrophoresis, for some reason, shows 2 or more bands. Those cases appear to be common enough to suggest an advantage in moving out of the liquid phase, where DNA secondary structures can cause problems, into the vapor phase, where there are no secondary structure effects. Repeating sequence lengths are used in genetic-linkage studies; a single mistake in a linkage study can be ruinous. Thus, MS appears to offer advantages.
Functional Genomics
I want to end this presentation at the level of gene function because that is really the heart of biology. The use of genome information is going to be extremely complicated, in that it takes us to the level of function, and function is much more difficult than just sequence information.
There are a number of reasons why function is difficult. Sequences that control gene-expression patterns are still a mystery, and we are rather impotent in dealing with them. Proteins are quite difficult compared with nucleic acids. Each protein is an individual; it has idiosyncratic properties. It can be hard to purify. Pathophysiology is
complex and is very species-dependent. The isolation of a human-disease gene does not mean that the same disease phenotype will result when it is put into a mouse. Genes do not work alone. There is a combinatorial problem of immense complexity to deal with in sorting out issues of function.
I will present a brief case history of 1 study of function. In the past, to understand the function of a single gene has required a scientific lifetime of effort. This case history was an attempt to perform a genetic manipulation of the simplest function that I can think of: asking a cell to die on demand. This example arose in the context of environmental control. The work was a collaboration with David Kaplan, who was at the Army Natick Laboratory but is now head of the Biotechnology Center at Tufts University. Our objective was genetic engineering of a microorganism, the soil bacterium Pseudomonas putida, so that, when it was released into the environment, it would metabolize aromatic hydrocarbons in a spill until the hydrocarbons were gone and then kill itself. We needed to find a "poison pill" that we could insert into the genome of this organism. The pill would have to be linked into the genetic-control circuitry of the organism, stay silent until the hydrocarbons were gone, and then be activated and kill the organism. Compared with the control networks that Leroy Hood described, this is very simple. We were working in a bacterium—1 cell—and asking it to do 1 thing.
P. putida has a large plasmid under the control of a promoter called PM, which is activated by the product of the xylS gene in the presence of aromatic hydrocarbons, like 3-methyl benzoate; so if the bacterium finds a munitions spill or the equivalent, this pathway is activated, and a set of catabolic enzymes is made that metabolize all those hydrocarbons. For the pill, we decided to use the protein streptavidin, which tightly binds the natural vitamin biotin.
Streptavidin is, in principle, a very effective poison because that is what nature designed it to be. Streptavidin is a natural antibiotic. It is made by the bacterium Streptomyces avidinii and secreted in an inactive form by that organism. It diffuses into the environment, becomes activated, and kills everything in the environment because it depletes the environment of biotin. Thus, the streptomyces has a competitive advantage.
The problem that one faces in trying to perform this simple killing function is that, before it is needed for suicide, the streptavidin concentration in these organisms must be effectively zero. If it is not zero, it will make the organism sick; there will then be a tendency for cells with mutations that inactivate the control system to grow preferentially, and the net result is that the pill will be disabled. Once the organism has finished its hydrocarbon meal, the streptavidin molecules are needed for suicide, and they should appear rapidly and kill the host before it has a chance to escape by mutating. Designing a regulating system that will perform in this way is hard.
The 3-dimensional structure of streptavidin was the first novel structure solved by the multiple-wavelength anomalous dispersion method, which is uniquely enabled by cyclotron radiation and makes DOE's synchrotron sources an absolutely essential resource for any kind of modem structural biology.
Streptavidin kills organisms because it interrupts a number of key metabolic pathways, including pyruvate metabolism and fatty acid biosynthesis; bacteria do not have a chance once this material is expressed inside them. The problem is how to control such a lethal poison. Figure 2 shows the simplest model that one can think of. The organism's natural control pathway, 3-methyl benzoate activating the xylS gene, is used to make an inhibitor to keep the streptavidin gene shut off. The cell should survive. When this pathway is turned off, streptavidin expression is no longer inhibited, and the cell should die.
This simple system does not work at all. The control is not nearly tight enough. So we tried a more-complicated system. William Studier, at the Brookhaven National Laboratory, developed what is the gold standard today in bacterial expression systems, a system sufficiently complicated that I will not try to go through all the steps here. It has many more steps than are shown in figure 2. Four processes are connected. The cells survive as long as they are growing on 3-methyl benzoate, but a cascade of events occurs and kills them if they are not growing on 3-methyl benzoate.
We dutifully engineered all of the Studier pieces into Pseudomonas putida, and it did not work at all. It is not tight enough. At that point, we were almost ready to throw in the towel. Fortunately, Cassandra Smith, a coworker on the project, had a clever suggestion: Make the control even fighter by not just operating on the protein level, but also by making antisense RNA. This is a fancy trick; the details would take quite a while to go through. Basically, what we have done is create redundant control pathways. Instead of having 1 pathway for each event, we have 2. This acts not only like a fall-safe system, but also like a quicker system, so that the cells survive as long
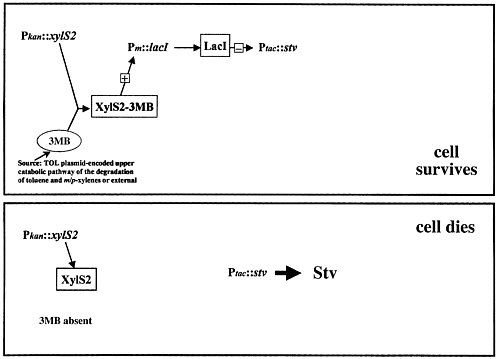
FIGURE 2. Suicide-control system: simplest possible model.
as they are metabolizing 3-methyl benzoate, or anything else like it, but they do kill themselves rapidly once they run out of hydrocarbon.
The actual mechanics are amusing, and the details are sketched in figure 3 for the cognoscenti. One clones the genes that will be expressed out of phase, head-to-head under promoters without terminators. Then, for example, when lacI is expressed, it also makes antisense strands against the streptavidin transcripts. When streptavidin is made, it makes antisense strands against lacI transcripts. By doing this kind of systems engineering, we have made nearly perfect on-off switches, and the system works well.
The killing is better by a factor of 10, better than anyone else has done before in any kind of suicide system. But it is still not good enough for environmental use; we still have some problems with mutagenic escape from suicide. In the next year, we plan to make the system even more complicated. We are going to introduce in parallel an entirely separate second suicide pill; if the organism tries to escape from 1 pill by mutating, it will still be subject to the other. The 2 pills must not have any components in common; if they do, a single mutation could rule them both out. We do not know whether this new system will work well enough to allow anyone to use the organism in the environment. I hope that that vignette provides the flavor of how complex life is, especially when one is dealing with living organisms at the level of function.
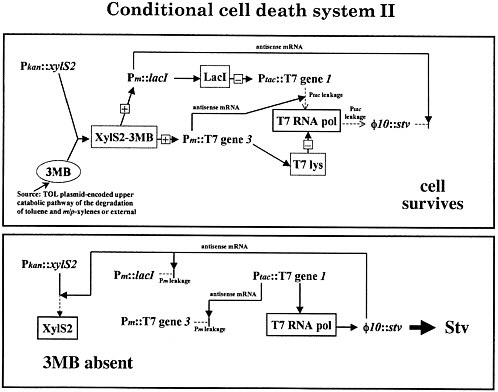
FIGURE 3. Suicide system: construct that was actually functional.
CONCLUSION
My goal here has been to show that we have come a long way very fast. What the genome project has done is open up a tremendous challenge. When the project was conceived, the notion was that we would have 100,000 genes to study in the year 2005, but we have 100,000 genes to study right now, and this is the enormous challenge that presents itself across a wide range of problems from information science through structural biology to plain biology.
Discussant
James Fickett
Bioinformatics Group
Smithkline Beecham Pharmaceuticals
King of Prussia, Pennsylvania
In the late 1970s, biologists were beginning to see that the new technology of sequencing DNA was going to affect every area of biology, and Walter Good at the Los Alamos National Laboratory was one of the people who saw that. He also saw that having the sequence data in one place and available electronically for mathematical analysis was going to make a huge difference in how well we could use those data. He began what later became the GenBank database. At that time, it was a pilot project called the Los Alamos Sequence Library1; Box 1 is from an early internal report at Los Alamos. The database was about a year old, and there were almost 0.5 million bases of DNA.
Now we get something like 1 million bases of new DNA-sequence data per day. Figure 1 does not show it, but the human sequences continue on the next page for a few lines—there were 10 or 20 human sequences in the database at that time. Now, we have about 50 million bases, and we are expecting another 200 million in the next year.
People often compare the human genome to a blueprint or a map of genetic information—a blueprint for the organism or a map of heredity. But a big difference is that in maps of the earth or in blueprints of buildings, the position of each item matters a lot. In the genome, to a first approximation, the location of a gene in the genome does not matter much. Many genes are independent units—not independent logically, but independent positionally—so in a sense the genome project is a project not to get the sequence, but to get the genes.
GRAIL, a program developed at the Oak Ridge National Laboratory,2 is the standard tool for finding the genes in DNA-sequence data. We can do a reasonable job of finding the approximate locations of genes. It still requires much experimental work to track down exactly where the genes are and a huge amount of work to find out their functions.
One important method for beginning to understand the function of genes is to see from the DNA sequence, if possible, under what conditions a given gene is expressed, when it is turned on, and when it is turned off. R. Tjian described the initiation complex that begins the transcription of DNA into an RNA copy of the gene.3 This process is a primary point of control of gene expression. No one knows how many transcription factors—that is, proteins that bind the DNA and control gene expression—there are. There might be tens of thousands. Genes are exquisitely controlled in many different contexts for precise expression just when they are needed.
In a related vein, to follow up on one of Charles Cantor's comments, one thing that computational methods have done fairly well is help us to locate the genes in the DNA sequence. We would also like to be able to locate the precise point labeled the core promoter, where the transcription of the gene begins. We have not done so well on that. There are a number of programs, including some that are fairly obvious and fairly simple. A colleague and I recently finished benchmarking these methods and found, unfortunately, that they do not perform well enough for practical use yet.4 We have a long way to go.
Biology, however, is a science of special cases, and the right way to solve the general problem of finding the beginnings of all genes might be to find the control points for particular classes of genes. For example, myogenin and mef2 are 2 transcription factors important in turning genes on in skeletal muscle. In the problem of trying to decode these regulatory regions, there is a great opportunity for deep collaboration between the experimental and the information and computational sciences. The interdisciplinary approach not only will pay off, but it is absolutely required. Both those proteins were known to be important in the regulation of muscle-specific genes, and they were known to interact. That is, if you grabbed one with a monoclonal antibody, you found that it dragged the other one along with it. They were known to interact, but the relevant pattern in the DNA—being able to look at the gene sequence and figure out whether the proteins were binding, and so on—was not known.
Figure 1 shows that we can make progress computationally to see what kinds of patterns are important. If you look at the myogenin-binding site in this enhancer, you will see that it is perfectly conserved; the mef2 site is
|
BOX 1 Directory of Sequences Two directories to the data bank are kept current and are available on-line. The short directory contains a brief definition of each sequence, usually confined to one line, and so is useful for direct examination. The long directory contains the entire heading for each sequence. The software option DIRECTORY allows it to be searched for the appearance of any character string. There follows first the short directory, then the long one.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

FIGURE 1 mef2 and myogenin: Spacing is conserved.
almost perfectly conserved in these 4 organisms, but in between, the sequence has diverged a great deal in evolutionary history. Nevertheless, the spacing between the sites is perfectly conserved in the first 3 cases, and there is a deletion in the last case that corresponds to 1 full turn of the helix.
There is a geometry here that is clearly important. If you look at the muscle transcriptional regulatory regions in general, you find that this geometry of spacing between the myoD family sites and mef2 family sites is important for muscle specificity and can be used to find some muscle-specific genes.5 If we combine the experimental studies of finding out where these sites are and the sequence-analysis studies of looking for the geometry of the patterns in the DNA, we will get to the point where we will be able to write computer programs that can look through the DNA and say, Here is a gene that is turned on in liver, here is a gene that is turned on in the atrium in the fetal heart, and so on.
That approach will help in the challenge of finding the function of all the new genes that are turning up in the genome-sequencing projects.
One of the biggest surprises in the genome project has been that, as we have uncovered thousands of new genes, we have found that about half of them do not look like anything we have ever seen before, and we have no clue as to what they do. Clearly, this will require an interdisciplinary approach.
REFERENCES
1. Cinkosky, MJ, Fickett, JW, Gilna, P, and Burks, C. Electronic data publishing and GenBank. Science 1991; 252: 1273-1277.
2. Xu, Y, and Uberbacher, EC. Automated gene identification in large-scale genomic sequences. J Computat Biol 1997; 4: 325-338.
3. Tjian, R. Molecular machines that control genes. Sci Am 1995; Feb: 54-61.
4. Fickett, JW, and Hatzigeorgiou, A.G. Eukaryotic promoter recognition. Genome Res 1997; 7(9):861-878.
5. Fickett, JW. Coordinate positioning of reef 2 and myogenin binding sites. Gene 1996; 172, GC19-GC32.
Discussant
Keith O. Hodgson
Department of Chemistry
Stanford University
Palo Alto, California
I would like to speak about one particular component of the Department of Energy (DOE) and Biological and Environmental Research (BER) program: structural biology. As we look toward the future, it is going to be at the disciplinary interfaces where exciting opportunities will develop and revolutionary advances will occur. For structural biology, this will be the interface between chemistry and biology that enables understanding of structure and its function at the molecular level. We have begun to see an evolution in this direction that is driven by a number of other activities, including the genome programs and an increased focus on molecular medicine. As we move into the 21st century, understanding structure and function at the molecular level will become increasingly important, challenging, and rewarding.
I do not have time to talk about what synchrotron radiation is and why it is important; suffice it to say that it is an important enabling technology that provides an extremely intense source of X-rays that can be used to investigate structure. Virtually all we know about the structure of molecules comes from studying the interaction of molecules with electromagnetic radiation. We learn about structure at the level of individual atoms in larger biologic molecules by using wavelengths of light that are comparable with the spacing of the atoms, and that means X-rays. Biologic materials are composed mainly of carbon, hydrogen, and oxygen, so they do not scatter or interact with X-rays strongly. Synchrotron radiation offers intensity increased by a factor of several powers of 10 over conventional X-ray sources (comparable with laser intensities, but in the X-ray region). This has revolutionized our ability to study atomic structure in large molecules with protein crystallography. Synchrotron radiation has also enabled substantial biologic applications of other techniques, such as X-ray absorption spectroscopy and small-angle scattering, which provide information complementary to that obtained with crystallography.
Synchrotron radiation is a polychromatic source, and its multiple wavelengths can be used to solve the phase problem in crystallography (which is necessary to visualize atomic structure). The extremely high intensity of synchrotron radiation allows an even higher level of resolution for biologic molecules.
I would like to discuss how this field has grown and how this has been enabled by the vision of the BER program. What is happening in this field worldwide? It is useful to look at trends in growth for new protein crystal structures. I have talked about X-ray crystallography, but there is also nuclear magnetic resonance (NMR), which is increasingly important for smaller structures. However, NMR has difficulty when the molecular weights get large, as in supermolecular structures, interaction among proteins, and interaction between proteins and DNA or large viruses. X-ray crystallography is an important technology for studying structure in these larger systems. One way of looking at the growth and influence of synchrotron radiation in this field is represented by Carl-Ivar Branden's compilation. He analyzed the number of new structures published in 3 journals—Nature, Science, and Structure—in 1992-1995. In Nature, for example, the number of new protein structures based on synchrotron radiation grew from 39% in 1992 to 73% in 1995. Similar growth was seen in the structure papers published in the other 2 journals. That is just one measure of the growth in the importance of synchrotron radiation in solving structures with X-ray crystallography. One finds similar growth trends when looking at the demand for access to the synchrotron facilities.
How has BER played a role in this growth and in meeting demand? In the United States, it supports more than half the funding in this field which provides for the facilities that enable structural biologists to have access to synchrotron radiation. BER funding has grown from a modest level in 1990 to more than $10 million in 1997. The National Institutes of Health (NIH) plays a complementary role in supporting beam lines and operations for structural-biology research at the synchrotrons through its National Center for Research Resources. Together, the support from these 2 agencies is what enables the community to do structural-biology research at these shared multiuser facilities in the United States.
There has been a substantial increase in the use of the 4 DOE-funded synchrotron facilities for structural-biology. The number of users has grown from about 2,000 in 1990 to about 3,000 in 1996, but the fraction of
structural biology or life-science users has grown from only 6-7% to almost 30% in the same period. In part, this is due directly to BER support of activities in the structural biology.
Similar trends are seen in industrial interest in structural molecular biology. There has been strong growth in the number of crystallographers and the number of pharmaceutical and biotechnology companies investing in this technology over the past 5-7 yr, and almost every major pharmaceutical company is investing in the development and support of a structural group. That also translates to increased demand and use of the synchrotron facilities because the same people want access for problems ranging from drag design to structures of modified enzymes with more-favorable industrial properties.
Structural molecular-biology research integrates many parts of the BER portfolio, including relationship to the genome and environmental fields, such as bioremediation. Structural biology cuts across a number of disciplines, components of which transcend many aspects of the portfolio of energy research. It certainly builds strongly on core competences of the DOE. DOE is the nation's premier provider of synchrotrons; the technology, the expertise, and the intellect resides mainly within DOE. DOE operates 4 of the major synchrotron facilities in the United States. It also operates the neutron programs, which I should note are also players in structural biology. Such programs are unique strengths of the national laboratories and provide essential capabilities to the nation's researchers in academic institutions, at the national laboratories, and in industry.
I want to point out 2 more things. One is a recent report from the Institute of Medicine that considered the importance of resource-sharing in biomedical research. The report pointed out the incentives and some of the disincentives for research-sharing. It noted the importance of resource-sharing as one moves toward more and more advanced technologies and pointed out, as a case study, that synchrotron resources are an excellent example.
Finally, Nature of May 1, 1997, carried a report by the Biomedical and Biological Sciences Research Council in the United Kingdom. Structural molecular biology was targeted as an important growth field. The analysis concluded that the United States is currently the leader in this field, with more than 50% of the world's output. It also identified synchrotron facilities as essential for the future.
It is a challenge to be a visionary, to think about what will make a serious difference, to see how the BER program can move forward in the best way. Structural biology can be one of the cornerstones on which the future of BER will be built.
Discussant
Dagmar Ringe
Department of Chemistry
Brandeis University
Waltham, Massachusetts
The next challenge for the postgenomic era is to determine function from structure. That might sound relatively simple, but it is not, as some examples will show.
Generally speaking, proteins are grouped into families. The families are based on function if that is the information available or on structure if that information is available. Can we put the 2 together? For example, 1 protein family that is well characterized is the globin family, of which myoglobin is a member. The members of this family have similar structures and similar active sites, and those that are involved in chemical transformations catalyze similar reactions.
We might expect to find other families that are unified by structure and function. For instance, the 2 enzymes D-amino acid aminotransferase 1 and branched-chain amino transferase2 are similar enough at the sequence level that it is expected that their 3-dimensional structures will be similar. That is indeed the case. The 3-dimensional structures look similar, and both enzymes catalyze the same reaction, a transformation called transamination. Ostensibly, if I am looking for a set of transaminases, all I have to do is look for this type of sequence. However, nature is not that simple. Another aminotransferase, L-aspartate aminotransferase, 3 bears no sequence similarity to D-aminotransferase, has no structural similarity to it, and yet catalyzes the same reaction by the same mechanism.
Those 2 enzymes do not resemble each other at all, nor is there any way to derive 1 structure from the other. In addition, it is not possible to predict the function of 1 from that of the other. That is the problem: The structure does not necessarily indicate the function, nor does the function dictate the structure.
A more striking example of the problem is provided by a comparison of 2 enzymes that have similar 3-dimensional structures. Aldose reductase reduces sugar to sorbitol and is implicated in the blindness that is often a complication accompanying diabetes.4 The second structure has a similar architecture but a completely different function: It is a racemase that is involved in cell-wall biosynthesis.5 Thus, even if it becomes possible to predict the structure from a sequence, it will be much more difficult, if not impossible, to determine the function.
We need better means of determining structure from sequence and ultimately of deducing function. There have been interesting advances in determining structure from sequence. These are computational and not yet totally accurate, but in some cases at least potentially useful. There are, however, 2 approaches that might be used to determine the function of a protein. They are based on a simple premise: If you can determine an interaction partner for a protein, very often you can determine its function. Consequently, we are going to explore means of determining how ligands interact with proteins. One of these technologies is called solvent mapping, and the other uses combinatorial chemistry. The former method relies on the availability of a 3-dimensional structure; the latter does not.
An enzyme can be represented as a space-filling model in which the protein looks like a solid object. Around this object is a halo of water molecules that covers much of the surface, surmised from biophysical studies and, in part, observed crystallographically. If a ligand is going to interact with this enzyme, it must displace some of the water molecules to reach the surface. In a crystallographic experiment, binding of ligands can be determined directly. However, if the ligand is unknown, then designing the experiment is much more difficult. One method of determining binding sites on the protein and some of the characteristics of the ligand that might associate with such a site is to use small probe molecules to map the surface of the protean. In a series of crystallographic experiments, the surface of the protein can be mapped by a series of organic molecules, representative of parts of a larger ligand. The result is a distribution of such probe molecules over the surface of the protein. These probes tend to cluster. In a series of experiments with elastase and several organic solvents, the probe molecules clustered at the active site in regions indicative of the sites at which side chains of a peptide would interact with the enzyme.6 This approach is amenable to computational methods, and a number of these have been used to map the surface of a protein by calculating the energy of interaction of a probe molecule with the surface.6,7
Thus, determining a structure and the positions of probe molecules, either experimentally or computationally, could make it possible to determine the types of molecules with which a protein interacts, and thereby its function.
A second method of determining function does not rely on 3-dimensional structure. Instead, it relies solely on the ability of a protein to interact with compounds in a library of diverse compounds. With today's synthetic strategies, large variants of a structural motif can be synthesized, and large numbers of structural motifs can be designed. Such libraries are used in the search for unique molecules that will interact with a specific target in the search for new drag leads. Ideally, such a library should cover as much diversity space as possible because the type of interaction that could prove to be advantageous cannot be predicted. In testing such a library against a number of targets, we have found that the interactions observed for any 1 target tend to cluster. For instance, if the targets are a particular receptor, the interactions observed occur with some types of compounds, not others, and not with compounds that interact with, for instance, tyrosine kinases. This result implies that a library of the proper diversity can be used to differentiate types of functions by looking at the types of interactions that an unknown protein undergoes. A relatively simple screening mechanism would let us determine the functions of proteins about which we know nothing.
In summary, 2 types of technologies could be useful in linking sequence to function. One depends on structural, the other on screening, methods. Both require some protein to work with. Obtaining structures of all proteins is not a practical goal. However, the structural method can be applied to proteins that do not yield to other methods. In addition, as structure-prediction methods improve, the method can be used with computers. Screening methods also require protein, and obtaining sufficient quantities is an in vitro challenge. Ultimately, refinements of these methods will be required if we are to turn sequence information into function information about the many gene sequences becoming available.
REFERENCES
1. Sugio S and others. Biochemistry 1995;34:9661-9.
2. Okada K and others. J Biochem 1997;121:637—41
3. Smith DL and others. Biochemistry 1989;28:8161-7.
4. Harrison DH and others. Biochemistry 1994;33:2011-20.
5. Shaw JP and others. Biochemistry 1997;36:1329-42.
6. Ringe D. Curr Opin Struct Biol 1995;5:825-9.
7. Miranker A and others. Proteins 1991;11:29-34.




















