

































































Below is the uncorrected machine-read text of this chapter, intended to provide our own search engines and external engines with highly rich, chapter-representative searchable text of each book. Because it is UNCORRECTED material, please consider the following text as a useful but insufficient proxy for the authoritative book pages.
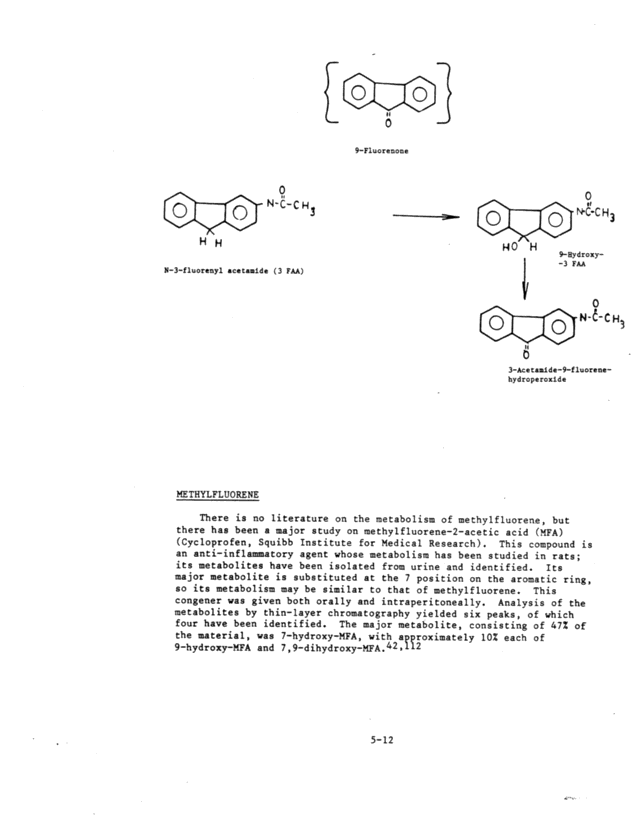
OH 0R . OH OH \ ~1, 2-Dlhydroxy 5-Me thylchrysene HO H3 OUCH ~7,8-Dihydro 5-Bydroxy-H >~ 9''10-Dlhydro-9, 10-tihydroxy FLUORENONE There appear to have been no direct studies on the metabolism of fluorenone~ but N-3-fluorenyl acetamide (3-FAA) yielded two metabolic products.] 2 Because the parent compound is carcinogenic, it appears that the derivatives are detoxification products. The authors sug- gested that metabolism of 3-FAA consists of two sequential reactions: the initial formation of 9-hydroxy-3-F M as an intermediate to the for- mation of 3-acetamido-9-fluorene hydroperoxide, which is then dehydrog- genated to form 9-oxo-3-FAA. Exposure of rainbow trout to a number of hydrocarbons showed no bioaccumulation of fluorenone; the compound is most likely metabolized to excretible products.65 However, no at- tempt was made to analyze any metabolic products. There is no litera- ture on the isolation and identification of fluorenone metabolites. S-11 7, 8-dihydroxy
- - o 9-Fluorenone o l o ~ N-C-cH" H H N-3-fluorenyl acetamide ( 3 FM) }£E: THYLFLUORENE (~1 ~ LO I ~0 H 1 o 3~ ~C-Cu3 9~Bydroxy- -3 FAA o 4~N-~-C H. 11 3-Acetamite-9-fluorene- hydroperoxide There is no literature on the metabolism of methylfluorene, but there has been a major study on methylfluorene-2-acetic acid (MFA) (Cycloprofen, Squibb Institute for Medical Research). This compound is an anti-inflammatory agent whose metabolism has been studied in rats; its metabolites have been isolated from urine and identified. Its major metabolite is substituted at the 7 position on the aromatic ring, so its metabolism may be similar to that of methylfluorene. This congener was given both orally and intraperitoneally. Analysis of the metabolizes by thin-layer chromatography yielded six peaks, of which four have been identified. The ma jar metabolite, consisting of 47% of the material, was 7-hydroxy-MFA, with approximately 10% each of 9-hydroxy-MFA and 7,9-dihydroxy-MFA.42~112 5-12 i ... ..
CH3 rot tie thylfluorene -_ I O [~ O |9H3 tie thylfl uorene- 2_caticacid HO~-C-OH OH ~ 7, 9-Dlhydroxy CYCLOPENrA [ Cd ] PYRENE - - O ~ C-tO H 7-llydroxy ~Lc i-OH 9-Nydroxy Cyc logenta [ cd ] pyrene has been ident i f fed as a component of carbon black.66, 9 Its metabolism has been studied in rat-liver micro- somes.67 The major metabolize isolated by HPLC has been identified as trane-3,4-dihydroxy-3,4-dihydrocyclopenta~ct~pyrene. Several came portents not yet well characterized consisted presumably of phenolic derivatives, as well as metabolites that appear to have saturation of the ethylene bridge. 5-13 I. .. .
~- - ~clopenta[cd]pyre~ - DIBENZOTHIOPHENE AND BENZOTHIOPHENE b~- - - - Dibenzothiophene and benzothiophene are biodegradable in both eutrophic and oligotrophic pond waters.20 Their major metabolite is 1,2-dihydro-1,2-dihydroxydibenzothiophene, with later ring degradation to benzothiophenedione. Benzothiophene and dibenzothiophene form a thioketone, a dihydrodiol (cis and bans isomers and a diketone). There have been no studies dealing with further metabolism. Enzymes other than microsomal monooxygenases may also be involved in the metabolic activation of PAHs. Eling et al.52 and Marnettl25 have shown that numerous xenobiotics, including the dihydrodiol metabolites of PAHs, can be cooxygenated during the oxygenation of arachidonic acid by prostaglandin synthetase. In the case of PAHs, when the dihydrodiols are generated, this novel pathway could lead to an alternative pathway for the formation of diol-epoxides. These studies have been done on in vitro model systems. The relevance of their pathway in viva is unknown. There is a suggestion in the literature that nitropyrenes are metabolized by bacteria, presumably via a nitroreductase, to produce a high mutagenicity; however, there has been no isolation or character- ization of metabolites. Because mammalian cells have much-reduced nitroreductase activity, this rationale has been used to explain the lack of activity in mammalian cells.62 5-14 - Trane-3, 4-dthydroxy '5~: 09H 3, 4-dihydrocyclopenta [ cd ] pyren. ~. .... ..
s Dibenzothiophene Benzo thiophene H OH OH ASH LC-H o - . a; it' S' r' o - 5-1S 1 OH S H ~5-0 ~-so ~S)~ Benzo thiophenet lone ILL .. -.
IN VIVO FORMATION AND DISAPPEARANCE OF PAH METABOLITE-DNA ADDUCTS . . . HISTORICAL PERSPECTIVE . Brookes and co-workers observed that 3H-labeled PAHs applied to the backs of mice30 or incubated with mouse-embryo celis48 resulted in covalent binding of radioactivity to DNA, RNA, and protein. Grover and Sims76 and Gelboin62 showed that PAHs require metabolic activa tion by mixed-function oxidases if they are to react covalently with cellular macromolecules. The interactions between PAH metabolites and nucleic acids have since received considerable attentiOn.63, 80,151,182 Identification of the reactive PAH metab olites that form adducts with DNA has been emphasized, because forma tion of these adducts is believed to be essential for tumor initiation, although interaction with RNA and protein may also be important. Initial attempts to identify the reactive metabolites that bound to DNA focused on the arene oxide intermediates proposed by Boyland,25 and especially on the K-region arene oxides, because, according to quantum mechanics) carcinogenic PAHs are distinguished by an electron- rich K region.l°, 53 They induce mall transformation of cells and are active in mutagenicity tests. ~ In addition, they are b li f pAHs70~72-74,104,163~170 and will bind to DNA in vitro.18 However, it became obvious that K-region epoxide-DNA adducts were not the adducts formed in viva between BaP metabolites and DNA.18 A similar conclusion was reached in studies with 7-MBA.17 Borgen et al.22 found that, in a microsomal activating system, the 7,8-diol BaP metabolite bound to DNA to a much greater extent than any other known dial or phenol. Sims et al.172 provided evidence that the BaP metabolite-DNA adduct formed by BaP metabolism by Syrian hamster-embryo cells in culture was chromatographically ~ dentical with an adduct formed by metabolism of BaP 7, 8-dio1 and proposed that t'ne reactive metabo1ite was a diol-epoxide (DE). Studies in various in vivo model systems such as cell cultures and organ explants 13,18,29,)1,79,93,172 and in in `'ivo skin, lung. liver, and forestomach of mice have shown that the major Ba? metabolit2-DNA adduct observed after exposure of these tissues to BaP is the (+~-BaP DE I-deoxyguanosine adduct. (+)-BaP DE I is apparently the major enzy- matic rnetabolite of --trams BaP 7,8-diol. ~ The adduct results from the interact ion of ~ + ~ -BaP DE I wi th two amino groups of guanine . The cis isomer (-~-BaP DE II is also formed enzymaticaily from (-~-trans-BaP 7,8-diol. 194 The --Bar DE II-deoxyguanosine adduct is formed to the same extent as the ~ +)-BaP DE I-deoxyguanos ine adduct in lung and liver of rabbits, as opposed to the results in mice (C. Bixler and M. W. Anderson, unpublished data). Structures of the BaP DE isomers are shown in Figure 5-5. Although the predominant by nding of BaP DE is to the 2-amino group of guani8e~ these 7 iol-epoxides can also bind to the N7 of guanine, 1 adenine, 9 , 9, 130, 1 5 and cytidinel75 and to phosphate real dues.00 ,108 5-16
Evidence is Crating that gthg; PAHs--e.g., 7-MBAg35~127 benzanthracene,1 ~ chrYsene 1 ,1 5-methylchrysene 1 dibenzanthracene,l90 3-Mc,l05,l}9 and DMgA16,48,91,132,1~9__are similarly converted to highly reactive diol-epoxides, which then interact with DNA in viva. All these diol-epoxides have a structural similarity. Jerina and Daly94 pointed out that the epoxide ring is in a bay region and suggested the term "bay-region diol-epoxides" for these highly reactive metabolites of PAHs. The bay-rezion diol-enoxides are mutagenic 89,96,115,117,126,136,174,185,186 have transforming activity in mammalian cells,843126 are carcinogenic in newborn mice3l~l00,l0l~ll7 and Chinese ham te 1 in cells 1173187 a d are initiators in cells of mouse skin.~55~&0~0~,ii5~7~75 The mutagenicity and carcinogenicity, combined with the observation that bay-region diol-epoxide-DNA adducts are the major adducts formed in viva in target tissue, have led to the hypothesis that bay-region diol-epoxide adducts are the ultimate carcinogens generated by metabolism of most PAHs.963115 However, it should be pointed out that many nonmetabolized PAHs that are termed carcinogenic either lack a bay-region benzene ring or contain nonreactive substitutes in this molecular region.80 In addition, PAH metabolite-DNA adduct formation in viva has been examined only for BaP, DMBA, and 3-MC, and these studies have concentrated on target tissues in mice (see Table 5-1~. PAH metabolites other than bay-region diol-epoxides can also bind to DNA. The K-region epoxide of BaP binds DNA coval~ntly.168~170 Incubation of BaP with microsomes in the presence of exogenous UNA results in a variety of BaP metabolite-DNA adducts.12~148 In particular, adducts are formed from further metabolism of 9-hydroxy- BaP, possibly the 4,5-epoxy-9-hydroxy-BaP metabolize. The major DNA adduct observed after exposure of hepatocytes in culture to BaP resulted from the further metabolism of 9-hydroxy-BaP.97 A BaP-phenol-oxide-DNA adduct was the major adduct observed in rat lung and liver after intravenous administration of BaP.23 Various structural modifications of PAH diol-epoxide metabolites do not inhibit binding to DNA.80,86,87 Dose-response relationships for formation of PAH metabolite-DNA adducts in target tissue would be helpful in the low-dose extrapolation problem for PAH carcinogenesis.7361 Many pharmacokinetic processes determine the extent of formation of PAH metabolite-DNA adducts in an organ after exposure of an animal to a PAH (see Figure 5-1~. Although most of these processes have not been completely characterized, some generalizations regarding the extent of adduct formation in viva can be made from recent reports (see Table 5-1~. Previous reviews of covalent binding of PAHs to DNA have not analyzed in viva adduct formation in detail.63~80~151~182 5-17 ~ . .. . ..
CHARACTERIZATION OF PAH METABOLITE-DNA ADDUCTS . . . Table 5-1 lists the studies concerned with the in viva formation of PAH metabolite-DNA adducts. Most of them used mice and BaP. HPLC analysis of BaP-deoxyribonucleoside adducts formed in lung, liver, and forestomach of A/HeJ mice after oral administration of BaP (3 mg/mouse) is shown in Figure 5-6.8~184 HPLC analysis is shown for DNA samples isolated by the hydroxylapatite and precipitation pro- cedures. Radioactivity elated in the water wash (water fraction, WF) and in early portions of the water:methanol gradient that varied from 407 to 70%. This uncharacterized early-etuting radioactivity was much higher in UNA samples isolated by precipitation than by the hydroxyl- apatite method (see Figure 5-6), although it was still substantial, especially in liver and forestomach, in samples isolated by the hydroxylapatite procedure. Three distinct peaks--I, II, and III in Figure 5-6--were observed id the gradient portion of the chroma- tography. The specific activity (picomoles per milligram of DNA) asso- ciat~d with these peaks is independent of the procedure used to isolate DNA. Peaks II and III have been identified as (+~-BaP DE I-deoxy- guanosine and BaP DE II-deoxyguanosine adducts, respectively.6 Peak I is probably generated from 9-hydroxy-BaP, although it could be a BaP DE I-deoxycytosine adduct. Small, late-eluting peaks were con- sistently observed, especially in lung samples (Figure 5-6~. They could be BaP DE I-deoxyadenosine or BaP 4,5-oxide adducts. Similar BaP metabolite-DNA adduct profiles were observed in lung, liver, and forestomach from ICR/Ha and C57BL/6J mice after oral administration of BaP.9~90 Eastman _ al.51 examined the in viva binding of BaP to DNA in lung,-liver, and kidney of Aroclor 1254-treated A/J mice after intravenous administration of BaP. The only identified adducts observed by Sephadex LH20 chromatography were BaP DE-DNA adducts. Early-eluting radioactivity was present in the chromatograph.51 Eastman and Bresnick50 and Eastman et al.51 used Sephadex LH20 chromatography to analyze the 3-MC metabolite-DNA adduct profile in lung and liver of several mouse strains after intravenous admin- istration of 3-MC (Figure S-7~. Two major 3-MC-deoxyribonucleoside adduct peaks were observed in Lung and liver of each mouse strain examined. HPLC analysis49 demonstrated seven 3-MC metabolite-DNA adduct peaks in lung and liver of C57BL/6J mice, with the two major adduct peaks corresponding to those observed by Sephadex LH20 chroma- tography.50351 Early-eluting peaks (Figure 5-7) were also present in the chromatography of these studies with 3-MC. Binding of 3-MC, BaP, and DMBA to DNA has been examined in skin of several mouse strains (Table 5-1~. In each study with BaP, the major adduct observed was BaP DE-deoxyguanosine. Sephadex LH20 chromatography revealed only one adduct peak. The HPLC adduct profile in skin was virtually identical with that in Figure 5-6 for lung, 5-18 ~ . .. . ..
forestomach, and liver.ll~l4'l07,l49 The adduct profile for 3-MC in mouse skin was the same in each strain examined, but was slightly different from that for lung and liver (Figure 5-7), in that three 3-MC metabolite-deoxyribonucleoside peaks were observed in the Sephadex LH20 chromatograph 2 f skin whereas only two peaks were observed for lung and liver. 9-51,130 Sephadex LH20 chromatography of DMBA-deoxyribonucleoside adducts in skin was similar in each mouse strain studied; three peaks were observed.150 Early-eluting peaks were also observed in the chromatography of these investigations of PAR metabolite-DNA adduct formation in mouse skin. The formation of DNA adducts of the carcinogen 15,16-dihydro- 11-methylcyclopentataiphenanthrene-17-one (11-me2t3~1ketone) was examined in liver, lung, and skin of TO mice.l, ~ HPLC analysis revealed eight 11-methylketone metabolite-DNA adduct peaks in each of the tissues.2 The major adduct was generated from the interaction between the anti-3,4-dihydro-3,4-trans-dihydroxy-1,2-dihydro-1,2- epoxide (diol-egoxide) metabolite of 11-methylketone and deoxy- guanosine.l,2,3 There is no major qualitative difference in the adduct pattern among the three tissues. The adduct profile for each of the three tissues was not substantially altered by the route of administration (intramuscular, topical, and intraperitoneal). In the studies with mice, the PAH metabolite-DNA binding profiles are very similar in all tissues of all strains examined. For BaP, the predominant characterized adduct is the BaP DE I-deoxyguanosine adduct. When HPEC analysis was used, a BaP DE II-DNA adduct was also observed, as well as an adduct probably generated from 9-hydroxy-BaP. There is also evidence of BaP DE-deoxyadenosine adducts, although in relatively small amounts. The DNA adduct profiles for 3-MC are the same in lung and liver of each mouse strain examined and only slightly different in skin. The pattern of DMBA metabolite-DNA adducts in skin is the same for all strains examined. The HPLC profiles for 11-methyl- ketone metabolite-DNA adducts are very similar in lung, liver, and skin, with the major adduct being a diol-epoxide metabolite- deoxyguanosine adduct. The in vivo formation of BaP metabolite-DNA adducts has recently been examined in male Sprague-Dawley rats and male New Zealand rabbits (Table 5-1~. In rats, BaP was administered intravenously at 1.0 and 10.0 ~mol/kg. Several chromatographically distinct nucleoside-bound adducts were3Observed in lung, whereas only one adduct was apparent in the liver.2 The predominant BaP-nucleoside adduct formed in vivo in rat lung and liver was chromatographically identical with adducts formed on further metabolism of BaP phenols, possibly because of the interaction of 9-hydroxy-BaP 4,5-oxide with DNA.6312323 The BaP DE adducts were not detected in rat liver, and only a relatively small amount was observed in rat lung. The BaP DE adducts in rat lung accounted for only 1.4% of total UNA binding and 3.3% of the adducts generated by BaP phenol~s). Thus, the in vivo BaP metabolite-DNA adduct proftles obtained in lung and liver of Sprague-Dawley rats are 5-19
distinctly different from those observed in various mouse strains. This is the only known case in which the BaP DE adduct is not the predominant BaP metabolite-DNA adduct formed in viva. In an examination of BaP metabolite-DNA adduct profiles in lung liver of male New Zealand rabbits, BaP was administered either orally or intraperitoneally at 50 mg/kg. The DNA adduct profiles were identical with those in mice (Figure 5-6), with one notable difference (Bixler and Anderson, unpublished data): In rabbits, there was approximately 757 as much BaP DE II-deoxyguanosine adduct as BaP DE I adduct, whereas in mice, there was only 10% as much BaP DE II adduct as BaP DE I adduct. This is the only known case in viva in which the BaP DE II-deoxyguanosine adduct approaches the BaP DE I adduct in amount. and It should be emphasized that, in these investigations of PAH metabolite-DNA adduct formation, large amounts of the UNA-associated -radioactivity chromatographed not as nucleoside-bound adducts, but rather as uncharacterized fast-eluting peaks (Figures 5-6 and 5-7~. Although Adriaenssens et al.3 showed that isolation of DNA by a hydroxylapatite procedure, instead of by the precipitation method, significantly reduces the amounts of these early-eluting peaks, the peaks still account for a large proportion (especially in liver and forestomach) of the total radioactivity eluted in chromatography. Some workers have ignored these early-eluting peaks by a pre-elution step with Sephadex LH20 chromatography (e.g., Figures 5-6 and 5-7~. These peaks are also observed in in vitro studies and in in viva model systems (see Boroujerdi et al.2 ). The radioactivity appears to reflect some tissue-specific reactions, such as those exhibited by the different patterns in lung and liver.50351 Eastman and Bresnick50 showed, by using borate-eluted Sephadex LH20 and DEAE-Sephadex chromatography, that early-eluting radioactivity contains numerous constituents. Studies that used [14C]BaP and [3H]BaP5~14l~l49 and the results of Eastman and Bresnick50 and Eastman et al.51 suggested that only a small amount of this radioactivity Is due to tritium exchange, whereas experiments of other investigatorsl5~159 suggested the opposite. The results of Eastman and Bresnick50 suggested that only a small amount of the radioactivity in the early peaks is related to oligonucleotides. Phosphoi~i~sters might contribute to the early-eluting radioactivity. ~ 1 In any case, because a considerable amount of radioactivity appears in the early- eluting peaks, their identification deserves further consideration. COMPARISON OF EXTENT OF PAH METABOLITE-DNA ADDUCT FORMATION BETWEEN TISSUES AND BETWEEN SPECIES _ . _ Specific activities (SAs), in picomoles per milligram of DNA, of PAH metabolite-DNA adducts have been determined in several tissues after administration of PAHs. Table 5-2 gives the SAs of BaP DE adducts in lung and liver of various mouse strains and New Zealand rabbits. The amounts of BaP DE adducts are very similar in lung and 5-20 ~ . ... ..
liver for each study reported in Table 5-2. Anderson et al.8 examined the BaP DE adducts in lung, liver, and forestomach of A/HeJ mice for oral administration of BaP at 2-1,350 ~mol/kg. The SAs of the BaP DE adducts in lung and liver were similar over the entire dose range and ranged over 3 orders of magnitude in this study. The SA of BaP DE adducts in forestomach of mice is very similar to that in lung and liver after oral administration of BaP.338390 The-similarity of the BaP DE adduct amounts in lung, liver, and forestomach is rather surprising, inasmuch as the disposition and rate of metabolism of BaP in these tissues are probably very dissimilar. The higher rate of BaP metabolism in liver is reflected in the greater total DNA binding and protein binding in liver, compared with lung and forestomach.8 The SAs of the BaP phenol-oxide adduct were also very similar in lung and liver of the Sprague-Dawley rat for each intravenous dose of BaP (Table 5-3~. As mentioned previously, this was the predominant adduct observed in lung and liver of this species.23 As with mice, the total DNA binding was significantly higher in liver. Eastman and Bresnick50 examined 3-MC metabolite-DNA adduct formation (Figure 5-7) in lung and liver of several mouse strains at several points after intravenous injection of 3-MC (12.6 mg/mouse). Mixed-function oxidases were induced with Aroclor 1254 24 h before injection of 3-MC. The amounts of adducts were significantly higher in lung than in liver in each mouse strain and at each time (Table 5-4~. Total DNA-associated radioactivity in liver is not significantly different from that observed in lung. Thus, the relative binding of 3-MC to DNA of lung and liver of Aroclor 1254-treated mice is distinctly different from that of BaP in untreated mice. The ratio of 3-MC-DNA binding in liver to that in lung is smaller than the ratio for BaP for both nucleoside-bound adducts and total DNA-associated radioactivity (Tables 5-3 and S-43. At present, BaP and 3-MC are the only PAHs for which the amounts of PAR metabolite-DNA adducts can be compared between lung and liver. It should be emphasized that the SAs for the in viva studies reported in Table 5-1 are calculated on the basis of the total DNA in the organ. These values for the BaP DE adducts (Table S-2) do not differentiate between lung and liver and therefore do not appear to offer any explanation for susceptibility of the lung and resistance of the liver to BaP-induced neoplasia in, for example, A/HeJ and A/J mice. However, it is likely that the amounts of adducts formed in different cell types vary considerably. This possibility has the greatest implication for organs, such as the lung, that contain a multitude of cell types. Although little is known about the localization of carcinogen-DNA adducts in lung, cytochrome P-450-dependent monooxygengse enzymes appear to be much more localized in lung than in liver.4~'lb5,l06 The conciliated bronchiolar epithelial (Clara) cells of rabbit lung have been identified as having high concentrations of these enzymes--a finding that correlates with the observed pulmonary toxicity of 4-ipomeanol, which is thought to 5-21 Hi; - . ,i, . ...
result from high covalent binding of a reactive metabolite to proteins in the Clara cells of a number of species.24 The Clara cells amount to only It of the pulmonary cells.)9 The high concentration of cytochrome P-450 in Clara cells may also be important in nitrosamine-induced pulmonary carcinogenesis.138 Because the average SAs of BaP DE-DNA adducts are similar in lung and liver, the above considerations suggest that adduct contents might be much higher in some pulmonary cell types than in hepatocytes. Also, the removal rates of the BaP DE-DNA adducts might vary considerably in different cell types in the lung. Examination of adducts in individual cell types might allow differentiation of tissues with respect to susceptibility and resistance to PAH-induced neoplasia. ~ Results of several studies allow comparisons to be made between amounts of PAH metabolite-DNA adducts in the same tissue from different species (strains). BaP DE adducts and 3-NC metabolite-nucleoside adducts (Figure 5-7) can be compared in lung and liver of different strains of mice under the same dosage regimen (Tables 5-2 and 5-4~. The BaP DE adduct amounts are very similar in lung of A/HeJ and C57BL/6J mice and 10 times smaller in ICR/Ha mice (Table 5-2~. A/HeJ mice exhibit high susceptibility, ICR/Ha moderate susceptibility, and C57BL/6J high resistance to BaP-induced pulmonary neoplasia. Obviously, the SAs of the BaP DE adducts do not differentiate between species in susceptibility to BaP-induced neoplasia. Also, the disappearance rates of BaP DE adducts are Similar in C57BL/6J and A/HeJ mice.Y In contrast, Eastman and Bresnick50 claimed that their results regarding 3-MC metabolite-nucleoside adducts formed and their disappearance rates do differentiate between 3-MC-induced pulmonary neoplasia in the various mice strains (Table 5-4~. It is hard to understand why adduct contents or their disappearance rates based on total organ DNA would differentiate between mouse strains on the basis of pulmonary susceptibility to one PAH and not another. Again, examination of SAs in individ~l~1 _, call ~..~ :_=, _ this dilemma. row ~.~ ~ ~ ·yyc;~ rI~l~,~lu urlraVe1 Phillips et al.150 examined the covalent binding of OMBA, 3-MC, and BaP to DNA in the skin of mice of various strains. Neither the amounts of PAH metabolite-DNA adducts nor their disappearance rates showed a correlation with the reported susceptibilities of the strains to PAH-induced skin carcinogenesis. Thus, the results of this study based on average cellular SAs of the organ are in agreement with the BaP, but not the 3-MC, study of pulmonary adducts in the different mouse strains.9~50390 Ashurst and Cohen 1 confirmed the results of Phillips _~4al.150 with HPLC analysis. Baer-Dubowska and Alexandrov examined the binding of BaP to skin of rats and mice under conditions known to initiate tumorigenesis in the skin of mice. The patterns of BaP metabolite-DNA adduct profiles were identical in the two species and very similar to those in Figure 5-6. The amounts of the BaP DE-deoxyguanosine adducts and total DNA-associated radio- activity were 3 times higher in mouse skin. The adduct difference probably does not differentiate between the mouse susceptibility and rat resistance to BaP-initiated skin tumorigenesis. 5-22 lo. . .
DOSE-RESPONSE RELATIONSHIPS OF PAH METABOLITE-DNA ADDUCTS . . . Although there have been a considerable number of in viva studies on PAH metabolite-DNA adduct formation (Table 5-1), there are only three reported dose-response~studies of amounts of PAH metabolite-DNA adducts. Phillips et al.l50 treated C57BL mice topically with DMBA at 0.025-1.0 ~mol/mouse. DMBA metabolite-DNA adducts in skin were determined by Sephadex LH20 chromatography. Three DMBA metabolite- deoxynucleoside adduct peaks were present at each dose, and the SAs plotted in Figure 5-8 are the sums of these three peaks. Pereira et al.149 examined the formation of epidermal BaP-DNA adducts in ICR/Ha mice at topically applied doses of 0.01-300 mg/mouse. The SAs in Figure S-9 are essentially linear with dose throughout the dose range; the log-log plots have slopes of approximately 1. Peak I in Figure 5-9 represents early-eluting peaks, and peak III in Figure 5-9 represents the BaP DE-deoxyguanosine adducts. Anderson et al.8 investigated the dose dependence of BaP metabolite-DNA adducts in the lung, liver, and forestomach of A/HeJ mice (Figure 5-10~. BaP was administered orally at 0.048-29.7 ~mol/mouse, and animals were sacrificed 48 h later. -The SAs plotted in Figure 5-10 are for the BaP DE-deoxyguanosine adducts. Similar dose-response curves are obtained for the early-eluting peaks (WF and IP in Figure 5-6) and the BaP-phenol-oxide adduct (Peak I in Figure 5-6~. The curves in Figure 5-10 are either linear with a slope greater than 1 and concave downward or linear with a slope of 1 and concave upward (Figures 5-8 and 5-9, respectively). This means that the percentage of the dose that becomes bound to DNA as BaP DE adducts decreases as the dose decreases in the tissues of the AJHeJ mice (Figure 5-10), whereas the percentage of the dose that becomes bound to epidermal DNA is constant or actually increases as dose decreases (Figures S-5 and 5-6, respectively). However, the values of the BaP DE adducts in lung and forestomach of A/HeJ mice at the lowest dose examined are only approximately 50t lower than those predicted by simple proportion from the adduct values at the highest dose (Figure 5-10~. Thus, the results of these dose-response studies do not reveal the existence of any threshold dose below which binding of PAH metabolites to DNA does not occur. EFFECT OF AHH INDUCERS ON PAH METABOLITE-DNA ADDUCT FORMATION . . . . _ The effect of AHH inducers on the in vivo binding of BaP to DNA has been examined in several tissg~s of various mouse strains.36390~184 In a study by Wilson et al.,1 adducts were dete~uined under conditions known to resulE in inhibition of BaP-induced pulmonary 5-23 ~;
neoplasia by 6-NF. Female A/HeJ mice were fed either a 6-NF diet (0.3% of diet) or a control diet for 16 d and then given BaP (6 mg/mouse). The animals were returned~to a control diet for 2 wk and then placed on either a 6-NF diet or a control diet for another 16 d. This regimen results in a 94t reduction in BaP-induced pulmonary adenomas.18I In the adduct study, ~ H]BaP (6 mg/mouse) was administered to one group (A) of animals, both control and 6-NF-treated, on the sixteenth day of the first NF feeding.184 Another group (B) of mice, control and S-NF-treated, were given unlabeled BaP (6 mg/mouse) on the sixteenth day of the first 6-NF feeding and t3H]BaP (6 mg/mouse) on the six- teenth day of the second S-NF feeding. Animals in both groups were re- turned to control diets after administration of [3H]BaP and sacrificed 48 h later. Table 5-5 gives the percentage decrease in BaP DE-DNA adduct formation in the lung and liver of 6-NF-treated mice. The decrease in the amount of BaP DE-DNA adducts in the lung, 86-93%, appears to correlate with the inhibition of pulmonary adenoma formation (947~.181,184 No BaP DE-DNA addicts were detected in the liver of 6-NF-treated mice. Wilson et al.~4 also examined the effects of two other AHH inducers, TODD and Aroclor 1254, on BaP-DNA adduct formation in lung and liver of A/HeJ mice. These inducers, like S-NF, markedly decreased the formation of the BaP DE adducts in lung and liver (Table 5-5~. It is interesting that TODD and Aroclor 1254 had little effect on or actually increased total DNA-associated radioactivity in the early-eluting peaks. In a similar study, Ioannou et al. investigated the effect of S-NF treatment on BaP DE adduct formation in ICR/Ha mice. 6-NF treatment also resulted in a significant reduction, 80-90%, in BaP DE addicts in lung, liver, and forestomach of this strain. Cohen et al.36 examined the effect of TCDO treatment on BaP DE-DNA adduct formation and BaP-induced tumor initiation in the skin of SENCAR and CD-1 mice. BaP DE adduct formation in skin was completely inhibited in both strains of mice, and BaP-initiated papilloma forma- tion was inhibited by 93~.36 Total DNA-associated radioactivity and covalent binding of BaP to skin protein were increased (Table 5-6~. The increase in total DNA-associated radioactivity was due to the increase in the amounts of the early-eluting peaks.36 Again, inhibition of BaP-induced tumor formation correlates with inhibition of BaP DE adduct formation in the target tissue, but not with total DNA- associated radioactivity on covalent binding of BaP to protein. In a continuation of their studies on the anticarcinogenic effects of TODD, DiGiovanni et al.44 showed that the time course for induction of epidermal AHH and UDPGT correlated with the time course for the inhibitory effects of TODD on tumor initiation with DMBA, BaP, and 3-MC. Thus, AHH inducers inhibit in vivo BaP DE-DNA adduct formation in every tissue of every mouse strain examined. The effects of AHH inducers on BaP DE-DNA adduct formation in viva contrast markedly with their effects in vitro, i.e., in lung and liver microsomes, isolated peripheral lung and liver tissue slices, and hepatocytes. Treatment of animals with AHH inducers stimulates the formation of BaP DE adducts in 5-24 ~... . ..
vitro (see Wilson et al.184 for references). The effect of AHH inducers on total DNA-associated radioactivity and on covalent binding of BaP to protein is similar in viva and in vitro. The reason for the disparity between the in viva and in vitro results for BaP DE adduct formation is unclear--in particular, the relationship between induction of AHH and decreased in viva BaP DE adduct formation. Induction of AHH has been postulated as a mechanism for the anti- carcinogenic action of a wide variety of ds 36~44~45~9o~l54'l8o~l8l~l84 The results of Wilson et al. 84 and Cohen et al.36 discussed above suggest that AHH inducers, such as OFF and TODD, inhibit BaP-induced neoplasia by reducing the amount of BaP DE-DNA adducts formed in the target tissue. The data of Pyerin et al.154 also suggest that prior AHH induction is a protective mechanism against DMBA-induced tumor initiation in mouse skin. The concept of a protective effect of prior treatment with AHH inducers against PAH-induced tumor initiation might appear to be incompatible with the suggestion that AHH inducibilit and PAH-induced tumor initiation may be causally related.64~03~33~55~43~47~78 Relationships between AHH inducibility and PAH-induced tumor susceptibility have been examined in a great variety of tissues and animals; including man. It was proposed that humans with intermediate and high AHH inducibility carry a higher risk for lung and laryngeal cancer than persons with low inducibility.103~178 However, these findings could not be confirmed.143 In various strains of mice, AHH inducibi-lity correlates well with susceptibility to PAH-induced carcinogenesis for some PARs but not for others.64~133~135~147 However, none of these studies allows any assessment-of whether AHH induction in the target tissue preceded PAH-induced tumor initiation. Even in cases in which positive trends exist between AHH inducibility and susceptibility to PAH-induced tumor initiation, prior treatment with AHH inducers will probably protect against tumor initiation by the PAR. Whether the prior induction of AHH in the target tissue is part of the protective mechanism is unclear. IN VIVO DISAPPEARANCE OF PAR METABOLITE-DNA ADDUCTS . . . Mutations and malignant transformations of cells by chemicals may be a consequence of DNA synthesis on parent-strand templates containing unexcised chemically induced lesions. Thus, the ability of a cell to repair the damaged DNA by an error-free pathway could constitute a critical protective mechanism against mutagenesis and carcino- genesisi633129 However, Feldman et al.55356 and Shinohara and Cerutti 7 reported the persistence of BaP DE-DNA adducts in human alveolar tumor cell A549 and secondary mouse embryo fibroblast, -respectively. Dipple and Roberts47 also noticed the persistence of 7-bromomethylbenz~aJanthracene metabolite-DNA adducts during replication in cell cultures. Cerutti et al.33334 observed that significant fractions of BaP metabolite-DNA adducts were still present in the DNA when the alkaline-elusion profile of the parent DNA had 5-25
returned to normal. The persistent lesions could result from the loss of excision repair during prolonged incubation or from the adducts' becoming part of apportion of DNA that cannot be repaired by the excision pathways. There have been only a few studies on the in viva disappearance of PAR metabolite-DNA adducts. Before these results are discussed, it should be emphasized that the interpretation of in vivo rates of disappearance of carcinogen-DNA adducts has several limitations. When the SAs of the adducts are based on total ONA content in an organ, the disappearance rates are average cellular values. The disappearance rate in the target cells could be masked by the rates in nontarget cells, especially if the target cells are only a small fraction of the total cells in the organ. In addition, in viva rates of disappearance of specific activities of adducts cannot be unequivocally equated with enzymatic excision rates, because cell turnover will also result in a decrease in the SA of the adduct. Eastman and Bresnick50 examined the disappearance of 3-MC metabolite-DNA adducts in lung and liver of several mouse strains (Table 5-4~. In each strain examined, the adduct decreased significantly faster in liver than in lung. The adducts are more persistent in the lungs of A/J and C3H/HeJ mice--strains susceptible to 3-MC-induced pulmonary adenomas--than in the lungs of highly resistant strains. Whether these differences in disappearance rates and initial amounts of 3-MC metabolite-DNA adducts are causally related to differences in tissue and species susceptibility to 3-MC-induced tumorigenesis is unclear. Anderson and Wilson9 examined the disappearance of BaP metabolite-DNA adducts in lung and liver of A/HeJ and C57BL/6J mice (Table 5-7~. The disappearance rates in A/HeJ were examined at oral doses of 0.011 and 6.0 mg/mouse. The amount of BaP DE-DNA adducts in lung and liver decreased monoexponentially with t ime at each BaP dose. Although the initial BaP DE-DNA adduct amount at the higher dose was more than 1,000 times larger than that at the lower dose in lung (liver), the half-life of BaP DE adducts in lung (liver) was similar at the two doses (Table 5-7~; thus, there is no apparent threshold dose for removal of BaP DE adducts in lung and liver of A/HeJ mice. BaP DE-DNA adducts had a biphasic decay in the lung and liver of C57BL/6J mice (BaP at 6 mg/mouse). Also, the BaP DE adducts in the lung and liver of C57BL/6J mice could be approaching a constant, nondecaying value. Examination of adduct amounts at longer times after the initial dose will be required to determine whether persistent adducts exist. The half-life of the adducts in the terminal phase was similar to that observed in the A/HeJ mice (Table 5-7~. In contrast with the results of Eastman and Bresnick50 with 3-MC, these data do not appear to offer any explanation for the tissue and species differences in susceptibility to BaP-induced neoplasia. 5-26 ~. ,.
t The in viva disappearance of 11-methylketone metabolite-DNA adducts was examined in lung, liver, and skin of TO mice for 14 d after initial treatment.2 Rate of DNA turnover was also examined over this same time span. Rates of disappearance of the major adducts in skin and lung could not be measured above the normal rate of DNA turnover, whereas, in the liver, adducts were removed rapidly (half-life, about 2.5 d) relative to the DNA turnover rate. Thus, enzymatic repair of adducts would be occurring in liver, whereas adducts might be persistent in some cell types of skin and lung.2 Again, whether these apparently persistent adducts of skin and lung are causally related to the initiation of neoplasia in these tissues by 11-methyl- ketone awaits further investigation. The disappearance of PAH metabolite-DNA adducts has also been examined in skin of mice. Phillips et al.150 studied the disappearance of DMBA metabolite-DNA adducts in skin of several mouse strains. The half-life of the adducts was 1-2 d in each strain examined. These results do not explain the strain difference in susceptibility to DMBA-induced neoplasia in skin of mice. Similar conclusions were obtained with BaP and 3-MC, although adduct amounts were examined at only two points.150 Rayman and Dipplel56 examined the formation and disappearance of 7-bromomethylbenz~ajanthracene and 7-bromomethyl-12-methylbenz~aJanthracene metabolite-DNA adducts in skin of Swiss S mice. The adduct amounts of the less carcinogenic 7-bromomethylbenz~aJanthracene were higher and required a longer time to reach maximums--4 d vs. 1 d--after a single topical application of 1 Amos. For both chemicals, the adducts decayed rapidly from the maximum, the half-lives being less than 24 h. The data do not differ- entiate between the carcinogenic potency of these two PAHs in mouse skin. Pelkonen et al.146 examined the disappearance of BaP metabolite-DNA adducts in skin and subcutaneous tissue of C3H and C57BL/6 mice. No strain difference~was observed in the disappearance in either strain of the BaP 4,5-oxide or the BaP DE adducts. Thus, rates of disappearance of the adducts do not differentiate between the C57BL/6 resistance and the C3H susceptibility to BaP-initiated subcutaneous fibrosarcomas. In summary, no generalization regarding these data on in viva disappearance of PAH metabolite-DNA adducts can be made now. In some investigations, extrapolation of the adduct-time curve suggests complete removal of the adducts, whereas adducts appear to persist in other studies. Similar results were obtained in studies of adduct removal in cell cultures. And adduct removal rates differ significantly between tissues susceptible and resistant to PAH-induced neoplasia in some cases and not in others. It is possible that the in vivo results on adduct disappearance rates based on total organ UNA content are misleading, in that PAH metabolite-DNA adducts might be persistent in some cells that constitute only a small fraction of the total cell population. 5-27
PAH METABOLITE-DNA ADDUCT AMOUNTS AS A MEASURE OF EFFECTIVE . _ ~ . . . BIOLOGIC DOSE OF SOME PAH TOXICITY . . . . It has been suggested that the amount of carcinogen-DNA binding is a measure of the effective dose of a carcinogen.113 This proposal is consistent with the somatic-mutation theory of tumor initiation by chemical carcinogens. Although the extent of carcinogen-DNA binding has been successfully used to rank a series of carcinogens, such as PAHs and alkylating agents, for carcinogenic potency,30337388 there have been few, if any, serious attempts to use the amount of carcinogen-DNA adducts formed in the target tissue as a predictive tool for low-dose extrapolation in carcinogenesis. Anderson et al.7 and Gehring and Blau suggested a general scheme for the incorporation of pharmacokinetics in low-dose risk estimation for chemical carcinogenesis. This section discusses the feasibility of using some measure of chemical-induced DNA damage as an effective biologic dose of a carcinogen. IN VITRO MUTAGENESIS Several recent studies have examined the extent of PAH-induced ONA damage in cells undergoing mutation and have attempted to relate the PAH-induced mutation frequencies to the amounts of PAH metabolite-DNA adducts. Wigley et al.1 and Newbold et al. 8 examined the mutagenicity of benz~aJanthracene, 3-MC, DMBA, BaP, and 7-MBA in a cell-mediated mutagenesis system, using BHK 21 cells to metabolize the PAH and Chinese hamster (V-79) cells as targets for mutation. The frequencies of PAH-induced mutation were not significantly different at equivalent amounts of PAH metabolite-DNA adducts. Yang et al.193 examined the induced-mutation frequency in normal fibroblasts and XP12BE cells as a function of the number of BaP DE-DNA adducts in the cells when they were released from confluence and plated for the expression of 6-thioguanine resistance. The mutation frequency is linearly related to the BaP DE I-DNA adduct amount when the cells were released from confluence.193 Fahl et al.54 examined the induction of Hist+ reverse-mutation frequencies by BaP DE I, BaP DE II, and 9-hydroxybenzota~pyrene in Salmonella typhimurium cells (TA 98 and TA 100) as a function of the number of bacterial DNA bases modified by the electrophilic BaP metabolites. The induced-mutation frequencies were linearly related to the DNA adduct amounts in each case (Figure 5-11~. Newbold et al.137 investigated the mutagenicity of BaP DE I and BaP DE II in V-79 cells as a function of BaP DE-DNA adduct amount in the V-79 cells. At sublethal doses of the diol-epoxides, the induced-mutation frequencies were linearly related to the DNA adduct amounts in the cells for both BaP DE I and BaP DE II. The relation of mutation frequency to DNA adduct amount, however, becomes exponential at toxic amounts of BaP DE-DNA adducts. Newbold and Brookes 36 also 5-28
observed a linear relationship between induced-mutation frequency and the concentrations of BaP DE I in the medium as long as that concentration was sublethal. Mutation induction by PAHs, as well as by other mutagens and UV radiation, appears to result from the cells' attempt to deal with the unexcised chemical-induced DNA lesions at the time of replication. Moreover, induced-mutation frequencies are linearly related, at least at sublethal doses of the mutagen, to the amounts of mutagen-DNA adducts present when the cells are undergoing replication. This linear relationship makes DNA adduct amounts a good measure of the effective biologic dose of mutagens in in vitro assay systems. Comparisons of the mutagenic potency of PAHs should be based on the slope of the curve relating induced-mutation frequency to PAH metabolite-DNA adduct amounts. CARCINOGENESIS In addition to the results obtained in in vitro mutagenic assay systems, the in vivo results discussed below suggest that PAH metabolite-DNA adduct amounts in a known target tissue are a good measure of the effective biologic dose of PAHs for initiation of neoplasia. Several investigations have shown a positive correlation between the carcinogenicity of a series of PAHs of widely differing carcinogenic potencies and their extent of reaction with DNA.30'32'68'88'150 This correlation was not observed with the binding of the reactive metabolite of the PAHs to protein and RNA. ~ Similarly, the binding of 6-propiolactone and similar alkylating agents to DNA, but n3Ot to RNA and protein, corresponds to their tumor-initiating potency. 7 Total DNA binding (picomoles of radioactivity associated with DNA per milligram of DNA) was used to rank the carcinogens for carcinogenic potency. Recent studies have suggested that amounts of specific carcinogen-DNA adducts should be used as a measure of effective dose, instead of total DNA binding. For example, for the nitrosamines and nitrosamides, correlation between carcinogenicity and nucleic acid alkylation has been observed only with 06 alkylation of guanine, and not with N7 alkylation of guanine, even though the latter alkylation is approximately 10 times greater than the former. ,144 Current evidence suggests that the bay-region diol-epoxides, such as BP DE, are the ultimate carcinogenic forms for most PAHs. The use of specific carcinogen-DNA adduct amounts, instead of total DNA binding, should improve the ability to rank a series of carcinogens. Studies with inhibitors of carcinogenesis have shown that tumor response chances quantitatively with PAH metabolite-DNA adduct amount.6'363903184 Swann et al.176 showed that changes in the incidence of dimethylnitrosam~ne-induced kidney tumors produced by 5-29
changes in the diet and by treatment with BaP correspond to the changes that these treatments produce in the alkylation of the target-tissue DNA by dimethylnitrosamine. Janss and Ben92 found a correlation between the amount of DMBA bound to DNA and the incidence of mammary tumors in rats of different ages. Several studies have shown that, although the time for appearance of the first tumor was not necessarily dose-dependent, the average time between the administration of a single dose of a carcinogen and the individual appearances of the several tumors was definitely dose-dependent. This increase in the average time for the development of tumors as the dose is lowered led Swann et al.176 to suggest that the time needed to achieve full malignancy is a function of the amount of the initial preneoplas tic lesion, i.e., the formation of carcinogen-DNA adducts. Most in viva studies of carcinogen-DNA adduct formation have used total DNA content in an organ to calculate the specific activities of the adducts. However, it is likely that the amounts of adducts formed vary considerably in different cell types. This possibility has the greatest implication for organs (such as the lung) that contain a multitude of cell types and in which the cytochrome P-450-dependent monooxygenase enzymes are localized to a few cell types.43~155~166 Obviously, the specific activities of carcinogen-DNA adducts in individual cell populations would be a better measure of the effective dose of a carcinogen than the values based on the total DNA content of the organ. In addition to the extent of carcinogen-induced DNA damage, the capacity of cells to repair such damage and the degree of cell replication are critical in the initiation of carcinogenesis. As with the measurements of SAs of carcinogen-DNA adducts, most in viva studies of repair of carcinogen-induced DNA damage have been based on total cell populations and thus represent average cellular values. Lewis and Swenbergll did study the differential repair of 06-methylguanine in the DNA of rat hepatocytes and nonparenchymal cells (NPCs) after administration of 1,2-dimethylhydrazine. The NPCs are the target cells in 1,2-dimethylhydrazine-induced liver neoplasia. Although the initial alkylation was similar in both cell types, the NPCs repaired 06-methylguanine more slowly than the hepatocytes. This led to a much greater accumulation of the promutagenic lesion in the target cells (NPCs). The rate of cell division was also much higher in the target cells.ll9 Thus, in this model system, carcinogen-DNA repair rates and cell division rates were definitely correlated with target-cell susceptibility. Such studies of PAHs are needed to identify the target cells for PAH-induced neoplasia in such organs as the lung. Carcinogen-DNA adduct amounts, their rates of removal, and cell turnover rates may not be able to explain the difference in organ and species susceptibility to chemical-induced neoplasia. Promotional 5-30
aspects of carcinogenesis might be required to explain these differences. Determination of whether the initiation characteristics--adduct amounts, DNA-repair rates, and cell turnover rates--can explain the species, strain, and organ differences in susceptibility to PAH-induced neoplasia will require more detailed examination of individual cllgl types in the organs, such as the studies by Lewis and Swenberg. ~ Even if the initiation characteristics cannot explain organ, species, and strain differences, this does not detract from the use of PAN metabolite-DNA adduct amounts as a measure of the effective biologic dose of a carcinogen in a known target tissue. For low-dose extrapolation of carcinogenic data, for the ranking of a series of similar carcinogens, and for determining the effect on neoplasia of pretreatments that alter the metabolism of a carcinogen, the results discussed above strongly suggest that specific PAR metabolite-DNA adduct amounts are a good measure of effective biologic dose. Adduct amounts in individual cell types of a target organ would probably be an even better measure of effective biologic dose. However, they are not now practical, because the separation of cell types is not generally feasible. We should attempt to incorporate DNA-repair and cell turnover rates into the effective biologic dose of a carcinogen. The use of these initiation characteristics as a measure of effective biologic dose has practical value, because they can usually be studied at doses much lower than those used in bioassay studies. TRANSCRIPTION AND REPLICATION Several s tudies have shown that the functions of ONA during transcription and replication are inhibited by the presence of carcinogen-DNA adducts on the ONA template (Grunberger and Weinstein77~. Mizusawa and Kakefudal: ~ concluded that the BaP DE I-DNA adduct inhibits chain elongation with little effect on initiation of NINA replication. These authors, Yamaura et al.,192 and Hsu et al. 7 suggested that elongation of the deoxypolynucleotide chain was terminated at each BaP DE I binding site of the template. Leffler et al.114 demonstrated a progressive inhibition of transcription with increasing amounts of BaP DE I adducts on the template. However, in contrast with the above-mentioned results on replication, Pulkrabek et al.152 concluded that, with some degree of frequency, RNA polymerase can bypass BaP DE I adducts in a template to permit continued chain elongation. They also showed that BaP DE I-modified plasmid DNA could not transfect a receptive Escherichia cold strain to antibiotic resistance. The modification of DNA by metabolites of aflatoxin and 2-acetamidofluorene inhibits replication and transcription on the modified template in a similar manner.77 Most of the detailed studies of the effects of carcinogen-modified DNA on transcription and replication have been performed in in vitro model systems. However, because the DNA adducts formed in viva after 5-31
exposure to BaP are, at least in most cases, the same as those which were used in the in vitro studies, these basic functions of DNA might also be affected after in viva exposure to BaP. Dose-response studies of formation of PAN metabolite-DNA adducts revealed that adducts will exist after environmental exposure to PAHs. Moreover, recent studies by Anderson et al. (unpublished data) have shown that BaP metabolite-DNA adducts are formed in many organs, both susceptible and nonsusceptible to BaP-induced neoplasia. BaP DE I-DNA adducts were observed in lung, liver, forestomach, brain, kidney, and colon of mice after oral exposure to BaP. Even if the environmental exposures to PAHs are too small to induce neoplasia, the formation of ONA adducts after such exposures could produce aberrations in the transcription (replication) of genetic information in many organs and perhaps lead to subtle toxic effects. Obviously, the amounts of DNA adducts constitute the appropriate measure of effective biologic dose for these · . . eons' aerations . 5-32 of .. .
c) a) a' 0 ce 0 ^ ~ - - 00 ON 0 ~ ~ ~ 0 0 0 ~ ~ ~ ~ ~ red ~ ~ ~ ~ cad us ~ us ~ ~ ~ ~ ~ O ~ ~ ret ~ ~. - ~ ~- ~ Lo en c) l o o o ~ · L, ~ >` ~1 co is co ~b a) a' a) ~·- x ·- ~o o o o.- ~3 so of . ~ . , o :^ ~ _ _ _ ^ U' ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~14 ~·- 0 ~ ~ a, ~ ~ a' 0 a) ~ a) u' ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~1 ·,1 a, a, . - .,4 · - ·,4 · - ·,4 ·- C) ~ ~ ~ ~ ~ ~ _ ~ _ ~ ~ l_ _ ~ _ _ ~ ~ _ ~ ~ ~^ - - - ~1 ~bo ~ o~ ~ os bo to ~ bo ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~o ¢ ~ ~ ~ ~ ~ ~ ~ ~ ~.~-.~-.-.~-~- ~ =~-.- ~ ~·- ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ .~ ~ . ¢ O ~ ~ ~ ~ ~ ~ ~ ~ ~co ~ co u~ u~ u~ co u~ c~ co u~ u~ ~ ~ cn co ~ ~a~ ~7 · u) · - Q S ~1 a) u~ ~ ~ C~ ~ ~ Ct ~ ~ Ct Ct C~ Ct C~ Ct ._ ~. ~ C) ~ C} C) C~ ~ C) ~ C) ~ C) ~ C ~- ~J·· · · · · · · · ·,4 ·- ·,! ·,~ ·,4 ·- ·,4 ·- ·,4 ·,4 ·~4 ·- · · -~- ·- · · o 0 ~00 0 0 ° ......... 0000000000 0 0 ~00 ·~ E~ ~ ~ ~ ~ E~ ~ ~ ~ E~ ~ E~ E~ ~ ~O 1 a U~ . 0 C~0 ~X ¢ ~. 0 . .0 . ~o 0 C ~· ~· - 0 0 E~4 ~1 ~ 1 1 ~. ~ 0 oo ~^ ~\ (~) ~C) C~_ ~0 ~ C ~oo ~0 ~0 0 0 0 _ 0 C ~0 C ~0 ~C~ ~C~ 0 ~ ~o~ 0 · - 0 ~......... ... ~........ u, ~ - O ~ ~ O O O O O O O O O O ~ O O O O ~ ~1 ~ ~ c~ t_ ~0 1 O e~ _ c~ c ~0 ~O C ~_ _ s~ . t_ 1 0 . ~ 0 0 ~ ~ a' ~ e S~ O C ~ ~ ~ 0 ~ . . : ~o o o ~ ~:^ , u ~ ~c l O ~ Gl b0 ~ tI, ¢ ~ ~ ~.- _ ~ ~ _., - cn 1 ¢ U) ~ ~ ~ ~O O ~ p: '_ p~ ~ ~ ~ ~· ~- <1) ~O ~IJ ~ ~^ ~ a .- I ^ ~ ~ ~a) ~ ~ C) ~¢ ~ :t C) ~- ~1 0 :~ 1 1 1 ¢ ~1 1 1~ : ~P4 ~P4 ~P ~ 1 1 ~ ~-_4 ~3 1 . .m ~ ~ ~ ~ ~ ~q ~ ~ ~ ~ ~ - ~ ~ ~ ~ ~ O X ~C ~O ~n O ~ _U' C ~^, _ ~CO ~ ~ ~ C ~u, 3 u)~- ~ O u ~cn ~ ~/D ¢ .- me ~ Cd 1 ~3 - ~.= 4 UO C-l ~ ~^ ~1- _ ^ ¢ ~ ~ 1 ¢ . ~C ~t_ _ ·~4 O a) ~a =~¢ ^ ~ ~ . :^ ~ :r: ~ a ~s~ I ~n 0 ~n ~ \~) ~= (~ ~- ~c ~ ~a ~c~ ~ ~s~ :: a ~ 0·- ~ a ,4 ~ ~ ~ ~ ~u~ ~ ,~ ~ ~ ce -5: _ -1 ~ ~ ,~ ~ct ~ C ~oO S" ~ 1 ~ c: ~ ~ ~ a: ~ _ ~ ~ ~ ~, - (,, ~ ~ ~ ~ ~ po pq ~ ~ ~ ~ X :~: ~ ~ ~ -~ -~ ~ - ~ r~ r~ ~ ~ Z ~ ~ ~ ~ r~ ~eo s~ 3 0 a u~ ~ u ~ u~ u~ ~ 3 ~ u~ ~ u~ ~ 0 0 0 ,1 ~ a ~u co ¢ <: ~ c~ ~ ¢ ¢ c~ ~c~ ~ c) :e ~ c: c~ C~ ~ C) c~ ~ 3 u: z c~ ~c 5-33 ~;
- ~ cd to to to on en ~ ~ ~ ~ ~ ~ ~ ~If - a ~ lo of ~ ~ ~ ~In ~ O ^ cO ~ ~ ·- e ·- ~ c' ¢ o E ·- ~ Z ~ U)~,1 =~= ~=~- e C) ~ ~0\ 00 U] ~ 0 0 0 0 V. C) z . . . ~U' ma ~boo ~ m ~ ~ ~ e ~ ^ ~3° ° ~ o a ~ en Go Go ~ ~ ~ o o ~ o O ~ O ~ ~ ~ ~ ~ 0 ~ ~ ~ O ~ ~ ~ ~ cn =m ~............. ~¢ <¢ u, ~ ~ ~ ~ o o o o ~ ~ o o o o o o o ~·- ~ Z c' ~ tn . - .cat e cn =: . o.- ............... ~·- a, ~ ~ ~ 1 ~C:, O O O O O O O O ~ ~ ~ O O P~ ~, tn ~ 3 ct L ~O~ e ~ ~ ~ ~ ~ ~ e ~ e ~ ~ ~ ~ e P~ ~ ~ P. ~ ~ ~ ~ p~ ~ ~ ~ p" p~ ~ ~ ~ ~ ~ · -· - O '_1 O ~ _ a: ~ ~^ a) O ~ ~ b0 E ~D ~ O ·,. ~ OC ~ ~U~ ~ l ~. ~l ~ .,/ ~ e ~ ~v u, O O ~ ~ O O O O O O O O O O O O . ' a) 0 O ~ 44 - ~ - ~ ~ ~ ~ ~· ~ ~ ¢ ~ e -- -~- ~ ~ cn ~ <: ~ ~ . O ~ S ~ ~ ~ . - ~ ~. - cn a) ~0 C ~ ~ ~ ~ e , - , e ~cn =.- O 0 CD :~. ·- · :d P~ s4 i4 3 tC ~a so ~ oo ~ oo ~ ~ ~ a' ~o ~ ~ ~o ~ oo ~ u, cs ~ ? ~ ~ ~ ? ~ ~ ~ ~ ~ ~ ~ ? ~ ? .,' ~ ~ =- - =~ - O O. - =- - · - ~ - =- - S E ~ ~ ~ ,_1 ~ ~ ~, ,~ ,.~ ~ ,_; ~ ,_1 ~ ,~ , ~v O O ~ · tn o~ ~E cn ~ ~ ~ U, ~ ~U' ~ 00 ,] v ~ ~ ~3 ~.- . .~.~.~- D ~ D ~o ~ X D ~ ~ ~ S /, C~ ~ ~ ~O I s~ ~ ~ s" ~ ~ ^ ·,1 O = ·- ~, ~ ~ ~a1 ~ ~ ~a ·-, ~ ~ ~ ~ D~ ~ ~ ~ ~ ~cn ~ a) =.~ ~ ~ c ~ ~ ~ ~ - - - ~ ~ca} ~ ~ u' a~, ~ =: ~ ~ F~ ~ ~ ~ ~ ~ 3 3 3 3 O ~ ~m o ~c~ ~ ~ U~ ~a) o aJ o ~ 3 :~ U) ¢ ~ ¢ ~ ~ ~ ~ C) ~ ¢ ¢ ¢ Z Z Z Z ~C) 5-34 C X · -
TABLE 5-3 In Viva Formation of BP Metabolite-DNA Adduces in Lung and Liver of Rats Doseja Tissue ~mol/kg Specific Activity,b fmol/m~ of DNA Total DNA Binding BP-pheno 1-ox ide- DNA Adduct . Lung 1.0133 + 11 55 e 5 + 6 ~ 0 Liver 1.0730 + 220 48.2 + 5.5 Lung 10.0 680 + 165 178 + 41 Liver 10 ~ 0 2 ~ 500 + 610 168 + 25 al.V. injec Lion. bDetennined 1 h af2t~r I.V. injection of [3H]BP. Data from BorouJerd~ et al. Mean + S.E. for three experiments. 5-35 A. . .
TABLE 5-4 Binding of 3-MC to 1)NA in Lung and Liver of Mice Specific Activity,b fmol/mg of DNA In Vivo Nucleoside Strain Tissue Exposure Timea TotalC Bound Adductc A/J Lung 4 h 113 44 .9 7 d 74 30.0 28 ~106 15.9 Liver 4b 135 5.5 7 d 96 5.5 28 d 96 0 C3H/HeJ Lung 4 h 76 17. 6 7 d 43 8.3 28 d 43 6.1 L iver 4 h 45 4 . 4 7 d 20 0.6 28 d 29 0 DBA/2J Lung 4 h 68 16. 3 7 d 35 6.3 28 d 24 1.1 Liver 4 h 64 4. 8 7 d 50 1.3 28 d 28 0 C57BL/63 Lung 4 h 173 16.1 7 d 103 2. 7 28 d 83 1.7 Liver 4 h 139 9.7 7 d 135 0 28 d 107 0 aTime of sacrifice after I.V. injection of .[3H]3-MC (12.6 mg/mouse). bData from Eastman and Bresnick.50 CMean of 6-10 mice (3-5 determinations). Fairly large individual variation was observed with S.D. of + 25Z of mean. dPeaks V and VI in Figure 5-4. Values obtained by multiplying ~ of radioactivity eluting from Sephadex LH20 as peaks V and VI by total DNA-as soc iated radioac t ivity . 5-36 ~. .
TABLE 5-5 Effect of AHH Inducters on In Viva Formation of BP DE-DNA Adducts and on Total DNA-Associated Radioactivity in Micea Specific ActivityC (pmol/mg of DNA) of Total BP DE-DNA Adducts DNA-Associated in Treated Mice, Radioactivity, Strain Treatmentb Tissue % of control % of control - A/HeJ S-NF (Group A) Lung 15 43 A/HeJ S-NF (Group A) Liver 0 67 A/HeJ S-NF (Group B) Lung ~46 A/HeJ 6-NF (Broup B.) Liver 0 48 A/HeJ TCDD Lung 5 73 A/HeJ TCDO Liver 0 182 A/HeJ Aroclor 1254 Lung 9 123 A/HeJ Aroclor 1254 Liver O 101 ICR/Ha 6-NF Lung 16 42 ICR/Ha S-NF Liver 7 S5 ICR/Ha 6-NF Forestomach 16 40 aUntreated or treated mice were killed 48 h after oral dose of [3H]BP (6 mg/mouse). DNA isolated from tissue was enzymatically digested, and deoxyribonucleosides were chromatographed on HPEC. Specific activity of BP DE-DNA adducts calculated from HPLC chromatogram. bMice were fed ~NF (3 mg/g of diet) for 2 wk before [3H]BP administra- tion. See text for discussion of Group A and B animals. Animals treated with TCDD (80 nmol/kg) 4 d before [3H]BP administration. Aroclor 1254-induced mice received inducer (500 mg/kg) 48 h before administration of [3H]BP. CData for A/HeJ mice from Wilson et al.184 Data for ICR/Ha mice from Ioannou et al.90 Zero means adduct not detected in treated animals. 5-37 ~; ~.. .
BP Dose,a Strain mg/mouse T ime b h ? DNA TABLE 5-6 Effects of Pretreatment with TODD on In Vivo Covalent Binding of BP to Mouse Epidermal DNA, RNA, and Protein Hydrocarbon Bound to Macromolecules (pmol/mg) in Treated Animals,C of control RNA Protein SENCAR 25 3 250 217 255 SENCAR 25 24 2 78 - 133 260 CD-1 50 24 318 100 242 aMice received single topical application of [3H]BP. Animals received single topical application of TODD ( 1 me in 0. 2 ml of acetone) or acetone 72 h before carcinogens bTime of sacrifice after application of BP. CData from Cohen et al . 36 - 5-38 Em .. .
TABLE 5-7 In Viva Disappearance Rates of BP DE-DNA Adducts in Lung and Liver of Mice Half-life for Dis P.O. Dose, appearance of BP DE Strain Tissue mg/mouse DNA Adducts,a days A/HeJ Lung 6.0 17 A/HeJ Lung 0.012 19 A/HeJ Liver 6.0 9 A/HeJ Liver 0.012 16 C57BL/6J Lung 6.0 19 ~ C57BL/6J Liver 6.0 14 aAnimals sacrificed at intervals of 10 h to 28 d after oral dose of [3H] BP. Specific activity of BP DE-DNA adducts decreased mono- exponentially in lung and liver of A/HeJ mice. There was biphasic decay of adducts in lung and liver of C57BL/6J mice . Hal f-life of terminal phase given here. Values are sums of BP DEI- and BP DEII- DNA adducts . Data from Anderson and Wilson. 9 5-39 . - D~. _'. '
- - A Q"- Am_ ,1_ .._ O C)= -_ ~ a) O ~ Q - TO ~ a, lo.- I_ - o lo a' O ~ _ C .0 ~ - ._ ~ O O id, . _._ ~ Cal 00_ ._ lo O _ _ O _ ~ _ m._ .~° E ~ II _ m~ ~o ~ , ~_ of__ -_ a_ To in ~ o lo. _ ~ .t ~ 0 3 . __ _ o , o X ~ 5-40 1 _ Cal 1_-- ~ 0 o Cat Cal - 1 ~i ..
- - ~ - Chemico I ·xposilreExposure Uptoke ~Uptake 1 I Non m crow l :~1 enzymes 1 enzymes . _ ~_ Detoxification No ~ damage ~ Repolr Voicer. I I mutt I - Biotronstonnolion ~ F~ Moc~romohc. hincl;ng Chem~eot binding ,~ I disk. I \? |? ? At_ , Mol'~nont Tra nsf ormation 1 -2 1 Repair Genetic lesions - 3 Corcinogenesis } 4 FIGURE 5-2 . Sequence of pos ~ ible events from exposure to · - care 1nogenes ~ s ~ . 5-41 in;
~ o- ~ ~ - ~- o 0~ o >: at an l:' i to ... ~ / Z' LL Cal Q - O O . CD \ I \ o ~ - C~ _ 1 o Wo ~- ~ I I ,. . .... to, ., ~ .w .. ~ . .... 1 >_, 1 XJ ,Oz~ to / ~= 5-42 i Lt) _ Z O V an ILL =) L1J . J Z cr Ct A A ~N ~= I C) A ~a, O V m ~ o o ID E o v o o C; o 1 Us Cal
@; HOW on 't ~ of H0~ I)- 7. a- dial OH H0~ OH FIGURE 5-4. Sequences in formation of highly electrophilic 7, 8-diol-9 ,10-epoxide. 5-43 Hi; 3,,. i. .
99~ HO"' fir FIGURE 5-S. Structures of BPDE; both isomers represent enant lamer ic mixtures . 5-44 ~ . '. .
Boo lL c, IL c, w Nina We 5-45 O N O lo 1° .0 ___-,' O o o CO o TO Se ~ .° ~ - o 0 0 ~0 2 ~ o 0 0 Pa C) o In ~ ~ . - ¢ o to :^ E an o ~ o C) u A; ,~ o C _ 1 of l C: 1 CL o 1 ~ ~ c; u cn .. o U. ^ 1 ~ ~ ~ on CC ~ ~ ho ^ It o ~ ~ o o . ·^ ~ ~C o ~ ·^ - v cn to ~ ~ o ~u o ~ ~ · co ~u E ~ X 0 o ~ ~ ~ ~C ~ C9 ~ ~ ~ ~ o~ .. . ~ so~ ^ 3 ~ u' U, ~ sq ~ ~ ¢ CO o ~ ^ C) o ~D tn ~ ~ o .^ C) ~ ~ ~. o ~ ~ o U . - 3 ~ o ~ o ~n E . ~ ~C) ~n~ X o o 3 ~ . ~ ~{: c' ~n ~ ^ o o 3= · '' o ~ · o.o . (V == ~ ~ ~ U, ~ ~ ~ . I,, ~ ~ ~ ~ tr: _ ~ ~ ~ ~ ~ et oo i4 ~ ~C ~ ~ 0 1 ~/ O D u~ O =' C~ - :^ X o~ ~ e E~ S4 _~ ~ 1 N 0 :~= O ^ tn 3 3 ~ ^ elD tJ ~ C~ X ~ ~ tn cn _ · ~ eY ~v . ~ ~ ,=: ~ s" 1 0 JJ ~ · CL ~ ~ ~ ^ 3 r: c~ ~c 0 0 _ ct ~ c; =: ~ 0 ~ E ~ O ~ ~ :: E ~ v ~ ~ c' ~ ~ 0 ~ ct ~ ~ ~ ~ 0 ~ ~ ~ u' c:
~or Al ~ i'i'260 !1i li !1 i )~ J i cp`~: A . ~ - ~ 20 ELUT10N V=u~E Id) on o FIGURE 5-7. Sephadex LH-20 column chromatography of d~oxyribonucleosites from DNA of mice treated with ~ H]-3MC. Chromatography was performed on 0.9 x 30-cm column eluted with 140~m1 gradient of 30~100% methanol in water. Lois is composite graph to indicate pose ible peaks of radioactivity obtainable. Peaks I to IV, early peaks obtainable from liver DNA. Peaks V ant VI, nucleoside- bound adducts that appear maximally in A/J lung DNA. A260' unmodified deoxyribonucleotide; arrow at bottom, position of elusion of marker, 4-~-nitrobenzyl~pyridine. Reprinted with permission from Eastman and Bresnick.49 t 5-46
An o 30 2 - ,- - ~ to o ~ / / 1= ~ an 1& ~ Mo! DMSA/Mouse FIGURE 5-8. Binding of 7,12-dimethylbenzrajanthracene to DNA of skin of C57BL mice 19 h after topical application. Binding determined from elusion profiles of DNA hydrolyeates prepared from treated skin. Reprinted with permission from Phillips et al.150 5-47 I" ....
000 _ (A} Pad ~lOO: I 111 bed m · HPLC ~ ~ . H~c . ~ 5~ LH- ~t 2 ; ~ ~ ~_ . 001 , Go' t ~ : , ·--1 ' ''' ''" 000t' ' '''''I ' At-. ~ &- -' ........ ........ 100 1000 001 01 ~ 10 100 ,000 0~ ~ ~P/Ha_} ~ t ' ~ ~t · - - --! 1 , ,, 001 01 1 t0 De ~ peso B P/~_1 FIGURE 5-9 . Dose dependence of BaP adduc t format ion in epidermal DNA. Groups of mice, 15-20 each were treated topically with 1, 2, 5, 10, 25, and 100 ng of []H]BP and sacrificed 24 h later. Enzymat ical ly digested epidermal DNA was chromatographed on Sephadex LH-30 or Waters Clg-pBondapak column. A, dose dependene of peak I formation; B. dose dependence of peak III formation. Reprinted wi th permiss ion from Pereira et al . 149 it. 5-48 lo. .. .
\ A: l o ~ En Is lo LL - \ \ cr lo - \ 8 \ \ ZJ : ~
looo 750 500 250 : _ TA100 TABS / / so - ° 5Yl-I · BP 9- phenol 100 200 30G No ElP Molecuics / Solmonel~a Genome FIGURE 5-11. Linear relation between BP DE I, BP DE II, and 9-hydroxybenzo~a~pyrene-induced mutation frequencies at histidine locus in Salmonella typhimurium strains TA 100 and TA 98 and BP metabolite-ONA adducts in bacterial cells. Data from Fahl et al.54 5-50 - . ...
REFERENCES 1. Abbott' P. J. 3 and M. M. Coombs. ONA adducts of the carcinogen, 15,16-dihydro-11-methylcyclopentata~phenanthren-17-one, in viva and In vitro: High pressure liquid chromatographic separation and partial characterization. Carcinogenesis 2:629-636, 1981. 2. Abbott, P. J., and F. Crew. Repair of DNA adducts of the carcin- ogen 15,16-dihydro-11-methylcyclopentata~phenanthren-17-one in mouse tissues and its relation to tumor induction. Cancer Res. 41:4115-4120, 1981. 3. Adriaenssens, P. I., C. J. Bixler, and M. W. Anderson. Isolation and quantitation of DNA-bound benzo~a~pyrene metabolites: Comparison of hydroxyapatite and precipitation procedures. Anal. Biochem. 123:162-169, 1982. 4. Aitio, A. Different elimination and effect on mixed function oxidase of 20-methylcholanthrene after intagastric and intra peritoneal administration. Res. Commun. Chem. Pathol. Pharmacol. 9:701-710, 1974. Alexandrov, K., P. Brookes, H. W. S. King, M. R. Osborne, and M. H. Thompson. Comparison of the metabolism of benzotaipyrene and binding to DNA caused by rat liver nuclei and microsomes. Chem. Biol. Interact. 12:269-277, 1976. Anderson, M. W., M. Boroujerdi, and A. G. E. Wilson. Inhibition in viva of the formation of adducts between metabolites of _ benzo a~pyrene and DNA by butylated hydroxyanisole. Cancer Res. 41:4309-4315, 1981. 7. Anderson, M. W., D. G. Hoel, and N. L. Kaplan. A general scheme for~the incorporation of pharmacokinetics in low-dose risk estimation for chemical carcinogenesis: Example--vinyl chloride. Toxicol. Appl. Pharmacol. 55:154-161, 1980. 8. Anderson, M. W., C. M. White, and P. I. Adriaenssens. Effect of butylated hydroxyanisole (BHA) on dose-response for benzo~a)- pyrene (BP) metabolite-DNA adducts in mice. Proc. Amer. Assoc. Cancer Res. 23:90, abstr. no. 355, 1982. 9. Anderson, M. W., and A. G. E. Wilson. Formation and disappear- ance In viva of benzo~a~pyrene (BP)-DNA adducts in lung and liver of A/HeJ and C57BL/6J mice. Proc. Amer. Assoc. Cancer Res. 22:89, abstr. no. 351, 1981. 10. Arcos, A. C., and M. F. Argus. Chemical Induction of Cancer. Structural Bases and Biological Mechanisms. Vol. IIa. New York: Academic Press, 1974. 387 pp. 11. Ashurst, S. W., and G. M. Cohen. In vivo formation of benzo~a~pyrene dial epoxide-deoxyadenosine adducts in the skin of mice susceptible to benzo~a~pyrene-induced carcinogenesis. Int. J. Cancer 27:357-364, 1981. Ashurst, S. W., and G. M. Cohen. Magnesium ions affect the quantitative but not the qualitative microsome mediated binding of benzo~aipyrene to DNA. Chem. Bio1. Interact. 28: 279-289, 1979. 5-51 ~ ... ...
13. Autrup, H., C. C. Harris, B. F. Trump, and A. M. Jeffrey. Metabolism of benzo~a~pyrene and identification of the major benzo~a~pyrene-DNA adducts in cultured human colon. Cancer Res. 38:3689-3696, 1978. 14. Baer-Dubowska, W., and K. Alexandrov. The binding of benzota] pyrene to mouse and rat skin DNA. Cancer Lett. 13:47-52, 1981 Baird, W. M., and P. Brookes. Isolation of the hydrocarbon- deoxyribonucleoside products from the DNA of mouse embryo cells treated in culture with 7-methylbenz~aJanthracene-3H. Cancer Res. 33:2378-2385, 1973. 16. Baird, W. M., and A. Dipple. Photosensitivity of DNA-bound 7,12-dimethylbenz~aJanthracene. Int. J. Cancer 20:427-431, 1977. 17. Baird, W. M., A. Dipple, P. L. Grover, P. Sims, and P. Brookes. Studies on the formation of hydrocarbon-deoxyribonucleoside products by the binding of derivatives of 7-methylbenz~a]- anthracene to DNA in aqueous solution and in mouse embryo cells in culture. Cancer Res. 33:2386-2392, 1973. 18. Baird, W. M., R. G. Harvey, and P. Brookes. Comparison of the cellular DNA-bound products of benzo~a~pyrene with the products formed by the reaction of benzotaipyrene-4,5-oxide with DNA. Cancer Res. 35:54-57, 1975. 19. Bock, F. G., and T. L. Dao. Factors affecting the polynuclear hydrocarbon level in rat mammary glands. Cancer Res. 21: 1024-1029, 1961. 20. Bohonos, N., T. W. Chou, and R. J. Spanggord. Some observa- tions on biodegradation of pollutants in aquatic systems. Jpn. J. Antibiot. 30(Suppl. ~ :275-285, 1977. (Inc. Medicus 19: 1516, 1978) 21. Booth, J., G. R. Keysell, K. Pal, and P. Sims. The metabolism of polycyclic hydrocarbons by cultured human lymphocytes. FEBS Lett . 43: 341-344, 1974 e 22. Borgen, A., H. Darvey, N. Castagnoli, T. T. Crocker, R. E. Rasmussen, and I. Y. Wang. Metabolic conversion of benzota]- pyrene by Syrian hamster liver microsomes and binding of metabolites to deoxyribonucleic acid. J. Med. Chem. 16:502-506, 1973. 23. Boroujerdi, M., H. C. Kung, A. G. E. Wilson, and M. W. Anderson. Metabolism and DNA binding of benzo~a~pyrene ~n vivo in the rat. Cancer Res. 41:951-957, 1981. 24. Boyd, M. R. Biochemical mechanisms in pulmonary toxicity of furan derivatives, pp. 71-101. In E. Hodgson, J. R. Bend, and R. M. Philpot, Eds. Reviews in Biochemical Toxicology. Vol. 2. New York: Elsevier/North Holland, 1980. 25. Boyland, E. The biological significance of metabolism of poly- cyclic compounds. Biochem. Soc. Symp. 5:40-54, 1950. 25. Boyland, E., M. Kimura, and P. Sims. Metabolism of polycyclic compounds. 26. The hydroxylation of some aromatic hydrocarbons by the ascorbic acid model hydroxylating system and by rat-liver microsomes. Biochem. J. 92:631-638, 1964. 15. 5-52
27. Boyland, E., and P. Sims. Metabolism of polycyclic compounds. 24. The metabolism of benz~aJanthracene. Biochem. J. 91:493 506, 1964. 31. 28. Boyland, E., and P. Sims. The metabolism of benz~aJanthracene and dibenz~a,hianthracene and their 5, 6-epoxy-5, 6-dihydro derivatives by rat-liver homogenates. Biochem. J. 97:7-16, 1965. 29. Brookes, P., H. W. S. King, and M. R. Osborne. The interaction of polycyclic hydrocarbons with DNA of mammalian cells in culture, pp. 43-49. In H. V. Gelboin and P. O. P. Ts'o, Eds. Polycyclic Hydrocarbons and Cancer. Vol. 2. Molecular and Cell Biology. New York: Academic Press, 1978. 30. Brookes, P., and P. D. Lawley. Evidence for the binding of poly nuclear aromatic hydrocarbons to nucleic acid of mouse skin: Relation between carcinogenic power of hydrocarbons and their binding to deoxyribonucleic acid. Nature (London) 202:781-784, 1964. Buening, M. K., P. G. Wislocki, W. Levin, H. Yagi, D. R. Thakker, H. Akagi, M. Koreeda, D. M. Jerina, and A. H. Conney. Tumori genicity of the optical enantiomers of the diastereomeric benzota~pyrene 7,8-diol-9,10-epoxides in newborn mice: Excep t i onal activity 0 f ~ + ~ - 7 6, 8 a -d ihyd roxy-9a , 1 On- epoxy-7,8,9,10 tetrahydrobenzotaipyrene. Proc. Natl. Acad. Sci. USA 7S :5358 536 1, 19 78 . 32. Buty, S. G., S. Thompson, and T. J. Slaga. The role of epidermal aryl hydrocarbon hydroxylase in the covalent binding of polycyclic hydrocarbon to DNA and its relationship to tumor initiation. Biochem. Biophys. Res. Commun. 70:1102-1108, 1976. 33. Cerutti, P. A., F. Sessions, P. V. Hariharan, and A. Lusby. Repair of DNA damage induced by benzota~pyrene diol-epoxides I and II in human alveolar tumor cells. Cancer Res. 38:2118-2124, 1978. 34. Cerutti, P., K. Shinohara, M.-L. Ide, and J. Remsen. Formation and repair of benzotaipyrene-induced DNA damage in mammalian cells, pp. 203-212. In H. V. Gelboin and P. O. P. Ts'o, Eds. Polycyclic Hydrocarbons and Cancer. Vol. 2. Molecular and Cell Biology. New York: Academic Press, 1978. 35. Chouroulinkov, I., A. Gentil, B. Tierney, P. Grover and P. Sims. The metabolic activation of 7-methylbenz~ajanthracene in mouse skin: High tumour-initiating activity of the 3,4-dihydrodiol. Cancer Lett. 3:247-253, 1977. 36.~ Cohen, G. M., W. M. Bracken, R. P. Iyer, D. L. Berry, J. K. Selkirk, and T. J. Slaga. Anticarcinogenic effects of 2,3,7,8- tetrachlorodibenzo-p-dioxin on benzo~a~pyrene and 7,12-dimethyl- benz~aJanthracene tumor initiation and its relationship to DNA binding. Cancer Res. 39:4027-4033, 1979. 37. Colburn, N. H., and R. K. Boutwell. The binding of S-propio- lactone and some related alkylating agents to ONA, RNA, and protein of mouse skin; relation between tumor-initiating power of alkylating agents and their binding to DNA. Cancer Res. 28: 653-660, 1968. 5-53
38. Coombs, M. M., A.-M. Kissonerghis, J. A. Allen, and C. W. Vose. Identification of the proximate and ultimate forms of the carcinogen, 15,16-dihydro-11-methylcyclopentata~phenanthren-17- one. Cancer Res. 39:4160-4165, 1979. 39. Creasia, D. A., J. K. Poggenburg, Jr., and P. Nettesheim. Elution of benzota~pyrene from carbon particles in the respira- tory tract of mice. J. Toxicol. Environ. Health 1:967-975, 1976. 40. Dao, T. L., F. G. Bock, and S. Crouch. Level of 3-methylchol- anthrene in mammary glands of rats after intragastric instilla- tion of carcinogen. Proc. Soc. Exp. Biol. Med. 102:635-638, 1959. 41. Daudel, P., M. Duquesne, P. Vigny, P. L. Grover, and P. Sims. Fluorescence spectral evidence that benzo~a~pyrene-DNA products in mouse skin arise from diol-epoxides. FEBS Lett. 57:250-253, 1975. 42. Dean, A. V., S. J. Lan, K. J. Kripalani, L. T. DiFazio, and E. C. Schreiber. Metabolism of the (my-, (hi-, (-~-enantiomers of ~-methylfluorene-2-acetic acid (Cicloprofen) in rats. Xenobiotica 7:549-560, 1977. Dees, J. H., L. D. Coe,-Y. Yasukochi, and B. S. Masters. Immuno fluorescence of NADPH-cyctochrome c (P-450) reductase in rat and minipig tissues injected with phenobarbital. Science 208:1473 1475, 1980. 44. DiGiovanni, J., D. L. Berry, G. L. Gleason, G. S. Kishore, and T. J. Slaga. Time-dependent inhibition by 2,3,7,8-tetrachloro- dibenzo-p-dioxin of skin tumorigenesis with polycyclic hydro- carbons. Cancer Res. 40:1580-1587, 1980. 45. DiGiovanni, J., T. J. Slaga, D. L. Berry, and M. R. Juchau. Inhibitory effects of environmental chemicals on polycyclic aromatic hydrocarbon carcinogenesis, pp. 145-168 In T. J. Slaga, Ed. Carcinogenesis--A Comprehensive Survey. Vol. 5. Modifiers of Chemical Carcinogenesis: An Approach to the Biochemical Mechanism and Cancer Prevention. New York: Raven Press, 1980. 46. Dipple, A., and J. A. Nebzydoski. Evidence for the involvement of a diol-epoxide in the binding of 7,12-dimethylbenz~a)- anthracene to UNA in cells in culture. Chem. Biol. Interact. 20:17-26, 1978. 47. Dipple, A., and J. J. Roberts. Excision of 7-bromomethyl- benz~aJanthracene-DNA adducts in replicating mammalian cells. Biochemistry 16:1499-1503, 1977. 48. Duncan, M., P. Brookes, and A. Dipple. Metabolism and binding to cellular macromolecules of a series of hydrocarbons by mouse embryo cells in culture. Int. J. Cancer 4:813-819, 1969. 49. Eastman, A., and E. Bresnick. Metabolism and DNA binding of 3-methylcholanthrene. Cancer Res. 39:4316-4321, 1979. 50. Eastman, A., and E. Bresnick. Persistent binding of 3-methyl- cholanthrene to mouse lung DNA and its correlation with susceptibility to pulmonary neoplasia. Cancer Res. 39:2400- 2405, 1979. 43. 5-54
51. Eastman, A., J. Sweetenham, and E. Breenick. Comparison of in viva and in vitro binding of polycyclic hydrocarbons to DNA. Chem. Biol. Interact. 23:345-353, 1978. 52. Eling, T. E., J. Boyd, G. Reed, R. Mason, and K. Sivarajah. Xenobiotic metabolism by prostaglandin endoperoxide synthetase. Drug Metab. Rev. (in prese) 53. Elson, L. A., F. Goulden, and F. L. Warren. The urinary partition of sulphur in rats treated with aromatic hydrocarbons, with special reference to growth retardation. Biochem. J. 39: 301-308, 1945. 54. Fahl, W. E., D. G. Scarpelli, and K. Gill. Relationship between benzota~pyrene-induced DNA base modification and frequency of reverse mutations in mutant strains of Salmonella typhimurium Cancer Res. 41:3400-3406, 1981. 55. Feldman, G., J. Remsen, K. Shinohara, and P. Cerutti. Excisa- bility and persistence of benzo~a~pyrene DNA adducts in epithelioid human lung cells. Nature 274:796-798, 1978. 56. Feldman, G., J. Remsen, T. V. Wang, and P. Cerutti. Formation and excision of covalent deoxyribonucleic acid adducts of benzotaipyrene 4,5-epoxide and benzo~a~pyrenediol epoxide I in human lung cells A549. Biochemistry 19:1095-1101, 1980. 57. Feron, V. J., D. DeJong, and M. A. H. Rijk. Clearance of benzo~a~pyrene from the respiratory tract of hamsters following its intratracheal instillation with or without ferric oxide. Zentralbl. Bakteriol. Parasitenkd. Infektionskr. Hyg., Abt. l:Orig., Reihe B. 163:441-447, 1976. (Chem. Abs. 86:84496c, 1977) 58. Flesher, J. W. Distribution of radioactivity in the tissues of rats after oral administration of 7,12-dimethylbenz~a]- anthracene-3H. Biochem. Pharmacol. 16:1821-1831, 1967. 59. Fulmer, J. n., and R. G. Crystal. The biochemical basis of pulmonary function, pp. 419-466 . In R. G. Crystal, Ed. The Biochemical Basis of Pulmonary Function. Lung Biology in Health and Disease. Vol. 2. New York: Marcel Dekker, 1976. 60. Gamper, H., T. Meehan, K. Straub, A. S.-C. Tung, and M. Calvin. Reactions of activated benzo~a~pyrene with DNA and RNA, pp. 51- 61. In H. V. Gelboin and P. O. P. Ts to, Ed~. Polycyclic Hydrocarbons and Cancer. Vol. 2. Molecular and Cell Biology. New York: Academic Press, 1978. 61. Gehring, P. J. , and G. E. Blau. Mechanisms of carcinogenesis: Dose response. J. Environ. Pathol. Toxicol. 1:163-179, 1977. 62. Gelboin, H. V. A microsome-dependent binding of benzo~a]- pyrene to DNA, pp. 175-182. In E. D. Bergmann and B. Pullman, Ed~. Physico-Chemical Mechanismn of Carcinogenesis. Proc. Internatl. Symposium, Jeru~alem, 21-25 October 1968. The Jerusalem Sympo~ia on Quantum Chemistry and Biochemistry I. Jerusalem: The Israel Academy of Sciences and Humanities, 1969. (distributed by Academic Press, N.Y.) 63. Gelboin, H. V. Benzo~a~pyrene metabolism, activation, and carcinogenesis: Role and regulation of mixed-function oxidases and related enzymes. Physiol. Rev. 60:1107-1166, 1980. 64. Gelboin, H. V. Cancer suaceptibility and -carcinogen metabolism. New Engl. J. Med. 297:384-386, 1977. 5-55
65. Gerhart, E. H., and R. M. Carlson. Hepatic mixed-function oxidase activity in rainbow trout exposed to several polycyclic aromatic compounds. Environ. Res. 17:284-295`, 1978. 66. Gold, A. Carbon black adsorbates: Separation and identifica- tion of a carcinogen and some oxygenated polyaromatics. Anal. Chem. 47: 1469-1472, 1975. 67. Gold, A., and E. Eisenstadt. Metabolic activation of cyclo- penta~cd~pyrene to 3,4-epoxycyclopenta~cd~pyrene by rat liver microsomes. Cancer Res . 40: 3940-3944, 1980. 68. Goshman, L.~M., and C. Heidelberger. Binding of tritium labe led polycyc 1 ic hydrocarbons to DNA o f mouse skin. Cancer Res. 27: 1678-1688, 1967. 69. Grimmer, G. Analysis of automobile exhaust condensates, : pp. 29-39. In U. Mohr, D. Schmabl, and L. Tomatis, Eds. Air Pollution and Cancer in Man. IARC Scientific Publications No. 16. Lyon, France: International Agency for Research on Cancer, 1977. 70. Grover, P. L. K-region epoxides of polycyclic hydrocarbons: Formation and further metabolism by rat-lung preparations. Biochem. Pharmacol. 23:333-343, 1974. 71. Grover, P. L., A. Hewer, K. Pal, and P. Sims. The involvement of a diol-epoxide in the metabolic activation of benzo~a~pyrene in human bronchial mucosa and in mouse skin. Int. J. Cancer 18: 1-6, 1976. 72. Grover, P. L., A. Hewer, and P. Sims. Epoxides as microsomal metabolites of polycyclic hydrocarbons. FEBS Lett. 18:76-80, 1971. 73. Grover, P. L., A. Hewer, and P. Sims. Formation of K-region epoxides as microsomal metabolizes of pyrene and benzota]- pyrene. Biochem. Pharmacol. 21:2713-2726, 1972. 74. Grover, P. L., A. Hewer, and P. Sims. K-region epoxides of polycyclic hydrocarbons: Formation and further metabolism of benz~aianthracene 5,6-oxide by human lung preparations. FEBS Lett. 34:63-68, 1973. 75. Grover, P. L., A. Hewer, and P. Sims. Metabolism of polycyclic hydrocarbons by rat-lung preparations. Biochem. Pharmacol. 23:323-332, 1974. 76. Grover, P. L., and P. Sims. Enzyme-catalysed reactions of polycyclic hydrocarbons with deoxyribonucleic acid and protein ~n vitro. Biochem. J. 110 :159-160, 1968. 77. Grunberger, D., and I. B. Weinstein. Biochemical effects of the modification of nucleic acids by certain polycyclic aromatic carcinogens. Prog. Nucleic Acid Res. Mol. Biol. 23:105-149, 1979. 78. Harper, K. H. The metabolism of pyrene. Brit. J. Cancer 11:499-507, 1957. 79. Harris, C. C., A. L. Frank, C. van Haeften, D. G. Kaufman, R. Connor, F. Jackson, L. A. Barrett, E. M. McDowell, and B. F. Trump. Binding of [3H]benzo~a~pyrene to DNA in cultured human bronchus. Cancer Res. 36:1011-1018, 1976. 5-56 ~ ....
Harvey, R. G. Carcinogenic hydrocarbons: Metabolic activation and the mechanism of cancer induction, pp. 439-468. In D. B. Walters, Ed. Safe Handling of Chemical Carcinogens, Mutagens, Teratogens and Highly Toxic Substances. Vol. 2. Ann Arbor, Mich.: Ann Arbor Science Publishers, Inc., 1980. 81. Hecht, S. S., E. LaVoie, R. Mazzarese, S. Amin, V. Bedenko, and D. Hoffman. 1,2-Dihydro-1,2-dihydroxy-5-methylchrysene, major activated metabolite of the environmental carcinogen 5-methylchrysene. Cancer Res. 38:2191-2194, 1978. 82. Hecht, S. S., E. LaVoie, R. Mazzarese, N. Hirota' T. Ohmori, and D. Hoffmann. Comparative mutagenicity, tumor-initiating activity, carcinogenicity, and In vitro metabolism of fluorinated 5-methylchrysene. J. Natl. Cancer Inst. 63:855-861, 1979. 83. Hecht, S. S., M. Loy, R. Mazzarese, and D. Hoffmann. On the carcinogenicity of 5-methylchrysene: Structure-activity studies and metabolism, pp. 119-130. In H. V. Gelboin and P. O. P. Tsto, Eds. Polycyclic Hydrocarbons and Cancer. Vol. Environment, Chemistry, and Metabolism. New York: Academic Press, 1978. 84. Heidelberger, C. Chemical carcinogenesis. Ann. Rev. Biochem. 44:79-121, 1975. Henry, M. C., and D. G. Kaufman. Clearance of benzota~pyrene from hamster lungs after administration on coated particles. J. Natl. Cancer Inst. 51:1961-1964, 1973. 86. Hsu, W.-T., R. G. Harvey, E. J. S. Lin, and S. B. Weiss. A bacteriophage system for screening and study of~biologically active polycyclic aromatic hydrocarbons and related compounds. Proc. Natl. Acad. Sci. USA 74:1378-1382, 1977. 87. Hsu, W.-T., E. J. Lin, R. G. Harvey, and S. B. Weiss. Mechanism of phage {X174 DNA inactivation by benzota]- pyrene-7,8-dihydrodiol-9,10-epoxide. Proc. Natl. Acad. Sci. USA 74:3335-3339, 1977. 88. Huberman, E., and L. Sachs. ~NA binding and its relationship to carcinogenesis by different polycyclic hydrocarbons. Int. J. Cancer 19:122-127, 1977. 89. Huberman, E., L. Sachs, S. K. Yang, and H. V. Gelboin. Identification of mutagenic metabolites of benzotaipyrene in mammalian cells. Proc. Natl. Acad. Sci. USA 73: 607-611, 1976. 90. Ioannou, Y. M., A. G. E. Wilson, and M. W. Anderson. Effect of butylated hydroxyanisole ~-angelica lactone, and S-naphtho- flavone on benzo(~)pyrene: DNA adduct formation ~n vivo in the forestomach, lung, and liver of mice. Cancer Res. 42: ll99-1204, 1982. 91. Ivanovic, V., N. E. Geacintov, A. M. Jeffrey, P. P. Fu, R. G. Harvey, and I. B. Weinstein. Cell and microsome mediated binding of 7,12-dimethylbenz~ajanthracene to DNA studied by fluorescence spectroscopy. Cancer Lett. 4:131-140, 1978. 92. Janss, D. H., and T. L. Ben. Age-related modification of 7,1' dimethylbenz~aJanthracene binding to rat mammary gland DNA. J. Natl. Cancer Inst. 60: 173-177, 1978. 5-57 ~. .
93. Jeffrey, A. M., I. B. Weinstein, K. W. Jennette, K. Grzeskowiak, K. Nakanishi, R. G. Harvey, H. Autrup, and C. Harris. Structures of benzo~a~pyrene-nucleic acid adducts formed in human and bovine bronchial explants. Nature - (London) 269:348-350, 1977. 94. Jerina, D. M., and J. W. Daly. Oxidation at carbon, pp. 13-32. In D. V. Parke and R. L. Smith, Eds. Drug Metabolism--From Microbe to Man; A Symposium. London: Taylor and Francis, 1977. (Chem. Abs. 88: 16697k, 1978) 95. Jerina, D. M., R. E. Lehr, H. Yagi, O. Hernandez, P. M. Dansette, P. G. Wislocki, A. W. Wood, R. L. Chang, W. Levin, and A. H. Conney. Mutagenicity of benzotaipyrene derivatives and the description of a quantum mechanical model which pre- dicts the ease of carbonium ion formation from dial epoxides, pp. 159-177. In F. J. de Serres, J. R. Fouts, J. R. Bend, and R. M. Philpot, Eds. In Vitro Metabolic Activation in Muta . genesis Testing. New York: Elsevier/North Holland Inc., 1976. 96. Jerina, D. M., H. Yagi, R. E. Lehr, D. R. Thakker, M. Schaefer- Ridder, J. M. Karle, W. Kevin, A. W. Wood, R. L. Chang, and A. H. Conney. The bay-region theory of carcinogenesis by polycyclic aromatic hydrocarbons, ppe 173-188. In H. V. Gelboin and P. O. P. Ts'o, Eds. Polycyclic Hydrocarbons and Cancer. Vol. 1. Environment, Chemistry, and Metabolism. New York: Academic Press, 1978. 97. Jernstrom, B., S. Orrenius, O. Undeman, A. Graslund, and A. Ehrenberg. Fluorescence study of ONA-binding metabolites of benzo~a~pyrene formed in hepatocytes is~lated from 3-methyl- cholanthrene-treated rats. Cancer Res. 38:2600-2607, 1978. 98. Kakefuda, T., and H. Yamamoto. Modification of DNA by benzota~pyrene diol epoxide I, pp. 63-74. In H. V. Gelboin and P. O. P. Ts'o, Eds. Polycyclic Hydrocarbons and Cancer. Vol. 2. Molecular and Cell Biology! New York: Academic Press, 1978. 99. Kakefuda, T., and H. Yamamoto. Modification of DNA by the benzota~pyrene metabolite diol-epoxide r-7,t-8-hydroxy-9,10-oxy- 7,8,9,10-tetrahydrobenzota~pyrene. ProcO Natl. Acad. Sci. USA 75:415-419, 1978. 100. Kapitulnik, J., W. Levin, A. H. Conney, H. Yagi, and D. M. Jerina. Benzota~pyrene 7,8-dihydrodiol is more carcinogenic than benzota~pyrene in newborn mice. Nature (London) 266: 378-380 , 1977. 101. Kapitulnik, J., P. G. Wislocki, W. Levin, H. Yagi, D. M. Jerina, and A. H. Conney. Tumorigenicity studies with diol-epoxides of benzo~a~pyrene which indicate that (+~-trans-76,8~-dihydroxy-9~, 10 ~epoxy-7,8,9,10-tetrahydrobenzota~pyrene is an ultimate carcinogen in newborn mice. Cancer Res. 38:354-358, 1978. 102. Kaplan, E., and H. R. Gutmann. Microsomal metabolism of aryl- amides by the rat and guinea pig--II. Oxidation of 3-fluor- enylacetamide at carbon atom 9 formation of 3-acetamido- 9-fluorenone. Biochem. Pharmacol. 28:1609-1614, 1979. 5-58
Kellermann, G., C. R. Shaw, and M. Luyten-Kellerman. Aryl hydrocarbon hydroxylase inducibility and bronchogenic carcinoma. New Engl. J. Med. 289:934-937, 1973. 104. Keysell, G. R., J. Booth, P. L. Grover, A. Hewer, and P. Sims. The formation of "K-region" epoxides as hepatic microsomal metabolites of 7-methylbenz~aJanthracene and 7,12-dimethyl- benz~aJanthracene and their 7-hydroxymethyl derivatives. Biochem. Pharmacol. 22:2853-2867, 1973. 105. King, H. W. S., M. R. Osborne, and P. Brookes. The metabolism and TUNA binding of 3-methylcholanthrene. Int. J. Cancer 20: 564-571, 1977. 106. King, H. W. S. , M. H. Thompson, and P. Brookes. The role of 9- hydroxybenzo~a~pyrene in the microsome mediated binding of benzo~a~pyrene to DNA. Int J. Cancer 18 :339-344, 1976. 107. Koreeda, M., P. D. Moore, P. G. Wislocki, W. Levin, A. H. Conney, H. Yagi, and D. M. Jerina. Binding of benzo~a~pyrene-7,8-diol- 9,lO-epoxides to DNA, RNA and protein of mouse skin occurs with high stereoselectivity. Science 199:778-781, 1978. 108. Koreeda, M., P. D. Moore, H. Yagi, H. J. C. Yeh, and D. M. Jerina. Alkylation of polyguanylic acid at the 2-amino group and phosphate by the potent mutagen (+~-76,8a-dihydroxy-9S, 106-epaxy-7,8,9,10-tetrahydrobenzota~pyrene. J. Amer. Chem. Soc. 98:6720-6722, 1976. 109. Kotin, P., H. L. Falk and R. Busser. Distribution, retention, and elimination of C14-3,4-benzpyrene after administration to mice and rats. J. Natl. Cancer Inst. 23:541-555, 1959. 110. Kouri, R. E. Genetic Differences in Chemical Carcinogenesis. Boca Raton, Fla.: CRC Press, 1980. 223 pp. 111. Kouri, R. E., G. Connolly, D. W. Nebert, and R. A. Lubet. The metabolism of dibenz~a,h~anthracene (DBA) by mouse hepatic microsomes and its relationship to DBA-induced tumor formation. Proc. Amer. Assoc. Cancer Res. 23:86, abstr. No. 337, 1982. 112. Lan, S. J., A. V. Dean, K. J. Kripalani, and A. I. Cohen. Metabolism of a-methylfluorene-2-acetic acid (Cicloprofen): Isolation and identification of metabolites from rat urine. Xenobiotica 8:121-131, 1978. 113. Lawley, P. D. Approaches to chemical dosimetry in mutagenesis and carcinogenesis: The relevance of reactions of chemical mutagens and carcinogens with DNA, pp. 1-36. In P. L. Grover, Ed. Chemical Carcinogens and DNA. Vol. 1. Boca Raton, Fla.: CRC Press, Inc., 1979. 114. Leffler, S., P. Pulkrabek, D. Grunberger, and I. B. Weinstein. Template activity of calf thymus ONA modified by a dihydrodiol epoxide derivative of benzota~pyrene. Biochemistry 16:3133-3136, 1977. 115. Lehr, R. E., H. Yagi, D. R. Thakker, W. Levin; A. W. Wood, A. H. Conney, and D. M. Jerina. The bay region theory of polycyclic aromatic hydrocarbon-induced carcinogenicity, pp. 231-241. In P. W. Jones and R. I. Freudenthal, Eds. Polynuclear Aromatic Hydro- carbons: Analysis, Chemistry, and Biology. Vol. 3. New York: Raven Press, 1978. 5-59 ~. ... .
116 . Levin, W., A. W. Wood, R. L. Chang, H. Yagi, H. D. Mah, D. M. Jerina, and A. H. Conney. Evidence for bay region ac t ivat ion of chrysene 1,2-dihydrodiol to an ultimate carcinogen. Cancer Res . 38: 1831-1834, 1978. 117. Levin, W., A. W. Wood, P. G. Wislocki, R. L. Chang, J. Kapitulnik, H. D. Mah , H. Yagi , D. M. Jerina, and A. H. Conney. Mutagenicity and carcinogenicity of benzota~pyrene and benzota]- pyrene derivatives, pp. 189-202. In H. V. Gelboin and P. 0. P. Ts'o, Eds. Polycyclic Hydrocarbons and Cancer. Vol. 1. Environment, Chemistry, and Metabolism. New York: Academic Press, 1978. 118. Lewis, J. G., and J. A. Swenberg. Differential repair of o6 mecny~guantne In ONA of rat hepatocytes and nonparenchymal cells. Nature (London) 288:185-187, 1980. 119. Lewis, J. G., and J. A. Swenberg. Effect of 1,2-dimethyl- hydrazine and diethylnitrosamine on cell replication and unscheduled DNA synthesis in target and nontarget cell popula- tions in rat liver following chronic administration. Cancer Res. 42:89-92, 1982. 120. Liebermann, C. Ueber Chrysen. Justus Liebigs Ann. Chem. 158: 299-315, 1871. 121. Loveless, A. Poss~ble relevance of 0-6 alkylation of deoxy guanosine to the mutagenicity and carcinogenicity of nitro- samines and nitrosamides. Nature 223:206-207, 1969. 122. MacNicoll, A. D., P. M. Burden, H. Rattle, P. L. Grover, and P. Sims. The formation of dihydrodiols in the chemical or enzymatic oxidation of dibenz~a,c~anthracene, dibenz~a,b]- anthracene and chrysene. Chem. Biol. Interact. 27:365-379, 1979. 123. MacNicoll, A.D., P. L. Grover, and P. Sims. The metabolism of a series of polycyclic hydrocarbons by mouse skin maintained in short-term organ culture. Chem. Biol. Interact. 29:169-188, 1980. 124. Margison, G. P., and P. J. O'Connor. Nucleic acid modifications by N-nitroso compounds, pp. 111-159. In P. L. Grover, Ed. Chemical Carcinogens and DNA. Vol. 1. Boca Raton, Fla.: CRC Press, Inc., 1979. 125. Marnett, L. J. Polycyclic aromatic hydrocarbon oxidation during prostaglandin biosynthesis. Life Sci. 29:531-546, 1979. 126. Marquardt, H., S. Baker, P. L. Grover, and P. Sims. Malignant transformation and mutagenesis in mammalian cells induced by vicinal diol-epoxides derived from benzota~pyrene. Cancer Lett. 3: 31-36, 1977. 127. Marquardt, H., S. Baker, B. Tierney, P. L. Grover, and P. Sims. The metabolic activation of 7-methylbenz (aJanthracene: The i nduc t ion 0 f mal ignant bans forma t ion and mu ta t ion in ma~a 1 fan cells by non-K-region dihydrodiols. Int. J. Cancer 19:828-833, 1977. 128. McCann, J., and B. N. Ames. Detection of mutagens in the Salmonella/microsome test: icals; Discussion. Proc. Natl. Acad. Sci. 5-60 carcLnogens as Assay of 300 chem USA 73:950-954, 1976. ~ . .. .
129. McCormick, J. J., and V. M. Maher. Effect of DNA repair on the cytotoxicity and mutagenicity of polycyclic hydrocarbon metabolites in human cells, pp. 221-232. In H. V. Gelboin and P. O. P. Ts'o, Eds. Polycyclic Hydrocarbons and Cancer. Vol. 2. Molecular and Cell Biology. New York: Academic Press, 1978. 130. Meehan, T., K. Straub, and M. Calvin. Benzotaipyrene dial epoxide covalently binds to deoxyguanosine and deoxyadenosine in DNA. Nature (London) 269:725-727, 1977. Mizusawa, H., and T. Kakefuda. Inhibition of ONA synthesis in vitro by binding of benzo~a~pyrene metabolite diol-epoxide I to DNA. Nature 279:75-77, 1979. 132. Moschel, R. C., W M. Baird, and A. Dipple. Metabolic activation of the carcinogen 7,12-dimethylbenz~aJanthracene for DNA bind- ing. Biochem. Biophys. Res. Commun. 76:1092-1098, 1977. 133. Nebert, D. W., S. A. Atlas, T. M. Guenthner, and R. E. Kouri. The _ locus: Genetic regulation of the enzymes which metabolize polycyclic hydrocarbons and the risk for cancer, pp. 345-390. In H. V. Gelboin and P. O. P. Ts'o, Eds. Polycyclic Hydrocarbons and Cancer. Vol. 2. Molecular and Cell Biology. New York: Academic Press, 1978. 134. Nebert, D. W., H. J. Eisen, M. Negishi, M. A. Lang, L. M. Hjelmeland, and A. B. Okey. Genetic mechanisms controlling the induction of polysubstrate monooxygenase (P-450) activities. Ann. Rev. Pharmacol. Toxicol. 21:431-462, 1981. 135. Nebert, D. W., and N. M. Jensen. The Ah locus: Genetic regula- tion of the metabolism of carcinogens, drugs, and other environ- mental chemicals by cytochrome P-450-mediated monooxygenases. CRC Crit. Rev. Biochem. 6:401-437, 1979. 136. Newbold, R. F., and P. Brookes. Exceptional mutagenicity of a benzota~pyrene diol epoxide in cultured mammalian cells. Nature (London) 261:52-54, 1976. 137. Newbold, Re F., P. Brookes, and R. G. Harvey. A quantitative comparison of the ~utagenicity of carcinogenic polycyclic hydro- carbon derivatives in cultured mammalian cells. Int. J. Cancer 24: 203-209, 1979. 138. Newbold, R. F., C. B. Wigley, M. H. Thompson, and P. Brookes. Cell-mediated mutagenesis in cultured Chinese hamster cells by carcinogenic polycyclic bydrocarbons : Nature and extent of the associated hydrocarbon-DNA reaction. Mutat. Res. 43:101-116, 1977. 139. Nordqvist, M., D. R. Thakker, K. P. Vyas, H. Yagi, W. Levin, D. E. Ryan, P. E. Thomas, A. H. Conney, and D. M. Jerina. Metabolism of chrysene and phenanthrene to bay-region diol epoxides by rat liver enzymes. Mol. Pharmacol. 19:168-178, 1981. 140. Osborne, M. R., R. G. Harvey, and P. Brookes. The reaction of trans-7,8-dihydroxy-anti-9,10-epoxy-7,8,9, 10-tetrahydrobenzo~a)- pyrene with UNA involves attack at the N/-position of guanine moieties. Chem. Biol. Interact. 20:123-130, 1978. 141. Osborne, M. R., M. H. Thompson, H. W. S. King, and P. Brookes. Retention of tritium during the binding of tritiated benzo~a)- pyrene to DNA. Int. J. Cancer 16:659-664, 1975. ~;
151. 142. Owens, I. S., and Nebert, D. W. Genetically-mediated UBPG trans- ferase induction by polycyclic aromatic compounds associated with the "Ah locus" in the mouse. Pharmacologist 17:217, abstr. no. 228, 1975. 143. Paigen, B., H. L. Gurtoo, E. Ward, J. Minowada, L. Houten, R. Vincent, N. B. Parker, and J. Vaught . Human aryl hydrocarbon bydroxylase and cancer risk, pp. 429-438. In P. W. Jones and R. I. Freudenthal, Eds. Carcinogenesis--A Comprehensive Survey. Vol. 3. Polynuclear Aromatic Hydrocarbons: Second International Symposium on Analysis, Chemistry, and Biology. New York: Raven Press, 1978. 144. Pegg, A. E. Formation and metabolism of alkylated nucleosides: Possible role in carcinogenesi~ by nitroso compounds and alkylat- ing agents. Adv. Cancer Res. 25:195-269, 1977. 145. Pelfrene, A. F. Experimental evaluation of the clearance of 3,4-benzo~a~pyrene in association with talc from hamster lungs. Amer. Ind. Hyg. Assoc. J. 37:706-710, 1976. 146. Pelkonen, O., A. R. Boobis, R. C. Levitt, R. E. Kouri, and D. W. Nebert. Genetic differences in the metabolic activation of benzota~pyrene in mire. Attempts to correlate tumorigenesis with binding of reactive intermediates to UNA and with mutagenesis In vitro. Pharmacology 18:281-293, 1979. 147. Pelkonen, O., A. R. Boobis, and D. W. Nebert. Genetic differ- ences in the binding of reactive carcinogenic metabolites to DNA, pp. 383-400. In P. W. Jones and R. I. Freudenthal, Eds. Carcinogenesis--A Comprehensive Survey. Vol. 3. Polynuclear Aromatic Hydrocarbons: Second International Symposium on Analysis, Chemistry, and Biology. New York: Raven Press, 1978. 148. Pelkonen, O., A. R. Boobis, H. Yagi, n. M. Jerina, and D. W. Nebert. Tentative identification of benzota~pyrene metabolite- nucleoside complexes produced ~n vitro by mouse liver micro- somes. Mol. Pharmacol. 14:306-322, 1978. 149. Pereira, M. A., F. J. Burns, and R. E. Albert. Dose response for benzo~a~pyrene adducts in mouse epidermal DNA. Cancer Res. 39:2556-2559, 1979. 150. Phillips, D. H., P. L. Grover, and P. Sims. The covalent binding of polycyclic hydrocarbons to ONA in the skin of mice of different strains. Int. J. Cancer 22:487-494, 1978. Phillips, D. H., and P. Sims. Polycyclic aromatic hydrocarbon metabolites: Their reactions with nucleic acids, pp. 29-57. In P. L. Grover, Ed. Chemical Carcinogens and DNA. Vol. II. Boca Raton, Fla.: CRC Press, Inc., 1979. 152. Pulkrabek, P., S. Leffler, D. Grunberger, and I. B. Weinstein. Modification of deoxyribonucleic acid by a diol epoxide of benzota] pyrene. Relation to deoxyribonucleic acid structure and conforma t ion and e f fe~ t s on bans fec t tonal ac t ivity . Biochemistry 18: 5128-5134, 1979. 153. Pullman, A., and B. Pullman. Cancerisation par les Substances Chimiques et Structure Moleculaire. Paris: Masson & Cie, 1955. 306 pp. 5-62 ~. .. .
154. Pyerin, W. G., H. A. Oberender, and E. Hecker. Extent of skin tumour initiation in mice by 7,12-dimethylbenz~aJanthracene and induction of arylhydrocarbon monooxygenase are not causally related. Cancer Lett. 10:155-162, 1980. 155. Pylev, L. N., F. J. C. Roe, and G. P. Warwick. Elimination of radioactivity after intratracheal instillation of tritiated 3,4-benzopyrene in hamsters. Brit. J. Cancer 23:103-115, 1969. 156. Rayman, M. P., and A. Dipple. Structure and activity in chemical carcinogenesis. Comparison of the reactions of 7-bromomethyl- benz~aJanthracene and 7-bromomethyl-12-methylbenz~aJanthracene with mouse skin deoxyribonucleic acid in vivo. Biochemistry 12: 1538-1542, 1973. 157. Rees, E. D., P. Mandelstam, J. Q. Lowry, and H. Lipscomb. A study of the mechanism of intestinal absorption of benzota]- pyrene. Biochim. Biophys. Acta 225:96-107, 1971. 158. Reznik-Schuller, H., and G. Reznik. Experimental pulmonary carcinogenesis. Int. Rev. Exp. Pathol. 20:211-281, 1979. 159. Santella, R. M., D. Grunberger, and I. B. Weinstein. DNA- benzotaipyrene adducts formed in a Salmonella typhimurium mutagenesis assay system. Mutat. Res. 61:181-189, 1979. 160. Santodonato, J., P. Howard, and D. Basu. Multimedia Health Assessment Document for Polycyclic Organic Matter. Report EPA-600/9-79-008. Prepared for U.S. Environmental Protection Agency. Syracuse, N.Y.: Center for Chemical Hazard Assessment, Syracuse Research Corp., 1979. 537 pp. 161. Schlede, E., R. Kuntzman, and A. H. Conney. Stimulatory effect of benzo~a~pyrene and phenobarbital pretreatment-on the biliary excretion of benzo~a~pyrene metabolites in the rat. Cancer Res. 30:2898-2904, 1970. 162. Schlede, E., R. Kuntzman, S. Haber, and A. H. Conney. Effect of enzyme induction on the metabolism and tissue distribution of benzo~a~pyrene. Cancer Res. 30:2893-2897, 1970. 163. Selkirk, J. K., E. Hube`~an, and C. Heidelberger. An epoxide is an intermediate in the microsomal metabolism of the chemical carcinogen, dibenz~a,h~anthracene. Biochem. Biophys. Res. Commun. 43:1010-1016, 1971. 164. Sellakumar, A., F. Stenback, and J. Rowland. Effects of different dusts on respiratory carcinogenesis in hamsters induced by benzo~a~pyrene and diethyloitrosamine. Eur. J. Cancer 12: 313-319, 1976. 165. Serabjit-Singh, C. J., T. R. Devereux, J. R. Fouts, R. M. Philpot, and C. G. Plopper. Rabbit pulmonary monooxygenase enzymes in tissue sections and in isolated cell fractions, pp. 451-454. In J. A. Gustafsson, J. Carlstedt-Duke, A. Mode, and J. Rafter, Eds. Biochemistry, Biophysics and Regulation of Cytochrome P-450. Developments in Biochemistry, Vol. 13. New York: Elsevier/North Holland Biomedical Press, 1980. 166. Serabjit-Singh, C. J., C. R. Wolf, R. M. Philpot, and C. G. Plopper. Cytochrome p-450: Localization in rabbit lung. Science 207:1469-1470, 1980. 5-63
167. Shinohara, K., and P. A. Cerutti. Excision repair of benzota]- pyrene-deoxyguanosine adducts in baby hamster kidney 21/C13 cells and in secondary mouse embryo fibroblasts C57BL/6J. Proc. Natl. Acad. Sci. USA 74:979-983, 1977. 168. Sims, P. Epoxides as reactive intermediates in aromatic hydro- carbon metabolism. Biochem. Soc. Trans. 3:59-62, 1975. 169. Sims, P. Qualitative and quantitative studies on the metabolism of a series of aromatic hydrocarbons by rat-liver preparations. Biochem. Pharmacol. 19:795-818, 1970. 170. Sims, P., and P. Grover. Epoxides in polycyclic aromatic hydro- carbon metabolism and carcinogenesis. Adv. Cancer Res. 20: 165-274, 1974. 171. Sims, P., P. L. Grover, T. Kuroki, E. Huberman, H. Marquardt, J. K. Selkirk, and C. Heidelberger. The metabolism of benz~a]- anthracene and dibenz~a,h~anthracene and their related "K-region" epoxides, cis-dihydrodiols and phenols by hamster embryo cells. Biochem. Pharmacol. 22:1-8, 1973. 172. Sims, P., P. L. Grover, A. Swaisland, K. Pal, and A. Hewer. Metabolic activation of benzo~a~pyrene proceeds by a diol- epoxide. Nature (London) 252:326-328, 1974. ' 173. Slaga, T. J., W. J. Bracken, G. Gleason, W. Levin, H. Yagi, D. M. Jerina, and A. H. Conney. Marked differences in the skin tumor-initiating activities of the optical enantiomers of the diastereomeric benzota~pyrene 7,8-diol-9,10-epoxides. Cancer Res. 39:67-71, 1979. 174. Slaga, T. J., E. Huberman, J. K. Selkirk, R. G. Harvey, and W. - M. Bracken. Carcinogenicity and mutagenicity of benz~a)- anthracene diols and diol-epoxides. Cancer Res. 38:1699-1704, 1978. 175. Straub, K. M., T. Meehan, A. L. Burlingame, and M. Calvin. Identification of the major adducts formed by reaction of benzo~a~pyrene diol epoxide with DNA ~n vitro. Proc. Natl. Acad. Sci. USA 74:5285-5289, 1977. 176. Swann, P. F., D. G. Kaufman, P. N. Magee, and R. Mace. Induc- tion of kidney tumours by a single dose of dimethylnitrosamine: Dose response and influence of diet and benzo~a~pyrene pretreat- ment. Brit. J. Cancer 41:285-294, 1980. 177. Thakker, D. R., W. Levin, H. Yagi, D. Ryan, P. E. Thomas, J. M. Karle, R. E. Lehr, D. M. Jerina, and A. H. Conney. Metabolism of benzotajanthracene to its tumorigenic 3,4-dihydrodiol. Mol. Pharmacol. 15:138-153, 1979. 178. Trell, E., R. Korsgaard, B. Hood, P. Kitzing, G. Norden, and B. G. Simonsson. Aryl hydrocarbon hydroxylase inducibility and laryngeal carcinomas. Lancet 2:140, 1976. (letter) 179. Vigny, P., M. Duquesne, H. Coulomb, B. Tierney, P. L. Grover, and P. Sims. Fluorescence spectral studies on the metabolic activation of 3-methylcholanthrene and 7,12-dimethylbenz~a]- anthracene in mouse skin. FEBS Lett. 82:278-282, 1977. 180. Wattenberg, L. W. Inhibitors of carcinogenesis, pp. 299-316. In A. C. Griffin and C. R. Shaw, Eds. Carcinogens: Identification and Mechanisms of Action. Symposium on Fundamental Cancer Research, 31st, M.D. Anderson Hospital and Tumor Institute, Houston, Texas, 1978. New York: Raven Press, 1979. 5-6-4 ~. .. ..
181. Wattenberg, L. W., and J. L. Leong. Inhibition of the carcino- genic action of benzo~a~pyrene by flavones. Cancer Res. 30: 1922-1925, 1970. 182. Weinstein, I. B., A. M. Jeffrey, S. Leffler, P. Pulkrabek, H. Yamasaki, and D. Grunberger. Interactions between polycyclic aromatic hydrocarbons and cellular macromolecules, pp. 3-36. In H. V. Gelboin and P. O. P. Ts'o, Eds. Polycyclic Hydrocarbons and Cancer. Vol. 2. Molecular and Cell Biology. New York: Academic Press, 1978. 183. Wigley, C. B., R. F. Newbold, J. Amos, and P. Brookes. Cell mediated mutagenesis in cultured Chinese hamster cells by poly- cyclic hydrocarbons: Mutagenic ity and DNA react ion related to carcinogenicity in a series of compounds. Int. J. Cancer 23: 691-696, 1979. 184. Wilson, A. G. E ., 11. C . Kung, M. Boroujerdi , and M. W. Anderson. Inhibition in viva of the formation of adducts between - metabolites of benzota~pyrene and DNA by aryl hydrocarbon hydroxylase inducers. Cancer Res. 41:3453-3460, 1981. 185. Wislocki, P. G., A. W. Wood, R. L. Chang, W. Levin, H. Yagi, 0. Hernandez, D. M. Jerina, and A. H. Conney. High mutagenicity and toxicity of a dial epoxide derived from benzota~pyrene. Biochem. Biophys. Res. Commun. 68:1006-1012, 1976. 186. Wood, A. W., R. L. Chang, W. Levin, R. E. Lehr, M. Schaefer- Ridder, J. M. Karle, D. M. Jerina, and A. H. Conney. Mutagenicity and cytotoxicity of benz~ajanthracene diol epoxides and tetrahydroepoxides: Exceptional activity of the bay region 1, 2-epoxides. Proc. Natl . Acad. Sci. USA 74: 2746- 2750, 1977. 187. Wood, A. W., W. Levin, R. L. Chang, R. E. Lehr, M. Schaefer- Ridder, J. M. Karle, D. M. Jerina, and A. H. Conney. Tumori- genicity of f ive dihydrodiols of benz (a~anthracene on mouse skin: Exceptional activity of benz (a~anthracene 3 ,4-dihydro- diol. Proc. Natl. Acad. Sci. USA 74: 3176-3179, 1977. 188. Wood, A. W., W. Levin, A. Y. H. Lu, D. Ryan, S. B. West, R. E. Lehr, M. Schaefer-Ridder, D. M. Jerina, and A. }I. Conney. Mutagenicity of metabolically activated benzo [a] anthracene 3 ,4- d;hydrodiol: Evidence for bay region activation of carcinogenic polycyc kc hydrocarbons . Biochem. Biophys . Res . Commun. 72: 680-686, 1976. 189. Wood, A. W., W. Levin, D. Ryan, P. E. Thomas, H. Yagi, H. D. Mah, D. R. Thakker, D. M. Jerina, and A. H. Conney. High mutagenicity of metabolically activated chrysene 1, 2-dibydro- diol: Evidence for bay region activation of chrysene. Biochem. Biophys. Res. Commun. 78:847-854, 1977. 190. Wood, A. W., W. Levin, P. E. Thomas, D. Ryan, J. M. Karle, H. Yagi, D. M. Jerina, and A. H. Conney. Metabolic activation of dibenzo~a,6)anthracene and its dihydrodiols to bacterial mutagens. Cancer Res. 38: L967-1973, 1978. 191. Yamagawa, R., and K. Ichikawa. Experimentelle Studie uber die Pathogenese der Epithelialgeschwulste. Mitt. a.d. med. Fakult. d. k. Univ. zu Tokyo 15:295-344, 1915-1916. (Inc. Medicus 14:294, 1916) 5-65
192. Yamaura, I., B. H. Rosenberg, and L. F. Cavalieri. The major adducts of cis and bans benzo [a] pyrene diol epoxides cause chain termination during DNA synthesis in vitro. Chem. Biol. Interact. 37: 171-180, 1981. 193. Yang, L. L., V. M. Maher, and J. J. McCormick. Error-free excision of the cytotoxic and mutagenic N2-deoxyguanosine DNA adduct formed in human fibroblasts by (+~-7S,8a-dihydroxy- 9~,10~-epoxy-7 ,-8 ,19, 10-tetrahydrobenzo [a] pyrene. Proc. Natl. Acad. Sci. USA 77: 5933-5937, 1980. 194. Yang, S. K., P. P. Roller, and H. V. Gelboin. Benzo[a]pyrene metabolism: Mechanism in the formation of epoxides, phenols, dihydrodiols, and the 7,8-diol-9,10-epoxides, pp. 285-301. In P. W. Jones and R. I. Freudenthal, Eds. Carcinogenesis--A Comprehensive Survey. Vol. 3. Polynuclear Aromatic Hydro- carbons: Second International Symposium on Analysis, Chemistry and Biology. New York: Raven Press, 1978. 5-66 =. .. .. ..
6 POLYCYCLIC AROMATIC HYDROCARBONS IN FOOD AND WATER AND THEIR METABOLISM BY HUMAN TISSUES This chapter deals with the relation of PAHs to human metabolism. Specifically, its purposes are to collate a large volume of literature dealing with the capacities of a number of human tissues to interact with and biotransform selected PAHs; to define, where possible, the effects of these compounds on human tissues; and to examine the principal sources of human exposure to PAHs through food and water. PAR METABOLISM BY HUMAN TISSUES The abilities of various human tissues to metabolize PAHs have been extensively studied, with emphasis on the chemical biotransformations that are catalyzed by tissues that can be readily sampled (such as blood cells, skin, and placenta) or that can be biopsied or cultured (such as fibroblasts, liver, and intestinal and tracheohronchial epithelium). The chemical biotransformations of selected PAHs that such tissues carry out are in general qualitatively similar to those demonstrated in animal tissues, although there are considerable species and organ differences in catalytic activities of relevant enzymes. These differences may be great enough to preclude comparative generalizations; and for the most part the relation between in vitro and in vivo enzymatic activities is unclear. Moreover, it is apparent from the findings reviewed in this chapter that the enzymatic capacity to biotransform PAHs to ultimate carcinogens in various tissues is not necessarily correlated with the demonstrated ability of PAHs to produce cancers in those tissues. SKIN That benzota]pyrene hydroxylase can be induced in cultured human skin was first demonstrated in l972.1° Foreskins from children who were circumcised 2-4 d after birth were shown to contain an enzyme that bydroxylates the carcinogen benzo~aipyrene (BaP), and induction of the enzyme (by a factor of 2-5) was demonstrated when the foreskins were cultured for 16 ~ in the presence of 10 AM henz~ajanthracene. Among a group of 13 skin samples studied, control enzymatic activities extended over a threefold range and were not correlated with race, age of mother, or medications given to mother or infant.2 The enzyme had an absolute requirement for NADER and molecular oxygen and was completely inhibited by CO; these findings suggested the involvement of a species of cytochrome P-450 in the bydroxylation reaction. The presence of this heme protein in low concentrations in cutaneous tissue was later 6-1 At; . ~. ... .
demonstrated by Bickers et al.22 Coal-tar products, which are widely used in dermatologic practice in conjunction with exposure to ultraviolet light (~.g., the Goeckerman regimen for psoriasis62963), were also shown to induce aryI hydrocarbon hydroxylase (AHH) signifi- cantly in patients with dermatologic disease where the coal tar was applied, but not in skin distant from the site of application.21 In concurrent studies in neonatal rats,21 although (as in humans) distant skin sites were unaffected by the coal-tar application, AHH activity in the livers of treated animals increased to more than 20 times the c,ytto] values. Among five identifiable constituents of coal tar studied for their AHH-inducing properties in human skin, BaP was the most potent; pyrene and anthracene also caused significant induction of the enzyme. In isolated cultured human hair follicles, Vermorken _ al.,185 using radioactive BaP as substrate, not only demonstrated the presence of the hydroxylase, but identified the formation of the 3-OH, 7,8-dihydro-7,8-dihydroxy, and 9,10-dihydro- 9,10-dihydroxy metabolites of this PAH. BaP clearly has cytotoxic effects on cultured human skin fibroblasts, although relatively high concentrations are required for cytotoxicity. Milo et al.123 studied the influence of the three carcinogenic PAHs, 7,12-dimethylbenzanthracene (7,12-DMBA), 3-methyl- cholanthrene (3-MC), and BaP on mixed-function oxidase activity, cell proliferation kinetics, and DNA damage in cultured fibroblasts. They found that only BaP, at 10 ~g/ml or higher, affected all the cellular metabolic characteristics examined. 7,12-DMBA at 6 ~g/ml or higher induced the mixed-function oxidase system and stimulated DNA synthesis; 3-MC at concentrations as high as l5 ~g/ml produced no significant cellular alterations. Similarly, 5-fluoro-7,12-DMBA, anthracene, and phenanthrene had no effects on these buman cells. The authors concluded that BaP alone could initiate all the biochemical events probably necessary to trigger transformation of human cells in v~tro. PAH-induced cytotoxicity to cultured human fibroblasts has also been demonstrated by Strniste and Brakel72 and Aust et al.8 In the former study, normal fibrohlasts and xeroderma pigmentosum (XP) cells were used, and BaP was "activated" by light radiation (near ultraviolet), rather than enzymes. Photoactivation (at 300-400 nm) of BaP produced at least three identifiable quinones (1,6-, 3,6-, and 6,12- isomers), as well as more hydrophilic products, depending on the duration of light exposure. Formation of these products was oxygen-dependent. The irradiation products led to several types of ONA *"Mixed-function oxidase" refers to the NADPH-dependent enzyme complex containing cytochrome P-450s in the membranes of the endoplasmic reticulum, which catalyzes the oxidation of numerous structurally diverse molecules, including drugs, steroid hormones, and carcinogens. 6-2
damage, with covalently bound hydrocarbons constituting the major lesion under all conditions studied. XP cells were more sensitive to damage than normal cells (by a factor of 1.7-2), and sensitivity increased by a factor of 10 when long-wavelength ultraviolet light was used. 7,12-D~BA, 3-MC, and BaP were also examined; the order of phototoxicity was 7,12-DMBA > BaP > benzote~pyrene > 3-MC. In the study of Aust et al.,8 a human epithelial cell-mediated cytotoxicity and mutagenicity assay system for BaP was developed with human fibroblasts as the target cells. Lethally x-irradiated human kidney-carcinoma epithelial cells were cocultivated with human XP skin fibroblasts (XP12BE) lacking excision-repair capability for BaP-DNA adducts. Under defined conditions, the frequency of mutation to 6-thioguanine resistance and PAH binding to DNA were shown to be concentration-dependent. Two principal BaP-DNA adduct peaks could be identified--a major peak consistent with an adduct standard synthesized from the anti-isomeric 7,8-dibydrodiol-9,10-epoxide of the hydrocarbon and a minor peak consistent with the syn-isomeric form of this metabolize. The results are consistent with those in other reports on BaP adducts formed in human explant tissue from lung,161 colon,l° esophagus,68 and bronchus,75 and they represent an advance in the development of sensitive assay systems for detecting biologic responses to human epithelial-cell activation of BaP. Direct neoplastic transformation of human fibroblasts by carcinogens has also' been demonstrated. In the study of Kakunaga,79 normal human adult fibroblasts exposed to the carcinogen 4-nitro- quinoline' 1-oxide underwent malignant transformation In a process requiring numerous cell divisions. When injected into athymic (nude) mice, the transformed cells produced solid tumors at the site of inoculation. Because it could not be metabolically activated by the target cells used, 3-MC was unable to effect transformation; the use of other PAHs and induced microsomes with high concentrations of cytochrome P-450 to activate 3-MC was not examined. Normal human foreskin cell populations were neoplastically transformed in studies by Milo and DiPaolo 24~125 with a number of non-PAH carcinogens; and treatment with a tumor promoter alone (phorbol ester) has been shown90 to induce neoplas tic transformation' in fibroblasts from humans genetically predisposed to cancer (familial polyposis of the colon). Thus' it can be inferred that cells already in an "initiated state" as a result of a genetic defect represent a novel fibroblast system that may provide a means for exploring separately the roles of initiators and promoters in carcinogenesis. Painterl35 used HeLa cells to develop a rapid screening test to detect agents that damage human DNA. The test measures thymidine uptake into the cells at various times (principally 1-2 h) after treatment with a presumptive carcinogen or mutagen. In this test system, BaP was inert unless metabolically activated by incubation with rat-liver microsomes. Brookes and Duncan26 compared the effects of PAHs on primary human embryo cells and HeLa cells. Fibroblasts from skin, lung, muscle, and gut were cultured and treated with 3H-labeled BaP and 7,12-DMBA. 6-3
Both hydrocarbons were metabolized in the cultures, 7,12-DMBA more slowly than BaP; among the cell types studied, lung fibroblasts metabolized the compounds more efficiently than others and retained this capacity well in subculture. The binding of BaP and 7,12-DMBA to DNA, RNA, and protein of these primary lung-derived fibroblasts was also studied (metabolism of each hydrocarbon exceeded 75% during the 48-96 h of treatment) and was found to be significantly greater for BaP than for 7,12-DMBA. The data in this study also established parallelisms, at least for BaP, between hydrocarbon binding to cellular macromolecules in fibroblast cultures derived from mouse embryos and those derived from human lung cells. Such parallelism is of more than casual interest, in view of the susceptibility of the mouse to hydrocarbon carcinogenesis and the known correlation between hydrocarbon-DNA binding and cancer-producing activity in mouse skin. The effects of pyrene and BaP in the human diploid fibroblast culture WI-38 were studied by Weinstein et al.188 Neither caused significant damage (compared with controls), as assessed by mitotic index or chromosomal breaks after 1-h pulse exposures. However, metabolic activation of BaP with microsomes resulted in a dramatic decrease in mitotic index and a significant increase in breakage. Microsomal incubation did not alter the inertness of pyrene in this test system. Freeman et al.60 have made interesting observations on comparative aspects of hydrocarbon metabolism in skin epithelial and fibroblast cultures. A comparison of the ability of epithelial-cell colonies and of fibroblast colonies from the same 13 subjects to metabolize BaP to a water-soluble form demonstrated clearly the markedly greater metabolizing capacity of epithelial cells. There was a 20-fold difference in this capacity of epithelial cells; within individual subjects, the ability of epithelial cells to metabolize the PAN exceeded that of fibroblasts by as much as a factor of 40. There appeared to be a major effect of culture age (6-55 d) on the ability of epithelial cells to metabolize BaP. A direct toxicity of BaP to normal human epithelial-cell cultures has been described by Dietz and Flaxman.5O This toxicity was reflected in a dramatic reduction in epidermal-cell outgrowth, a decrease in mitotic indexes, a loss of the well-ordered cell relationships, and the early appearance of giant cells ranging in diameter from 100 to 200 ym. In a clinical study that clearly could not be carried out today, Cottini and Mazzone45 (in 1939) applied a 1% solution of BaP daily (up to 120 d) to the skin of 26 normal subjects and patients with various dermatologic disorders and examined the gross and histologic consequences. The sequential epidermal changes, of which gross pigmentation and verrucae were the most frequent, and histologic alterations (which regressed within several months when treatment was terminated) led the authors to conclude that "benzopyrene, if applied to human skin for protracted periods, would be' carcinogenic as it is in animals." 6-4 .. .. . .
The metabolism of benz~ajanthracene, 7,12-DMBA, and BaP by human mammary epithelial-cell aggregates in culture has been investigated by Grover et al.66 with nonneoplastic tissue obtained from eight patients undergoing reduction mammoplasty. All three PAHs were metabolized to water-soluble and organic-solvent-soluble products; the latter included K-region and non-K-region dihydrodiols. The major dihydrodiols detected as metabolites of the parent PAHs were the 8,9-dihydrodiols of BaP and 7,12-DMBA and the 9,10-dihydrodiol of BaP. The hydrocarbons bound to the proteins and DNA of the epithelial cells, but there were wide differences between different PAHs in extent of binding between tissue preparations from different patients. Some of the PAH-deoxyribonucleoside adducts formed from 7,12-DMBA and BaP appeared to have been produced through reactions of bay-region diol-epoxides with DNA, but little reaction with DNA was detected in tissue preparations treated with BaP. Unscheduled DNA synthesis induced by DNA-damaging chemicals has been measured in nonreplicating human fibroblasts by autoradiographic methods that are not readily applicable to organotypic epithelial-cell cultures. To evaluate the range of chemical sensitivity and DNA-repair responses of human skin epithelial cells, Lake et al.95 developed a semiquantitative in vitro method for measuring unscheduled DNA synthesis in normal foreskin epithelial cells. On serial subculture of organotypic primary skin cultures, the unscheduled DNA synthesis response elicited by 3-MC decreased in parallel with the ability of cells to metabolize PAHs to water-soluble metabolizes. The working hypothesis was that procarcinogens that are efficiently activated by human skin-specific metabolism will be detected with unscheduled DNA synthesis as an end point. LIVER AND INTESTINE l Obana et al.l30 analyzed quantitatively and qualitatively the PAH content of samples of human liver and fatty tissue. Six samples of liver and 10 samples of fat were obtained at autopsy from 10 persons who died of unstated causes (although the tissues were reported to be "free from cancers. Smoking habits, occupations, etc., were not described. The tissue samples analyzed were quite large (40-120 g), and the PAHs were determined without complex pretreatment. Table 6-1 shows the analytic results for liver, and Table 6-2 the comparable data for fat tissue. Note that PAH concentrations are expressed as parts per trillion (ppt), not parts per billion (ppb), and are in general extremely low. Nevertheless, the data indicate that, on the average, the PAH concentration in liver was one-third that in fat. Pyrene had t'ne highest concentration, followed by anthracene. Although the number of samples was small, no sex or age differences were evident. The known carcinogens benz~ajanthracene and dibenz~ah~anthracene were not detected in either tissue. However, BaP was detected in small amounts (20 ppt) in both liver and fat. This finding should be compared with that of Tomingas et al.,178 who detected BaP at 1-15,000 ppb in 6-5 a. A. . ..
human bronchial-carcinoma tissue (24 samples). Obana et al.130 called attention to the fact that the PAHs in the human tissues they examined were different, in both concentration and composition, from the PAHs that had been identified in marine samples. For example, pyrene was found in oysters at 7-52 ppb and in Wakame seaweed at 12-41 ppb; the comparable figures for BaP were 0.3-2.6 ppb and 0.6-9 ppb, respectively. Mor'eover, although pyrene was the most abundant PAH in all cases, the next most abundant in the human tissues was anthracene, whereas in the marine samples the next most abundant were benzoteipyrene and benzotbifluoranthene. These qualitative and quantitative distinctions, especially the marked concentration differences between nontumorous130 and tumorous tissues178 and between a common food source in the area and the human tissue samples, need to be recalled in evaluating the importance of the food content of PAHs, as well as the role of malignant pathology, when trying to determine the significance of the body or tissue burden of these hydrocarbons. The liver contains the highest concentration of cytochrome P-450 in the body. The activity of the pathway by which heme, the prosthetic group of this heme protein, is synthesized can' tee greatly induced by a host of foreign chemicals and can approach the rates of heme synthesis in erythroid cells; and the enzymatic capacity of hepatic cells to carry out the biotransformations that characterize the great variety of PAH metabolites formed in vitro, and probably in viva, has been well defined. Only selected PAH transformations catalyzed by liver cells are reviewed here, with some emphasis on the relationships of PAH- 'and drug-metabolizing capabilities and on recent data indicating that carcinogen metabolism may be increased by direct actions on relevant membrane-bound enzymes, as well as by the conventionally assumed process of increased de nova synthesis of enzyme protein, i.e., Induction. Dybing et al.54 have examined the in vitro metabolism and metabolic activation of several carcinogenic PAHs in subcellular fractions from seven human livers. The patients all suffered from total cerebral infarction and were serving as potential kidney donors (maintained temporarily by life-support systems) at the Huddinge University Hospital in Sweden. At the appropriate time, liver extirpation was perfo..~`ed; within 20 min after the procedure, perfusion had been completed and the tissue frozen in liquid nitrogen. This study may mimic the enzymatic properties of human liver cells in the living subject as closely as experimentally possible, other than by direct biopsy or surgery in a living patient. Because of the unique source of the tissue studied, some of the data merit recording here. Microsomal cytochrome P-450 content (seven livers) was 0.16-0.60 nmol/mg of protein, with a mean + S.D. of 0.36 + 0.15, and ASH activity averaged 175 + 138 pmol/mg of protein per minute; one sample had a value of 483 pmol/mg. These activities are approximately the same as those in liver microsomes of untreated mice and rats. ASH activities expressed per nanomole of cytochrome P-450 varied by a factor of 2.8 among the seven liver samples. 6-6
Conney and co-workers27~28)4o,4l,73~8l,82~94,l56,l57,l74 conducted a series of studies of direct liver-cell metabolism of carcinogens and compared such metabolism with that of drugs. They established that carcinogen metabolism may be increased not only through enzyme induction, but through enzyme activation as well. They compared the oxidative metabolism of BaP with that of antipyrine, hexobarbital, coumarin, zoxazolamine, and 7-ethoxycoumarin in 32 adult liver samples obtained at autopsy some 8-20 h after death. When enzyme activity for one substrate was plotted against enzyme activity for a second substrate for each of the 32 liver samples, significant correlations were found. For example, for BaP paired against anti- pyrine, hexobarbital, zoxazolamine, and coumarin, the correlation coefficients were 0.85, 0.72, 0.69, and 0.57, respectively. Some drug-drug metabolizing activities also showed a high correlation (e.g., antipyrine and hexabarbital, r = 0.79; antipyrine and coumarin, r = 0.72), whereas in other instances, metabolizing capacities did not have a high correlation, e.g., carcinogen vs. drug (BaP and 7-ethoxycoumarin, r = 0.35) and drug vs. drug (e.g., hexobarbital and 7-ethoxycoumarin, r ~ 0.37~. The findings raise the possibility that an in viva drug-metabolism assay (e.g., a plasma disappearance-rate study of a suitable test drug) might predict some carcinogen- metabolizing capabilities of a person and suggest the presence in humans of multiple monooxygenase systems for the substrates studied, as well as their heterogeneous distribution in the population. Individual differences in the rates of metabolism of BaP (7-fold) in these and other liver samples studied41 were considerable, although they did not approach the known species differences40 in rates of metabolism of drugs. The effects of PAH administration on the metabolic disposition of specific carcinogens such as BaP, have not been studied in humans, but Schlede et al.15 ,1 7 recently examined the metabolic disposition of radiolabeled BaP in rats, and the results probably can be extrapolated to humans. Pretreatment of rats with unlabeled BaP greatly increased the plasma disappearance rate of a tritiated dose of the same compound given intravenously; the effect was especially marked during the first 5 min after the intravenous dose of the radiolabeled material, and the increased rate lasted for at least 6 h. This effect of pretreatment with the compound was paralleled by a lower concentration of [3H]BaP in brain, liver, and fatty tissues; similar but more varied results were observed in lung tissue . These influences of BaP pretreatment on a later intravenous dose of the 3H-labeled chemical were also observed when the radiolabeled PAH was administered orally. 3-MC and 7,12-DMBA pretreatment of animals produced comparable effects on the metabolic disposition and tissue contents of radiolabeled BaP. Pyrene, anthracene, and phenobarbital had little or no such effect on the in viva disposition of this compound. In other studies, the biliary excretion of [14CiBaP was shown to be increased by pretreatment with the unlabeled compound; however, no increase in excretion into bile of the 14C-labeled metabolites of BaP was observed after prior administration of this PAH. These findings suggest that conversion of 6-7
BaP to its metabolites may be the rate-limiting step in the biliary excretion of the compound. Phenobarbital had no effects on the pharma- cokinetics (plasma disappearance) of [14C]BaP and its metabolites; this drug might stimulate the conjugation of hydroxylated BaP derivatives before their excretion into bile. The relevance of these findings to humans is related not only to the (probably) qualitatively similar pharmacokinetics of such a chemical as BaP--particularly its extensive excretion via bile--but also to the potentially important use of selected drugs, singly or multiply, to increase disposal of PAHs and their metabolites from the body by stimulating conjugation and biliary excretion and by increasing otherwise innocuous metabolic biotransformations. Prough et al. 143 have also studied BaP metabolism in human liver, kidney, and lung, and they characterized the metabolites formed by HPLC techniques. Tissue samples were obtained within 1-5 h after death, and assays were completed within the succeeding 2-4 h. In the analysis of metabolites formed from BaP, quinones, three classes of dihydrodiols, and two classes of phenols were categorized. Table 6-3 summarizes the rates of formation of these metabolites by tumor, liver, kidney, and lung microsomes and presents comparable data in rodents. There was a very large variation in the human metabolism of BaP, compared with that demonstrated in studies carried out concurrently in rodents. That might reflect, as the authors noted, either the controlled environment of the animals studied or a genetic variation in humans. In the human liver, activities for metabolite formation were substantially lower than those in rat microsomal fractions, and there were significant differences in the BaP-metabolite profiles. A greater proportion of benzene-ring metabolites was formed by human lung microsomes than by human liver and kidney microsomes (or rodent lung microsomes). The relative increase in the 9,10-dihydrodiol product, as well as some increase in the 7,8-dibydrodiol metabolite, accounted for the larger portion of this difference among lung, liver, and kidney microsomes. There is an apparent biologic inconsistency in these findings: although the human lung is the principal site of PAR tumorigenesis, as the authors observed, the 9,10-dihydrodiol product is suggested to have little biologic activity on further metabolism, whereas other tissues, such as the liver, formed large concentrations of the 7,8-dihydrodiol, a "proximate carcinogen.'' Nevertheless, the findings are important, providing not only data on comparative rates of formation of metabo- lites constituting the "HPLC profiles" in man and rodents, but also intertissue metabolic profiles of BaP biotransformation for three major organ systems in man. Thus, they extended the earlier findings of Selkirk et al.160 on liver cells and lymphocytes. A major advance in defining the role of the liver in PAN metabolism and the factors that regulate liver monooxygenase activity has been the demonstration that hepatic microsomal oxidation of specific substrates can be directly increased in vitro, in addition to their DroDerty of being induced in viva, by various chemical treatments.27~28~73~174 Conney and colleagues have shown that 7, 8-benzof lavone added to homogenates of human liver samples can increase the rate of BaP 6-8 lo. ....
hydroxylation by a factor of up to 11. Benzoflavone also increased some drug hydroxylations substantially, although benzoflavone at concentrations lower by a factor of about 100 paradoxically inhibited these reactions. Marked individuality for activating and inhibiting effects of benzoflavone were noted, and no significant effects on the oxidation of some drug substrates (e.g., coumarin and hexobarbital) were observed. The enhancing effect of 7,8-benzoflavone on BaP oxidation was shown to extend to o ther f lavonoids, such as f lavone, nob i le t in, and tangeritin. Related compounds--such as apigenin, chrysin, fisetin, flavonone, galargin, hesperitin, kaempferal, morin, myricitin, naringerin, and quercetin--inhibited BaP oxidation. The stimulatory effect of 7,8-benzoflavone on BaP 9,10-dihydrodiol oxidation to bay-region epoxides was also studied and shown to have significant species-specific characteristics. With untreated hamster microsomes, more than 60% of the total metabolites of the hydrocarbon were bay-region diol-epoxides, whereas human liver formed less than it of such metabolites. Addition of 7,8-benzoflavone to the microsomal incubations dramatically stimulated the formation of these metabolites in human (and rabbit) liver microsomes. The stimulatory effects of flavonoids on hydrocarbon oxidation have recently been shown to occur in vivo as well,l°° so the biologic importance of this phenomenon for the intact host is likely to be considerable. The mechanism of flavonoid activation of BaP hydroxylation has recently been explored in detail by Huang et al.73 The flavone stimulates the NADPH reduction of cytochrome P-450, but not that of cytochrome c, by NADPH-cytochrome c reductase; this finding supports the idea that the catalytic sites for these substrates of the reductase are different. Other evidence that these catalytic sites are different has also recently been provided by the studies of Yoshinaga et al.197-200 Metabolic transformation of PAHs and their binding to cellular macromolecules in cultured human gut tissues have been described. Harris et al.68 examined the metabolic fate of BaP and several other compounds in cultured esophogeal explants from eight patients, six of whom had esophageal carcinomas. Metabolism of the 3H-labeled PAH to water-soluble ~etabolites varied among the eight patients over the range of 1-68% of total metabolism; the variation found within a single case, however, in relation to different anatomic segments of the esophagus (proximal, middle, and distal) was quite narrow--2%. In spite of the 68-fold variation in metabolism among subjects, the patterns of conjugates formed from metabolites in general were qualitatively similar: sulfate esters, 21-55%; glucuronide conjugates, 7-37%; and glutathione conjugates, 24-66%. Most of the radioactivity of the organic-solvent-soluble metabol ites of BaP cochromatographed with authentic metabolites of this compound, including its proximate carcinogenic, (-~-trans-7 , 8-dihydro-7,8-dihydroxy, derivative. Despite the predominant occurrence of esophageal cancer in the distal segment, the patterns of metabolites formed in all segments of the organ were similar. Binding of 3H-labeled PAHs to ONA and protein was detected in all eight cases, with binding to protein greater than to ONA in each 6-9 .~. .. .
instance. Binding among the eight cases varied 99-fold, and at least three hydrocarbon-DNA adducts, including the specific guanine adduct, were recognized. The adducts appeared identical with those previously found in human colon and bronchus; and the patterns of both metabolism and adduct formation with BaP were analogous to those found in experimental animals susceptible to the carcinogenic action of PAHs. Autrup et al.,9 in an earlier and less detailed but essentially similar study, had reported comparable data based on human colon explants of tumor-free tissue. The binding of labeled BaP to cellular protein was several times higher than that to DNA; however, hydrocarbon binding to DNA correlated with tissue AHH activity (r = 0.87), whereas no such correlation existed for protein binding. DNA binding (BaP, picomoles bound per 10 ma) among seven tissue samples varied over a 25-fold range; variation within subjects was small. In an extension of this work, Autrup et al.ll studied the comparative metabolism of BaP (and aflatoxin B1) and hydrocarbon-DNA adduct formation in cultured human and rat colon explants. Adduct formation (in 103 cases) varied over a 125-fold range in the tumor samples and in the same subject over a 3- to 10-foId range in different segments of the organ. A number of hydrocarbon-DNA adducts were identified, and both qualitative and quantitative rat-human differences were demonstrated. Although overall BaP metabolism was similar in rat and human colon tissue, the ratio of organic-solvent-soluble to water-soluble metabolites was higher in the human; sulfate esters predominated in rat colon, whereas equivalent quantities of sulfate esters and glutathione conjugates were formed in the human tissue; and hydrocarbon-DNA binding was distinctly greater in human colon, although, as noted, there was marked variability in adduct formation within a given subject. The comparative hydration of styrene 7,8-oxide, octane 1,2-oxide, naphthalene l, 2-oxide, phenanthrene 9,10-oxide ? benz~aJanthracene 5,6-oxide, 3-MC 11,12-oxide, dibenz~ah~anthracene 5,6-oxide, and BaP 4,5-, 7,8-, 9,10-, and 11,12-oxides to their dihydrodiols was investigated in microsomes from nine human liver samples obtained at autopsy.80 The substrate specificity of the epoxide hydratase in human liver ~nicrosomes was very similar to that of the epoxide hydratase in rat liver microsomes. Phenanthrene 9,10-oxide was the best substrate for the human and rat epoxide hydratases, and dibenz~ah~antoracene 5,6-oxide and BaP 11,12-oxide were the poorest substrates. Plotting epoxide hydratase activity obtained with one substrate against epoxide hydratase activity for another substrate for each of the nine human livers revealed excellent correlations for all combinations of the 11 substrates studied (r = 0.87-0.99~. The data suggest the presence in human liver of a single epoxide hydratase with broad substrate specificity, although the results do not exclude the possible presence in human liver of several epoxide hydratases that are nder similar regulatory control. 6-10
