
9. References
Ada35 Adams, B. A., and Holmes, E. L., J. Soc. Chem. Ind. 54, 1 ( 1935).
Ahr76 Ahrens, H., Kaffrell, N., Trautmann, N., and Herrmann, G., Phys. Rev. C 14, 211 ( 1976).
All86 Allardyce, B. W., in Nuclei Off the Line of Stability, R. A. Meyer and D. S. Brenner, Eds. (Amer. Chem. Soc. Pub., Washington, D.C., 1986), p. 406.
Als86 Alstad, J. Björnstad, T., and Skarnemark, G., Nucl. Instrum. Methods Phys. Res. A244, 490 ( 1986).
Ami68 Amiel, S., in Nuclear Chemistry, Vol. 1, L. Yaffe, Ed. (Academic Press, New York, 1968), p. 251.
Amp64 Amphlett, C. B., Inorganic Ion Exchanges (Elsevier, Amsterdam, 1964).
And57 Andersson, G., Tove, P. A., Jung, B., and Svensson, I. B., Nucl. Phys. 3, 493 ( 1957).
And69 Andersson, C., Andersson, S. O., Liljenzin, J. O., Reinhardt, H., and Rydberg, J., Acta Chem. Scand. 23, 2781 ( 1969).
And81 Andrl, R. A., Novick, V. J., and Greenwood, R. C., Nucl. Instrum. Methods 186, 153 ( 1981).
Arm85 Armbruster, P., Agarwal, Y. K., Brüchle, W., Brügger, M., Dufour, H., Gäggeler, H., Hessberger, F. P., Hofmann, S., Lemmertz, P., Münzenberg, G., Poppensieker, K., Reisdorf, W., Schädel, M., Schmidt, K. H., Schneider, J. H. R., Schneider, W. F. W., Sümmerer, K., Vermeulen, D., Wirth, G., Ghiorso, A., Gregorich, K. E., Lee, D., Leino, M., Moody, K. J., Seaborg, G. T., Welch, R. B., Wilmarth, P., Yashita, S., Frink, C., Greulich, N., Herrmann, G., Hickmann, U., Hildebrand, N., Kratz, J. V., Trautmann, N., Fowler, M. M., Hoffman, D. C., Daniels, W. R., von Gunten, H. R., and Dornhöfer, H., Phys. Rev. Lett. 54, 406 ( 1985).
Arn53 Arnott, D. G., and Wells-Cole, J., Nature 171, 269 ( 1953).
Aro70 Aronsson, P. O., Ehn, E., and Rydberg, J., Phys. Rev. Lett. 25, 590 ( 1970).
Aro74a Aronsson, P. O., Johansson, B. E., Rydberg, J., Skarnemark, G., Alstad, J., Bergersen, B., Kvåle, E., and Skarestad, M., J. Inorg. Nucl. Chem. 36, 2697 ( 1974).
Aro74b Aronsson, P. O., Skarnemark, G., and Skarestad, M., Inorg. Nucl. Chem. Lett. 10, 499 ( 1974).
Aro74c Aronsson, P. O., Skarnemark, G., and Skarestad, M., J. Inorg. Nucl. Chem. 35, 1689 ( 1974).
Aro74d Aronsson, P. O., Skarnemark, G., Kvåle, E., and Skarestad, M., Inorg. Nucl. Chem. Lett. 10, 753 ( 1974).
Ate55 Aten Jr., H. W., and DeVries-Hamerling, T., Physica 21, 544 ( 1955).
Aum74 Aumann, D. C., and Weismann, D., Nucl. Instrum. Methods 117, 459 ( 1974).
Ays73 Aystö, J., and Valli, K., Nucl. Instrum. Methods 111, 531 ( 1973).
Ays74a Aystö, J., Hillebrand, S., Hellmuth, K-H., and Valli, K., Nucl. Instrum. Methods 120, 163 ( 1974).
Ays74b Aystö, J., Pummalainen, P., and Valli, K., Nucl. Instrum. Methods 115, 65 ( 1974).
Ays76 Aystö, J., Rantala, V., Valli, K., Hillebrand, S., Kortelahti, M., Eskola, K., and Raunemaass, T., Nucl. Instrum. Methods 139, 325 ( 1976).
Bäc65 Bächmann, K., Radiochim. Acta 4, 124 ( 1965).
Bäc66 Bächmann, K., Radiochim. Acta 6, 62 ( 1966).
Bäc75 Bächmann, K., and Matschoss, V., J. Inorg. Nucl. Chem. 37, 15 ( 1975).
Bäc76a Bächmann, K., Matschoss, V., Rudolph, J., Steffen, A., and Tsalas, S., Nucl. Instrum. Methods 139, 343 ( 1976).
Bäc76b Bächmann, K., and Rudolph, J., J. Radioanal. Chem. 32, 243 ( 1976).
Bai62 Bailey, R. A., U.S. Atomic Energy Commission Report, NAS-NS-3106 ( 1962).
Bak80 Baker, J. D., Gehrke, R. J., Greenwood, R. C., Meikrantz, D. H., and Novick, V. J., J. Inorg. Nucl. Chem. 42, 1547 ( 1980).
Bak81 Baker, J. D., Gehrke, R. J., Greenwood, R. C., and Meikrantz, D. H., Radiochim. Acta 28, 51 ( 1981).
Bak82 Baker, J. D., Gehrke, R. J., Greenwood, R. C., and Meikrantz, D. H., J. Radioanal. Chem. 74, 117 ( 1982).
Bar86 Barth, H. G., Barber, W. E., Lochmüller, C. H., Majors, R. E., and Regnier, F. E., Anal. Chem. 58, 211R ( 1986).
Bar88 Barth, H. G., Barber, W. E., Lochmüller, C. H., Majors, R. E., and Regnier, F. E., Anal. Chem. 60, 387R ( 1988).
Bec82 Becker, H. W., Buchmann, L., Gorres, J., Kettner, K. U., Krawinkel, H., Rolfs, C., Schmalbrock, P., Trautvetter, H. P., and Vlieks, A., Nucl. Instrum. Methods 198, 277 ( 1982).
Bec96 Becquerel, H., C. R. Acad. Sci., Paris 122, 420 ( 1896).
Ben81 Benedict, M., Pigford, T. H., and Levi, H. W. (McGraw-Hill, New York, 1981), p. 466.
Bib-1870 Bible, King James version (William W. Harding, Philadelphia, 1870).
Bjö77a Björnstad, T., Kvåle, E., Skarnemark, G., Aronsson, P. O., Kaffrell, N., Trautmann, N., and Stender, E., J. Inorg. Nucl. Chem. 39, 1107 ( 1977).
Bjö77b Björnstad, T., Kvåle, E., Skarnemark, G., and Aronsson, P. O., J. Inorg. Nucl. Chem. 39, 1929 ( 1977).
Bjö81 Björnstad, T., Nucl. Instrum. Methods 188, 375 ( 1981).
Bög74a Bögl, W., and Bächmann, K., Inorg. Nucl. Chem. Lett. 10, 697 ( 1974).
Bög74b Bögl, W., and Bächmann, K., Inorg. Nucl. Chem. Lett. 10, 805 ( 1974).
Bög74c Bögl, W., and Bächmann, K., Radiochem. Radioanal. Lett. 17, 239 ( 1974).
Bög74d Bögl, W., Büttner, K., and Bächmann, K., Radiochem. Radioanal. Lett. 18, 315 ( 1974).
Bög74e Bögl, W., Bächmann, K., and Büttner, K., Radiochim. Acta 21, 33 ( 1974).
Bög75a Bögl, W., and Bächmann, K., J. Inorg. Nucl. Chem. 37, 1115 ( 1975).
Bög75b Bögl, W., Bächmann, K., and Matschss, V., J. Inorg. Nucl. Chem. 37, 1557 ( 1975).
Bög76 Bögl, W., and Bächmann, K., J. Radioanal. Chem. 34, 223 ( 1976).
Bro80 Brodén, K., Persson, H., Rydberg, J., and Skarnemark, G., Nucl. Instrum. Methods 195, 539 ( 1980).
Bro81 Brodén, K., Skarnemark, G., Björnstad, T., Eriksen, D., Haldorsen, I., Kaffrell, N., Stender, E., and Trautmann, N., J. Inorg. Nucl. Chem. 43, 765 ( 1981).
Brü82 Brüchle, W., and Herrmann, G., Radiochim. Acta 30, 1 ( 1982).
Brü85 Brügger, M., Hildebrand, N., Karlewski, T., Trautmann, N., Mazumdar, A. K., and Herrmann, G., Nucl. Instrum. Methods Phys. Res. A234, 218 ( 1985).
Brü88 Brüchle, W., Schädel, M., Scherer, U. W., Kratz, J. V., Gregorich, K. E., Lee, D., Nurmia, M., Chasteler, R. M., Hall, H. L., Henderson, R. A., and Hoffman, D. C., Inorg. Chim. Acta 146, 267 ( 1988).
Cab75 Cabot, C., Deprun, C., Gauvin, H., Le Beyec, Y., and Lefort, M., Nucl. Instrum. Methods 125, 397 ( 1975).
Cam53 Campbell, E. C., and Nelson, F., Phys. Rev. 91, 499A ( 1953).
Cam56 Campbell, E. C., and Nelson, F., J. Inorg. Nucl. Chem. 3, 233 ( 1956).
Car67 Cartwright, P. F. S., Newman, E. J., and Wilson, D. W., Analyst 82, 663 ( 1967).
Cas81a Cassidy, R. M., in Trace Analysis, Vol. 1, J. F. Lawrence, Ed. (Academic Press, New York, 1981), p. 148.
Cas81b Cassidy, R. M., in Trace Analysis, Vol. 1, J. F. Lawrence, Ed. (Academic Press, New York, 1981), p. 121.
Cho56a Choppin, G. R., Harvey, B. G., and Thompson, S. G., J. Inorg. Nucl. Chem. 2, 66 ( 1956).
Cho56b Choppin, G. R., and Silva, R. J., J. Inorg. Nucl. Chem. 3, 153 ( 1956).
Cle74 Clearfield, A., Nancollas, G. H., and Blessing, R. H., in Ion Exchange and Solvent Extraction, Vol. 5, J. A. Marinsky and Y. Marcus, Eds. (Marcel Dekker, New York, 1974).
Clu71 Clumpner, J. A., J. Chem. Phys. 55, 5042 ( 1971).
Col71 Coleman, C. F., and Roddy, J. W., in Solvent Extraction Reviews, Vol. 1, Y. Marcus, Ed. ( 1971), p. 63.
Cor51a Coryell, C. D., and Sugarman, N. (Eds.), Radiochemical Studies: The Fission Products, National Nuclear Energy Series, Division IV, Vol. 9, Books 1, 2 and 3 ( McGraw-Hill, New York, 1951).
Cor51b Coryell, C. D., and Sugarman, N. (Eds.), Radiochemical Studies: The Fission Products, National Nuclear Energy Series, Division IV, Vol. 9, Books 1, 2, and 3 (McGraw-Hill, New York, 1951).
Cur98a Curie, P., and Curie, S. Mme., Compt. Rend. 127, 175 ( 1898).
Cur98b Curie, P., Curie, S. Mme., and Bemont, G., Compt. Rend. 128, 1215 ( 1898).
Dau73 Dautet, H., Gujrathi, S., Wiesehahn, W. J., D'Auria, J. M., and Pate, B. D., Nucl. Instrum. Methods 107, 49 ( 1973).
Dau77 D'Auria, J. M., in Proc. Isotope Separator On-Line Workshop held at Brookhaven National Laboratory, 1977, Report No. BNL-50847 ( 1977), p. 237.
Dau86 D'Auria, J. M., in Nuclei Off the Line of Stability, R. A. Meyer and D. S. Brenner, Eds. (Amer. Chem. Soc. Pub., Washington, D.C., 1986), p. 432.
De70a De, A. K., Khopkar, S. M., and Chalmers, R. A., Solvent Extraction of Metals (Van Nostrand, London, 1970), p. 57.
De70b De, A. K., Khopkar, S. M., and Chalmers, R. A., Solvent Extraction of Metals (Van Nostrand, London, 1970).
De70c De, A. K., Khopkar, S. M., and Chalmers, R. A., Solvent Extraction of Metals (Van Nostrand, London, 1970), p. 215.
De70d De, A. K., Khopkar, S. M., and Chalmers, R. A., Solvent Extraction of Metals (Van Nostrand, London, 1970), p. 201.
De70e De, A. K., Khopkar, S. M., and Chalmers, R. A., Solvent Extraction of Metals (Van Nostrand, London, 1970), p. 165.
De70f De, A. K., Khopkar, S. M., and Chalmers, R. A., Solvent Extraction of Metals (Van Nostrand, London, 1970), p. 158.
Del67 del Marmol, P., and Néve de Mévergnies, M., J. Inorg. Nucl. Chem. 29, 273 ( 1967).
Den66 Denig, R., Trautmann, N., and Herrmann, G., Z. Anal. Chem. 216, 41 ( 1966).
Den67 Denschlag, H. O., and Gordus, A. A., Z. Anal. Chem. 226, 62 ( 1967).
Dev62 DeVoe, J. R., U.S. Atomic Energy Commission Report No. NAS-NS-3108 ( 1962).
Die83 Dienstbach, F., and Bächmann, K., Radiochim. Acta 34, 215 ( 1983).
Dil51 Dillard, C. R., Adams, R. M., Finston, H., and Turkevich, A., in Radiochemical Studies: The Fission Products, National Nuclear Energy Series, Division IV, Vol. 9, Book 2, C. D. Coryell and N. Sugarman, Eds. (McGraw-Hill, New York, 1951), p. 624.
Dom83 Domanov, V. P. Khyubener, Z., Shalaevskii, M. R., Timokhin, S. N., Petrov, D. V., and Zvara, I., Sov. Radiochemistry (Eng. Tr.) 25, 23 ( 1983).
Dor72 Dorfner, K., Ion Exchangers. Properties and Applications (Ann Arbor Science, Ann Arbor, 1972).
Dor90 Dorsey, J. G., Foley, J. P., Cooper, W. T., Barford, R. A., and Barth, H. G., Anal. Chem. 62, 324R ( 1990).
Dro56 Dropesky, B. J., and Schardt, A. W., Phys. Rev. 102, 426 ( 1956).
Eic79 Eichler, B., Gäggeler-Koch, H., and Gäggeler, H., Radiochim. Acta 26, 193 ( 1979).
Eic82 Eichler, B., and Zvara, I., Radiochim. Acta 30, 233 ( 1982).
Elc79 Elchuk, S., and Cassidy, R. M., Anal. Chem. 51, 1434 ( 1979).
Eng79 Engelhardt, H., High Performance Liquid-Chromatography Chemical Laboratory Practice , translated from German by G. Gutnikov (Springer Verlag, 1979).
Eve83 Everaerts, F. M., Mikkers, F. E. P., Verheggen, Th. P. E. M., and Vacik, J., in J. Chromatog. Library, Vol. 22A, “Fundamentals and Applications of Chromatographic and Electrophoretic Methods,” Part A, E. Heftmann, Ed. (Elsevier, Amsterdam, 1983), p. A331.
Fel69 Felton, H., J. Chromatog. Sci. 7, 13 ( 1969).
Fel73 Feldstein, H., and Amiel, S., Nucl. Instrum. Methods 113, 181 ( 1973).
Fle56 Flegenheimer, J., and Seelman-Eggelbert, W., Proc. Intern. Conf. on the Peaceful Uses of Atomic Energy 7, 152 ( 1956).
Fol69 Folger, H., Kratz, J. V., and Herrmann, G., Radichem. Radioanal. Lett. 1, 185 ( 1969).
Fra78 Franz, G., and Herrmann, G., J. Inorg. Nucl. Chem. 40, 945 ( 1978).
Fre59 Freiser, H., and Morrison, G. H., Ann. Rev. Nucl. Sci. 9, 221 ( 1959).
Fri61 Friedli, W., and Schumacher, E., Helv. Chim. Acta 44, 1839 ( 1961).
Fri77 Fritz, J. S., Pure Appl. Chem. 49, 1547 ( 1977).
Gäg86 Gäggeler, H., Eichler, B., Greulich, N., Herrmann, G., and Trautmann, N., Radiochim. Acta 40, 137 ( 1986).
Gau73 Gauvin, H., Hahn, R. L., Le Beyec, Y., Lefort, M., and Livet, J., Nucl. Phys. A208, 360 ( 1973).
Geh82a Gehrke, R. J., Greenwood, R. C., Baker, J. D., and Meikrantz, D. H., Radiochim. Acta 31, 1 ( 1982).
Geh82b Gehrke, R. J., Greenwood, R. C., Baker, J. D., and Meikrantz, D. H., Z. Phys. A306, 363 ( 1982).
Ghi55 Ghiorso, A., Harvey, B. G., Choppin, G. R., Thompson, S. G., and Seaborg, G. T., Phys. Rev. 98, 1518 ( 1955).
Ghi58 Ghiorso, A., Sikkeland, J., Walton, J. R., and Seaborg, G. T., Phys. Rev. Lett. 1, 18 ( 1958).
Ghi74 Ghiorso, A., Nitschke, J. M., Alonso, J. R., Alonso, C. T., Nurmia, M., Seaborg, G. T., Hulet, E. K., and Lougheed, R. W., Phys. Rev. Lett. 33, 1490 ( 1974).
Gid65 Giddings, J. C., Dynamics of Chromatography, Part I (Marcel Dekker, 1965).
Gil85 Gill, R. L., and Piotrowski, A., Nucl. Instrum. Methods Phys. Res. A234, 213 ( 1985).
Gil86 Gill, R. L., in Nuclei Off the Line of Stability, R. A. Meyer and D. S. Brenner, Eds. (Amer. Chem. Soc. Pub., Washington, D.C., 1986), p. 420.
Gir70 Girardi, F., Pietra, R., and Sabbioni, E., J. Radioanal. Chem. 5, 141 ( 1970).
Gla36 Glazebrook, H. H., and Pearson, T. G., J. Amer. Chem. Soc. 58, 1777 ( 1936).
Gla37 Glazebrook, H. H., and Pearson, T. G., J. Amer. Chem. Soc. 59, 567 ( 1937).
Gor55 Gordon, L., Anal. Chem. 27, 1704 ( 1955).
Gor59 Gordon, L., in Comprehensive Analytical Chemistry, Vol. IA, C. L. Wilson and E. W. Wilson, Eds. (Elsevier, New York, 1959), p. 530.
Gra73 Grapengiesser, B., and Rudstam, G., Radiochim. Acta 20, 85 ( 1973).
Gre63a Greendale, A. E., and Love, D. L., Anal. Chem. 35, 632 ( 1963)
Gre63b Greendale, A. E., and Love, D. L., Anal. Chem. 35, 1712 ( 1963).
Gre63c Greendale, A. E., and Love, D. L., Nucl. Instrum. Methods 23, 209 ( 1963).
Gre73 Green, H., Talanta 20, 139 ( 1973).
Gre82 Greenwood, R. C., Gehrke, R. J., Baker, J. D., and Meikrantz, D. H., Radiochim. Acta 30, 57 ( 1982).
Gre83 Greenwood, R. C., Gehrke, R. J., Baker, J. D., Meikrantz, D. H., and Reich, C. W., Phys. Rev. C27, 1266 ( 1983).
Gri85 Griffin, H. C., and Rengan, K., Nucl. Instrum. Methods Phys. Res. A241, 511 ( 1985).
Hag62 Hagebo, E., Kjelberg A., and Pappas, A. C., J. Inorg. Nucl. Chem. 24, 117 ( 1962).
Hah39 Hahn, O., and Strassmann, F., Naturwissenschaften 27 (11), 89 ( 1939).
Hah40 Hahn, O., and Strassmann, F., Naturwissenschaften 28, 54 ( 1940).
Ham82 Hamilton, R. J., and Sewell, P. A., Introduction to High Performance Liquid Chromatography, second edition (Chapman and Hall, London, 1982).
Ham84 Hamel, L. A., Chauvin, J., Lessard, L., and Jeremie, H., Nucl. Instrum. Methods Phys. Res. 228, 9 ( 1984).
Ham90 Hampton, C. V., and McHarris, Wm. C., J. Radioanal. Nucl. Chem. 142, 183 ( 1990).
Han79 Hansen, P. G., Ann. Rev. Nucl. Part. Sci. 29, 69 ( 1979).
Har47 Harris, D. H., and Tompkins, E. R., J. Amer. Chem. Soc. 69, 2792 ( 1947).
Har75 Hardy, J. C., Schmeing, H., Geiger, J. S., and Graham, R. L., Nucl. Phys. A246, 61 ( 1975).
Har86 Hardy, J. C., in Nuclei Off the Line of Stability, R. A. Meyer and D. S. Brenner, Eds. (Amer. Chem. Soc. Pub., Washington, D.C., 1986), p. 414.
Hel66 Helfferich, F., in Ion Exchange, Vol. 1, J. A. Marinsky, Ed. (Marcel Dekker, New York, 1966), p. 65.
Hel75 Hellmuth, K. H., and Valli, K., Nucl. Instrum. Methods 125, 99 ( 1975).
Hen81 Henry, E. A., Lien III, O. G., and Meyer, R. A., in Proc. Fourth Intern. Conf. on Nuclei Far from Stability (Helsingor, Denmark, 1981), CERN Report 81-09, p. 334.
Her69 Herrmann, G., and Denschlag, H. O., Ann. Rev. Nucl. Sci. 19, 1 ( 1969).
Her82 Herrmann, G., and Trautmann, N., Ann. Rev. Nucl. Part. Sci. 32, 117 ( 1982).
Hic80 Hickmann, U., Greulich, N., Trautmann, N., Gäggeler, H., Gäggeler-Koch, H., Eichler, B., and Herrmann, G., Nucl. Instrum. Methods 174, 507 ( 1980).
Hir64 Hirschfelder, J. O., Curtiss, C. F., and Bird, R. B., Molecular Theory of Gases and Liquids (John Wiley, New York, 1964), p. 372.
Hof39 Hoffman, J. I., and Lundell, G. E. F., J. Research 22, 465 ( 1939).
Hof66 Hoffman, D. C., Michelsen, O. B., and Daniels, W. R., Ark. Fys. 36, 211 ( 1966).
Hof88 Hoffman, D. C., Henderson, R. A., Gregorich, K. E., Bennett, D. A., Chasteler, R. M., Gannett, C. M., Hall, H. L., Lee, D. M., Nurmia, M. J., Cai, S., Agarwal, R., Charlop, A. W., Chu, Y. Y., Seaborg, G. T., and Silva, R. J., J. Radioanal. Nucl. Chem. 124, 135 ( 1988).
Hog66 Hogfeldt, E., in Ion Exchange, Vol. 1, J. A. Marinsky, Ed. (Marcel Dekker, New York, 1966), p. 139.
Hor67 Horvath, C. G., Preiss, B. A., and Lipsky, S. R., Anal. Chem. 39, 1422 ( 1967).
Hor77 Horwitz, E. P., Bloomquist, C. A. A., and Delphin, W. H., J. Chromatog. Sci. 15, 41 ( 1977).
Hub67 Huber, J. R. K., and Hulsman, Y. A. R. J., Anal. Chim. Acta 38, 405 ( 1967).
Hub69 Huber, J. F. K., J. Chromatog. Sci. 7, 85 ( 1969).
Hüb80a Hübener, S., and Zvara, I., Radiochim. Acta 27, 157 ( 1980).
Hüb80b Hübener, S., Radiochem. Radioanal. Lett. 44, 79 ( 1980).
Hüb82 Hübener, S., and Zvara, I., Radiochim. Acta 31, 89 ( 1982).
Hul80 Hulet, E. K., Lougheed, R. W., Wild, J. F., Landrum, J. H., Nitschke, J. M., and Ghiorso, A., J. Inorg. Nucl. Chem. 42, 79 ( 1980).
Hul83 Hulet, E. K., Radiochim. Acta 32, 7 ( 1983).
Ima80 Imanishi, N., Fujiwara, I., and Nishi, T., J. Nucl. Sci. Technol. (Japan) 17, 935 ( 1980).
Irg74 Irgolic, K. J., and Kudchadker, M. V., in Selenium, R. A. Zingaro and W. C. Cooper, Eds. (Van Nostrand-Reinhold, New York, 1974), p. 708.
Irv76 Irving, H. M. N. H., J. Radioanal. Chem. 33, 287 ( 1976).
Irv82 Irving, H. M. N. H., in Treatise on Analytical Chemistry, Part I, Vol. 5, second edition, P. J. Elving, E. Grushka, and I. M. Kolthoff, Eds. (Wiley, New York, 1982), p. 508.
Iye63 Iyer, R. H., Mathews, C. K., Ravindran, N., Rengan, K., Singh, D. V., Ramaniah, M. V., and Sharma, H. D., J. Inorg. Nucl. Chem. 25, 456 ( 1963).
Jak62 Jakovac, Z., and Pucar, Z., J. Chromatog. 7, 380 ( 1962).
Jos88 Jost, D. T., Gäggeler, H. W., Vogel, C. H., Schädel, M., Jäger, E., Eichler, B., Gregorich, K. E., and Hoffman, D. C., Inorg. Chim. Acta 146, 255 ( 1988).
Jun71 Junglas, H., MacFarlane, R. D., and Fares, Y., Phys. Rev. Lett. 27, 556 ( 1971).
Jun76 Jungclas, H., Wilhelm, H. G., Westmeier, W., Kornahl, W., Fares, Y., Molzahn, D., Brandt, R., and Wollnik, H., Nucl. Instrum. Methods 137, 93 ( 1976).
Kau61 Kaufman, H. C., Handbook of Organic Compounds (Van Nostrand, New York, 1961), p. 1489.
Kaw82 Kawade, K., Yamamoto, H., Amano, H., Hanada, M., Katoh, T., Okano, K., Kawase, Y., and Fujiwara, I., Nucl. Instrum. Methods 200, 417 ( 1982).
Kel77 Keller, Jr., O. L., and Seaborg, G. T., Ann. Rev. Nucl. Sci. 27, 139 ( 1977).
Kel84 Keller, Jr., O. L., Radiochim. Acta 37, 169 ( 1984).
Ken23 Kendall, J., and Crittenden, E. D., Proc. Natl. Acad. Sci. U.S.A. 9, 75 ( 1923).
Ken25 Kendall, J., and Clarke, B. L., Proc. Natl. Acad. Sci. U.S.A. 11, 393 ( 1925).
Ken26 Kendall, J., Jette, E. R., and West, Wm., J. Amer. Chem. Soc. 48, 3114 ( 1926).
Ket47 Ketelle, B. H., and Boyd, G. E., J. Amer. Chem. Soc. 69, 2800 ( 1947).
Kir69 Kirkland, J. J., J. Chromatog. Sci. 7, 7 ( 1969).
Kla69 Klapisch, R., Ann. Rev. Nucl. Sci. 19, 33 ( 1969).
Kla74 Klapisch, R., in Nuclear Spectroscopy and Reactions, Part A, J. Cerny, Ed. (Academic Press, New York, 1974), p. 273.
Kle54 Kleinberg, J., U.S. Atomic Energy Commission Report LA-1721 (1954).
Kle75 Klein, G., Kaffrell, N., Trautmann, N., and Herrmann, G., Inorg. Nucl. Chem. Lett. 11, 511 ( 1975).
Kof51 Kofoed-Hansen, O., and Nielsen, K. O., K. Dan. Vidensk. Selsk. Mat. Fys. Medd. 26 (7) ( 1951).
Kol32 Kolthoff, I. M., J. Phys. Chem. 36, 860 ( 1932).
Kol36 Kolthoff, I. M., J. Phys. Chem. 40, 1027 ( 1936).
Kol39a Kolthoff, I. M., and O'Brien, A. S., J. Amer. Chem. Soc. 61, 3409 ( 1939).
Kol39b Kolthoff, I. M., and O'Brien, A. S., J. Amer. Chem. Soc. 61, 3414 ( 1939).
Kol39c Kolthoff, I. M., and O'Brien, A. S., J. Chem. Phys. 7, 401 ( 1939).
Kol71 Kolarik, Z., in Solvent Extraction Reviews, Vol. 1, Y. Marcus, Ed. ( 1971), p. 1.
Kos74 Kosanke, K. L., McHarris, Wm. C., and Warner, R. A., Nucl. Instrum. Methods 115, 151 ( 1974).
Kra65 Kraak, W., and Walz, G. O., J. Chromatog. 20, 197 ( 1965).
Kra70 Kratz, J. V., and Herrmann, G., J. Inorg. Nucl. Chem. 32, 3713 ( 1970).
Kra73 Kratz, K. L., and Herrmann, G., Radiochem. Radioanal. Lett. 13, 385 ( 1973).
Kra78 Kratz, K. L., Radiochim. Acta 25, 1 ( 1978).
Kra86 Kratz, J. V., Trautmann, N., and Herrmann, G., in Nuclei Off the Line of Stability, R. A. Meyer and D. S. Brenner, Eds. (Amer. Chem. Soc. Pub., Washington, D.C., 1986), p. 482.
Kra89 Kratz, K. V., Zimmermann, H. P., Scherer, U. W., Schädel, M., Brüchle, W., Gregorich, K. E., Gannett, C. M., Hall, H. L., Henderson, R. A., Lee, D. C., Leyba, J. D., Nurmia, M. J., Hoffman, D. C., Gäggeler, H., Jost, D., Baltensperger, U., Ya Nai-Qi, Türler, A., and Lisnert, Ch., Radiochim. Acta 48, 121 ( 1989).
Kus61 Kusaka, Y., and Meinke, W. W., U.S. Atomic Energy Commission Report, NAS-NS-3104 ( 1961).
Lan42 Langer, A., J. Chem. Phys. 10, 321 ( 1942).
Lan43 Langer, A., J. Chem. Phys. 11, 11 ( 1943).
Lan50 Langer, A., Anal. Chem. 22, 1288 ( 1950).
Lee73 Lee, J. K., Barker, J. A., and Abraham, F. F., J. Chem. Phys. 58, 3166 ( 1973).
Lev51 Levinger, J. S., Meiners, E. P., Sampson, M. B., Snell, A. H., and Wilkinson, R. G., in Radiochemical Studies: The Fission Products, National Nuclear Energy Series, Vol. 9, Book 2, C. D. Coryell and N. Sugarman, Eds. ( 1951), p. 603.
Lie81 Lien III, O. G., Steveson, P. C., Henry, E. A., Yaffe, R. P., and Meyer, R. A., Nucl. Instrum. Methods 185, 351 ( 1981).
Lie82 Lien III, O. G., Ph.D. thesis, University of California, Davis ( 1982).
Lin73 Lin, C. C., and Wahl, A. C., J. Inorg. Nucl. Chem. 35, 1 ( 1973).
Lin82 Lin, J., Rengan, K., and Meyer, R. A., Radiochem. Radioanal. Lett. 50, 399 ( 1982).
Liv38 Livingood, J. J., and Seaborg, G. T., Phys. Rev. 54, 391 ( 1938).
Mac74a MacFarlane, R. D., and McHarris, Wm. C., in Nuclear Spectroscopy and Reactions, Part 1, J. Cerny, Ed. (Academic Press, New York, 1974), p. 243.
Mac74b Macias, E. S., Head, R. E., Hseuh, H. C., and Zalusky, M. R., Nucl. Instrum. Methods 122, 365 ( 1974).
Mai61 Mair, B. J., in Treatise on Analytical Chemistry, Part I, Vol. 3, I. M. Kolthoff and P. J. Elving, Eds. (Interscience, New York, 1961), p. 1469.
Maj82 Majors, R. E., Barth, H. G., and Lochmuller, C. H., Anal. Chem. 54, 323R ( 1982).
Maj84 Majors, R. E., Barth, H. G., and Lochmuller, C. H., Anal. Chem. 56, 300R ( 1984).
Mar47 Marinsky, J. A., Glendenin, L. E., and Coryell, C. D., J. Amer. Chem. Soc. 69, 2781 ( 1947).
Mar69 Marcus, Y., and Kertes, A. S., Ion Exchange and Solvent Extraction of Metal Complexes (Interscience, New York, 1969).
Mar82a Marhol, M., in Wilson and Wilson's Comprehensive Analytical Chemistry, Vol. XIV, G. Svehla, Ed. (Elsevier, Amsterdam, 1982), p. 24.
Mar82b Marhol, M., in Wilson and Wilson's Comprehensive Analytical Chemistry, Vol. XIV, G. Svehla, Ed. (Elsevier, Amsterdam, 1982), p. 33.
Mar82c Marhol, M., in Wilson and Wilson's Comprehensive Analytical Chemistry, Vol. XIV, G. Svehla, Ed. (Elsevier, Amsterdam, 1982), p. 110.
Mar82d Marhol, M., in Wilson and Wilson's Comprehensive Analytical Chemistry, Vol. XIV, G. Svehla, Ed. (Elsevier, Amsterdam, 1982), p. 490.
Mas71 Massart, D. L., Radiochemical Techniques Series Report No. NAS-NS-3113 ( 1971).
May61 Mayer, J. E., and Mayer, M. G., Statistical Mechanics (John Wiley, New York, 1961), p. 295.
Maz76 Mazumdar, A. K., Wagner, H., Walcher, W., and Lund, T., Nucl. Instrum. Methods 139, 319 ( 1976).
Maz80 Mazumdar, A. K., Wagner, H., Krömer, G., Walcher, W., Brügger, M., Stender, E., Trautmann, N., and Lund, T., Nucl. Instrum. Methods 174, 183 ( 1980).
Maz81 Mazumdar, A. K., Wagner, H., Walcher, W., Brügger, M., and Trautmann, N., Nucl. Instrum. Methods 186, 131 ( 1981).
McM40 McMillan, E., and Abelson, P. H., Phys. Rev. 57, 1185 ( 1940).
Mei49a Meinke, W. W., U.S. Atomic Energy Commission Report AECD-2738 ( 1949).
Mei49b Meinke, W. W., U.S. Atomic Energy Commission Report AECD-2738 ( 1949), p. 268.
Mei55 Meinke, W. W., and Sunderman, D. N., Nucleonics 13 (12), 58 ( 1955).
Mei80 Meikrantz, D. H., U.S. Department of Energy Report EGG-PHYS-5260 ( 1980).
Mei81 Meikrantz, D. H., Gehrke, R. J., McIsaac, L. D., Baker, J. D., and Greenwood, R. C., Radiochim. Acta 29, 93 ( 1981).
Mey80 Meyer, R. A., and Henry, E. A., in Proc. Workshop on the Nuclear Spectroscopy of Fission Products, T. von Egidy, Ed. (Grenoble, 1979) (Institute of Physics, London, 1980), p. 59.
Mey90 Meyer, R. A., J. Radioanal. Nucl. Chem. 142, 135 ( 1990).
Mic75 Michl, H., in Chromatography, a Laboratory Handbook of Chromatographic and Electrophoretic Methods, third edition, E. Heftmann, Ed. (Van Nostrand, New York, 1975), p. 282.
Min82a Minczewski, J., Chwastowska, J., and Dybezynski, R., Separation and Preconcentration Methods in Inorganic Trace Analysis (Ellis Harwood, Chichester, 1982), p. 188.
Min82b Minczewski, J., Chwastowska, J., and Dybezynski, R., Separation and Preconcentration Methods in Inorganic Trace Analysis (Ellis Harwood, Chichester, 1982), Chap. 5.
Min82c Minczewski, J., Chwastowska, J., and Dybezynski, R., Separation and Preconcentration Methods in Inorganic Trace Analysis (Ellis Harwood, Chichester, 1982), p. 228.
Min82d Minczewski, J., Chwastowska, J., and Dybezynski, R., Separation and Preconcentration Methods in Inorganic Trace Analysis (Ellis Harwood, Chichester, 1982), p. 216.
Min82e Minczewski, J., Chwastowska, J., and Dybezynski, R., Separation and Preconcentration Methods in Inorganic Trace Analysis (Ellis Harwood, Chichester, 1982), p. 66.
Mit85 Mitchell, J. W., in Inorganic Chromatographic Analysis, J. C. MacDonald, Ed. (John Wiley, 1985), p. 327.
Moo60 Moore, F. L., U.S. Atomic Energy Commission Report No. NAS-NS-3101 ( 1960).
Moo87 Moody, K. J., Brüchle, W., Brügger, M., Gäggeler, H., Haefner, B., Schädel, M., Sümmerer, K., Tetzlaff, H., Gerrmann, G., Kaffrell, N., Kratz, J. V., Rogowski, J., Trautmann, N., Skålberg, M., Skarnemark, G., Alstad, J., Fowler, M. M., Z. Phys. A328, 417 ( 1987).
Mor57 Morrison, G. F., and Freiser, H., Solvent Extraction in Analytical Chemistry (John Wiley, New York, 1957).
Mot79 Motl, O., and Novotny, L., in Laboratory Handbook of Chromatographic and Allied Methods, O. Mikes, Ed. (Ellis Harwood, Chichester, England, 1979), p. 150.
Nac56 Nachod, F. C., and Schubert, J. (Eds.), Ion Exchange Technology (Academic Press, New York, 1956).
Nai84 Nair, A. G. C., Srivastava, A., Srivastava, B. K., Prakash, S., and Ramaniah, M. V., J. Radioanal. Nucl. Chem. 82, 263 ( 1984).
Nau54 Naumann, R. A., and Gerhart, J. B., Phys. Rev. 96, 1452 ( 1954).
Nie55 Nielsen, A. E., J. Colloid Sci. 10, 576 ( 1955).
Nie64 Nielsen, A. E., Kinetics of Precipitation (MacMillan, New York, 1964).
Nie83 Nielsen, A. E., in Treatise on Analytical Chemistry, Part 1, Vol. 3, second edition, I. M. Kolthoff and P. J. Elving, Eds. (John Wiley, New York, 1983), p. 269.
Nov80 Novgorodov, A. F., Adilbish, M., Zaitseva, N. G., Kowalew, A., and Kovacs, Z., J. Radioanal. Chem. 56, 1 ( 1980).
Nov81 Novik, S. I., Björnstad, T., Alstad, J., Brodén, K., and Skarnemark, G., Nucl. Instrum. Methods 185, 175 ( 1981).
Nuh72 Nuh, F. M., Slaughter, D. R., Shihab-Eldin, A., and Prussin, S. G., Radiochim. Acta 17, 149 ( 1972).
Nuh75 Nuh, F. M., Slaughter, D. R., Prussin, S. G., Kratz, K. L., Franz, H., and Herrmann, G., Phys. Lett. B53, 435 ( 1975).
Ohy72 Ohyoshi, A., Ohyoshi, E., Tamai, T., Takemi, H., and Shinagawa, M., J. Nucl. Sci. Technol. 9, 658 ( 1972).
Oka81 Okano, K., Kawase, Y., Kawade, K., Yamamoto, H., Hanada, M., Katoh, T., and Fujiwara, I., Nucl. Instrum. Methods 186, 115 ( 1981).
Oro55 O'Rourke, J. D., and Johnson, R. A., Anal. Chem. 27, 1699 ( 1955).
Osc82 Oscik, J., Adsorption (Ellis Harwood, Chichester, England, 1982).
Ove51 Overstreet, R., and Jacobson, L., in Radiochemical Studies: The Fission Products, National Nuclear Energy Series, Division IV, Vol. 9, Book 2, C. D. Coryell and N. Sugarman, Eds. (McGraw-Hill, New York, 1951), p. 621.
Pat70a Paterson R., An Introduction to Ion Exchange (Hayden, London, 1970), p. 90.
Pat70b Paterson R., An Introduction to Ion Exchange (Hayden, London, 1970).
Pel42 Peligot, E., Ann. Chim. Phys. 5 (3), 7 ( 1842); quoted by H. M. N. H. Irving in Treatise on Analytical Chemistry, Part I, Vol. 5, P. J. Elving, E. Grushka, and I. M. Kolthoff, Eds. ( 1982), p. 506.
Pep57a Peppard, D. F., Mason, G. W., Maier, J. L., and Driscoll, W. J, J. Inorg. Nucl. Chem. 4, 334 ( 1957).
Pep57b Peppard, D. F., Moline, S. W., and Mason, G. W., J. Inorg. Nucl. Chem. 4, 344 ( 1957).
Pep71 Peppard, D. F., Ann. Rev. Nucl. Sci. 21, 365 ( 1971).
Per89 Persson, H., Skarnemark, G., Skålberg, M., Alstad, J., Liljenzin, J. O., Bauer, G., Haberberger, F., Kaffrell, N., Rogowski, J., and Trautmann, N., Radiochim. Acta 48, 177 ( 1989).
Pio84 Piotrowski, A., Gill, R. L., and McDonald, D. C., Nucl. Instrum. Methods Phys. Res. 224, 1 ( 1984).
Pos61 Poskanzer, A. M., and Foreman, Jr., B. M., J. Inorg. Nucl. Chem. 16, 323 ( 1961).
Pru79 Prusik, Z., in Laboratory Handbook of Chromatographic and Allied Methods, O. Mikes, Ed. (Ellis Horwood, Chichester, 1979), p. 649.
Pry79 Pryde, A., and Gilbert, M. T., Applications of High Performance Liquid Chromatography (Chapman and Hall, London, 1979).
Puc60 Pucar, Z., and Jakovac, Z., J. Chromatog. 3, 477 ( 1960).
Puc62 Pucar, Z., and Jakovac, Z., J. Chromatog. 9, 106 ( 1962).
Puu73 Puumalainen, P., Aystö, J., and Valli, K., Nucl. Instrum. Methods 112, 485 ( 1973).
Qur69 Qureshi, I. H., McClendon, L. T., and LaFleur, P. D., National Bureau of Standards, Technical Note No. 458 ( 1969), p. 70.
Raj75 Rajcsanyi, P. M., and Rajcsanyi, E., High-Speed Liquid Chromatography, Chromatographic Science Series, Vol. 6 (Marcel Dekker, 1975).
Rav79 Ravn, H. L., Phys. Rpts. 54, 201 ( 1979).
Rei50 Reid, J. C., and Calvin, M., J. Amer. Chem. Soc. 72, 2948 ( 1950).
Rei66 Reichenberg, D., in Ion Exchange, Vol. 1, J. A. Marinsky, Ed. (Marcel Dekker, New York, 1966), p. 227.
Rei67 Reinhardt, H., and Rydberg, J., in Solvent Extraction Chemistry, Proc. Intern. Conf. held at Gothenburg, Sweden, 1966, D. Dyrssen, J. O. Liljenzin, and J. Rydberg, Eds. (North-Holland, Amsterdam, 1967), p. 612.
Rei69 Reinhardt, H., and Rydberg, J., Acta Chem. Scand. 23, 2773 ( 1969).
Ren64 Rengan, K., and Meinke, W. W., Anal. Chem. 1 ( 1964).
Ren65 Rengan, K., and Pierson, W. R., J. Inorg. Nucl. Chem. 27, 2113 ( 1965).
Ren66 Rengan, K., Ph.D. dissertation submitted to the University of Michigan ( 1966), p. 75.
Ren68 Rengan, K., and Griffin, H. C., J. Inorg. Nucl. Chem. 30, 2887 ( 1968).
Ren76 Rengan, K., and Griffin, H. C., Radiochem. Radioanal. Lett. 24, 1 ( 1976).
Ren77 Rengan, K., and Griffin, H. C., Radiochem. Radioanal. Lett. 29, 253 ( 1977).
Ren82a Rengan, K., Lin, J., Massey, T. N., Zendel, M., and Meyer, R. A., Radiochem. Radioanal. Lett. 50, 385 ( 1982).
Ren82b Rengan, K., Lin, J., and Meyer, R. A., Radiochem. Radioanal. Lett. 50, 393 ( 1982).
Ren82c Rengan, K., Lin, J., Zendel, M., and Meyer, R. A., Nucl. Instrum. Methods 197, 427 ( 1982).
Ren82d Rengan, K., Lin, J., and Meyer, R. A., Radiochem. Radioanal. Lett. 51, 339 ( 1982).
Ren85a Rengan, K., in Artificial Radioactivity, special lectures delivered at the International Symposium held at Pune, January 1985, K. Narayana Rao and H. C. Arnikar, Eds. (Tata McGraw-Hill, New Delhi, 1985), p. 61.
Ren85b Rengan, K., Lin, J., Lim, T., Harrell, J., and Meyer, R. A., Nucl. Instrum. Methods Phys. Res. A242, 160 ( 1985).
Ren86a Rengan, K., in Proc. Symp. on Nuclei Off the Line of Stability, Vol. II, Chicago, 1985, R. A. Meyer and D. G. Brenner, Eds. (Clark University Press, 1986), p. S96.
Ren86b Rengan, K., and Griffin, H. C., J. Radioanal. Nucl. Chem. 98, 255 ( 1986).
Ren90a Rengan, K., J. Radioanal. Nucl. Chem. 142, 173 ( 1990).
Ren90b Rengan, K., in Exotic Nuclear Spectroscopy, W. C. McHarris, Ed. (Plenum Press, 1990), p. 609.
Ric34 Rice, F. O., and Glasebrook, A. L., J. Amer. Chem. Soc. 56, 2381 ( 1934).
Ric71 Riccato, M. T., Radiochim. Acta 15, 3 ( 1971).
Rie70a Rieman III, W., and Walton, H. F., Ion Exchange in Analytical Chemistry (Pergamon Press, 1970), p. 1.
Rie70b Rieman III, W., and Walton, H. F., Ion Exchange in Analytical Chemistry (Pergamon Press, 1970), p. 14.
Rie70c Rieman III, W., and Walton, H. F., Ion Exchange in Analytical Chemistry (Pergamon Press, 1970), p. 54.
Roe86 Roeckl, E., in Nuclei Off the Line of Stability, R. A. Meyer and D. S. Brenner, Eds. (Amer. Chem. Soc. Pub., Washington, D.C., 1986), p. 439.
Rot73 Rothbart, H. L., in An Introduction to Separation Science, B. L. Karger, L. R. Snyder, and C. Horrath, Eds. (John Wiley, 1973), p. 337.
Rud73 Rudstam, G., and Grapengiesser, B., Radiochim Acta 20, 97 ( 1973).
Rud80 Rudolph, J., and Bächmann, K., Radiochim. Acta 27, 105 ( 1980).
Rud81 Rudstam, G., Aagraard, P., Hoff, P., Johansson, B., and Zwicky, H. V., Nucl. Instrum. Methods, 186, 365 ( 1981).
Run81 Runser, D. J., Maintaining and Troubleshooting HPLC Systems —A User's Guide (John Wiley, New York, 1981).
Rut00 Rutherford, E., Philos. Mag, Series 5, XLIX, 1 ( 1900).
Rut03 Rutherford, E., and Soddy, F., Philos. Mag. 5, 576 ( 1903).
Ryd69 Rydberg, J., J. Acta Chem. Scand. 23, 647 ( 1969).
Ryd80 Rydberg, J., Persson, H., Aronsson, P. O., Selme, A., and Skarnemark, G., Hydrometallurgy 5, 273 ( 1980).
Sam63 Samuelson, O., Ion Exchange Separations in Analytical Chemistry (John Wiley, New York, 1963).
Sat52 Sato, T. R., Diamond, H., Norris, W. P., and Strain, H. H., J. Amer. Chem. Soc. 74, 6154 ( 1952).
Sch57 Schumacher, E., and Streiff, H. J., Helv. Chim. Acta 40, 234 ( 1957).
Sch58 Schumacher, E., and Streiff, H. J., Helv. Chim. Acta 41, 824 ( 1958).
Sch60 Schumacher, E., and Friedli, W., Helv. Chim. Acta 43, 1706 ( 1960).
Sch69 Schüssler, H. D., Grimm, W., Weber, M., Tharun, U., Denschlag, H. O., and Herrmann, G., Nucl. Instrum. Methods 73, 125 ( 1969).
Sch72 Schüssler, H. D., and Herrmann, G., Radiochim. Acta 18, 123 ( 1972).
Sch73 Schmidt-Ott, W-D., Mlekodaj, R. L., and Bingham, C. A., Nucl. Instrum. Methods 108, 13 ( 1973).
Sch75 Schmidt-Ott, W-D., Nucl. Instrum. Methods 130, 177 ( 1975).
Sch76 Schmeing, H., Koslowsky, V., Wightman, M., Hardy, J. C. MacDonald, J. A., Faestermann, T., Andrews, H. R., Grieger, J. S., and Grahman, R. L., Nucl. Instrum. Methods 139, 335 ( 1976).
Sch77a Schädel, M., Trautmann, N., and Herrmann, G., Radiochim. Acta 24, 27 ( 1977).
Sch77b Schmidt-Ott, W. D., von Dincklage, R. D., and Hiller, W. J., Nucl. Instrum. Methods 141, 553 ( 1977).
Sch78 Schädel, M., Brüchle, W., Haefner, B., Kratz, J. V., Schorstein, W., Trautmann, N., and Herrmann, G., Radiochim. Acta 25, 111 ( 1978).
Sch79 Schwedt, G., Chromatographia 12, 613 ( 1979).
Sch88a Scherer, U. W., Kratz, J. V., Schädel, M., Brüchle, W., Gregorich, K. E., Henderson, R. A., Lee, D., Nurmia, M., and Hoffman, D. C., Inorg. Chim. Acta 146, 249 ( 1988).
Sch88b Schädel, M., Brüchle, W., and Haefner, B., Nucl. Instrum. Methods Phys. Res. A264, 308 ( 1988).
Sch89 Schädel, M., Brüchle, W., Jäger, E., Schimpf, E., Kratz, J. V., Scherer, U. W., and Zimmermann, H. P., Radiochim. Acta 48, 171 ( 1989).
Sea46a Seaborg, G. T., McMillan, E., Kennedy, J. W., and Wahl, A. C., Phys. Rev. 69, 366 ( 1946).
Sea46b Seaborg, G. T., Wahl, A. C., and Kennedy, J. W., Phys. Rev. 69, 367 ( 1946).
Sea58 Seaborg, G. T., The Transuranium Elements (Addison Wesley, Reading, MA, 1958), p. 149.
Sea82 Seaborg, G. T., in Actinides in Perspective, N. M. Edelstein, Ed. (Pergamon Press, New York, 1982), p. 1
Sem83 Semkow, T., and Wahl, A. C., J. Radioanal. Chem. 79, 93 ( 1983).
Shu62a Shukla, S. K., and Adloff, J. P., J. Chromatog. 8, 501 ( 1962).
Shu62b Shukla, S. K., and Adloff, J. P., J. Chromatog. 9, 455 ( 1962).
Shu71 Shukla, J. P., Chandrasekharan, E. S., and Rengan, K., Radiochem. Radioanal. Lett. 6, 307 ( 1971).
Shu72 Shukla, J. P., Chandrasekharan, E. S., and Rengan, K., Sep. Sci. 7, 193 ( 1972).
Sic80 Siczek, A. A., Meisenhelder, J. H., Bernstein, G. J., and Steindler, M. J., Radiochim. Acta 27, 51 ( 1980).
Sil70a Silva, R., Sikkeland, J., Nurmia, M., and Ghiorso, A., Inorg. Nucl. Chem. Lett. 6, 733 ( 1970).
Sil70b Silva, R., Harris, J., Nurmia, M., Eskola, K., and Ghiorso, A., Inorg. Nucl. Chem. Lett. 6, 871 ( 1970).
Sil77 Silva, R. J., Trautmann, N., Zendel, M., Dittner, P. F., Stender, E., and Ahrens, H., Nucl. Instrum. Methods 147, 371 ( 1977).
Ska76 Skarnemark, G., Stender, E., Trautmann, N., Aronsson, P. O., Björnstad, T., Kaffrell, N., Kvåle, E., and Skarestad, M., Radiochim. Acta 23, 98 ( 1976).
Ska77a Skarnemark, G., Aronsson, P. O., Björnstad, T., Kvåle, E., Kaffrell, N., Stender, E., and Trautmann, N., J. Inorg. Nucl. Chem. 39, 1487 ( 1977).
Ska77b Skarnemark, G., dissertation for the Degree of Doctor of Technology, Chalmers University of Technology, Göteborg, Sweden ( 1977).
Ska80 Skarnemark, G., Aronsson, P. O., Brodén, K., Rydberg, J., Björnstad, T., Kaffrell, N., Stender, E., and Trautmann, N., Nucl. Instrum. Methods 175, 323 ( 1980).
Ska83 Skarnemark, G., Brodén, K., Yun, M., Kaffrell, N., and Trautmann, N., Radiochim. Acta 33, 97 ( 1983).
Ska86a Skarnemark, G., Brodén, K., Kaffrell, N., Prussin, S. G., Trautmann, N., Rengan, K., Eriksen, D., Kusnezov, D. F., and Meyer, R. A., Z. Phys. A323, 407 ( 1986).
Ska86b Skarnemark, G., Skålberg, M., Alstad, J., and Björnstad, T., Physica Scripta 34, 597 ( 1986).
Ska90 Skarnemark, G., Alstad, J., Kaffrell, K., and Trautmann, N, J. Radioanal. Nucl. Chem. 142, 145 ( 1990).
Smi56 Smith, H. L., and Hoffman, D. C., J. Inorg. Nucl. Chem. 3, 243 ( 1956).
Smi72 Smith, I., and Feinberg, J. G., Paper and Thin Layer Chromatography and Electrophoresis, second edition (Longman, London, 1972), p. 114.
Sny75 Snyder, L. R., in Chromatography—A Laboratory Handbook of Chromatographic and Electrophoretic Methods , E. Heffman, Ed. (Van Nostrand, New York, 1975), p. 46.
Sny79 Snyder, L. R., and Kirkland, J. J., Introduction to Modern Liquid Chromatography, second edition (John Wiley, New York, 1979).
Sod05 Soddy, F., “Annual Progress Reports to Chemical Society,” reproduced in Trenn, T. J., Radioactivity and Atomic Theory, Vol. 1. (Halsted Press, 1975), p. 263.
Sod14 Soddy, F., Ann. Prog. Report Chem. Soc. 10, 266 ( 1914).
Spe47a Spedding, F. H., Voigt, A. F., Gladrow, E. M., and Sleight, N. R., J. Amer. Chem. Soc. 69, 2777 ( 1947).
Spe47b Spedding, F. H., Voigt, A. F., Gladrow, E. M., Sleight, N. R., Powell, J. E., Wright, J. M., Butler, T. A., and Figard, P., J. Amer. Chem. Soc. 69, 2786 ( 1947).
Sta64 Stary, J., The Solvent Extraction of Metal Chelates, Vol. 1 (Pergamon Press, Oxford, 1964).
Sta84 Stachel, J., Kaffrell, N., Trautmann, N., Skarnemark, G., and Eriksen, D., Z. Phys. A316, 105 ( 1984).
Ste61a Stevenson, P. C., and Nervik, W. E., U.S. Atomic Energy Commission Report NAS-NS-3020 ( 1961).
Ste61b Stevenson, P. C., and Nervik, W. E., U.S. Atomic Energy Commission Report NAS-NS-3020 ( 1961), p. 155.
Ste61c Stevenson, P. C., and Nervik, W. E., U.S. Atomic Energy Commission Report NAS-NS-3020 ( 1961), p. 156.
Ste78 Stevenson, P. C., private communication ( 1978).
Ste80 Stender, E., Trautmann, N., and Herrmann, G., Radiochem. Radioanal. Lett. 42, 291 ( 1980).
Str73 Strelow, F. W. E., in Ion Exchange and Solvent Extraction, Vol. 5, J. A. Marinsky and Y. Marcus, Eds. (Marcel Dekker, New York, 1973), p. 121.
Str74 Strickert, R. G., Amiel, S., and Wahl, A. C., Inorg. Nucl. Chem. Lett. 10, 129 ( 1974).
Sug47 Sugarman, N., J. Chem. Phys. 15, 544 ( 1947).
Sun55 Sunderman, D. N., U.S. Atomic Energy Commission Report AECU-3159 ( 1955).
Sun57 Sunderman, D. N., and Meinke, W. W., Anal. Chem. 29, 1578 ( 1957).
Syn90 Synovec, R. E., Johnson, E. L., Moore, L. K., and Renn, C. N., Anal. Chem. 62, 357R ( 1990).
Tal86 Talbert, Jr., W. L., and Bunker, M. E., in Nuclei Off the Line of Stability, R. A. Meyer and D. S. Brenner, Eds. (Amer. Chem. Soc. Pub., Washington, D.C., 1986), p. 426.
Tal87 Talbert, Jr., W. L., and Bunker, M. E., in Nuclear Structure, Reactions, and Symmetries, R. A. Meyer and V. Paar, Eds. (World Scientific Pub. Pte., Singapore, 1987), p. 645.
Tet86 Tetzlaff, H., Hermann, G., Kaffrell, N., Kratz, J. V., Rogowski, J., Trautmann, N., Skålberg, M., Skarnemark, G., Alstad, J., Fowler, M. M., Moody, K. J., Brüchle, W., Gaggeler, H., Schädel, M., Sümmerer, K., J. Less-Common Metals, 441 ( 1986).
Tho50 Thompson, H. S., J. Roy. Agr. Soc. Engl. 11, 68 ( 1850), quoted in Rieman III, W., and Breyer, A. C., in Treatise on Analytical Chemistry, Part I, Vol. 3, I. M. Kolthoff and P. J. Elving, Eds.(Interscience, 1961), p. 1521.
Tho54 Thompson, S. G., Harvey, B. G., Choppin, G. R., and Seaborg, G. T., J. Amer. Chem. Soc. 76, 6229 ( 1954).
Tom47 Tompkins, E. R., Khym, J. X., and Cohn, W. E., J. Amer. Chem. Soc. 69, 2769 ( 1947).
Tom68 Tomlinson, L., and Hurdus, M. H., J. Inorg. Nucl. Chem. 30, 1125 ( 1968).
Tom71 Tomlinson, L., and Wahl, A. C., J. Inorg. Nucl. Chem. 33, 3609 ( 1971).
Tom73 Tomlinson, L., and Hurdus, M. H., Radiochim. Acta 17, 199 ( 1973).
Tra72 Trautmann, N., Kaffrell, N., Behlich, H. W., Folger, H., Herrmann, G., Hubscher, D., and Ahrens, H., Radiochim. Acta 18, 86 ( 1972).
Tra75 Trautmann, N., Aronsson, P. O., Björnstad, T., Kaffrell, N., Kvåle, E., Skarestad, M., Skarnemark, G., and Stender, E., Inorg. Nucl. Chem. Lett. 11, 729 ( 1975).
Tra76a Trautmann, N., and Herrmann, G., J. Radioanal. Chem. 32, 533 ( 1976).
Tra76b Trautmann, N., Kaffrell, N., Ahrens, H., and Dittner, P. F., Phys. Rev. C 13, 872 ( 1976).
Tra81 Trautmann, N., Greulich, N., Hickmann, U., Kaffrell, N, Stender, E., Zendel, M., Gäggler, H., Brodén, K., Skarnemark, G., and Eriksen, D., in Proc. Fourth Intern. Conf. on Nuclei Far from Stability (Helsingor, Denmark, 1981), Report CERN 81-09, p. 727.
Tra85 Trautmann, N., in Artificial Radioactivity, special lectures delivered at the International Symposium held at Pune, January 1985, K. Narayana Rao and H. C. Arnikar, Eds. (Tata McGraw-Hill, New Delhi 1985), p. 12.
Tsu83 Tsuneyoshi, T., Yamamoto, M., and Otozai, K., Radiochim. Acta 33, 135 ( 1983).
Vin81 Vine, E. N., and Wahl, A. C., J. Inorg. Nucl. Chem. 43, 877 ( 1981).
Wal67a Walton, A. G., The Formation and Properties of Precipitates (Interscience, 1967), p. 98.
Wal67b Walton, A. G., The Formation and Properties of Precipitates (Interscience, 1967).
Wal67c Walton, A. G., The Formation and Properties of Precipitates (Interscience, 1967), p. 143.
Wal80 Walton, H. F., Anal. Chem. 52, 15R ( 1980).
Was66 Wasowicz, S., and Rutkowski, S., Chem. Anal. (Warsaw) 11, 603 ( 1966), quoted in reference Min82e.
Way50 Way, T. J., J. Roy. Agr. Soc. Engl. 11, 313 ( 1850),quoted in Rieman III, W., and Breyer, A. C., Treatise on Analytical Chemistry, Part I, Vol. 3, I. M. Kolthoff and P. J. Elving, Eds.(Interscience, 1961), p. 1521.
Wea74 Weaver, B., Ion Exchange and Solvent Extraction 6, 189 ( 1974).
Weg69 Wegener, P. P., in Nonequilibrium Flows, P. P. Wegener, Ed. (Yale University, 1969), Chap. 4.
Wei75 Weiffenbach, C., Gujrathi, S. C., Lee, J. K. P., and Houdayer, A., Nucl. Instrum. Methods 125, 245 ( 1975).
Wei81 Weis, M., and Denschlag, H. O., J. Inorg. Nucl. Chem. 43, 437 ( 1981).
Wei87 Weis, M., Ahrens, H., Denschlag, H. O., Fariwar, M., Herrmann, G., and Trautmann, N., Radiochim. Acta 42, 201 ( 1987).
Wes69 Westgaard, L., Rudstam, G., and Jonsson, O. C., J. Inorg. Nucl. Chem. 31, 3747 ( 1969).
Wes91 Westgaard, L., and Westgaard, J., Med. Tusand. Tak. 28, 10 ( 1991).
Whi61 White, J. C., and Ross, W. J., U.S. Atomic Energy Commission Report No. NAS-NS-3101 ( 1961).
Wie72 Wien, K., Fares, Y., and MacFarlane, R. D., Nucl. Instrum. Methods 103, 181 ( 1972).
Wie73 Wiesehahn, W. J., D'Auria, J. M., Dautet, H., and Pate, B. D., Can. J. Phys. 51, 2347 ( 1973).
Wie74 Wiesehahn, W. J., D'Auria, J. M., and Irwin, J. C., Nucl. Instrum. Methods 114, 401 ( 1974).
Wie75a Wieme, R. J., in Chromatography, a Laboratory Handbook of Chromatographic and Electrophoretic Methods, third edition, E. Heftmann, Ed. (Van Nostrand, New York, 1975), p. 228.
Wie75b Wiesehahn, W. J., Bischoff, G., and D'Auria, J. M., Nucl. Instrum. Methods 124, 221 ( 1975).
Wie75c Wiesehahn, W. J., Bischoff, G., and D'Auria, J. M., Nucl. Instrum. Methods 129, 187 ( 1975).
Wil74 Wilhelm, H. G., Junglas, H., Wollnik, H., Snider, D. F., Brandt, R., and Lust, K. H., Nucl. Instrum. Methods 115, 419 ( 1974).
Wol75 Wollnik, H., Wilhelm, H. G., Robig, G., and Junglas, H., Nucl. Instrum. Methods 127, 539 ( 1975).
Wol76 Wollnik, H., Nucl. Instrum. Methods 139, 311 ( 1976).
Wol77 Wollnik, H., Porstendorfer, J., Robig, G., and Wilhelm, H. G., Nucl. Instrum. Methods 144, 247 ( 1977).
Yam80 Yamamoto, H., Ikeda, Y., Kawade, K., Katoh, T., and Nagahara, T., J. Inorg. Nucl. Chem. 42, 1539 ( 1980).
Yam81 Yamamoto; H., Ikeda, Y., Kawade, K., Katoh, T., and Nagahara, T., J. Inorg. Nucl. Chem. 43, 855 ( 1981).
Zen78 Zendel, M., Stender, E., Trautmann, N., and Herrmann, G., Nucl. Instrum. Methods 153, 149 ( 1978).
Zen80 Zendel, M., Trautmann, N., and Herrmann, G., J. Inorg. Nucl. Chem. 42, 1387 ( 1980).
Zen81 Zendel, M., Trautmann, N., and Herrmann, G., Radiochim. Acta 29, 17 ( 1981).
Zen87 Zendel, M., Prussin, S. G., Rengan, K., Massey, T. N., and Meyer, R. A., Nucl. Instrum. Methods Phys. Res. A256, 180 ( 1987).
Zir74a Zirnheld, J. P., Nucl. Instrum. Methods 120, 171 ( 1974).
Zir74b Zirnheld, J. P., Nucl. Instrum. Methods 120, 179 ( 1974).
Zir79 Zirnheld, J. P., Schutz, L., and Wohn, F. K., Nucl. Instrum. Methods 158, 409 ( 1979).
Zol70 Zolotov, Yu. A., Extraction of Chelate Compounds (Ann Arbor-Humphrey Science, Ann Arbor, 1970).
Zva66 Zvara, I., Chuburkov, Yu. T., Tsaletka, R., Zvarova, T. S., Shalaevskii, M. R., and Shilov, B. V., Sov. J. At. En. 21, 709 ( 1966).
Zva69 Zvarova, T. S., and Zvara, I., J. Chromatog. 44, 604 ( 1969).
Zva70a Zvara, I., Chuburkov, T. Yu., Belov, V. Z., Buklanov, G. V., Zakhvataev, B. B., Zvarova, T. S., Maslov, O. D., Caletka, R., and Shalaevsky, M. R, J. Inorg. Nucl. Chem. 32, 1885 ( 1970).
Zva70b Zvarova, T. S., and Zvara, I., J. Chromatog. 49, 290 ( 1970).
Zva71 Zvara, I., Belov, V. Z., Chelnokov, L. P., Domanov, V. P., Hussonois, M., Korotkin, Yu. S., Schegolev, V. A., and Schalayevsky, M. R., Inorg. Nucl. Chem. Lett. 1, 1109 ( 1971).
Zva72 Zvara, I., Belov, V. Z., Domanov, V. P., Korotkin, Yu. S., Chelnokov, L. P., Shalaevskii, M. R., Schegolev, V. A., and Yussonnua, M., Sov. Radiochemistry (Eng. Tr.) 14, 115 ( 1972).
Zva75 Zvara, J., Keller, Jr., O. L., Silva, R. J., and Tarrant, J. R., J. Chromatog. 103, 77 ( 1975).
Zva76 Zvara, I., Belov, V. Z., Domanov, V. P., and Shalaevskii, M. R., Sov. Radiochemistry (Eng. Tr.) 18, 328 ( 1976).
Zva85 Zvara, I., Radiochim. Acta 38, 95 ( 1985).
Zyc83 Zychor, I., Rykaczcwski, K. Ahrens, H., Folger, H., Kurcewicz, W., Sümmerer, K., Kaffrell, N., and Trautmann, N., Radiochim. Acta 33, 1 ( 1983).
1. Introduction
In this part of the monograph, we present ultrafast radiochemical separation procedures that are available from the literature. Figure 48 shows the elements for which procedures are included. Table 12 provides a summary and key to the procedures. Each individual listing gives a short summary of the separation procedure and references the source article.
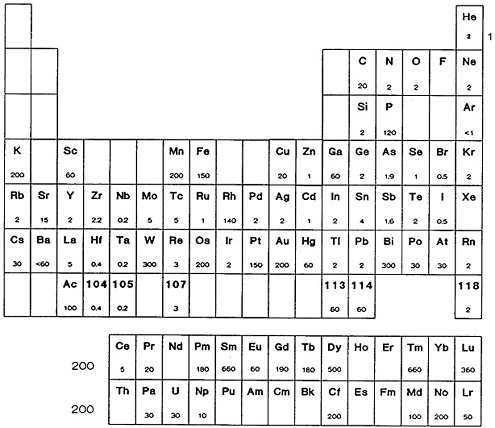
Figure 48. Elements for which fast chemistry procedures are included in Part II. The number below the element symbol gives the separation time (in seconds) for the fastest general procedure. The number on the right of the noble gas group and on the left of the lanthanides and actinides is the time reported for the generic procedure for that group.
Table 12. List of ultrafast radiochemical separation procedures.
|
Element |
Proc. # |
Mode |
Technique |
Time |
|
Actinides |
An-1 |
Autobatch |
Gas chromatography |
Few min |
|
Actinium |
Ac-1 |
Batch |
Extraction |
3 min |
|
Actinium |
Ac-2 |
Batch |
Extraction, ion exchange |
100 s |
|
Antimony |
Sb-1 |
Autobatch |
Volatilization |
2.7 s |
|
Antimony |
Sb-2 |
Batch |
Volatilization |
5.4 s |
|
Antimony |
Sb-3 |
Batch |
Volatilization |
10 s |
|
Antimony |
Sb-4 |
Batch |
Volatilization |
6 s |
|
Antimony |
Sb-5 |
Autobatch |
Volatilization |
1.6 s |
|
Antimony |
Sb-6 |
Batch |
Extraction |
~1 min |
|
Antimony |
Sb-7 |
Batch |
Volatilization |
10 to 15 s |
|
Antimony |
Sb-8 |
Batch |
Volatilization |
~10 s |
|
Antimony |
Sb-9 |
Batch |
Electrolysis |
10 to 20 s |
|
Antimony (As) |
Sb-10 |
Batch |
Volatilization |
<10 s |
|
Antimony (for Te) |
Sb-11 |
Autobatch |
Volatilization |
7.3 s |
|
Antimony |
Sb-12 |
Batch |
Plating, chemical |
<1 min |
|
Antimony |
Sb-13 |
Batch |
Volatilization |
Few s |
|
Argon |
Ar-1 |
Batch |
Condensation |
<1 s |
|
Arsenic |
As-1 |
Autobatch |
Volatilization |
2.5 s |
|
Arsenic |
As-2 |
Autobatch |
Volatilization |
5 s |
|
Arsenic |
As-3 |
Batch |
Volatilization |
5.A s |
|
Arsenic |
As-4 |
Continuous |
Adsorption, extraction |
Few s |
|
Arsenic |
As-5 |
Batch |
Volatilization |
10 s |
|
Arsenic |
As-6 |
Continuous |
Extraction |
3 or 4 s |
|
Arsenic |
As-7 |
Autobatch |
Volatilization |
1.9 s |
|
Arsenic |
As-8 |
Batch |
Volatilization |
~10 s |
|
Arsenic |
As-9 |
Batch |
Electrolysis |
10 to 20 s |
|
Arsenic (Sb) |
As-10 |
Batch |
Volatilization |
<10 s |
|
Arsenic |
As-11 |
Continuous |
Extraction |
1.0 s |
|
Astatine |
At-1 |
Batch |
Volatilization |
~30 s |
|
Astatine |
At-2 |
Batch |
Extraction |
Few min |
|
Astatine |
At-3 |
Batch |
Volatilization |
<30 s |
|
Astatine |
At-4 |
Batch |
Extraction |
90 s |
|
Barium |
Ba-1 |
Batch |
Electrophoresis |
<1 min |
|
Barium |
Ba-2 |
Continuous/batch |
Extraction |
<1 min |
|
Barium |
Ba-3 |
Batch |
Precipitation |
Few min |
|
Barium |
Ba-4 |
Batch |
Precipitation |
Few min |
|
Barium (from Cs) |
Ba-5 |
Continuous |
Ion exchange |
Few s |
|
Barium (from La) |
Ba-6 |
Batch |
Precipitation |
3 to 4 s |
|
Bismuth |
Bi-1 |
Batch |
Extraction |
5 min |
|
Bismuth (+Po) |
Bi-2 |
Batch |
Precipitation |
4 min |
|
Bromine |
Br-1 |
Batch |
Volatilization |
Few s |
|
Bromine |
Br-2 |
Batch/continuous |
Hot atom, gas phase |
10 s |
|
Bromine |
Br-3 |
Batch |
Volatilization |
8 s |
|
Bromine |
Br-4 |
Continuous |
Adsorption |
~1 s |
|
Bromine |
Br-5 |
Batch |
Electrophoresis |
40 s |
|
Bromine |
Br-6 |
Continuous/batch |
Hot-atom reaction |
2 s |
|
Bromine |
Br-7 |
Continuous |
Volatilization |
~1 s |
|
Bromine |
Br-8 |
Batch |
Hot-atom reaction |
0.5 s |
|
Bromine |
Br-9 |
Continuous |
Extraction |
~5 s |
|
Bromine |
Br-10 |
Batch |
Gas chromatography |
Few min |
|
Bromine |
Br-11 |
Batch |
Volatilization |
Few s |
|
Bromine |
Br-12 |
Batch |
Extraction |
30 s |
|
Bromine |
Br-13 |
Batch |
Volatilization |
<1 min |
|
Bromine |
Br-14 |
Batch |
Extraction |
100 s |
|
Bromine |
Br-15 |
Batch |
Distillation |
20 to 25 s |
|
Cadmium |
Cd- 1 |
Continuous |
Thermochromatography |
~1 s |
|
Cadmium |
Cd-2 |
Batch |
Extraction |
5 to 10 s |
|
Cadmium |
Cd-3 |
Continuous |
Extraction |
~2 s |
|
Cadmium |
Cd-4 |
Batch |
Thermochromatography |
Few s |
|
Cadmium |
Cd-5 |
Batch |
Extraction |
2 min |
|
Californium |
Cf-1 |
Batch |
Ion exchange |
Few min |
|
Carbon |
C-1 |
Batch |
Precipitation |
<1 min |
|
Carbon |
C-2 |
Batch |
Precipitation |
20 s |
|
Carbon |
C-3 |
Batch |
Volatilization |
Few min |
|
Cerium |
Ce-1 |
Batch |
Extraction |
~5 min |
|
Cerium |
Ce-2 |
Batch |
Electrophoresis |
2 to 3 min |
|
Cerium |
Ce-3 |
Continuous |
Extraction |
~5 s |
|
Cerium |
Ce-4 |
Continuous |
Extraction |
10 to 20 s |
|
Cerium |
Ce-5 |
Continuous |
Extraction |
<5 s |
|
Cerium |
Ce-6 |
Batch |
Electrophoresis |
6.5 min |
|
Cerium |
Ce-7 |
Batch |
Extraction |
2 min |
|
Cesium |
Cs- 1 |
Batch |
Electrophoresis |
~40 s |
|
Cesium |
Cs-2 |
Batch |
Precipitation |
3.5 min |
|
Cesium |
Cs-3 |
Batch |
Precipitation |
<3 min |
|
Cesium |
Cs-4 |
Batch |
Precipitation |
~30 s |
|
Copper |
Cu-1 |
Continuous |
Extraction |
20 s |
|
Copper |
Cu-2 |
Batch |
Plating, chemical |
~1 min |
|
Copper |
Cu-3 |
Batch |
Precipitation |
~1 min |
|
Copper |
Cu-4 |
Batch |
Precipitation |
4 to 5 min |
|
Copper |
Cu-5 |
Batch |
Plating, chemical |
2 min |
|
Dysprosium |
Dy-1 |
Autobatch |
Extraction, ion exchange |
<9 min |
|
Europium |
Eu-1 |
Batch |
Extraction |
1 min |
|
Gadolinium |
Gd-1 |
Autobatch |
Extraction, ion exchange |
3.2 min |
|
Gallium |
Ga-1 |
Batch |
Extraction |
Few min |
|
Gallium |
Ga-2 |
Batch |
Extraction |
~1 min |
|
Germanium |
Ge-1 |
Autobatch |
Volatilization |
10 s |
|
Germanium |
Ge-2 |
Autobatch |
Volatilization |
~5 s |
|
Germanium |
Ge-3 |
Continuous |
Adsorption, extraction |
Few s |
|
Germanium |
Ge-4 |
Batch |
Distillation |
25 s |
|
Germanium |
Ge-5 |
Batch/autobatch |
Distillation |
25 s |
|
Gold |
Au-1 |
Batch |
Extraction |
~4 min |
|
Gold |
Au-2 |
Batch |
Extraction |
Few min |
|
Hafnium |
Hf-1 |
Continuous |
Thermochromatography |
~0.4 s |
|
Hafnium |
Hf-2 |
Batch |
Extraction, precipitation |
5 min |
|
Hafnium |
Hf-3 |
Autobatch |
Ion exchange |
10 to 20 s |
|
Halogens |
Hal-1 |
Batch/continuous |
Emanation |
~1 s |
|
Halogens |
Hal-2 |
Batch/continuous |
Hot-atom reaction |
~2 s |
|
Helium |
He-1 |
Batch |
Emanation |
2 s |
|
Indium |
In-1 |
Batch |
Extraction |
5 to 10 s |
|
Indium |
In-2 |
Continuous |
Extraction |
~2 s |
|
Indium |
In-3 |
Batch |
Extraction |
Few s |
|
Iodine |
I-1 |
Batch |
Volatilization |
Few s |
|
Iodine |
I-2 |
Continuous |
Hot/atom reaction |
2 s |
|
Iodine |
I-3 |
Batch |
Hot/atom reaction |
0.5 s |
|
Iodine |
I-4 |
Continuous |
Extraction |
~5 s |
|
Iodine |
I-5 |
Batch |
Gas chromatography |
~1 min |
|
Iodine |
I-6 |
Batch |
Exchange |
52 s |
|
Iodine |
I-7 |
Batch |
Exchange |
2 s |
|
Iodine |
I-8 |
Batch |
Volatilization |
~30 s |
|
Iodine |
I-9 |
Batch |
Extraction |
28 s |
|
Iodine |
I-10 |
Batch |
Extraction |
28 s |
|
Iodine |
I-11 |
Batch |
Volatilization |
~10 to 15 s |
|
Iridium |
Ir-1 |
Batch |
Thermochromatography |
3 to 5 min |
|
Iridium |
Ir-2 |
Batch |
Volatilization |
Few s |
|
Iridium |
Ir-3 |
Batch/continuous |
Ion exchange |
Few s |
|
Iron |
Fe-1 |
Batch |
Precipitation |
3 to 5 min |
|
Iron |
Fe-2 |
Batch |
Extraction |
Few min |
|
Iron |
Fe-3 |
Batch |
Ion exchange |
150 s |
|
Krypton |
Kr-1 |
Batch |
Emanation |
Few s |
|
Krypton |
Kr-2 |
Batch |
Volatilization |
<1 min |
|
Lanthanides |
Ln-1 |
Autobatch |
Extraction, ion exchange |
<3 min |
|
Lanthanides |
Ln-2 |
Batch |
Electrophoresis |
~5 min |
|
Lanthanides |
Ln-3 |
Autobatch |
Gas chromatography |
Few min |
|
Lanthanides |
Ln-4 |
Batch |
Extraction |
15 to 20 s |
|
Lanthanum |
La-1 |
Batch |
Electrophoresis |
3 min |
|
Lanthanum |
La-2 |
Continuous |
Extraction |
10 to 20 s |
|
Lanthanum |
La-3 |
Continuous |
Extraction |
<5 s |
|
Lanthanum |
La-4 |
Batch |
Electrophoresis |
40 s |
|
Lanthanum |
La-5 |
Continuous |
Extraction, ion exchange |
~4 min |
|
Lanthanum |
La-6 |
Batch |
Electrophoresis |
4.5 min |
|
Lanthanum |
La-7 |
Batch |
Precipitation |
Few min |
|
Lanthanum |
La-8 |
Batch |
Precipitation |
1.7 min |
|
Lawrencium |
Lr-1 |
Batch |
Extraction |
50 s |
|
Lawrencium |
Lr-2 |
Batch |
Extraction |
3 min |
|
Lawrencium |
Lr-3 |
Batch |
Ion exchange |
6 min |
|
Lawrencium |
Lr-4 |
Autobatch |
Cation exchange (HPLC) |
160 s |
|
Lawrencium (from Md) |
Lr-5 |
Autobatch |
Extraction (HPLC) |
1 to 2 min |
|
Lead |
Pb-1 |
Batch/continuous |
Ion exchange |
Few s |
|
Lead |
Pb-2 |
Batch |
Ion exchange |
3 s |
|
Lead |
Pb-3 |
Continuous/batch |
Extraction |
<1 min |
|
Lead |
Pb-4 |
Batch |
Extraction |
5 min |
|
Lutetium |
Lu-1 |
Batch |
Extraction, precipitation |
6 min |
|
Manganese |
Mn-1 |
Batch |
Precipitation |
Few min |
|
Manganese |
Mn-2 |
Batch |
Precipitation |
Few min |
|
Mendelevium |
Md-1 |
Batch |
Ion exchange |
~5 min |
|
Mendelevium |
Md-2 |
Autobatch |
Cation exchange (HPLC) |
200 s |
|
Mendelevium (from Lr) |
Md-3 |
Autobatch |
Extraction (HPLC) |
1 to 2 min |
|
Mercury |
Hg-1 |
Batch |
Thermochromatography |
3 to 5 min |
|
Mercury |
Hg-2 |
Batch |
Volatilization |
1 to 5 min |
|
Mercury |
Hg-3 |
Batch |
Precipitation |
Few min |
|
Mercury |
Hg-4 |
Batch |
Volatilization |
<1 min |
|
Molybdenum |
Mo-1 |
Autobatch |
Extraction |
5 s |
|
Molybdenum |
Mo-2 |
Batch |
Thermochromatography |
3 to 5 min |
|
Molybdenum |
Mo-3 |
Batch |
Precipitation |
~2 min |
|
Molybdenum |
Mo-4 |
Batch |
Precipitation |
2 min |
|
Neon |
Ne-1 |
Batch |
Adsorption |
Few s |
|
Neptunium |
Np-1 |
Continuous |
Extraction |
10 s |
|
Neptunium |
Np-2 |
Autobatch |
Extraction, ion exchange |
2.7 min |
|
Niobium |
Nb-1 |
Autobatch |
Adsorption |
2.2 to 2.4 s |
|
Niobium |
Nb-2 |
Continuous |
Extraction |
~9 s |
|
Niobium |
Nb-3 |
Autobatch |
Adsorption |
2.2 s |
|
Niobium |
Nb-4 |
Batch |
Extraction |
<1 min |
|
Niobium |
Nb-5 |
Continuous |
Thermochromatography |
0.1 to 0.2 s |
|
Niobium |
Nb-6 |
Batch |
Thermochromatography |
<3 min |
|
Niobium |
Nb-7 |
Batch |
Recoil |
Few s |
|
Niobium |
Nb-8 |
Autobatch |
Adsorption |
~5 s |
|
Niobium |
Nb-9 |
Continuous |
Thermochromatography |
12 s |
|
Nitrogen |
N-1 |
Batch |
Volatilization |
5 to 10 s |
|
Nitrogen |
N-2 |
Batch |
Emanation |
8 s |
|
Nitrogen |
N-3 |
Batch |
Volatilization |
Few s |
|
Nobelium |
No-1 |
Batch |
Ion exchange |
Few min |
|
Noble gases |
Ng-1 |
Batch |
Emanation |
Few s |
|
Noble gases |
Ng-2 |
Batch/continuous |
Emanation |
~1 s |
|
Osmium |
Os-1 |
Batch |
Thermochromatography |
3 to 5 min |
|
Osmium |
Os-2 |
Batch |
Thermochromatography |
<5 min |
|
Osmium |
Os-3 |
Batch |
Distillation |
Few min |
|
Oxygen |
O-1 |
Batch |
Sorption |
~1 min |
|
Oxygen |
O-2 |
Batch |
Sorption |
Few s |
|
Palladium |
Pd- 1 |
Autobatch |
Extraction |
135 s |
|
Palladium |
Pd-2 |
Autobatch |
Extraction |
<1 min |
|
Palladium |
Pd-3 |
Batch |
Plating, chemical |
~2 s |
|
Phosphorus |
P-1 |
Batch |
Adsorption |
2 min |
|
Platinum |
Pt-1 |
Batch |
Precipitation |
2 to 3 min |
|
Polonium |
Po-1 |
Batch |
Plating, chemical |
~1 min |
|
Polonium |
Po-2 |
Batch |
Extraction |
<1 min |
|
Polonium (+Bi) |
Po-3 |
Batch |
Precipitation |
4 min |
|
Polonium |
Po-4 |
Batch |
Volatilization |
<30 s |
|
Potassium |
K-1 |
Batch |
Precipitation |
Few min |
|
Potassium |
K-2 |
Batch |
Precipitation |
Few min |
|
Praseodymium |
Pr-1 |
Batch |
Electrophoresis |
3 min |
|
Praseodymium |
Pr-2 |
Continuous |
Extraction |
10 to 20 s |
|
Praseodymium |
Pr-3 |
Batch |
Electrophoresis |
3 min |
|
Praseodymium |
Pr-4 |
Batch |
Extraction |
~5 min |
|
Praseodymium |
Pr-5 |
Batch |
Electrophoresis |
6.5 min |
|
Promethium |
Pm-1 |
Autobatch |
Extraction, ion exchange |
<3 min |
|
Protactinium |
Pa-1 |
Batch |
Extraction |
~30 s |
|
Protactinium |
Pa-2 |
Batch |
Extraction |
~2 min |
|
Protactinium |
Pa-3 |
Autobatch |
Extraction |
2.3 min |
|
Radon |
Rn-1 |
Batch |
Volatilization |
40 s |
|
Radon |
Rn-2 |
Continuous |
Condensation |
Few s |
|
Rhenium |
Re-1 |
Batch |
Thermochromatography |
<3 min |
|
Rhenium |
Re-2 |
Batch |
Thermochromatography |
3 to 5 min |
|
Rhenium |
Re-3 |
Batch |
Thermochromatography |
~3 min |
|
Rhenium |
Re-4 |
Batch |
Thermochromatography |
~5 min |
|
Rhenium |
Re-5 |
Continuous |
Thermochromatography |
<3 s |
|
Rhenium |
Re-6 |
Batch |
Extraction |
Few min |
|
Rhodium |
Rh-1 |
Batch |
Distillation |
140 s |
|
Rubidium |
Rb-1 |
Batch |
Recoil |
Few min |
|
Rubidium |
Rb-2 |
Batch |
Precipitation |
3 min |
|
Rubidium |
Rb-3 |
Batch |
Precipitation |
~1 min |
|
Rubidium |
Rb-4 |
Batch |
Precipitation |
Few s |
|
Ruthenium |
Ru-1 |
Continuous |
Volatilization |
1 s |
|
Ruthenium |
Ru-2 |
Batch |
Extraction |
40 to 50 s |
|
Ruthenium |
Ru-3 |
Autobatch |
Extraction |
8.3 s |
|
Ruthenium |
Ru-4 |
Batch |
Distillation |
~60 s |
|
Ruthenium |
Ru-5 |
Batch |
Distillation |
~5 s |
|
Ruthenium |
Ru-6 |
Batch |
Volatilization |
~5 s |
|
Ruthenium |
Ru-7 |
Batch |
Distillation |
30 to 40 s |
|
Ruthenium |
Ru-8 |
Continuous |
Extraction |
5 to 6 s |
|
Ruthenium |
Ru-9 |
Batch |
Thermochromatography |
3 min |
|
Ruthenium |
Ru-10 |
Batch |
Distillation |
~1 min |
|
Ruthenium |
Ru-11 |
Batch |
Distillation |
~3 min |
|
Samarium |
Sm-1 |
Autobatch |
Extraction, ion exchange |
~11 min |
|
Scandium |
Sc-1 |
Batch |
Extraction |
~1 min |
|
Selenium |
Se-1 |
Autobatch |
Volatilization, extraction |
5 s |
|
Selenium |
Se-2 |
Batch |
Exchange |
1.0 to 2.3 s |
|
Selenium |
Se-3 |
Batch |
Volatilization |
4 to 8 s |
|
Selenium |
Se-4 |
Continuous |
Thermal decomposition |
1 to 2 s |
|
Selenium |
Se-5 |
Continuous |
Adsorption |
~1 s |
|
Selenium |
Se-6 |
Batch |
Electrophoresis |
<1 min |
|
Selenium |
Se-7 |
Batch |
Precipitation |
45 to 50 s |
|
Selenium (Te) |
Se-8 |
Batch |
Volatilization |
<10 s |
|
Selenium |
Se-9 |
Batch |
Volatilization |
~5 s |
|
Silicon |
Si-1 |
Continuous |
Condensation |
Few s |
|
Silver |
Ag-1 |
Batch |
Exchange |
14 to 29 s |
|
Silver |
Ag-2 |
Autobatch |
Exchange |
4.1 s |
|
Silver |
Ag-3 |
Batch |
Plating, chemical |
~3 s |
|
Silver |
Ag-4 |
Batch |
Exchange |
~20 s |
|
Silver |
Ag-5 |
Batch |
Exchange |
~20 s |
|
Silver |
Ag-6 |
Batch |
Extraction |
5 to 10 s |
|
Silver |
Ag-7 |
Continuous |
Extraction |
~2 s |
|
Silver |
Ag-8 |
Batch |
Ion exchange |
Few min |
|
Silver |
Ag-9 |
Batch |
Recoil |
<1 min |
|
Silver |
Ag-10 |
Batch |
Precipitation |
<1 min |
|
Silver |
Ag-11 |
Batch |
Plating, chemical |
~2 s |
|
Strontium |
Sr-1 |
Batch |
Electrophoresis |
<1 min |
|
Strontium |
Sr-2 |
Batch |
Precipitation |
3 min |
|
Strontium |
Sr-3 |
Batch |
Exchange |
Few min |
|
Strontium |
Sr-4 |
Batch |
Precipitation |
~15 s |
|
Strontium |
Sr-5 |
Batch |
Precipitation |
~1.7 min |
|
Tantalum |
Ta-1 |
Continuous |
Thermochromatography |
0.1 to 0.2 s |
|
Tantalum |
Ta-2 |
Batch |
Extraction |
3 to 5 min |
|
Tantalum |
Ta-3 |
Autobatch |
Extraction |
10 to 20 s |
|
Technetium |
Tc-1 |
Batch |
Exchange |
5 s |
|
Technetium |
Tc-2 |
Batch |
Volatilization |
20 to 30 s |
|
Technetium |
Tc-3 |
Autobatch |
Extraction |
2.5 s |
|
Technetium |
Tc-4 |
Autobatch |
Volatilization |
1 s |
|
Technetium |
Tc-5 |
Continuous |
Extraction |
7 s |
|
Technetium |
Tc-6 |
Autobatch |
Extraction |
7.5 s |
|
Technetium |
Tc-7 |
Continuous |
Extraction |
7 s |
|
Technetium |
Tc-8 |
Continuous |
Extraction |
5 s |
|
Technetium |
Tc-9 |
Batch |
Precipitation |
5 to 6 s |
|
Tellurium |
Te-1 |
Autobatch |
Volatilization |
5.0 s |
|
Tellurium |
Te-2 |
Batch |
Exchange |
<1 min |
|
Tellurium |
Te-3 |
Continuous |
Thermal decomposition |
1 to 2 s |
|
Tellurium |
Te-4 |
Batch/continuous |
Volatilization |
~20 s |
|
Tellurium (Se) |
Te-5 |
Batch |
Volatilization |
<10 s |
|
Tellurium |
Te-6 |
Batch |
Precipitation |
Few min |
|
Terbium |
Tb-1 |
Autobatch |
Extraction, ion exchange |
<3 min |
|
Thallium |
Tl-1 |
Batch |
Extraction |
5 s |
|
Thallium |
Tl-2 |
Batch |
Ion exchange |
~2 s |
|
Thallium |
Tl-3 |
Batch |
Thermochromatography |
2 to 3 min |
|
Thallium |
Tl-4 |
Batch |
Precipitation |
5 min |
|
Thulium |
Tm-1 |
Batch |
Extraction chromatography |
11 min |
|
Tin |
Sn-1 |
Batch |
Volatilization |
~10 s |
|
Tin |
Sn-2 |
Batch |
Volatilization |
45 s |
|
Tin |
Sn-3 |
Batch |
Extraction |
~20 s |
|
Tin |
Sn-4 |
Batch |
Extraction |
45 s |
|
Tin |
Sn-5 |
Batch |
Precipitation |
~45 s |
|
Tin |
Sn-6 |
Batch |
Volatilization |
4 s |
|
Tin |
Sn-7 |
Batch |
Extraction |
Few min |
|
Tin |
Sn-8 |
Batch |
Extraction |
3 min |
|
Tin |
Sn-9 |
Batch |
Exchange |
2 min |
|
Tungsten |
W-1 |
Batch |
Thermochromatography |
~5 min |
|
Tungsten |
W-2 |
Autobatch |
Ion exchange |
10 to 20 s |
|
Uranium |
U-1 |
Continuous |
Extraction |
<1 min |
|
Uranium |
U-2 |
Continuous |
Extraction |
<1 min |
|
Uranium |
U-3 |
Batch |
Extraction |
1.4 min |
|
Uranium |
U-4 |
Batch |
Extraction |
Few min |
|
Uranium |
U-5 |
Batch |
Extraction |
~30 s |
|
Xenon |
Xe-1 |
Batch |
Emanation |
Few s |
|
Xenon |
Xe-2 |
Batch |
Emanation |
Few s |
|
Yttrium |
Y-1 |
Batch |
Extraction |
100 s |
|
Yttrium |
Y-2 |
Autobatch |
Ion exchange |
10 s |
|
Yttrium |
Y-3 |
Batch |
Extraction |
Few min |
|
Yttrium |
Y-4 |
Batch |
Extraction |
Few s |
|
Yttrium |
Y-5 |
Batch |
Precipitation |
40 s |
|
Yttrium |
Y-6 |
Batch |
Precipitation |
Few min |
|
Yttrium |
Y-7 |
Batch |
Ion exchange |
Few min |
|
Z = 104 |
104-1 |
Autobatch |
Extraction |
2 to 3 min |
|
Z = 104 |
104-2 |
Continuous |
Thermochromatography |
~0.4 s |
|
Z = 104 |
104-3 |
Continuous |
Volatilization |
<1 s |
|
Z = 104 |
104-4 |
Batch |
Ion exchange |
1 min |
|
Z = 105 |
105-1 |
Continuous |
Thermochromatography |
0.1 to 0.2 s |
|
Z = 105 |
105-2 |
Continuous |
Thermochromatography |
12 s |
|
Z = 105 |
105-3 |
Autobatch |
Extraction(HPLC) |
55 s |
|
Z = 107 |
107-1 |
Continuous |
Thermochromatography |
<3 s |
|
Z = 113, 114 |
113, 114-1 |
Continuous/batch |
Extraction |
<1 min |
|
Z = 118 (112 and 114) |
118-1 |
Continuous |
Condensation |
Few s |
|
Zinc |
Zn-1 |
Continuous |
Thermochromatography |
~1 s |
|
Zinc |
Zn-2 |
Batch |
Ion exchange |
~1 min |
|
Zinc |
Zn-3 |
Batch |
Extraction |
~1 min |
|
Zinc |
Zn-4 |
Batch |
Ion exchange |
1 to 2 min |
|
Zirconium |
Zr-1 |
Autobatch |
Extraction |
2.2 s |
|
Zirconium |
Zr-2 |
Continuous |
Extraction |
~7 s |
|
Zirconium |
Zr-3 |
Autobatch |
Extraction |
4.0 s |
|
Zirconium |
Zr-4 |
Batch |
Thermochromatography |
<3 min |
|
Zirconium |
Zr-5 |
Batch |
Precipitation |
Few min |
2. Procedures by Element
An-1
Actinides
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Gas chromatography
PRODUCTION MODE: Autobatch
REFERENCE: Greulich, N., Hickmann, U., Trautmann, N., and Herrmann, G., “Fast preparation and gas-chromatographic separation of lanthanide and actinide hexafluoroacetylacetonates,” Z. Anal. Chem. 323, 839–845 ( 1986).
PROCEDURE: The nuclear-reaction products were loaded onto a column of Chromosorb G coated with a mixture of hexafluoroacetylacetone (HFA) and trioctylphosphine oxide (TOPO). The column containing the actinide complexes was injected into the GC. A 2-m Chromosorb G column was used. Only trivalent actinides volatilized. Americium was volatilized around 200°C. For further details, please see procedure Ln-3 under “Lanthanides,” Greulich, N., Hickmann, U., Trautmann, N., and Herrmann, G.
Ac-1
Actinium
SEPARATION TIME: 3 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Chu, Y. Y., and Zhou, M. L., “Identification of 233Ac,” Phys. Rev. C 28, 1379–1381 ( 1983).
PROCEDURE: Uranium targets (50–300 mg/cm2) irradiated for 3 to 8 min with 28-GeV protons were dissolved, adjusted to 8M in HCl, and extracted with HDEHP in toluene (50%). Thorium was extracted by HDEHP, leaving actinium in the aqueous phase. Extractions were repeated to remove thorium completely. Actinium was further purified by HDEHP, TTA extractions, and anion exchange. This procedure specifically achieves separation of actinium from thorium in a short time.
AC-2
Actinium
SEPARATION TIME: 100 s
SEPARATION TECHNIQUE: Extraction, ion exchange
PRODUCTION MODE: Batch
REFERENCE: Chayawattanangkur, K., Herrmann, G., and Trautmann, N., “Heavy isotopes of 229–232Ac,” J. Inorg. Nucl. Chem. 35, 3061–3073 ( 1973).
PROCEDURE: Irradiated thorium salt was dissolved in a mixture of α-hydroxyisobutyric acid (HIB) and its ammonium salt, mixed with Dowex-50W X8 resin, and filtered. Actinium was retained by the resin along with yttrium, lanthanides, alkali, and alkaline earths. The resin was washed with NH4Cl (5%), and then with HIB (1M, pH 3.6); actinium was then eluted with HIB (1M, pH 4.65). The solution containing actinium was passed through a layer of Voltalef coated with HDEHP, which retained actinium. The Voltalef layer was washed with HCl (0.05M); actinium eluted with HCl (2M) and coprecipitated with Fe(OH)3.
Sb-1
Antimony
SEPARATION TIME: 2.7 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Autobatch
REFERENCE: Rudolph, W., Kratz, K. L., and Herrmann, G., “Half-lives, fission yields, and neutron-emission probabilities of neutron-rich antimony isotopes,” J. Inorg. Nucl. Chem. 39, 753–758 ( 1977).
PROCEDURE: From the sample dissolved in HCl (12M), the hydrides of antimony, selenium, tellurium, and arsenic were volatilized by a burst of nascent hydrogen generated by the addition of zinc powder. H2Se and H2Te were absorbed on quartz wool coated with 0.5M NaOH. The gas containing AsH3 and SbH3 was passed through KClO3 – HCl (9M) solution, which decomposes the hydrides and oxidizes arsenic and antimony to (V). The solution was passed through a layer of plastic powder coated with HDEHP, which retains only antimony. Also refer to Kratz, K. L., et al., Nucl. Phys. A317, 335–362 (1979). Also see procedure Sc-1 under “Selenium,” Kratz, J. V., and Herrmann, G., J. Inorg. Nucl. Chem. 32, 3713–3723 (1970).
Sb-2
Antimony
SEPARATION TIME: 5.4 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Tomlinson, L., and Hurdus, M. H., “Delayed neutron precursors. II. Antimony and arsenic precursors separated chemically,” J. Inorg. Nucl. Chem. 30, 1649–1661 ( 1968).
PROCEDURE: Uranium solution in HCl–H2SO4 containing germanium, arsenic, selenium, bromine, tin, tellurium, and iodine carriers and thiourea was used for irradiation. A stream of helium flowed through the irradiation chamber. During irradiation, hydrogen was generated by the addition of zinc. The hydrides generated, carried by the helium, were passed through a furnace maintained at a temperature below 650°C. Antimony deposited in the furnace was counted.
Sb-3
Antimony
SEPARATION TIME: 10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Greendale, A. E., and Love, D. L., “Rapid radiochemical procedure for antimony and arsenic,” Anal. Chem. 35, 632–635 ( 1963).
PROCEDURE: See procedure As-5 under “Arsenic,” Greendale, A. E., and Love, D. L., Anal. Chem. 35, 632–635 (1963).
Sb-4
Antimony
SEPARATION TIME: 6 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Braun, H., Denschlag, H. O., Izak-biran, T., and Lauppe, W., “Absolute gamma-ray line intensities and branching ratios in mass chain 133,” Radiochim. Acta 36, 95–102 ( 1984).
PROCEDURE: The irradiated uranium solution was mixed with HCl (12 M) and zinc powder to produce volatile hydrides of arsenic, antimony, selenium, and tellurium. The products were passed through a trap containing glass wool soaked in NaOH (0.5M); this trap retained the hydrides of selenium and tellurium. Most of the AsH3 and SbH3 passed through. A trap of glass wool coated with AgNO3 retained antimony. The gamma-ray measurements showed no interference from the decay of the isotopes of arsenic or its daughter products. Low-level contamination from tellurium was observed.
Sb-5
Antimony
SEPARATION TIME: 1.6 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Autobatch
REFERENCE: Meyer, R. A., and Henry, E. A., “Rapid, automated nuclear chemistry,” Nuclear Spectroscopy of Fission Products, T. von Egidy (Ed.) (The Institute of Physics, Bristol, 1979), p. 59–103.
PROCEDURE: The fission product solution, after removal of krypton and xenon by purging with N2, was treated with sodium borohydride to produce volatile hydrides. The hydrides produced were passed through a CaSO4 trap to remove selenium and tellurium. A trap containing KOH in ethanol removed SbH3 selectively while allowing AsH3 to pass through. Also see procedure Sb-11 under “Antimony,” Hicks, H. G., et al., Phys. Rev. C 27, 2203–2216 (1983).
Sb-6
Antimony
SEPARATION TIME: ~1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Pappas, A. C., “New antimony isotopes in fission,” Phys. Rev. 81, 299A ( 1951).
PROCEDURE: Irradiated uranium solution was mixed with antimony carrier and treated with Cl2 water to oxidize antimony to Sb(V). SbCl5 was extracted with isopropyl ether. The ether phase was washed with HCl – Cl2 solution. Antimony was stripped by reducing antimony to Sb(III) with N2H4 solution containing NaNCS. The aqueous solution was washed with isopropyl ether.
Sb-7
Antimony
SEPARATION TIME: 10 to 15 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Troutner, D. E., Wahl, A. C., and Ferguson, R. L., “Independent fission yield of 127Sb,” Phys. Rev. 134, B1027–B1029 ( 1964).
PROCEDURE: A modification of the procedure of Greendale and Love was used for the separation of antimony from fission products. Irradiated uranium solution was mixed with Sb(III) carrier in H2SO4. The resulting solution, which had 4 mg/mL of antimony and 30% H2SO4, was poured onto hot zinc granules. The SbH3 produced was swept by nitrogen gas and passed through a Br2– HCl solution. The SbH3 was decomposed and retained by this solution. Also see procedure As-5 under “Arsenic,” Greendale, A. E., and Love, D. L., Anal. Chem. 35, 632–635 (1963). Also refer to Fowler, M. M., and Wahl, A. C., J. Inorg. Nucl. Chem. 36, 1201–1212 (1974).
Sb-8
Antimony
SEPARATION TIME: ~10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: del Marmol, P., and Néve de Mévergnies, M., “Investigation of delayed neutron precursors of As, Sb, and Ge,” J. Inorg. Nucl. Chem. 29, 273–279 ( 1967).
PROCEDURE: See procedure As-8 under “Arsenic,” del Marmol, P., and Néve de Mévergnies, M., J. Inorg. Nucl. Chem. 29, 273–279 (1967).
Sb-9
Antimony
SEPARATION TIME: 10 to 20 s
SEPARATION TECHNIQUE: Electrolysis
PRODUCTION MODE: Batch
REFERENCE: Tomlinson, L., and Hurdus, M. H., “Delayed neutron precursors. I. Antimony and arsenic precursors separated by electrolysis,” J. Inorg. Nucl. Chem. 30, 1125–1138 ( 1968).
PROCEDURE: The hydrides of arsenic and antimony were generated in an electrolytic cell placed in the beam port of a reactor. The cell contained a solution of uranium and arsenic, germanium, tin, and antimony carriers. A current of 104 ± 4 A was passed through the cell for about 2 s, and the hydrides generated were swept by a flow of helium gas. The gases were passed through a furnace maintained at the required temperature for the selective decomposition of the hydrides. At 480°C, stibine decomposed, while a temperature of 890 °C was required for the decomposition of arsine. Tin hydride was removed when gases flowed through a CaSO4 tube before the furnace. Also see Tomlinson, L., J. Inorg. Nucl. Chem. 28, 287–301 (1966), and Anal. Chim. Acta 31, 545–551 (1964) and 32, 157–164 (1965).
Sb-10
Antimony (As)
SEPARATION TIME: <10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Naeumann, R., Folger, H., and Denschlag, H. O., “Determination of the nuclear charge distribution in the mass 132 chain from thermal neutron fission of 235U and 233U,” J. Inorg. Nucl. Chem. 34, 1785–1797 ( 1972).
PROCEDURE: See procedure Te-5 under “Tellurium,” Naeumann, R., et al., J. Inorg. Nucl. Chem. 34, 1785–1797 (1972).
Sb-11
Antimony (for Te)
SEPARATION TIME: 7.3 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Autobatch
REFERENCE: Hicks, H. G., Landrum, J. H., Henry, E. A., Meyer, R. A., Brandt, S., and Paar, V., “Population of 133I from the beta decay of fission product 133Teg and the cluster vibration model,” Phys. Rev. C 27, 2203–2216 ( 1983).
PROCEDURE: Krypton and xenon were purged from a fission-product solution by a stream of N2 gas. The volatile hydrides were generated using sodium borohydride. The gas was passed through a Drierite trap to remove tellurium and tin and then through a NaOH (0.5M) trap to remove selenium. The last trap of glass wool, which was soaked in a solution of KOH in methanol, removed antimony along with arsenic. After allowing sufficient time for the decay of the required antimony isotopes, the glass wool was washed with water and mixed with a boiling solution of (NH4)2S containing tellurium and antimony carriers. Tellurium was precipitated as the element by the addition of Na2SO3. Also see procedure Sb-5 under “Antimony,” Meyer, R. A., and Henry, E. A., Nuclear Spectroscopy of Fission Products, T. von Egidy (Ed.) (The Institute of Physics, Bristol, 1979), p. 59–103.
Sb-12
Antimony
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Plating, chemical
PRODUCTION MODE: Batch
REFERENCE: Der Mateosian, E., Goldhaber, M., Muehlause, C. O., and McKeown, M., “Multiple nuclear isomerism,” Phys. Rev. 72, 1271–1272 ( 1947).
PROCEDURE: This procedure was used to confirm that the activity produced by exposure to neutrons was indeed due to an isotope of antimony. The irradiated antimony metal was dissolved in HCl. Elemental antimony was plated on iron. After removal of antimony, tin was plated out on zinc.
Sb-13
Antimony
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Delucchi, A. A., Greendale, A. E., and Strom, P. O., “Cumulative fission yield and half-life of 134Sb,” Phys. Rev. 173, 1159–1165 ( 1968).
PROCEDURE: Uranium solution containing Sb(III) and Mo(VI) carriers was irradiated and mixed with 30% H2SO4 (4 mL). The solution was passed through a reaction chamber containing zinc and maintained at 1300°C. The gases generated were carried by a stream of N2 through a Drierite tube and then into a hot zone in a quartz tube. The stibine decomposed and deposited antimony in the elemental form. The antimony was collected on the quartz tube and on a glass filter located downstream.
Ar-1
Argon
SEPARATION TIME: <1 s
SEPARATION TECHNIQUE: Condensation
PRODUCTION MODE: Batch
REFERENCE: Hardy, J. C., Esterl, J. E., Sexctro, R. G., and Cerny, J., “Isospin purity and delayed-proton decay: N-17 and Ar-33,” Phys. Rev. C 3, 700–718 ( 1971).
PROCEDURE: Carbon disulfide vapor was irradiated with He-3 and swept with helium gas through a dry-ice trap; the carbon disulfide condensed to a liquid. The argon isotopes produced passed to the counting chamber.
As-1
Arsenic
SEPARATION TIME: 2.5 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Autobatch
REFERENCE: Kratz, J. V., Franz, H., and Herrmann, G., “Delayed neutrons from As isotopes 84As, 85As, and 86As,” J. Inorg. Nucl. Chem. 35, 1407–1417 ( 1973).
PROCEDURE: Zinc powder was added to a fission product solution in HCl containing arsenic, selenium, antimony, and tellurium carriers to produce a burst of nascent hydrogen and the hydrides of the carrier elements. The hydrides were allowed to flow through a tube containing quartz wool coated with a saturated solution of KOH in ethanol; this trap removed the hydrides of selenium, antimony, and tellurium efficiently. The AsH3 flowing out of the KOH trap was decomposed in a column of firebricks coated with AgNO3. Also see Franz, H., et al., Phys. Rev. Lett. 33, 859–862 (1974). Also refer to procedure Sc-1 under “Selenium,” Kratz, J. V., and Herrmann, G., J. Inorg. Nucl. Chem. 32, 3713–3723 (1970).
AS-2
Arsenic
SEPARATION TIME: 2.5 to 5 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Autobatch
REFERENCE: Kratz, J. V., Franz, H., and Herrmann, G., “Delayed neutrons from As isotopes 84As, 85As and 86As,” J. Inorg. Nucl. Chem. 35, 1407–1417 ( 1973).
PROCEDURE: The fission product solution in HCl, containing arsenic, selenium, antimony, and tellurium carriers, was mixed with zinc powder to generate hydrides of the carrier elements. The gases generated were passed through a tube containing quartz wool coated with a saturated solution of KOH in ethanol; this trap retained the hydrides of selenium, antimony, and tellurium while AsH3 passed through. AsH3 was decomposed by passing it through a solution of KClO3 in HCl (9M); the solution was then filtered through a layer of inert plastic powder coated with HDEHP, which retained germanium, bromine, antimony, and iodine contaminants. The arsenic in the filtrate was counted. Also see Kratz, J. V., and Herrmann, G., Proceedings of the 3rd Symposium on the Physics and Chemistry of Fission, IAEA publication, Vol. 2 (Rochester, 1973), p. 95; Kratz, J. V., et al., Nucl. Phys. A250, 13–37 (1973); and Franz, H., et al., Phys. Rev. Lett. 33, 859–862 (1974).
As-3
Arsenic
SEPARATION TIME: 5.4 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Tomlinson, L., and Hurdus, M. H., “Delayed neutron precursors. II. Antimony and arsenic precursors separated chemically,” J. Inorg. Nucl. Chem. 30, 1649–1661 ( 1968).
PROCEDURE: Uranium solution in HCl – H2SO4 containing germanium, arsenic, selenium, bromine, tin, antimony, tellurium, and iodine carriers and thiourea was irradiated. A flow of helium was maintained through the irradiation vessel. During irradiation, the hydrides were generated by the addition of zinc to the irradiation solution. The hydrides, carried by the helium, were passed through a furnace maintained at 620°C to remove antimony. Particulate antimony was removed by a silica-wool trap. Arsenic was decomposed by a second furnace at 950°C.
As-4
Arsenic
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Extraction, adsorption
PRODUCTION MODE: Continuous
REFERENCE: Zendel, M., Trautmann, N., and Herrmann, G., “Chemical reactions in a gas-jet recoil-transport system (II): continuous separation procedures for germanium and arsenic from fission products, ” Radiochim. Acta 29, 17–20 ( 1981).
PROCEDURE: Recoiling fission fragments were thermalized and carried by a stream of HCl – N2 (1 to 20, respectively). The gas was passed through a quartz-wool trap, which retained the fission products carried by clusters. The gas was then passed through a quartz spiral coated inside with silver and kept at ~800°C. The halogens formed nonvolatile products with silver and were retained in the spiral. The gas was then passed through a trap filled with polystyrene beads saturated with HDEHP, which retained arsenic almost quantitatively. Germanium in the gas coming out of the polystyrene trap was adsorbed by charcoal. The detector was set to measure the activities of the polystyrene-HDEHP trap.
As-5
Arsenic
SEPARATION TIME: 10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Greendale, A. E., and Love, D. L., “Rapid radiochemical procedure for antimony and arsenic,” Anal. Chem. 35, 632–635 ( 1963).
PROCEDURE: Mixed fission-product solution in 30% H2SO4, containing arsenic and antimony carriers, was dropped onto zinc powder (10–20 mesh) kept at 100°C. The hydrides generated were passed through a Drierite tube and then passed through a furnace at 600°C. Antimony hydride decomposed at this temperature, and metallic antimony was retained by a sintered-glass filter located outside the furnace. The gas flowing through the filter was passed through a quartz tube heated to a reddish glow. Arsenic hydride decomposed at this temperature and deposited as elemental As in the cooler portions of the tube.
As-6
Arsenic
SEPARATION TIME: 3 or 4 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Skarnemark, G., Brodén, K., Yun, M., Kaffrell, N., and Trautmann, N., “Rapid continuous-separation procedures for arsenic and ruthenium from complex reaction-product mixtures,” Radiochim. Acta 33, 97–100 ( 1983).
PROCEDURE: The recoiling fission products were stopped in a mixture of HCl – N2 (1:20) and dissolved in a solution of HCl (3M) – HI (1M). After degassing, the solution was passed on to a SISAK system. In the first centrifuge, arsenic was extracted with chloroform. Traces of krypton and xenon were also extracted. In the second centrifuge, arsenic was back-extracted with HCl (0.1M). The organic phase does not accumulate longer-lived arsenic isotopes. The measurements can be performed with the arsenic in the organic or aqueous phase. Also see procedure As-4 under “Arsenic,” Zendel, M., et al., Radiochim. Acta 29, 17–20 (1981).
As-7
Arsenic
SEPARATION TIME: 1.9 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Autobatch
REFERENCE: Meyer, R. A., and Henry, E. A., “Rapid, automated, nuclear chemistry,” Nuclear Spectroscopy of Fission Products, T. von Egidy (Ed.) (The Institute of Physics, Bristol, 1979), p. 59–103.
PROCEDURE: For details of the procedure, see procedure Sb-5 under “Antimony,” Meyer, R. A., and Henry, E. A. The AsH3 generated by sodium borohydride was not retained by the CaSO4 and NaOH traps. Finally, AsH3 was decomposed by a glass-wool trap containing HCl (9M) and Br2. Also see Henry, E. A., Lien III, O. G., and Meyer, R. A, CERN report No. 81-09, p. 334–338.
As-8
Arsenic
SEPARATION TIME: ~10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: del Marmol, P., and Néve de Mévergnies, M., “Investigation of delayed neutron precursors of As, Sb, and Ge,” J. Inorg. Nucl. Chem. 29, 273–279 ( 1967).
PROCEDURE: Irradiated uranium solution (containing suitable carriers) in 30% H2SO4 was transferred to a vessel containing zinc powder kept at 100°C. The hydrides produced were pumped through a Drierite tube and selectively decomposed by being passed through two quartz furnaces kept at selected temperatures. The temperature required for decomposition for antimony (600–750°C) < germanium < arsenic (1000°C). To collect arsenic, the first furnace was kept at 600 to 750°C and the second furnace at 1000°C; arsenic was collected on a quartz-wool plug at the end of the second furnace. Also see del Marmol, P., J. Inorg. Nucl. Chem. 30, 2873–2880 (1988).
As-9
Arsenic
SEPARATION TIME: 10 to 20 s
SEPARATION TECHNIQUE: Electrolysis
PRODUCTION MODE: Batch
REFERENCE: Tomlinson, L., and Hurdus, M. H., “Delayed neutron precursors. I. Antimony and arsenic precursors separated by electrolysis,” J. Inorg. Nucl. Chem. 30, 1125–1138 ( 1968).
PROCEDURE: See procedure Sb-9 under “Antimony,” Tomlinson, L., and Hurdus, M. H., J. Inorg. Nucl. Chem. 30, 1125–1138 (1968). Also see Tomlinson, L., J. Inorg. Chem. 28, 287–301 (1966); Anal. Chim. Acta 31, 545–551 (1964); and Anal. Chim. Acta 32, 157–164 (1965).
As-10
Arsenic (Sb)
SEPARATION TIME: <10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Naeumann, R., Folger, H., and Denschlag, H. O., “Determination of the nuclear charge distribution in the chain 132 from thermal neutron fission of 235U and 233U,” J. Inorg. Nucl. Chem. 34, 1785–1797 ( 1972).
PROCEDURE: See procedure Te-5 under “Tellurium,” Naeumann, R., et al., J. Inorg. Nucl. Chem. 34, 1785–1797 (1972).
As-11
Arsenic
SEPARATION TIME: 1.0 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Hickmann, U., Greulich, N., Trautmann, N., and Herrmann, G., “Chemical reactions in a gas-jet recoil-transport system (III): rapid separation of arsenic from fission products,” Radiochim. Acta 55, 57–59 ( 1991).
PROCEDURE: Nitrogen gas was passed through a Teflon vessel containing HF gas kept at 267 K; the gas mixture that came out of the Teflon vessel contained 37 vol% HF. The gas mixture was passed through a Teflon-coated 235U target chamber in the beam port of a reactor. The gas from the outlet of the target chamber carrying the fission products was passed through a quartz-wool trap that retained the clusters carrying nonvolatile fission products. The gas mixture then passed through a second trap of quartz powder freshly coated with silver and kept at 620 K, and then through a third trap of polytrifluoromonochlorethylene beads coated with di-(2-ethylhexyl)phosphoric acid (HDEHP). The HDEHP trap retained arsenic.
At-1
Astatine
SEPARATION TIME: ~30 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Hyde, E. K., and Ghiorso, A., “The alpha-branching of AcK and the presence of astatine in nature, ” Phys. Rev. 90, 267–270 ( 1953).
PROCEDURE: This procedure was used for quickly separating astatine from francium, lead, and bismuth. The starting material was purified francium, evaporated on a platinum disk. The disk was mounted in a chamber equipped with an electrically heated filament and a collector disk. By selection of appropriate current settings, the platinum disk was heated to a specific temperature where only astatine volatilized and deposited in the collector disk.
At-2
Astatine
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., Chemical Procedures Used in Bombardment Work at Berkeley, U.S. Atomic Energy Commission report AECD-2738 ( 1949), p. 248.
PROCEDURE: This procedure was developed by Barton for the separation of astatine from irradiated 203Bi or thorium metal. The irradiated target was dissolved in concentrated HCl or H2SO4. Astatine was extracted with diisopropyl ether. The organic phase was separated and used for counting.
At-3
Astatine
SEPARATION TIME: <30 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Thoresen, P. E., Asaro, F., and Perlman, I., “Concerning isomers of 208At, 206At, and 204At,” J. Inorg. Nucl. Chem. 26, 1341–1347 ( 1964).
PROCEDURE: The irradiated bismuth or lead oxide targets were heated with a gas torch, and the volatilized polonium and astatine were collected on a platinum plate. Astatine was selectively volatilized by heating the platinum plate to 450°C and deposited on another plate. Less than 1% of polonium accompanied the astatine. Also see procedure Po-4 under “Polonium,” Perlman, I., Asaro, F., Ghiorso, A., Larsh, A., and Latimer, R., Phys. Rev. 127, 917–922 (1962).
At-4
Astatine
SEPARATION TIME: 90 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Barton, Jr., G. W., Ghiorso, A., and Perlman, I., “Radioactivity of astatine isotopes,” Phys. Rev. 82, 13–19 ( 1951).
PROCEDURE: The irradiated bismuth target was dissolved, and the final solution was adjusted to 12M in HCl. The astatine was reduced to elemental state by the addition of ferrous sulfate. Astatine was extracted with diisopropyl ether. The ether phase was washed with dilute HCl and then evaporated on a platinum plate.
Ba-1
Barium
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Tamai, T., Takada, J., Matsushita, R., and Kiso, Y., “Half-life and gamma-ray energies of 143Ba and 144Ba,” Inorg. Nucl. Chem. Lett. 9, 973–979 ( 1973).
PROCEDURE: Irradiated uranium solution and the carrier mixture (barium + strontium) were spotted on the chromatographic paper wetted with the supporting electrolyte (1 × 10−2M nitrilotriacetic acid, pH 4.2). On the cathodic side, 1 cm from the sample, K2CrO4 (1 × 10−2M) solution was spotted. A potential gradient of 5000 V/15 cm was applied for 20 s. The barium position, previously determined by a tracer run, was cut and counted.
Ba-2
Barium
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous/batch
REFERENCE: Langrock, E. J., Bazarkina, T. V., and Czosnowska, W., “Procedure for selective solvent extraction of superheavy elements 113+ and 1142+ by use of crown ethers,” Radiochim. Acta 30, 229–231 ( 1982).
PROCEDURE: See procedure 114-1 under “Z = 114,” Langrock, E. J., et al., Radiochim. Acta 30, 229–231 (1982).
Ba-3
Barium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Preiss, I. L., and Strudler, P. M., “New neutron-deficient barium isotopes,” J. Inorg. Nucl. Chem. 24, 589–592 ( 1962).
PROCEDURE: Irradiated tin or indium foils were dissolved in HCl in the presence of barium and other suitable holdback carriers. Barium was precipitated as nitrate with fuming HNO3 or as sulfate with H2SO4 (0.5M). Impurities were estimated to be less than 5%. In some experiments, cesium was milked by dissolving the Ba(NO3)2 and precipitating cesium from alcohol solution as cesium chloroplatinate.
Ba-4
Barium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: D'Auria, J. M., Bakhru, H., and Preiss, I. L., “Deformation in the light barium isotopes: isomeric states of 125–127Ba,” Phys. Rev. 172, 1176–1186 ( 1968).
PROCEDURE: Indium or antimony targets were dissolved in a solution of 1:1 HCl:HNO3; if aluminum catcher foils were used, the aluminum foils were dissolved in hot, concentrated HCl. Barium and appropriate holdback carriers were added, and Ba(NO3)2 was precipitated. The precipitate was centrifuged and dissolved in a minimum volume of buffer solution; the barium reprecipitated as chromate or sulfate.
Ba-5
Barium (from cesium)
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Continuous
REFERENCE: Ruddy, F., and Pate, B. D., “The decay of 0.3-s 136Bam,” Nucl. Phys. 69, 471–476 ( 1965).
PROCEDURE: Cesium parent activity was chemically purified and sorbed on a bed of thallous phosphotungstate on a filter paper. The filter paper was washed continuously with 0.5M HCl to remove the barium daughter activity growing from cesium parent. The bed retained the cesium quantitatively. A pump was used to circulate HCl continuously.
Ba-6
Barium (from lanthanum)
SEPARATION TIME: 3 to 4 s
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Runnalls, N. G., Troutner, D. E., and Ferguson, R. L., “Charge distribution in fission: fractional cumulative fission yields of 143Ba and 144Ba from thermal-neutron-induced fission of 235U,” Phys. Rev. 179, 1188–1193 ( 1969).
PROCEDURE: Uranium solution containing lanthanum carrier was irradiated, and the solution was transferred to a fritted-glass filter. The filter held a solution of concentrated ammonia and barium carrier. The precipitated La(OH)3 was filtered. 1.1% of the barium coprecipitated with lanthanum; 0.4% of the lanthanum was in the filtrate.
Bi-1
Bismuth
SEPARATION TIME: 5 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., Chemical Procedures Used in Bombardment Work at Berkeley, U.S. Atomic Energy Commission report AECD-2738 ( 1949), p. 242.
PROCEDURE: See procedure Pb-4 under “Lead,” Meinke, W. W., U.S. AEC report AECD-2738, 1949, p. 240.
Bi-2
Bismuth (+Po)
SEPARATION TIME: 4 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Spiess, F. N., “Alpha-emitting isomer: 211Po,” Phys. Rev. 94, 1292–1299 ( 1954).
PROCEDURE: See procedure Po-3 under “Polonium,” Spiess, F. N., Phys. Rev. 94, 1292–1299 (1954).
Br-1
Bromine
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Perlow, G. J., and Stehney, A. F., “Halogen delayed-neutron activities,” Phys. Rev. 113, 1269–1272 ( 1959).
PROCEDURE: The uranium solution in HNO3 (1M) containing NaBrO3 (0.5M) was spread as a thin film over glass beads. After irradiation, a burst of air and Br2 vapor was sucked through the vessel containing uranium solution. The gas was then passed through a container of CCl4-coated glass beads. Br2 was retained by CCl4. Also see Chrysochoides, N. G., Anoussis, J. N., Mitsonias, C. A., and Perricos, D. C., J. Nucl. Energy 25, 551 (1971).
Br-2
Bromine
SEPARATION TIME: 10 s
SEPARATION TECHNIQUE: Hot atom, gas phase
PRODUCTION MODE: Batch/continuous
REFERENCE: Silbert, M. D., and Tomlinson, R. H., “Reaction of fission-recoil Br with methane,” Radiochim. Acta 5, 217–227 ( 1966).
PROCEDURE: The fission products recoiling from a uranium oxide target underwent hot-atom reaction with CH4 in the target chamber, forming organic halides. After irradiation, the gas in the chamber was swept for 2 to 5 s and the system was flushed for an additional 5 s with CH4. The gas flowing from the target chamber was filtered through a glass-wool plug and passed through a tube containing AgNO3-coated C-22 firebricks maintained at 50°C. The trap removed iodine activities. The gas was then allowed to flow through a molecular-sieve trap, which retained the Br activities. The molecular-sieve trap was counted. Also see Silbert, M. D., and Tomlinson, R. H., Radiochim. Acta 5, 217–227 (1966).
Br-3
Bromine
SEPARATION TIME: 8 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Nuh, F. M., Slaughter, D. R., Shihab-Eldin, A. A., and Prussin, S. G., “A rapid radiochemical procedure for fission-product Br,” Radiochim. Acta 17, 149–153 ( 1972).
PROCEDURE: The irradiated uranium solution was mixed with Br− and I− carriers; a solution of NaMnO4 was added, followed by the rapid addition of H2SO4 (15M) at 80°C. The heat generated led to vigorous boiling of the mixture, while Br− was oxidized to Br2. An air stream carried the Br2 through a series of traps; the first two contained H2SO4 (8M) – NaMnO4 to remove traces of iodine; the third trap, of polyethylene shavings at 80 to 85°C, removed RuO4. Bromine was collected in a trap of AgNO3 – Na2S2O5. The AgBr precipitate was removed by rapid filtration and washed with HNO3 (dilute). Also see Slaughter, D. R., Nuh, F. M., and Prussin, S. G., J. Inorg. Nucl. Chem. 38, 1753–1759 (1976).
Br-4
Bromine
SEPARATION TIME: ~1 s
SEPARATION TECHNIQUE: Adsorption
PRODUCTION MODE: Continuous
REFERENCE: Rengan, K., Lin, J., and Meyer, R. A., “Continuous gas-phase separation of Br-fission products with half-lives of 600 ms to 56 s,” Radiochem. Radioanal. Lett. 50, 393–398 ( 1982).
PROCEDURE: Fission fragments were allowed to recoil through 2.7 mg/cm 2 of aluminum foil that selectively stopped the heavy fragments. The fragments coming through the foil were thermalized in C2H4 – N2 stream. The nonvolatile fission products carried by C2H4 clusters were removed by a quartz-wool trap; selenium was removed by quartz wool coated with Ag – AgNO3. Bromine fission products were adsorbed in a charcoal trap. The volatile noble gases flowed through.
Br-5
Bromine
SEPARATION TIME: 40 s
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Tamai, T., Takada, J., and Matsushita, R., “A rapid, electrophoretic separation for short-lived Br in fission products,” Ann. Rep. Research, Reactor Inst., Kyoto Univ. 11, 48–56 ( 1978).
PROCEDURE: A chromatographic paper was wetted with supporting electrolyte (1 × 10−2M H2SO3, pH 1.9). Irradiated uranium solution was spotted on the chromatographic paper, and a voltage gradient of 5000 W15 cm was applied for 20 s. The position of Br−, BrO3−, earlier determined by a tracer run, was cut and counted.
Br-6
Bromine
SEPARATION TIME: 2 s
SEPARATION TECHNIQUE: Hot-atom reaction
PRODUCTION MODE: Batch/continuous
REFERENCE: Lundan, A., “On the decay of Br isotopes 86Br and 87Br,” Z. Physik 236, 403–409 ( 1970).
PROCEDURE: UO2 was irradiated in a chamber filled with CH4 to a pressure of 2 to 3 atm. Recoiling fission-product bromine and iodine reacted with the CH4 to form CH3Br and CH3I. A flow of CH4 maintained through the chamber carried the products formed to counting position. Bromine and iodine were collected in AgNO3.
Br-7
Bromine
SEPARATION TIME: ~1 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Continuous
REFERENCE: Ray, P. K., and Kenney, E. S., “Delayed neutron spectrum from 87Br created in thermal fission of 235U,” Nucl. Instr. Methods 134, 559–564 ( 1976).
PROCEDURE: An acid solution of uranium was irradiated and bromine-fission products were carried away from the irradiation vessel by a stream of Br2 – N2 gas mixture. The gas was passed through a mixture of HNO3 – NaBrO3 to remove iodine. The bromine was held in another trap by CCl4 along with krypton and xenon. Delayed neutron activity of the CCl 4 trap was measured. Also see procedure Br-1 under “Bromine,” Perlow, G., and Stehney, A. F., Phys. Rev. 113, 1269–1272 (1959), and procedure I-1 under “Iodine,” Perlow, G., and Stehney, A. F., Phys. Rev. 113, 1269–1272 (1959).
Br-8
Bromine
SEPARATION TIME: 0.5 s
SEPARATION TECHNIQUE: Hot-atom reaction
PRODUCTION MODE: Batch
REFERENCE: Kratz, K. L., “Independent fission yields and neutron-emission probabilities of short-lived halogen isotopes,” Radiochim. Acta 25, 1–7 ( 1978).
PROCEDURE: Thin UO2 targets were irradiated in CH4-filled capsules. After the irradiation, the gas containing the fission products was swept out of the capsule with CH4. The gas was allowed to flow through an aerosol filter and then through two traps; the first trap contained AgNO3 on silica gel at 300°C, and it retained the iodine. The second trap contained silica gel coated with AgCl and kept at 70 to 80°C, and it retained the bromine. Also see Kratz, K. L., and Herrmann, G., Radiochem. Radioanal. Lett. 13, 385–390 (1973), and Nucl. Phys. A229, 179–188 (1974).
Br-9
Bromine
SEPARATION TIME: ~5 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Brodén, K., Skarnemark, G., Björnstad, T., Eriksen, D., Haldorsen, I., Kaffrell, N., Stender, E., and Trautmann, N., “Rapid, continuous-separation procedures for Zr, Nb, Tc, Br, and I from complex reaction-product mixtures,” J. Inorg. Nucl. Chem. 43, 765–771 ( 1981).
PROCEDURE: Recoiling fission fragments thermalized and transported by a KCl aerosol were dissolved in H2SO4 (0.1M) – NaHSO3 (0.01M) solution containing Br− and I− carriers. The solution was degassed, mixed with NaNO2, and passed to a SISAK-2 system. In the first step, the solution was contacted with CHCl3 to remove iodine. The aqueous phase was mixed with KBrO3 (0.1M) – HNO3 (4M) solution, and in the second stage bromine was extracted with CCl 4. In the third step, bromine was stripped with NaHSO3 (0.2M) – H2SO4 (0.1M) solution. The bromine in the aqueous phase could be counted or exchanged with preformed AgCl layers and the AgCl layers counted.
Br-10
Bromine
SEPARATION TIME: 1 min
SEPARATION TECHNIQUE: Gas chromatography
PRODUCTION MODE: Batch
REFERENCE: Denschlag, H. O., and Gordus, A. A., “Gas-chromatographic technique for rapid isolation of U-fission products, ” Z. Anal. Chem. 226, 62–71 ( 1967).
PROCEDURE: UO2 deposited on aluminum was irradiated in a vial filled with CH4. The recoiling halogens formed CH3Br and CH3I. After the irradiation, the gaseous content of the vial was transferred to a GC column. As a stationary phase, Chromosorb-P or Dow silicone oil 550 was used. Helium was used as the carrier gas at a flow rate of 200 mL/min; depending on the experiment, the column was operated at a temperature between 20 and 180°C. The counting of bromine or iodine could be started within 1 min.
Br-11
Bromine
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Perlow, G. J., and Stehney, A. F., “Delayed neutrons from 15.5-s 88Br,” Phys. Rev. 107, 776–780 ( 1957).
PROCEDURE: Uranium solution in 1M HNO3 and containing KBrO3 was irradiated. Air containing Br2 vapor was bubbled through the solution after the irradiation. The gases were mixed with I2 vapor and passed through a solution of KBrO3. I2, along with the active I in the stream, was converted to IO4− and retained in the KBrO3 solution. The gas was then bubbled through CCl4 in front of the detector. The bromine was retained by the CCl4.
Br-12
Bromine
SEPARATION TIME: 30 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Sugarman, N., “Determination of the ranges of the fission fragments emitting delayed neutrons. Chemical identification of the 4.51-s delayed neutron activity, ” J. Chem. Phys. 15, 544–551 ( 1947).
PROCEDURE: Irradiated uranyl nitrate solution, in Na2CO3 (2M) containing Br− and I− carriers was mixed with Na2CO3 (2M) solution containing Br− and BrO3−. The solution was treated with NaOCl to oxidize I− to IO3−. The solution was added to a separatory funnel containing HNO3 (6M). The Br2 liberated was extracted with CCl4. The CCl4 layer was separated and used for counting.
Br-13
Bromine
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Creutz, E. C., Delsasso, L. A., Sutton, R. B., White, M. G., and Barkas, W. H., “Radioactivity produced by proton bombardment of Br and I,” Phys. Rev. 58, 481–486 ( 1940).
PROCEDURE: Irradiated lead bromide was heated in a stream of krypton. The gases were passed through a trap cooled by a mixture of alcohol and solid CO2 and then through a second trap cooled by liquid air. Free bromine condensed in the first trap, while krypton condensed in the second trap.
Br-14
Bromine
SEPARATION TIME: 100 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Levinger, J. S., Meiners, E. P., Sampson, M. B., Snell, A. H., and Wilkinson, R. G., “Chemical isolation of 56-s Br and 23-s I delayed-neutron activities, ” Radiochemical Studies: The Fission Products, Coryell, C. D., and Sugarman, N. (Eds.), National Nuclear Energy Series, Vol. 9, Book 2 ( 1951), p. 603.
PROCEDURE: Irradiated uranyl nitrate solution was mixed with HNO3 (8M) containing KClO3, Br−, and I−. The Br2 and I2 were liberated and extracted with CCl4. The CCl4 layer was transferred to a second separatory funnel containing KNO 2 solution acidified with HNO3. The Br2 was reduced to Br− and remained in the aqueous phase. The aqueous layer was used for counting. Also see Snell, A. H., et al., Phys. Rev. 72, 545–549 (1947).
Br-15
Bromine
SEPARATION TIME: 20 to 25 s
SEPARATION TECHNIQUE: Distillation
PRODUCTION MODE: Batch
REFERENCE: Williams, E. T., and Coryell, C. D., “Decay of 54-s 86Br and 55-s 87Br,” Phys. Rev. 144, 945–951 ( 1966).
PROCEDURE: Irradiated uranyl nitrate was transferred to a distillation flask containing Br(−) and I(−) carriers and H2SO4. Hot KMnO4 solution was added to the flask, and the elemental bromine produced was swept out using a stream of air. The bromine was reduced to Br −, precipitated as AgBr, filtered, washed, and mounted for counting.
Cd-1
Cadmium
SEPARATION TIME: ~1 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Rudstam, G., Aagaard, P., Hoff, P., Johansson, B., and Zwicky, H. O., “Chemical separation combined with an ISOL-system,” Nucl. Instr. Methods 186, 365–379 ( 1981).
PROCEDURE: An ion beam of selected mass entered through a narrow channel and impinged on a hot-tungsten filament. The volatile components were released into the gaseous phase and entered the thermochromatographic column. The temperature gradient of the column was selected carefully to suit the deposition temperature (Td) of the element of interest. A catcher foil kept at Td of the desired element collected it, and the detectors kept around it measured the activity. Elements that are less volatile were deposited in the vicinity of the foil, while more volatile elements left the column. For the separation of cadmium and zinc from neighboring elements, the foil was kept at 130°C.
Cd-2
Cadmium
SEPARATION TIME: 5 to 10 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Semkow, T., and Wahl, A. C., “Extraction of Ag(I), Cd(II), In(III), Sn(II), Sn(IV), Sb(III), and U(VI) from aqueous solutions by ketone solutions using single-step batch and continuous SISAK methods,” J. Radioanal. Chem. 79, 93–101 ( 1983).
PROCEDURE: See procedure In-1 under “Indium,” Semkow, T., and Wahl, A. C., J. Radioanal. Chem. 79, 93–101 (1983). Cadmium was extracted along with indium from the aqueous solution by a methyl isobutyl ketone-cyclohexanone mixture.
Cd-3
Cadmium
SEPARATION TIME: ~2 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Semkow, T., and Wahl, A. C., “Extraction of Ag(I), Cd(II), In(III), Sn(II), Sn(IV), Sb(III), and U(VI) from aqueous solutions by ketone solutions using single-step batch and continuous SISAK methods,” J. Radioanal. Chem. 79, 93–101 ( 1983).
PROCEDURE: See procedure In-2 under “Indium,” Semkow, T., and Wahl, A. C., J. Radioanal. Chem. 79, 93–101 (1983). The organic solvents extracted 97 to 99% of the cadmium. Also see Robinson, L., et al., Phys. Rev. C 31, 1334–1339 (1985).
Cd-4
Cadmium
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Fogelberg, B., Nagarajan, T., and Grapengiesser, B., “Levels and transition probabilities in 124In as observed in the decay of 124Cd,” Nucl. Phys. A 230, 214–220 ( 1974).
PROCEDURE: This procedure was used for the separation of cadmium from indium after mass separation using an on-line mass separator. The beam obtained after mass separation was allowed to strike a tungsten filament maintained at a temperature of 2000°C. The neutral atoms released were allowed to enter a heated quartz thermochromatographic column. The indium atoms were deposited near the entrance of the column; cadmium atoms were deposited near the cooler regions of the column. The decontamination factor obtained for cadmium from indium was about 100.
Cd-5
Cadmium
SEPARATION TIME: 2 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Gleit, C. E., and Coryell, C. D., “Decay of 49-min 118Cd and 5.1-s 118In,” Phys. Rev. 122, 229–231 ( 1961).
PROCEDURE: The irradiated uranium foil was dissolved in a mixture of concentrated HCl (2 mL)-concentrated HNO3 (0.2 mL). The solution was neutralized with KOH and diluted with 2M KOH solution (10 mL) containing NH2OH·HCl (1 g) and KNaC4H4O6·2H2O (Rochelle salt, 0.5 g). Cadmium, indium(III), silver, and palladium(II) carriers were added to the solution, and an extraction with CHCl 3 (20 mL) containing dithizone (2 mg) was performed. Cadmium was selectively extracted; it was back-extracted with HCl (0.5M). For solid sources, the cadmium was precipitated as (Ph4As)2CdCl4.
Cf-1
Californium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Silva, R. J., Hahn, R. L., Toth, K. S., Mallory, M. L., Bemis, Jr., C. E., Dittner, P. F., and Keller, O. L., “New isotopes 241Cf and 240Cf,” Phys. Rev. C 2, 1948–1951 ( 1970).
PROCEDURE: The californium atoms produced were allowed to recoil and deposit on a platinum disk coated with ammonium chloride. The disk was washed, and the solution was loaded on a Dowex 50 x 12 column maintained at 800°C. Ammonium α-hydroxyisobutyrate was used to elute californium. For additional details, see G. R. Choppin, B. G. Harvey, and S. G. Thompson, J. Inorg. Nucl. Chem. 2, 66 (1956).
C-1
Carbon
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Sherr, R., Muether, H. R., and White, M. G., “A new 20-s activity induced in B by high-energy protons,” Phys. Rev. 74, 1239–1240A ( 1948).
PROCEDURE: Carbon activities produced during irradiation of elemental boron or boron compounds such as boric acid can be partially removed as CO2 by blowing a gas through the irradiation cell. The gas was bubbled through a solution of barium hydroxide. Carbon activities were carried by the precipitate of BaCO3. The precipitate was filtered and washed.
C-2
Carbon
SEPARATION TIME: 20 s
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Sherr, R., Muether, H. R., and White, M. G., “Radioactivity of 10C and 14O,” Phys. Rev. 75, 282–292 ( 1949).
PROCEDURE: The radioactive gas produced during the proton bombardment of boron (as borax, H3BO3, or CaF2·BF3) was dissolved in barium hydroxide solution. Barium carbonate was precipitated by the addition of Na2CO3 solution. The precipitate was filtered and counted.
C-3
Carbon
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Barkas, W. H., “Some new reactions in light nuclei with high-energy protons,” Phys. Rev. 56, 287 ( 1939).
PROCEDURE: Nitrogen was bombarded with protons and the recoils were collected on paper. The paper was burned in the presence of oxygen, and the gas was bubbled through Ca(OH)2 solution. Carbon was finally precipitated as CaCO3.
Ce-1
Cerium
SEPARATION TIME: ~5 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Hoffman, D. C., Michelsen, O. B., and Daniels, W. R., “Beta decay of some Ce isotopes far to the neutron-rich side of stability, ” Arkiv. Fysik 36, 211–220 ( 1966).
PROCEDURE: The procedure involved extraction of Ce(III) into HDEHP followed by back-extraction with a mixture of H2O2 – 11M HNO3. Cerium was oxidized to (IV) with bromate and passed through a column of Hostaflon C2 powder equilibrated with HDEHP. The Ce(IV) was retained on the column. The praseodymium daughter activity was removed continuously by washing with HNO3 (6M).
Ce-2
Cerium
SEPARATION TIME: 2 to 3 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Yamamoto, H., Ikeda, Y., Kawade, K., Katoh, T., and Nagahara, T., “Decay properties of 145Ce and 146Ce,” J. Inorg. Nucl. Chem. 42, 1539–1546 ( 1980).
PROCEDURE: Irradiated uranium solution mixed with lanthanum, cerium, and praseodymium carriers was spotted on a chromatographic paper previously wetted with the supporting electrolyte (4 × 10−3M nitrilotriacetic acid, pH 1.8). An electrical potential of 4000 V/10 cm was applied for 1 min for 145Ce; for 146Ce a potential of 3000 V/10 cm was applied for 2 min. The cerium zone was identified by color reaction with Arsenazo.
Ce-3
Cerium
SEPARATION TIME: ~5 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Trautmann, N., Aronsson, P. O., Björnstad, T., Kaffrell, N., Kvale, E., Skarstad, M., Skarnemark, G., and Stender, E., “The combination, of the gas-jet recoil technique with the fast-chemical, on-line separation system SISAK,” Inorg. Nucl. Chem. Lett. 11, 729–735 ( 1975).
PROCEDURE: Recoiling fission products were thermalized in a C2H4 – N2 mixture and transported to a degassing unit; the fission products were transferred to HNO3 (1.25M) at 90°C. The solution was passed to a SISAK system, where in the first centrifuge yttrium, zirconium, niobium, and molybdenum, as well as some Br2 and I2, were extracted by di-(2-ethylhexyl) phosphoric acid (HDEHP) (2M) in Shellsol-T. Cerium was then oxidized to (IV) by the addition of HNO3 – H2SO4 – K2Cr2O7 and extracted by HDEHP (0.2M) in Shellsol-T. In the third centrifuge, cerium was stripped by H2SO4 – sulfamic acid – H2O2. In the final step, cerium was reoxidized to (IV) and adsorbed on a PVC column coated with HDEHP.
Ce-4
Cerium
SEPARATION TIME: 10 to 20 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Aronsson, P. O., Skarnemark, G., and Skarestad, M., “Short-lived isotopes of La, Ce, and Pr studied by SISAK technique, ” J. Inorg. Nucl. Chem. 36, 1689–1696 ( 1974).
PROCEDURE: Fission products formed in the target uranyl sulfate complex adsorbed on Dowex-1 X8 were continuously eluted with (NH4)2SO4 solution (pH 5) at 85°C. Cerium was oxidized with a mixture of HNO3 – H2SO4 – K2Cr2O7 and transferred to a SISAK system. In the first centrifuge, cerium was extracted with di-(2-ethylhexyl)phosphoric acid (HDEHP) (0.3 M) in kerosene. Cerium was stripped in the next centrifuge with a mixture of HNO3 – sulfamic acid – H2O2. The final step involved reoxidizing cerium to (IV) and passing it through a column of PVC coated with HDEHP, which retains cerium. The oxidizing solution continuously removed the accumulating daughter products. Also see Aronsson, P. O., et al., Inorg. Nucl. Chem. Lett. 10, 753–762 (1974).
Ce-5
Cerium
SEPARATION TIME: <5 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Skarnemark, G., Aronsson, P. O., Brodén, K., Rydberg, J., Björnstad, T., Kaffrell, N., Stender, E., and Trautmann, N., “An improved system for fast, continuous chemical separations (SISAK-2) in nuclear spectroscopic studies,” Nucl. Instr. Methods 171, 323–328 ( 1980).
PROCEDURE: The SISAK-2 system is described. The system, with specially designed, high-speed centrifuges of low hold-up volumes, allows liquid phases within 1 s. For details of cerium chemistry, see procedure Ce-4 under “Cerium,” Aronsson, P. O., et al., J. Inorg. Nucl. Chem. 36, 1689–1696 (1974).
Ce-6
Cerium
SEPARATION TIME: 6.5 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Ohyoshi, A., Ohyoshi, E., Tamai, T., and Shinagawa, M., “Short-lived isotopes of La, Ce and Pr in neutron irradiated U,” J. Inorg. Nucl. Chem. 34, 3293–3302 ( 1972).
PROCEDURE: See procedure La-7 under “Lanthanum,” Ohyoshi, A., et al., J. Inorg. Nucl. Chem. 34, 3293–3302 (1972). To achieve cerium separation, 86 to 93 V/cm was applied for 180 s. The cerium zone was located by color reaction with Arsenazo III.
Ce-7
Cerium
SEPARATION TIME: 2 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Hoffman, D. C., and Daniels, W. R., “Some short-lived isotopes of cerium and praseodymium,” J. Inorg. Nucl. Chem. 26, 1769–1793 ( 1964).
PROCEDURE: The irradiated uranyl nitrate was dissolved in HClO4 containing HNO3 and evaporated to dryness to remove volatile products. The residue was then taken up in HNO3 (11M) containing NaBrO3, Ce(III), and yttrium carriers. The solution was extracted with pre-equilibrated methyl isobutyl ketone (MIBK). The MIBK layer was washed twice with HNO3 (11M) containing bromate and carriers of yttrium, zirconium, and niobium. A third wash was performed with HNO3 (11M) – NaBrO3 – yttrium solution. The praseodymium daughter activities were milked periodically from the organic layer.
Cs-1
Cesium
SEPARATION TIME: ~40 s
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Tamai, T., Takada, J., Matsushita, R., and Kiso, Y., “Direct measurement of gamma-ray energies and half-life of 141Cs separated by paper electrophoresis,” J. Nucl. Sci. Technol. 9, 378–380 ( 1972).
PROCEDURE: Irradiated uranium solution was spotted on a chromatographic paper wetted with the supporting electrolyte, citric acid (1 × 10 −2M, pH 9.0). The electrophoretic separation was started 30 s after the end of irradiation. A potential gradient of 5000 V/15 cm was applied for 10 s. The cesium portion on the paper strip, as determined by an earlier tracer experiment, was cut and counted.
Cs-2
Cesium
SEPARATION TIME: 3.5 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: D'Auria, J. M., and Preiss, I. L., “Decay scheme of 123Cs,” Nucl. Phys. 84, 37–48 ( 1966).
PROCEDURE: The irradiated indium foil was dissolved in a 2:1 solution of HCl:HNO3. Cesium was precipitated from the solution as the chloroplatinate in the presence of appropriate hold-back carriers. The precipitate was filtered, washed, and mounted for counting. In other experiments, the irradiated foil was dissolved as above, and barium was precipitated as nitrate. The Ba(NO3)2 was dissolved, and cesium was milked from the solution by precipitation as chloroplatinate.
Cs-3
Cesium
SEPARATION TIME: <3 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Kalkstein, M. I., and Hollander, J. M., “New chain 126Ba – 126Ce,” Phys. Rev. 96, 730–734 ( 1954).
PROCEDURE: The irradiated indium metal or oxide was dissolved in the presence of barium carrier, and BaCl2 was precipitated with ether – HCl reagent. The BaCl2 was dissolved, and the barium was reprecipitated as nitrate with fuming HNO3. The Ba(NO3)2 was dissolved, and cesium was milked by precipitation as cesium cobaltinitrite. Barium impurities were removed from cesium by dissolving the cobaltinitrite in fuming HNO3 and performing several barium scavenges.
Cs-4
Cesium
SEPARATION TIME: ∼30 s
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Fritze, K., “Half-lives of 141Cs, 142Cs, and 143Cs,” Can. J. Chem. 40, 1344–1349 ( 1962).
PROCEDURE: The irradiated uranyl nitrate solution was mixed with cesium carrier and transferred to the membrane-filter assembly containing HClO4 (20%). The cesium perchlorate formed was filtered and used for further processing.
Cu-1
Copper
SEPARATION TIME: 20 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Aronsson, P. O., Johansson, B. E., Rydberg, J., Skarnemark, G., Alstad, J., Bergersen, B., Kvale, E., and Skarestad, M., “SISAK—A new technique for rapid, continuous (radio)chemical separations, ” J. Inorg. Nucl. Chem. 36, 2397–2403 ( 1974).
PROCEDURE: Continuous separation of copper from zinc was achieved using the SISAK system. The irradiated ZnCl2 solution (pH 3.0 to 3.5) was passed on to the first centrifuge, where copper was extracted into a 5% solution of LIX-64 (a mixture of a dialkyl hydroxime and 2-hydroxy, 5-alkylbenzophenone oxime) in kerosene. The organic phase was passed on to the second centrifuge, where it was washed with H2SO4 (pH 2) to remove nickel. The copper in the organic phase was back-extracted with HCl (0.5M) in the last centrifuge. The copper was reduced to elemental state and collected on an iron-wool column in front of the detector.
Cu-2
Copper
SEPARATION TIME: ~1 min
SEPARATION TECHNIQUE: Plating, chemical
PRODUCTION MODE: Batch
REFERENCE: Lindner, L., and Brinkman, G. A., “60Zn and 61Zn,” Physica 21, 747–748 ( 1955).
PROCEDURE: Irradiated nickel foils were dissolved in HNO3, and the acid concentration was reduced. Copper was deposited spontaneously from the solution on zinc dust. This procedure was used to milk copper daughter product from a short-lived zinc parent.
Cu-3
Copper
SEPARATION TIME: ~1 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Lindner, L., and Brinkman, G., A., “60Zn and 61Zn,” Physica 21, 747–748 ( 1955).
PROCEDURE: Irradiated nickel was dissolved in HNO3 and mixed with copper carrier. Copper was reduced to Cu(I) by the addition of sulfurous acid and precipitated as thiocyanate. This procedure was used for the milking of copper daughter products from short-lived zinc parents.
Cu-4
Copper
SEPARATION TIME: 4 to 5 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., Chemical Procedures Used in Bombardment Work at Berkeley, U.S. Atomic Energy Commission report AECD-2738 ( 1949), p. 76.
PROCEDURE: The procedure was developed by R. C. Lilly for the separation of copper from irradiated nickel. The irradiated nickel foil was dissolved in hot 6M HNO3 (0.5 mL) and evaporated to dryness. The residue was taken up in a few drops of concentrated HCl and taken to dryness. The process was repeated. Copper and cobalt carrier solutions were added, followed by concentrated NH4HSO3 (0.5 mL) and NH4SCN (0.5 mL). The mixture was warmed and the precipitate was filtered. After washing, the precipitate was mounted for counting.
Cu-5
Copper
SEPARATION TIME: 2 min
SEPARATION TECHNIQUE: Plating, chemical
PRODUCTION MODE: Batch
REFERENCE: Ward, T. E., Ihochi, H., and Meason, J. L., “New isomer: 68Cum,” Phys. Rev. 188, 1802–1806 ( 1969).
PROCEDURE: Irridiated zinc oxide or gallium metal was dissolved and the solution adjusted to 1 to 2M in HCl. Iron filings or iron metal was added to the solution. Copper produced in the nuclear reaction was reduced to the metallic state and was deposited on the iron.
Dy-1
Dysprosium
SEPARATION TIME: <9 min
SEPARATION TECHNIQUE: Extraction, ion exchange
PRODUCTION MODE: Autobatch
REFERENCE: Gehrke, R. J., Greenwood, R. C., Baker, J. D., and Meikrantz, D. H., “Identification of a new isotope, 168Dy,” Z. Phys. A306, 363–365 ( 1982).
PROCEDURE: See procedure Ln-1 under “Lanthanides,” Baker, J. D., et al., J. Radioanal. Chem. 74, 117–124 (1982).
Eu-1
Europium
SEPARATION TIME: 1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Malan, H. P., and Munzel, H., “Rapid radiochemical separations. II. Separation of europium from other lanthanides,” Radiochim. Acta 5, 20–23 ( 1966).
PROCEDURE: Irradiated Sm2O3 was dissolved in HCl (1M) containing europium and samarium carriers and then mixed with a solution of NaOH (1M) – EDTA (disodium salt, 0.1M). The resulting solution, at pH 1, was transferred to a separatory funnel containing sodium amalgam (0.1%) and shaken vigorously for 5 s. Barium amalgam (0.5%) was added to the funnel, and the amalgam phase was transferred to a second separatory funnel containing HCl (12M) flushed with N2. A white precipitate containing NaCl, BaCl2·2H2O, and EuCl2·2H2O was formed. The precipitate was filtered, washed with alcohol, and used. The europium yield was ~50%. The separation factor with respect to samarium was 103; for other lanthanides, the separation factor was higher by a factor of 10 or more. Also see Malan, H. P., and Mungel, H., Radiochim. Acta 5, 24–28 (1966).
Gd-1
Gadolinium
SEPARATION TIME: 3.2 min
SEPARATION TECHNIQUE: Extraction, ion exchange
PRODUCTION MODE: Autobatch
REFERENCE: Gehrke, R. J., Greenwood, R .C., Baker, J .D., and Meikrantz, D. H., “A new isotope 163Gd; comments on the decay of 162Gd,” Radiochim. Acta 31, 1–5 ( 1982).
PROCEDURE: See procedure Ln-1 under “Lanthanides,” Baker, J. D., et al., J. Radioanal. Chem. 74, 117–124 (1982).
Ga-1
Gallium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Livingood, J. J., and Seaborg, G. T., “Radioactive isotopes of iron,” Phys. Rev. 54, 51–55 ( 1938).
PROCEDURE: The irradiated iron was dissolved in a mixture of HCl and HNO3 containing gallium, manganese, and cobalt carriers. The iron was reduced to Fe(II) by the addition of mercury. The solution was adjusted to 6M in HCl and gallium was extracted with ether. Iron in the aqueous solution was extracted with ether after the oxidization to Fe(III) with HNO3.
Ga-2
Gallium
SEPARATION TIME: ~1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Crasemann, B., “Decay of 64Ga and 65Ga,” Phys. Rev. 90, 995–996 ( 1953).
PROCEDURE: The irradiated zinc foil or ZnO was dissolved, and the solution was made 6M in HCl. Gallium was extracted with ether; the ether layer was washed with 6M HCl. The ether was evaporated onto a counting plate.
Ge-1
Germanium
SEPARATION TIME: 10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Autobatch
REFERENCE: Kratz, J. V., Franz, H., Kaffrell, N., and Herrmann, G., “Gamma-ray emissions from 80-86As isotopes,” Nucl. Phys. A250, 13–37 ( 1975).
PROCEDURE: The fission-product solution was mixed with HCl (12M) containing germanium carrier and K2S2O8. N2 gas was bubbled through the solution, and the volatilized GeCl4 was absorbed in HCl (0.2M) containing arsenic carrier. Arsenic was separated from the solution after sufficient decay of germanium.
Ge-2
Germanium
SEPARATION TIME: ~5 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Autobatch
REFERENCE: del Marmol, P. and Van Tigchelt, H., “A fast radiochemical procedure for separating Ge from fission products, ” Radiochim. Acta 17, 52–54 ( 1972).
PROCEDURE: Irradiated uranium solution was mixed with HCl containing germanium carrier; the final HCl concentration adjusted to 9 to 11 M. Cl2 was gas-bubbled through the solution and the gas passed through Drierite and zinc powder. Germanium retained by the zinc powder was counted. Also see del Marmol, P., et al., J. Inorg. Nucl. Chem. 29, 273–279 (1967) and Nucl. Phys. A194, 140–160 (1972).
Ge-3
Germanium
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Extraction, adsorption
PRODUCTION MODE: Continuous
REFERENCE: Zendel, M., Trautmann, N., and Herrmann, G., “Chemical reactions in a gas-jet recoil-transport system (II): continuous separation procedure for Ge and As from fission products,” Radiochim. Acta 29, 17–20 ( 1981).
PROCEDURE: See procedure As-4 under “Arsenic,” Zendel, M., et al., Radiochim. Acta 29, 17–20 (1981). The detector was set up to record activities in the charcoal trap.
Ge-4
Germanium
SEPARATION TIME: 25 s
SEPARATION TECHNIQUE: Distillation
PRODUCTION MODE: Batch
REFERENCE: Robertson, R. G. H., and Austin, S. M., “Germanium-64,” Phys. Rev. Lett. 29, 130–133 ( 1972).
PROCEDURE: Irradiated zinc was dissolved in concentrated HCl containing KClO3. The GeCl4 was distilled under vacuum at room temperature and allowed to deposit in a cold trap in front of a detector. Also see Porile, N. T., Phys. Rev. 112, 1954–1958 (1958).
Ge-5
Germanium
SEPARATION TIME: 25 s
SEPARATION TECHNIQUE: Distillation
PRODUCTION MODE: Batch/autobatch
REFERENCE: Robertson, R. G. H., and Austin, S. M., “Neutron-deficient isotopes Ge-64 and Ge-65,” Phys. Rev. C 9, 1801–1812 ( 1974).
PROCEDURE: Irradiated zinc metal was dissolved in concentrated HCl containing KClO3. The GeCl4 was distilled under vacuum at room temperature; the GeCl4 was deposited in a cold trap (cooked by a mixture of dry ice and alcohol) in front of a detector. The actual distillation time was 15 s. After counting, the cold trap was cleaned by flushing with alcohol and drying with a stream of nitrogen. A schematic diagram of the apparatus is given in the paper.
Au-1
Gold
SEPARATION TIME: ~4 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., Chemical Procedures Used in Bombardment Work at Berkeley, U.S. Atomic Energy Commission report AECD-2738 ( 1949), p. 228.
PROCEDURE: The procedure was developed by S. G. Thompson and J. O. Rasmussen for purifying irradiated gold. Gold foil, after irradiation, was dissolved in aqua regia (6M, i.e., 2M HNO3 – 4M HCl), and hold-back carriers were added for mercury, platinum, iridium, and osmium. Gold was extracted with ethyl acetate; the organic phase was washed once or twice with aqua regia (6M). The organic phase was evaporated to prepare the counting sample.
Au-2
Gold
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Rasmussen, Jr., J. O., Thompson, S. G., and Ghiorso, A., “Alpha-radioactivity in the 82-neutron region,” Phys. Rev. 89, 33–48 ( 1953).
PROCEDURE: The irradiated gold or platinum targets were dissolved in hot, concentrated aqua regia. The solution was adjusted to 6M in acid. Gold was extracted with ethyl acetate. The organic phase was washed twice with 2M HCl.
Hf-1
Hafnium
SEPARATION TIME: ~0.4 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Zvara, I., Belov, V. Z., Chelnokov, L. P., Domanov, V. P., Hussonois, M., Korotkin, Yu. S., Schegolev, V. A., and Shalayevsky, M. R., “Chemical separation of kurchatovium,” Inorg. Nucl. Chem. Lett. 7, 1109–1116 ( 1971).
PROCEDURE: See procedure 104-2 under “Z = 104,” Zvara, I., et al., Inorg. Nucl. Chem. Lett. 7, 1109–1116 (1971). Also see Zvara, I., et al., Soviet Radiochemistry (Eng. Tr.) 14, 115–118 (1972).
Hf-2
Hafnium
SEPARATION TIME: 5 min
SEPARATION TECHNIQUE: Extraction, precipitation
PRODUCTION MODE: Batch
REFERENCE: Zychor, I., Rykaczewski, K., Ahrens, H., Folger, H., Kurcewicz, W., Summerer, K., Kaffrell, N., and Trautmann, N., “Hf and Lu isomers produced in heavy-ion collisions of 7.6 MeV/u 40Ar, 8.5 MeV/u 84Kr, and 8.5 MeV/u 136Xe on natural W targets,” Radiochem. Radioanal. Lett. 33, 1–2 ( 1983).
PROCEDURE: Irradiated tungsten was dissolved in a mixture of concentrated HNO3 – concentrated HF – H2O2. Boric acid was added to precipitate WO3, and the mixture was filtered. Hafnium and lutetium were extracted from the filtrate by TBP (absorbed on a fine-grained carrier) and stripped with HCl (1M). The HCl solution was then passed through a layer of Voltalef containing HDEHP, which retained hafnium and lutetium. The layer was washed with HCl; hafnium was removed with HF (0.25M) and coprecipitated with Fe(OH)3 in the presence of boric acid. Lutetium was left in the HDEHP layer.
Hf-3
Hafnium
SEPARATION TIME: 10 to 20 s
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Autobatch
REFERENCE: Bruchertseifer, H., Eichler, B., Estevez, J., and Zvara, I., “Fast, continuous, radiochemical isolation of the short-lived isotopes of hafnium, tantalum and tungsten produced by heavy-ion-induced reactions, ” Radiochim. Acta 47, 41–46 ( 1989).
PROCEDURE: The nitrogen gas containing KCl aerosol thermalized and carried the recoiling nuclear-reaction products. The stream was passed through a nuclear filter that retained the KCl particles. The flow was diverted to another identical filter unit while the KCl carrying the reaction products was dissolved in 0.5M HF and passed through a Wolfatit KPS resin column (cation exchanger, 0.5-cm i.d., 1- to 2-cm height; 0.3 to 1.7- mL/min). The resin retained lanthanides; hafnium passed through. The activity of the effluent was recorded.
Hal-1
Halogens
SEPARATION TIME: ~1 s
SEPARATION TECHNIQUE: Emanation
PRODUCTION MODE: Batch/continuous
REFERENCE: Ahrens, H., Patzelt, P., and Herrmann, G., “The half-lives of 91Br, 95Kr, 140I, 141I, and 144Xe,” J. Inorg. Nucl. Chem. 38, 191–192 ( 1976).
PROCEDURE: Volatile nuclides emanating from uranyl stearate were carried by N2 gas. The daughter products were collected on stainless-steel grids kept at a negative potential. The half-lives were deduced by using daughter activities in grids at different distances.
Hal-2
Halogens
SEPARATION TIME: ~2 s
SEPARATION TECHNIQUE: Hot-atom reaction
PRODUCTION MODE: Batch/continuous
REFERENCE: Lundan, A., and Anttila, K., “An apparatus for the production and fast separation of gaseous fission products,” Nucl. Instr. Methods 79, 333 ( 1970).
PROCEDURE: A UO2 target was placed in a chamber filled with CH4. Recoiling bromine and iodine form CH3Br and CH3I. The methyl halides formed, along with krypton and xenon, are swept by a stream of CH4. A silica gel coated with AgNO3 retained most of the bromine and iodine.
He-1
Helium
SEPARATION TIME: 2 s
SEPARATION TECHNIQUE: Emanation
PRODUCTION MODE: Batch
REFERENCE: Sommers, Jr., H. S., and Sherr, R., “Activity of 16N and 6He,” Phys. Rev. 69, 21–30 ( 1946).
PROCEDURE: Be(OH)2 in high-emanating form was prepared by precipitating beryllium from ammoniacal solution. The precipitate was washed repeatedly until free from ammonia. The solid was air dried and used after removal of fine particles. The material was kept in a container filled with H2 gas and irradiated with fast neutrons obtained from the bombardment of beryllium with deuterons. By manipulation of solenoid valves, the H2 gas containing 6He was transported to a container in the counting position. Counting was started 2 s after the end of the irradiation.
In-1
Indium
SEPARATION TIME: 5 to 10 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Semkow, T., and Wahl, A. C., “Extraction of Ag(I), Cd(II), In(m), Sn(II), Sn(IV), Sb(III), and U(VI) from aqueous solutions by ketone solutions using single-step batch and continuous SISAK methods,” J. Radioanal. Chem. 79, 93–101 ( 1983).
PROCEDURE: Aqueous solution containing indium and tin activities was made 0.1M in KI and 0.05M in H2SO4. The solution was extracted with 85 vol% methyl isobutyl ketone (MIBK) – 15 vol% cyclohexanone(CHO) containing 5 × 10−4M I2. More than 99% of In(III) was extracted in 1 s and less than 0.1% of Sn(IV) was extracted. Sn(IV) was extracted from the aqueous solution using a 0.05M solution of 2,3-dimercapto-propanol-1 (BAL, British anti-Lewisite) in 85 vol% MIBK – 15 vol% CHO. It was found necessary to keep the solutions free from O2 and I2. A 10-s extraction was performed. Tin can be back-extracted by shaking the organic phase for 2 min with 0.5M HCl – 0.1M KF – I2 solution. This procedure provides a fast indium-tin separation.
In-2
Indium
SEPARATION TIME: ~2 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Semkow, T., and Wahl, A. C., “Extraction of Ag(I), Cd(II), In(III), Sn(II), Sn(IV), Sb(III), and U(VI) from aqueous solutions by ketone solutions using single-step batch and continuous SISAK methods,” J. Radioanal. Chem. 79, 93–101 ( 1983).
PROCEDURE: The aqueous solution containing the actvities and 0.1M in KI and 0.05M in H2SO4 was passed onto the SISAK system. The organic extractant was a solution 85 vol% methyl isobutyl ketone (MIBK) and 15 vol% cyclohexanone (CHO) containing 5 × 10−4M I2. Indium was extracted into the organic phase with ~50% efficiency. Ag(I) and Cd(II) were extracted nearly quantitatively, while Sn(IV) remained in the aqueous phase. Also see Robinson, L., et al., Phys. Rev. C31, 1334–1339 (1985), and Semkow, T. M., et al., Phys. Rev. C30, 93–101 (1984).
In-3
Indium
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Gleit, C. E., and Coryell, C. D., “Decay of 49-min 118Cd and 5.1-s 118In,” Phys. Rev. 122, 229–231 ( 1961).
PROCEDURE: This procedure was used for milking indium from cadmium. From fission products, cadmium was extracted with dithizone in CHCl 3. Cadmium was then back-extracted with HBr, the solution was made 8M in HBr. Indium was extracted as HInBr4 with bis(2-chloroethyl) ether from the HBr solution. Cadmium remained in the aqueous phase as CdBr4−. Indium may be back-extracted from the ether solution with dilute HCl.
I-1
Iodine
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Perlow, G. J., and Stehney, A. F., “Halogen delayed-neutron activities,” Phys. Rev. 113, 1269–1272 ( 1959).
PROCEDURE: Uranium was dissolved in H2SO4 (0.1M) containing Fe(III) (0.3M) and Fe(II) (0.2M), and coated on glass beads in a vessel. The solution also contained Br− holdback carrier. After irradiation, air saturated with I2 vapor was sucked through the vessel. The air was then passed through a vessel containing glass beads coated with CCl4. The CCl4 retained the I2.
I-2
Iodine
SEPARATION TIME: 2 s
SEPARATION TECHNIQUE: Hot-atom reaction
PRODUCTION MODE: Continuous
REFERENCE: Lundan, A., “On the decay of bromine isotopes 86Br and 87Br,” Z. Physik 236, 403–409 ( 1970).
PROCEDURE: See procedure Br-6 under “Bromine,” Lundan, A., Z. Physik 236, 403–409 (1970).
I-3
Iodine
SEPARATION TIME: 0.5 s
SEPARATION TECHNIQUE: Hot-atom reaction
PRODUCTION MODE: Batch
REFERENCE: Kratz, K. L., “Independent fission yields and neutron-emission probabilities of short-lived halogen isotopes,” Radiochim. Acta 25, 1–7 ( 1978).
PROCEDURE: See procedure Br-8 under “Bromine,” Kratz, K. L., Radiochim. Acta 25, 1–7 (1978). Also see Kratz, K. L., and Herrmann, G., Radiochem. Radioanal. Lett. 13, 385–390 (1973).
I-4
Iodine
SEPARATION TIME: ~5 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Brodén, K., Skarnemark, G., Björnstad, T., Eriksen, D., Haldorsen, I., Kaffrell, N., Stender, E., and Trautmann, N., “Rapid continuous-separation procedures for Zr, Nb, Tc, Br, and I from complex reaction-product mixtures,” J. Inorg. Nucl. Chem. 43, 765–771 ( 1981).
PROCEDURE: Fission fragments were thermalized and transported by a KCl aerosol. In the dissolution chamber, the fission products were dissolved in NaHSO3 (0.01M) – H2SO4 (0.1M) solution containing I− carrier, then degassed, mixed with NaNO2 (to a final concentration of ~0.05M), and passed to a SISAK-2 system. In the first centrifuge, iodine was extracted with CCl4; in the second stage, iodine was stripped with NaHSO3 (0.2M) in H2SO4 (0.1M). The small krypton and xenon contaminants were removed in the third step by an extraction with CHCl3. The aqueous phase was counted for iodine.
I-5
Iodine
SEPARATION TIME: ~1 min
SEPARATION TECHNIQUE: Gas chromatography
PRODUCTION MODE: Batch
REFERENCE: Denschlag, H. O., and Gordus, A. A., “Gas-chromatographic technique for rapid isolation of U-fission products, ” Z. Anal. Chem. 226, 62–71 ( 1967).
PROCEDURE: See procedure Br-10 under “Bromine,” Denschlag, H. O., and Gordus, A. A., Z. Anal. Chem. 226, 62–71 (1967).
I-6
Iodine
SEPARATION TIME: 52 s
SEPARATION TECHNIQUE: Exchange
PRODUCTION MODE: Batch
REFERENCE: Ohm, H., Zendel, M., Prussin, S. G., Rudolph, W., Schroder, A., Kratz, K. L., Ristori, C., Pinston, J. A., Monnand, E., Schüssler, F., and Zirnheld, J. P., “Beta-delayed neutrons and high-energy gamma rays from decay of 137iodine,” Z. Phys. A 296, 23–33 ( 1980).
PROCEDURE: The fission-product halogens were exchanged with a preformed AgI precipitate by passing the fission-product solution through a layer of AgI. The iodine was later separated from Br by an oxidation-reduction-extraction cycle. For details on exchange, see Eckhardt, W., Herrmann, G., and Schüssler, H. D., Z. Analyt. Chem. 226, 71 (1967).
I-7
Iodine
SEPARATION TIME: 2 s
SEPARATION TECHNIQUE: Exchange
PRODUCTION MODE: Batch
REFERENCE: Denschlag, H. O., “Independent yields of 135I in the thermal neutron fission of 235U. The half-life of 135Te,” J. Inorg. Nucl. Chem. 31, 1873–1882 ( 1969).
PROCEDURE: Irradiated uranyl nitrate solution containing iodide, Te(IV), Sn(II), Sb(III), oxalic acid, and sulfite was passed through a preformed AgCl layer. The fission products halogens exchanged with AgCl and were retained. The layer was washed with a HNO3 solution (0.5M) containing oxalic acid (10%). The AgCl was then reduced to Ag; iodine in the solution was oxidized with NaNO2 (in the presence of HNO3) and extracted with an organic solvent. The I2 was deduced with sulfite and AgI precipitated for counting. The initial separation of iodine from the precursors was achieved in 2 s. Also see Denschlag, H. O., and Qaim, S. M., J. Inorg. Nucl. Chem. 33, 3649–3661 (1971).
I-8
Iodine
SEPARATION TIME: ~30 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Nuh, F. M., Slaughter, D. R., Prussin, S. G., Kratz, K. L., Franz, H., and Herrmann, G., “The gamma-ray competition with neutron emission in the decay of 137I,” Phys. Lett. B53, 435–438 ( 1975).
PROCEDURE: Hot (130°C) 15M sulfuric acid was added to a mixture of fission products containing iodide carrier, volatilized molecular iodine was collected in a bisulfite solution within 10 s after the end of the irradiation. The solution was allowed to stand for 5 s, and then krypton and xenon activities were removed by bubbling air through the solution. Iodine was finally precipitated as AgI for counting. For further details, see procedure Br-3 under “Bromine,” Nuh, F. M., et al., Radiochim. Acta 17, 149–153 (1972).
I-9
Iodine
SEPARATION TIME: 28 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Snell, A. H., Levinger, J. S., Meiners, Jr., E. P., Sampson, M. B., and Wilkinson, R. G., “Studies of delayed neutrons. II. Chemical isolation of the 56-s and the 23-s activities,” Phys. Rev. 72, 545–549 ( 1947).
PROCEDURE: Irradiated uranyl nitrate solution was mixed with Br− and I− carriers and HCl. The iodine was oxidized with NaNO2 solution, and the I2 produced was extracted with CCl4. The CCl4 layer was taken for counting.
I-10
Iodine
SEPARATION TIME: 28 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Levinger, J. S., Meiners, E. P., Sampson, M. B., Snell, A. H., and Wilkinson, R. G., “Chemical isolation of 56-s Br and 23-s I delayed-neutron activities, ” Radiochemical Studies: The Fission Products, National Nuclear Energy Series, Vol. 9, Coryell, C. D., and Sugarman, N. (Eds.), Book 2 ( 1951), p. 603.
PROCEDURE: A uranyl nitrate solution containing KI, KBr, and HCl was irradiated in a separatory funnel along with CCl4. After irradiation, NaNO2 solution was added to the funnel, and the iodine liberated was extracted with CCl4. The CCl4 layer was separated and counted.
I-11
Iodine
SEPARATION TIME: ~10 to 15 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Delucchi, A. A., and Greendale, A. E., “Fission yields of several iodine isotopes and half-life and fission yield of 135Te,” Phys. Rev. C 1, 1491–1497 ( 1970).
PROCEDURE: This procedure was used to release fission-product iodine. The target was a solution of uranium peroxide in periodic acid prepared by melting a mixture of the two in a weight ratio of 1:14, respectively. The material in powder form was mixed with activated carbon and irradiated. After irradiation, the periodic acid was decomposed by heating, and the iodine released was passed through a heated, sintered-glass frit and collected on a column of glass beads wetted with CCl4. The column was washed with CCl4; the iodine was stripped with an aqueous solution of NaHSO3. The aqueous solution was used for counting.
Ir-1
Iridium
SEPARATION TIME: 3 to 5 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayar, B., Novgorodov, A. F., Vocilka, I., and Zaitseva, N. G., “Rapid gas-thermochromatographic separation of radioactive elements. II. Au as a universal target for the rapid producing of radioactive Re, Os, Ir, and Hg isotopes,” Radiochem. Radioanal. Lett. 19, 43–53 ( 1974).
PROCEDURE: See procedure Re-2 under “Rhenium,” Bayar, B., et al., Radiochem. Radioanal. Lett. 19, 43–53 (1974). Also see Bayer, B., et al., Soviet Radiochemistry (Eng. Tr.) 16, 333–338 (1974).
Ir-2
Iridium
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Naumann, R. A., and Gerhart, J. B., “5.6-s 191mIr following 191Os decay,” Phys. Rev. 96, 1452–1453 ( 1954).
PROCEDURE: Irradiated osmium was converted to ammonium perosmiate, (NH4)2OsO5, and dissolved in NH4OH. The solution was dried on platinum foil and flamed. Osmium was volatilized, and iridium was left in the foil.
Ir-3
Iridium
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch/continuous
REFERENCE: Campbell, E. C., and Nelson, F., “Rapid ion-exchange techniques for radiochemical separations,” J. Inorg. Nucl. Chem. 3, 233–242 ( 1956).
PROCEDURE: This procedure was developed for the separation of short-lived Ir daughter products from parent osmium. Purified osmium activity was prepared in the chemical form OsCl62− and adsorbed on the anion exchanger Dowex-1 from HCl (6M) solution. Ir daughter activity was eluted with a small volume of HCl (6M). Also see procedure Pb-1 under “Lead,” Campbell, E. C., and Nelson, F., J. Inorg. Nucl. Chem. 3, 233–242 (1956).
Fe-1
Iron
SEPARATION TIME: 3 to 5 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., Chemical Procedures Used in Bombardment Work at Berkeley, U.S. Atomic Energy Commission report AECD-2738 ( 1949), p. 49.
PROCEDURE: The procedure was developed by R. C. Lilly for the separation of iron from irradiated nickel. Irradiated nickel foil was dissolved in hot, concentrated HNO3 containing cobalt, copper, and iron carriers, and iron was precipitated as the hydroxide. The mixture was centrifuged, the precipitate dissolved in a few drops of concentrated HCl, and carriers of cobalt, copper, and nickel were added to the solution. Iron was reprecipitated as the hydroxide and filtered. The precipitate was filtered, dried, and mounted for counting.
Fe-2
Iron
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Livingood, J. J., and Seaborg, G. T., “Radioactive isotopes of iron,” Phys. Rev. 54, 51–55 ( 1938).
PROCEDURE: The irradiated iron was dissolved in a mixture of HCl and HNO3 containing MnCl2, CoCl2, and NaH2PO4 as holdback carriers. After dissolution, the solution was adjusted to 6M in HCl and extracted with ether. The extract was washed with 6M HCl.
Fe-3
Iron
SEPARATION TIME: 150 s
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Strain, J. E., and Ross, W. J., “Reactions between 14.7-MeV neutrons and Ni-64,” J. Inorg. Nucl. Chem. 28, 2075–2080 ( 1966).
PROCEDURE: Irradiated nickel in 8M HCl solution was slurried with 2 g of Dowex-1 (in Cl− form), filtered, and washed with 8M HCl. Cobalt and iron carriers were added to the resin; the mixture was then slurried with water and filtered. The filtrate was then mixed with H2O2 and NH4OH, and the iron was precipitated as the hydroxide; the Fe(OH)3 precipitate was separated from the cobalt complex by filtration.
Kr-1
Krypton
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Emanation
PRODUCTION MODE: Batch
REFERENCE: Wolfsberg, K., “Nuclear charge distribution in fission: fractional yields of krypton and xenon isotopes from thermal-neutron fission of 233U and 239Pu and from 14-MeV neutron fission of 235U and 238U,” Phys. Rev. 137, B929–B935 ( 1965).
PROCEDURE: This procedure was used for determination of the fission yields of krypton and xenon isotopes. The fissile target, in the form of a thin film, was covered with barium or praseodymium stearate and irradiated. The wall of the irradiation chamber was lined with filter paper. Fission products recoiling from the target collected in the stearate. Krypton and xenon were quantitatively emanated from the stearate. The decay products of krypton and xenon were deposited on the filter-paper lining of the wall. Fission yields of krypton and xenon isotopes were determined by analysis of the decay products. Also see procedure Ng-1 under “Noble gases,” Wahl, A. C., J. Inorg. Nucl. Chem. 6, 263–277 (1958).
Kr-2
Krypton
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Creutz, E. C., Delsasso, L. A., Sutton, R. B., White, M. G., and Barkas, W. H., “Radioactivity produced by proton bombardmem of Br and I,” Phys. Rev. 58, 481–486 ( 1940).
PROCEDURE: See procedure Br-13 under “Bromine,” Creutz, E. C., et al., Phys. Rev. 58, 481–486 (1940).
Ln-1
Lanthanides
SEPARATION TIME: <3 min
SEPARATION TECHNIQUE: Extraction, ion exchange
PRODUCTION MODE: Autobatch
REFERENCE: Baker, J. D., Gehrke, R. J., Greenwood, R. C., and Meikrantz, D. H., “Advanced system for the separation of rare earth fission products, ” J. Radioanal. Chem. 74, 117–124 ( 1982).
PROCEDURE: Fission products, transported by NaCl aerosol and collected in Mylar film, were dissolved in hot HNO3 (4M) and passed through an extraction column to achieve separation of lanthanides (Ln) from other fission products. A column packed with Vydac C8 resin saturated with dihexyldiethylcarbamoylmethylphosphonate was used. The lanthanides were eluted with α-hydroxyisobutyric acid, AHIB (1M, pH 5.0), after washing the non-lanthanides with HNO3. The individual lanthanides were separated using an Aminex A-9 resin column operated at 95°C. A gradient elution technique with AHIB was used (0.65M, pH 3.6 to 0.95M, pH 4.8). The procedure can be tailored to elute any specific lanthanide within 3 min from the end of fission-product collection. Also see DOE report EGG-Phys-5269 (1980), CERN report 81-09, and Baker, J. D., et al., Radiochim. Acta 28, 51–54 (1981).
Ln-2
Lanthanides
SEPARATION TIME: ~5 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Ohyoshi, E., Ohyoshi, A., Tamai, T., and Shinagawa, M., “Rapid separation of fission-product lanthanides and yttrium by electromigration, ” J. Nucl. Sci. Technol. 8, 444–449 ( 1971).
PROCEDURE: An irradiated uranium solution containing the appropriate carriers was extracted with CCl4 containing Br2 to remove certain nuclides soluble in CCl4. The aqueous phase was spotted on a chromatographic paper wetted with the supporting electrolyte (4 × 10−3M nitrilotriacetic acid, pH 2.0). A potential gradient of 75 V/cm was applied for 4 min. The zones were located by color reaction: Arsenazo III for lanthanides, oxine for yttrium, and sodium rhodizonate for strontium.
Ln-3
Lanthanides
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Gas chromatography
PRODUCTION MODE: Autobatch
REFERENCE: Greulich, N., Hickmann, U., Trautmann, N., and Herrmann, G., “Fast-preparation and gas-chromatographic separation of lanthanide and actinide hexafluoroacetylacetonates,” Z. Anal. Chem. 323, 839–845 ( 1986).
PROCEDURE: The KCl-loaded N2 gas carrying the fission products was passed through a quartz-wool plug (qw) for a selected time. The qw was on top of a Chromosorb G column coated with a mixture of hexafluoroacetylacetone (HFA) and trioctylphosphine oxide (TOPO). The fission products were then washed with an aqueous phase onto the column. The column containing the lanthanide complexes was injected into the GC. The carrier gas N 2 was loaded with HFA. A 2-m Chromosorb G column with a temperature gradient of 150 to 2500°C was found to be advantageous. The heavier lanthanides had lower retention times. Conditions can be adjusted for the quick separation of a specific lanthanide element.
Ln-4
Lanthanides
SEPARATION TIME: 15 to 20 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Ramaswami, A., Oak, M. S., and Satya Prakash, “Cumulative yields of short-lived rare-earth fission products in the spontaneous fission of 252Cf,” Radiochim. Acta 54, 163–164 ( 1991).
PROCEDURE: Recoiling fission products from a thin 252Cf source were collected using NH4Cl pellet as a catcher. The pellet was dissolved in a minimum amount of HCl (0.1M) and passed through a column of Celite coated with di-(2-ethylhexyl)phosphoric acid (HDEHP). The column was washed with HCl (0.15M); only lanthanides were retained by the column under these conditions. Gamma-ray spectra of the column were obtained using an HPGe detector. The chemical yield was found to be 98 ± 2%.
La-1
Lanthanum
SEPARATION TIME: 3 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Yamamoto, J., Ikeda, Y., Kawade, K., Katoh, T., and Nagahara,T., “Decay studies of 143La and 147Pr,” J. Inorg. Nucl. Chem. 43, 855–865 ( 1981).
PROCEDURE: Irradiated uranium solution containing the appropriate carriers was spotted on chromatographic paper wetted with the supporting electrolyte (4 × 10−3M nitrilotriacetic acid, pH 1.8). A field of 300 V/cm was applied. The migration time was 2 min. The zones of lanthanum and praseodymium were located by the color reaction with Arsenazo III.
La -2
Lanthanum
SEPARATION TIME: 10 to 20 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Aronsson, P. O., Skarnemark, G., and Skarestad, M., “Short-lived isotopes of La, Ce, and Pr studied by the SISAK technique, ” J. Inorg. Nucl. Chem. 36, 1689–1696 ( 1974).
PROCEDURE: Fission products formed in the target uranyl sulfate complex adsorbed on a Dowex-1 X8 column were continuously eluted with (NH 4)2SO4 solution (pH 5) at 85°C. The eluent was mixed with HNO3 (0.25M) to adjust the pH to 1.4 and then passed on to a SISAK system. The trivalent lanthanides were extracted into HDEHP (0.25M) in kerosene in the first centrifuge. In the second centrifuge, they were stripped by HNO3 – H2SO4 – K2Cr2O7. The aqueous solution containing lanthanum was passed through a column containing PVC coated with HDEHP for further decontamination. Lanthanum in the aqueous solution was measured. Also see Skarnemark, G., et al., Nucl. Instrum. Methods 171, 323–328 (1980).
La-3
Lanthanum
SEPARATION TIME: <5 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REPERENCE: Skarnemark, G., Aronsson, P. O., Brodén, K., Rydberg, J., Björnstad, T., Kaffrell, N., Stender, E., and Trautmann, N., “An improved system for fast, continuous chemical separations (SISAK-2) in nuclear spectroscopic studies,” Nucl. Instr. Methods 171, 323–328 ( 1980).
PROCEDURE: For information about SISAK-2 system see procedure Ce-5 under “Cerium,” Skarnemark, G., et al., Nucl. Instr. Methods 171, 323–328 (1980). For details about chemical separation see procedure La-2 under “Lanthanum,” Aronson, P. O., et al., J. Inorg. Nucl. Chem. 36, 1689–1696 (1974).
La-4
Lanthanum
SEPARATION TIME: 40 s
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Ikeda, Y., Yamamoto, H., Kawade, K., Katoh, T., and Nagahara, T., “Decay study of 144La,” J. Phys. Soc. Japan 47, 1389–1394 ( 1979).
PROCEDURE: Irradiated uranium solution containing the appropriate carriers was spotted on the chromatographic paper wetted with the supporting electrolyte (4 × 10−3M nitrilotriacetic acid, pH 1.9). An electrical field of 5000 V/10 cm was applied for 20 s. The lanthanum zone was cut and counted. Also see procedure Ln-2 under “Lanthanides,” Ohyoshi, E., et al., J. Nucl. Sci. Technol. 8, 444–449 (1971) and procedure La-1 under “Lanthanum,” Yamamoto, H., et al., J. Inorg. Nucl. Chem. 43, 855–865 (1981).
La-5
Lanthanum
SEPARATION TIME: ~4 min
SEPARATION TECHNIQUE: Extraction, ion exchange
PRODUCTION MODE: Continuous
REFERENCE: Björnstad, T., Kvale, E., Skarnemark, G., Aronsson, P. O., Kaffrell, N., Trautmann, N., and Stender, E., “Decay properties of 143La,” J. Inorg. Nucl. Chem. 39, 1107–1111 ( 1977).
PROCEDURE: Fission products thermalized in a C2H4 – N2 mixture were mixed with HNO3 (pH 1.4), degassed, and passed on to a SISAK system. In the first centrifuge, lanthanides were extracted with HDEHP (0.3M) in kerosene, leaving barium, including 143Ba, in aqueous solution. The aqueous solution was passed to a second centrifuge through a 15-s delay unit, and the lanthanum grown was extracted into HDEHP. The organic phase was passed to a third centrifuge through a 200-s delay unit, and lanthanum was stripped with a HNO 3 – H2SO4 – K2Cr2O7 solution, leaving cerium grown from short-lived lanthanum in the HDEHP phase. The aqueous solution was passed through a Dowex-50W column, which retained lanthanum.
La-6
Lanthanum
SEPARATION TIME: 4.5 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Ohyoshi, A., Ohyoshi, E., Tamai, T., and Shinagawa, M., “Short-lived isotopes of La, Ce, and Pr in neutron irradiated uranium, ” J. Inorg. Nucl. Chem. 34, 3293–3302 ( 1972).
PROCEDURE: Irradiated solution [0.1 mL of 1 × 10−2M UO2(NO3)2] was mixed with a carrier solution containing lanthanum, cerium, and praseodymium and spotted on a paper strip set in a migration cell containing CCl4 as coolant. Nitrtlotriacetic acid at a pH of 2.0 was used as supporting electrolyte. A voltage of 86 to 93 V/cm was applied for 90 s. The lanthanum zone was located by color reaction with Arsenazo III. Also see Ohyoshi, A., et al., J. Nucl. Sci. Technol. 9, 658–661 (1972).
La-7
Lanthanum
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Preiss, I. L., Strudler, P. M., and Wolfgang, R., “Lanthanum isotopes in a possible new region of nuclear deformation, ” Phys. Rev. 129, 1284–1285 ( 1963).
PROCEDURE: Irradiated indium foils were dissolved in hot, concentrated HCl containing cesium, barium, and lanthanum carriers. Barium was precipitated as BaSO4 by the addition of a stoichiometric amount of 0.5M H2SO4 and centrifuged. LaF3 was precipitated from the supernate by the addition of NaF. The precipitate was filtered, washed, and used for counting or dissolved in a 4:1 mixture of HNO3:H3BO3 and used for the milking of barium by BaSO4 precipitation.
La-8
Lanthanum
SEPARATION TIME: 1.7 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Sheline, R. K., Sikkeland, T., and Chanda, R. N., “Experimental observations of a new region of nuclear deformation, ” Phys. Rev. Lett. 7, 446–449 ( 1961).
PROCEDURE: The irradiated palladium foil was dissolved in hot aqua regia and mixed with barium and lanthanum carriers. LaF3 was precipitated, filtered, washed with dilute HF, and used for counting.
Lr-1
Lawrencium
SEPARATION TIME: 50 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Silva, R., Sikkeland, T., Nurmia, M., and Ghiorso, A., “Tracer chemical studies of Lr,” Inorg. Nucl. Chem. Lett. 6, 733–739 ( 1970).
PROCEDURE: The recoiling products were collected on a platinum foil coated with NH4Cl. The products were dissolved in 100 µL of monochloroacetic acid – sodium acetate buffer at a pH of 3.05 and transferred to a test tube containing 100 mL of 0.2M 2-thenoyltrifluoroacetone (TTA) in methyl isobutyl ketone. Lawrencium was extracted by the TTA. The organic phase was separated, evaporated, flamed, and used for counting.
Lr-2
Lawrencium
SEPARATION TIME: 3 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Hoffman, D. C., Henderson, R. A., Gregorich, K. E., Bennett, D. A., Chasteler, R. M., Gannett, C. M., Hall, H. L., Lee, D. M., Nurmia, M. J., Cai, S., Agarwal, R., Charlop, A. W., Chu, Y. Y., Seaborg, G. T., and Silva, R. J., “Atom-at-a-time radiochemical separations of the heaviest elements: lawrencium chemistry,” J. Radioanal. Nucl. Chem. 124, 135–144 ( 1988).
PROCEDURE: The nuclear reaction products were carried by KCl aerosol and deposited on a metal foil. The deposit was dissolved in a buffer solution (monochloroacetic acid or acetic acid) and extracted with 0.1M 2-thenoyltrifluoroacetone (TTA) in methyl isobutyl ketone. The TTA phase was evaporated on a platinum plate and used for alpha counting. The extractions were carried out using buffers of different pH (1.86 to 5.5). The results showed that extraction of lawrencium was complete at a pH of 3.15.
Lr-3
Lawrencium
SEPARATION TIME: 6 min
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Hoffman, D. C., Henderson, R. A., Gregorich, K. E., Bennett, D. A., Chasteler, R. M., Gannett, C. M., Hall, H. L., Lee, D. M., Nurmia, M. J., Cai, S., Agarwal, R., Charlop, A. W., Chu, Y. Y., Seaborg, G. T., and Silva, R. J., “Atom-at-a-time radiochemical separations of the heaviest elements: lawrencium chemistry,” J. Radioanal. Nucl. Chem. 124, 135–144 ( 1988).
PROCEDURE: The nuclear reaction products were carried by KCl aerosol and were deposited on a metal foil. The deposit was taken up in HCl (0.05M) and loaded on a Dionex-cation exchange resin column (0.2-cm i.d. and 4.5 cm long, 10 µm resin) with appropriate lanthanide tracers. The lanthanides and actinides were eluted with ammonium α-hydroxyisobutyrate (0.05M, pH 4.47). Lawrencium was found to elute fairly close to the position of erbium.
Lr-4
Lawrencium
SEPARATION TIME: 160 s
SEPARATION TECHNIQUE: Cation exchange (HPLC)
PRODUCTION MODE: Autobatch
REFERENCE: Bruchle, W., Schädel, M., Scherer, U. W., Kratz, J. V., Gregorich, K. E., Lee, D., Nurmia, M., Chasteler, R. M., Hall, H. L., Henderson, R. A., and Hoffman, D. C., “The hydration enthalpies of Md(+3) and Lr(+3),” Inorg. Chim. Acta 146, 267–276 ( 1988).
PROCEDURE: The chemical separation was performed using the microprocessor-controlled Automated Rapid Chemistry Apparatus (ARCA). The KCl-loaded gas jet deposited the reaction products on a quartz frit. At the end of the collection time, the frit was washed with 300 mL of 0.05M α-hydroxyisobutyric acid (AHIB) at a pH of 4.85. The solution was loaded onto an Aminex A6 cation-exchange column (2 mm × 60 mm; 17.5-µm particle size) kept at 80°C. The activities were eluted with AHIB (0.12M, pH 4.85). Ten fractions were collected on tantalum disks (volume increased from 80 to 390 µL), evaporated, and counted. Lawrencium and mendelevium eluted between 120 and 160 s and 150 and 200 s, respectively.
Lr-5
Lawrencium (from mendelevium)
SEPARATION TIME: 1 to 2 min
SEPARATION TECHNIQUE: Extraction (HPLC)
PRODUCTION MODE: Autobatch
REFERENCE: Scherer, U. W., Kratz, J. V., Schädel, M., Bruchle, W., Gregorich, K. E., Henderson, R. A., Lee, D., Nurmia, M., and Hoffman, D. C., “Lawrencium chemistry: no evidence for oxidation states lower than 3+ in aqueous solution,” Inorg. Chim. Acta 146, 249–254 ( 1988).
PROCEDURE: The separation was performed using an Automated Rapid Chemistry Apparatus (ARCA) controlled by a microprocessor. The solution containing lawrencium and mendelevium was treated with a reducing agent (V2+, Cr2+, or NH3OH+) and passed through a column containing HDEHP. Lawrencium remained in the trivalent state and was retained on the column; mendelevium was reduced to the (2+) state and was present in the eluate. For further details, see procedure Md-3 under “Mendelevium,” Scherer, U. W., et al., Inorg. Chim. Acta 146, 249–254 (1988).
Pb-1
Lead
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch/continuous
REFERENCE: Campbell, E. C., and Nelson, F., “Rapid ion-exchange techniques for radiochemical separations,” J. Inorg. Nucl. Chem. 3, 233–242 ( 1956).
PROCEDURE: This procedure was developed for the separation of short-lived lead isotopes from parent bismuth. Purified bismuth activity in HCl (0.5M) solution was adsorbed on the anion exchanger Dowex-1. The resin was kept on a sintered-glass filter. Pb daughter activity was eluted with a small volume of HCl (0.5M). An apparatus was also set up for continuous elution of lead. Also see Campbell, E. C., and Nelson, F., Phys. Rev, 91, 499A (1953).
Pb-2
Lead
SEPARATION TIME: 3 s
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Stockendal, R., McDonell, J. A., Schmorak, M., and Bergstrom, I., “Nuclear isomerism in odd Pb isotopes,” Arkiv. Fysik 11, 165–188 ( 1957).
PROCEDURE: This procedure was used to separate lead from the bismuth parent. A solution containing bismuth was adjusted to 0.3M in HCl and passed through a Dowex-1 column kept at 82°C. Under these conditions, bismuth was sorbed strongly by Dowex-1, and lead could be eluted with HCl (0.3M).
Pb-3
Lead
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous/batch
REFERENCE: Langrock, E. J., Bazarkina, T. V., and Czosnowska, W., “Procedure for selective solvent extraction of superheavy elements 113+ and 114+ by use of crown ethers,” Radiochim. Acta 30, 229–231 ( 1982).
PROCEDURE: See procedure 114-1 under “Z = 114,” Langrock, E. J., et al., Radiochim. Acta 30, 229–231 (1982).
Pb-4
Lead
SEPARATION TIME: 5 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., Chemical Procedures Used in Bombardment Work at Berkeley, U.S. Atomic Energy Commission report AECD-2738 ( 1949), p. 240.
PROCEDURE: This procedure was developed by Karraker for the milking of lead from polonium. The parent polonium was kept in the solvent medium of TBP 20% in dibutyl ether. Lead was extracted with HCl (6 M). The HCl phase was washed with a TBP – ether mixture. Bismuth follows lead in the HCl phase. Bismuth and lead carriers were added, and bismuth was precipitated as BiOCl following the procedure of Neumann (U.S. AEC report AECD-2738, 1949, p. 238).
Lu-1
Lutetium
SEPARATION TIME: 6 min
SEPARATION TECHNIQUE: Extraction, precipitation
PRODUCTION MODE: Batch
REFERENCE: Zychor, I., Rykaczewski, K., Ahrens, H., Folger, H., Kurcewicz, W., Summerer, K., Kaffrell, N., and Trautmann, N., “Hf and Lu isomer produced in heavy-ion collisions of 7.6-MeV/u 40Ar, 8.5-MeV/u 84Kr, and 8.5-MeV/u 136Xe on natural W targets,” Radiochem. Radioanal. Lett. 33, 1–2 ( 1983).
PROCEDURE: For details, see procedure Hf-2 under “Hafnium,” Zychor, I., et al., Radiochem. Radioanal. Lett. 33, 1–2 (1983). After the removal of hafnium, the HDEHP layer was washed with HCl (7M) containing boric acid. The wash solution contained lutetium; lutetium was coprecipitated with Fe(OH)3.
Mn-I
Manganese
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Cohen, B. L., Charpie, R. A., Handley, T. H., and Olson, E. L., “57Mn,” Phys. Rev. 94, 953–954 ( 1954).
PROCEDURE: Irradiated iron was dissolved, and the medium was converted to concentrated HNO3. Manganese was precipitated as MnO2 by the addition of KClO3. The precipitate was purified by dissolving in an acid solution of H2O2 and reprecipitating as MnO2.
Mn-2
Manganese
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Livingood, J. J., and Seaborg, G. T., “Radioactive Mn isotopes,” Phys. Rev. 54, 391–397 ( 1938).
PROCEDURE: Irradiated samples (iron, chromium, and vanadium metal or chromium oxide) were dissolved in a mixture of HNO3 and HCl. Appropriate carriers were added, and the solution was adjusted to be 16M in HNO3. Manganese was precipitated as MnO2 by the addition of KClO3 to the boiling solution. MnO2 was redissolved in an acid solution of H2O2 and reprecipitated from HNO3 solution as before.
Md-1
Mendelevium
SEPARATION TIME: ~5 min
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Ghiorso, A., Harvey, B. G., Choppin, G. R., Thompson, S. G., and Seaborg, G. T., “New element Md, atomic number 101,” Phys. Rev. 98, 1518–1519 ( 1955).
PROCEDURE: The recoiling atoms from the target were collected on a gold foil. The foil was dissolved in aqua regia and the gold was removed by extraction with ethyl acetate. The aqueous solution was passed through a Dowex-1 column for complete removal of gold and other impurities, and washed with HCl (6 M). The eluent was evaporated and loaded on a Dowex-50 column. The separation of actinides was achieved by elution with ammonium α-hydroxyisobutyrate. The elution position of mendelevium was predetermined with tracer lanthanides. For further details, see Thompson, S. G., et al., J. Amer. Chem. Soc. 76, 6229–6236 (1954).
Md-2
Mendelevium
SEPARATION TIME: 200 s
SEPARATION TECHNIQUE: Cation exchange (HPLC)
PRODUCTION MODE: Autobatch
REFERENCE: Brüchle, W., Schädel, M., Scherer, U. W., Kratz, J. V., Gregorich, K. E., Lee, D., Nurmia, M., Chasteler, R. M., Hall, H. L., Henderson, R. A., and Hoffman, D.C., “The hydration enthalpies of Md(3+) and Lr(3+),” Inorg. Chim. Acta 146, 267–276 ( 1988).
PROCEDURE: The separation was achieved using an Aminex A6 cation-exchange column; α-hydroxyisobutyric acid was used as eluant. The chemistry was performed with an Automated Rapid Chemistry Apparatus (ARCA) controlled by a microprocessor. For details, see procedure Lr-4 under “Lawrencium,” Brüchle, W., et al., Inorg. Chim. Acta 146, 267–276 (1988).
Md-3
Mendelevium (from lawrencium)
SEPARATION TIME: 1 to 2 min
SEPARATION TECHNIQUE: Extraction (HPLC)
PRODUCTION MODE: Autobatch
REFERENCE: Scherer, U. W., Kratz, J. V., Schädel, M., Bruchle, W., Gregorich, K. E., Henderson, R. A., Lee, D., Nurmia, M., and Hoffman, D. C., “Lawrencium chemistry: no evidence for oxidation states lower than 3+ in aqueous solution,” Inorg. Chim. Acta 146, 249–254 ( 1988).
PROCEDURE: The separation was performed using an Automated Rapid Chemistry Apparatus (ARCA) controlled by a microprocessor. The KCl-loaded gas jet deposited the nuclear reaction products on a quartz frit. The deposit was dissolved in 0.3M HCl containing a reducing agent (0.01M V2+, 0.01M Cr2+, or 0.1M NH3OH+). The solution was first passed through a column of Zn(Hg) at 80 °C (celite at 100°C for NH3OH+) and then through a column of HDEHP (kept at 80°C) dispersed on an inert medium. Under the conditions of the experiment, only trivalent actinides (in this experiment, Lr3+) remained on the HDEHP column; the divalent actinides (in this experiment, Md2+) were in the eluate.
Hg-1
Mercury
SEPARATION TIME: 3 to 5 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayar, B., Novgorodov, A. F., Vocilka, I., and Zaitseva, N. G., “Rapid gas-thermochromatographic separation of radioactive elements. II. Au as a universal target for the rapid producing of radioactive Re, Os, Ir, and Hg isotopes,” Radiochem. Radioanal. Lett. 19, 43–53 ( 1974).
PROCEDURE: See procedure Re-2 under “Rhenium,” Bayar, B., et al., Radiochem. Radioanal. Lett. 19, 43–53 (1974).
Hg-2
Mercury
SEPARATION TIME: 1 to 5 min
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., Chemical Procedures Used in Bombardment Work at Berkeley, U.S. Atomic Energy Commission report AECD-2738 ( 1949), p. 232.
PROCEDURE: The procedure was developed by R. W. Fink and S. G. Thompson for the separation of mercury from irradiated gold or platinum. The target, after irradiation, was introduced into a chamber for vaporizing mercury. A thin platinum disk, cooled by water, was used for collecting mercury. The target was heated with a Bunsen flame. Thallium activities, if present, will follow mercury. Also see Fink, R. W., and Wiig, E. O., J. Am. Chem. Soc. 74, 2457–2460 (1952).
Hg-3
Mercury
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Smith, W. G., and Hollander, J. M., “Radiochemical study of neutron-deficient chains in the noble metal region,” Phys. Rev. 98, 1258–1265 ( 1955).
PROCEDURE: The irradiated gold metal was dissolved in aqua regia and diluted; mercury carrier was added. From the solution, gold was extracted with amyl acetate. Mercury was precipitated from the aqueous phase by the addition of SnCl2. For establishing genetic relationships, gold daughter activities were separated by periodic extraction of gold with amyl acetate.
Hg-4
Mercury
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Rasmussen, Jr., J. O., Thompson, S. G., and Ghiorso, A., “Alpha-radioactivity in the 82-neutron region,” Phys. Rev. 89, 33–48 ( 1953).
PROCEDURE: The irradiated gold foil was heated in a special steel chamber. A platinum plate, cooled by circulating water on the back, was kept just above the gold foil. Mercury was deposited on the platinum.
Mo-1
Molybdenum
SEPARATION TIME: 5 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Tittel, G., Kaffrell, N., Trautmann, N., and Herrmann, G., “Direct identification of 103–107Mo by a rapid chemical-separation procedure,” J. Inorg. Nucl. Chem. 39, 2115–2119 ( 1977).
PROCEDURE: The irradiated uranium or plutonium solution was mixed with HCl (5M) containing NaF and molybdenum and tellurium carriers. The mixture was reduced with Na2S2O4 containing NH4SCN. Molybdenum was reduced to (V), and tellurium was precipitated. After filtering the tellurium, the solution containing Mo(V) was passed through a layer of Voltalef coated with a 1:1 mixture of isoamyl alcohol and n-butylacetate. The molybdenum retained by this layer was eluted with H2SO4 – K2Cr2O7 and collected in Na2S2O4 – NH4SCN. The solution was passed through a second layer of coated Voltalef, washed with H2SO4 (3M), and the Voltalef layer was transferred for counting.
Mo-2
Molybdenum
SEPARATION TIME: 3 to 5 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayer, B., Novgorodov, A. F., Vocilka, I., and Zaitseva, N. G, “Fast gas-thermochromatographic separation of molybdenum neutron-deficient isotopes from AgCl,” Radiochem. Radioanal. Lett. 35, 109–120 ( 1978).
PROCEDURE: Irradiated AgCl was introduced into a furnace maintained at 630°C. A mixture of O2 – HCl gas (20 to 10% and 80 to 90%, respectively) was passed through the system. The gas from the furnace was passed through a long tube with a thermal gradient. Molybdenum deposits in the region that is at 40 to 100°C. Molybdenum deposited here contained less than 0.1% of other radioactive nuclides produced.
Mo-3
Molybdenum
SEPARATION TIME: ~2 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Duffield, R. B., and Knight, J. D, “Radioactivity of 91Mo and 93Mo,” Phys. Rev. 76, 573–574 ( 1949).
PROCEDURE: This procedure was designed to separate molybdenum from niobium activities. From the solution containing molybdenum (as molybdate) and niobium activities, iron hydroxide was precipitated. The precipitate carried niobium activities, leaving molybdenum in solution. Ammonium phosphomolybdate was precipitated from the supernatant solution after acidification.
Mo-4
Molybdenum
SEPARATION TIME: 2 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Orth, C. J., and Smith, R. K., “Short-lived isotopes of niobium and zirconium from fission,” J. Inorg. Nucl. Chem. 15, 4–17 ( 1960).
PROCEDURE: This procedure was used for milking molybdenum from niobium. Niobium was precipitated as hydrousoxide by the addition of hot, concentrated nitric acid to the fission-product solution containing Mo(VI), Te(IV), Te(VI), and Zr(IV) holdback carriers. The precipitate was dissolved in concentrated HNO3 - HF mixture; Mo(VI) carrier and bromine water were added, and niobium was precipitated by the addition of NH4OH. The mixture was centrifuged, and the supernatant solution was used for the assay of molybdenum.
Ne-1
Neon
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Adsorption
PRODUCTION MODE: Batch
REFERENCE: Dropesky, B. J., and Schardt, A. W, “Decay of the new nuclide 24Ne,” Phys. Rev. 102, 426–433 ( 1956).
PROCEDURE: After irradiation, neon gas was passed through a trap containing activated carbon cooled in dry ice. Pure neon collected in the counting cell. Impurities produced during irradiation were adsorbed by the carbon.
Np-1
Neptunium
SEPARATION TIME: 10 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Tetzlaff, H., Herrmann, G., Kaffrell, N., Kratz, J. V., Rogowski, J., Trautmann, N., Skålberg, M., Skarnemark, G., Alstad, J., Fowler, M. M., Moody, K. J., Brüchle, W., Gäggeler, H., Schädel, M., and Summerer, K., “Continuous separation and identification of neutron-rich neptunium isotopes from heavy-ion reactions by means of the centrifuge system ‘SISAK,”' J. Less-Common Metals 122, 441–444 ( 1986).
PROCEDURE: A KCl-loaded argon gas jet carried the reaction products to a SISAK system. The products were dissolved in a mixture of HCl (0.6M) – TiCl3 (0.3%), degassed and fed into the first centrifuge unit. In this unit, uranium, thorium, and some fission products were extracted with HDEHP (5%) in CCl4; neptunium, which was reduced to the trivalent state by TiCl3, was not extracted. The aqueous phase emerging from the first unit was mixed with HNO3 (6M) containing oxalic acid, tartaric acid, and potassium hydrogentartrate. The neptunium was oxidized to tetravalent state by HNO3 and was extracted with HDEHP (7%) in CCl4 in the second centrifuge unit. In the last unit, neptunium was back-extracted with H3PO3 (15%).
Np-2
Neptunium
SEPARATION TIME: 2.7 min
SEPARATION TECHNIQUE: Extraction, ion exchange
PRODUCTION MODE: Autobatch
REFERENCE: Moody, K. J., Brüchle, W., Brügger, M., Gäggeler, H., Haefner, B., Schädel, M., Summerer, K., Tetzlaff, H., Herrmann, G., Kaffrell, N., Rogowski, J., Trautmann, N., Skålberg, M., Skarnemark, G., Alstad, J., and Fowler, M. M., “New nuclides: 243Np and 244Np,” Z. Phys. A 328, 417–422 ( 1987).
PROCEDURE: The experiments were performed with the microprocessor-controlled Automated Rapid Chemical Apparatus (ARCA). The products accumulated for 4 min, were dissolved in HNO3, and were loaded onto an anion-exchange column; the neptunium was eluted with a solution of HCl – HF. The eluted neptunium, after conversion to a HCl medium, was loaded onto a column of tri-n-octylamine supported on an inert solid. Finally, neptunium was eluted with HCl containing a reducing agent.
Nb-1
Niobium
SEPARATION TIME: 2.2 to 2A s
SEPARATION TECHNIQUE: Adsorption
PRODUCTION MODE: Autobatch
REFERENCE: Weis, M., and Denschlag, H. O., “Fractional independent yields of 99Y, 99Zr,99mNb, and 99Nb in the thermal-neutron fission of 235U,” J. Inorg. Nucl. Chem. 43, 437–444 ( 1981).
PROCEDURE: Irradiated uranium solution in HNO3 (0.5M) containing tartaric acid and ascorbic acid was filtered through a preformed AgCl layer and washed with HNO3. The filtrate and the wash were mixed with concentrated HNO3 containing tartaric acid and zirconium, ruthenium, cesium, barium, and lanthanum carriers; the resulting solution (10 to 12M in HNO3) was filtered through three fiberglass filters and washed with HNO 3 (12M) containing tartaric acid. Niobium was retained by the fiberglass filters.
Nb-2
Niobium
SEPARATION TIME: ~9 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Brodén, K., Skarnemark, G., Björnstad, T., Eriksen, D., Haldorsen, I., Kaffrell, N., Stender, E., and Trautmann, N, “Rapid, continuous-separation procedures for Zr, Ni, Tc, Br, and I from complex reaction-product mixtures,” J. Inorg. Nucl. Chem. 43, 765–771 ( 1981).
PROCEDURE: See procedure Zr-2 under “Zirconium,” Brodén, K., et al., J. Inorg. Nucl. Chem. 43, 765–771 (1981).
Nb-3
Niobium
SEPARATION TIME: 2.2 s
SEPARATION TECHNIQUE: Adsorption
PRODUCTION MODE: Autobatch
REFERENCE: Ahrens, H., Kaffrell, N., Trautmann, N., and Herrmann, G., “Decay properties of neutron-rich niobium isotopes,” Phys. Rev. C 14, 211–217 ( 1976).
PROCEDURE: Uranium solution in HNO3 (0.1M) containing SO2 and holdback carriers for halogens, alkalies, alkaline earths, lanthanides, zirconium, and technetium was irradiated. The irradiated solution was passed through two layers of preformed AgCl, and the layers were washed with HNO3 (0.1M). The filtrates and the washings were collected in concentrated HNO3. The resulting solution, which was 10M in HNO3, was passed through two fiberglass filters. The niobium adsorbed on the filter was washed with HNO3 (10M), and the filters were transferred for counting. For better decontamination from zirconium, the niobium was eluted with HF (0.5M) – HNO3 (15M) mixture, boric acid was added, and it was readsorbed on the filter. This procedure took an additional 2.2 s.
Nb-4
Niobium
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Herzog, W., Trautmann, N., Denig, R., and Herrmann, G., “The decay of 31-s 98Zr, 2.9-s 98Nb, and 51-min 98Nb,” Z. Phys. A 276, 393–402 ( 1976).
PROCEDURE: Irradiated uranium solution was passed through a layer of preformed AgCl to remove halogen activities. The filtrate, adjusted to 8M in HNO3, was passed through a layer of plastic grains coated with TBP. Zirconium and uranium were retained by the TBP, and 2.9-s 98Nb was eluted with 8M HNO3. Also see procedure Zr-3 under “Zirconium,” Trautmann, N., et al., Radiochim. Acta 18, 86–101 (1972).
Nb-5
Niobium
SEPARATION TIME: 0.1 to 0.2 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Zvara, I., Belov, V. Z., Domanov, V. P., and Shalaevskii, M. R., “Chemical isolation of nilsbohrium as ekatantalum in the form of the anhydrous bromide. II. Experiments with a spontaneously fissioning isotope of nilsbohrium,” Soviet Radiochemistry (Eng. Tr.) 18, 328–334 ( 1976).
PROCEDURE: See procedure 105-1 under “Z = 105,” Zvara, I., et al., Soviet Radiochemistry (Eng. Tr.) 18, 328–334 (1976). Also see Belov, V. Z., et al., Soviet Radiochemistry (Eng. Tr.) 17, 87–92 (1975).
Nb-6
Niobium
SEPARATION TIME: <3 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayar, B., Vocilka, I., Zaitseva, N. G., and Novgorodov, A. F., “Fast gas-thermochromatographic separation of radioactive elements. IV. Extraction of neutron-deficient zirconium and niobium isotopes from irradiated silver chloride melt,” Radiochem. Radioanal. Lett. 34, 63–74 ( 1978).
PROCEDURE: See procedure Zr-4 under “Zirconium,” Bayar, B., et al., Radiochem. Radioanal. Lett. 34, 63–74 (1978). Also see Bayer, B., et al., Soviet Radiochemistry (Eng. Tr.) 16, 345–351 (1974).
Nb-7
Niobium
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Recoil
PRODUCTION MODE: Batch
REFERENCE: Mathur, H. B., and Hyde, E. K., “Radiations of 90Mo and the isomeric states of 90Nb,” Phys. Rev. 98, 79–84 ( 1955).
PROCEDURE: An intense, carrier-free sample of 90Mo was kept very close to a sodium iodide crystal. The aluminum cover of the crystal was kept at a potential of –500 V with respect to the sample. The 90Mo sample was removed, and the niobium daughter activity deposited on the aluminum cover of the crystal was counted.
Nb-8
Niobium
SEPARATION TIME: ~5 s
SEPARATION TECHNIQUE: Adsorption
PRODUCTION MODE: Autobatch
REFERENCE: Weis, M., Ahrens, H., Denschlag, H. O., Fariwar, M., Herrmann, G., and Trautmann, N., “Rapid sorption of niobium on glass surfaces,” Radiochim. Acta 42, 201–203 ( 1987).
PROCEDURE: The sorption of niobium on glass-fiber filters was investigated as a function of acid medium. The best sorption was obtained from HNO3 medium. The solution (3 mL) containing carrier-free niobium and zirconium carrier in 10M HNO3 was passed through five consecutive layers of glass-fiber filters in 2 s. The filters were then washed with 7 mL of the 10M HNO3. The efficiency for the retention of niobium was found to be 94%, and 2% zirconium contamination was present. The zirconium contamination could be reduced by a factor of 2 by the addition of complexing agents like F−, C2O4−, citric acid, and tartaric acid; however, the retention of niobium decreased.
Nb-9
Niobium
SEPARATION TIME: 12 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Nai-Qi, Ya., Jost, D. T., Baltensperger, U., and Gäggeler, H. W., “The SAPHIR gas-jet and a first application to an on-line separation of niobium,” Radiochim. Acta 47, 1–7 ( 1989).
PROCEDURE: Helium loaded with KCl aerosol carried the fission products from the target chamber. The gas was mixed with HBr and allowed to enter an on-line chromatography tube. The reaction products were stopped at a quartz plug maintained at 1000°C. The volatile bromides and oxybromides generated entered an isothermal part of the tube maintained at 300 to 400°C. The volatile products leaving the tube entered a reclustering chamber, then passed through a few-meter-long capillary and collected on a glass-fiber filter. An HPGe detector recorded the activity of the filter. The separation procedure is planned to be applied for element 105.
N-1
Nitrogen
SEPARATION TIME: 5 to 10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Novey, T. B., Knight, J. D., Schover, D. S., and Elliott, N., “Chemical identification of the 8-s activity found in pile-irradiated oxygen compounds,” National Nuclear Energy Series, Vol. 9, “Radiochemical Studies: The Fission Products,” Coryell, C. D., and Sugarman, N. (Eds.), Book 3 ( 1951), p. 1910.
PROCEDURE: Irradiated nitrite salt was treated with hydrazine in acid solution. Nitrogen that was present was converted to nitrogen gas while the oxygen was converted to water. The N2 gas was counted.
N-2
Nitrogen
SEPARATION TIME: 8 s
SEPARATION TECHNIQUE: Emanation
PRODUCTION MODE: Batch
REFERENCE: Sommers, Jr., H. S., and Sherr, R., “Activity of 16N and 6He,” Phys. Rev. 69, 21–30 ( 1946).
PROCEDURE: Emanation from Be(OH)2 was used. See procedure He-1 under “Helium,” Sommers, Jr., H. S., and Sherr, R., Phys. Rev. 69, 21–30 (1946). The target chamber pressure was adjusted for 8-s delivery time.
N-3
Nitrogen
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Alvarez, L., “N-17, a delayed neutron emitter,” Phys. Rev. 75, 1127–1132 ( 1949).
PROCEDURE: An aqueous solution of NH4F was irradiated with deuterons, and a stream of helium was passed through. The gas was passed through two ascarite tubes to trap CO 2. The stream was then passed through hot CuO and then through two ascarite traps. The purified gas was used for counting.
No-1
Nobelium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Fields, P. R., Friedman, A. M., Milsted, J., Atterling, H., Forsling, W., Holm, L. W., and Astrom, B., “Production of the new element 102,” Phys. Rev. 107, 1460–1462 ( 1957).
PROCEDURE: Recoiling products from a target were collected on a Tygon catcher foil. The foil was ignited on a platinum plate, and the residue was dissolved. The solution was loaded onto a Dowex-50 column, and the actinides were separated using ammonium α-hydroxyisobutyrate as eluent. The eluent in the predetermined position was collected and counted. For further details, see Thompson, S. G., et al., J. Amer. Chem. Soc. 76, 6229–6236 (1954), and Choppin, G. R., et al., J. Inorg. Nucl. Chem. 2, 66 (1956).
Ng-1
Noble Gases
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Emanation
PRODUCTION MODE: Batch
REFERENCE: Wahl, A. C., “Nuclear-charge distribution in fission: cumulative yields of short-lived krypton and xenon isotopes from the thermal-neutron fission of 235U,” J. Inorg. Nucl. Chem. 6, 263–277 ( 1958).
PROCEDURE: Barium stearate and uranyl stearate were found to emanate noble gases with high efficiency. This property was used to determine the cumulative yields of short-lived krypton and xenon isotopes. Also see Wahl, A. C., and Daniels, W. R., J. Inorg. Nucl. Chem. 6, 278 (1958). See procedure Kr-1 under “Krypton,” Wolfsberg, K., Phys. Rev. 137, B929–B935 (1965).
Ng-2
Noble Gases
SEPARATION TIME: ~1 s
SEPARATION TECHNIQUE: Emanation
PRODUCTION MODE: Batch/continuous
REFERENCE: Ahrens, H., Patzelt, P., and Herrmann, G., “The half-lives of 91Br, 95Kr, 140I, 141I, and 144Xe,” J. Inorg. Nucl. Chem. 38, 191–192 ( 1976).
PROCEDURE: See procedure Hal-1 under “Halogens,” Ahrens, H., et al., J. Inorg. Nucl. Chem. 38, 191–192 (1976).
Os-1
Osmium
SEPARATION TIME: 3 to 5 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayar, B., Novgorodov, A. F., Vocilka, I., and Zaitseva, N. G., “Rapid gas-thermochromatographic separation of radioactive elements. II. Gold as a universal target for the rapid producing of radioactive rhenium, osmium, iridium, and mercury isotopes,” Radiochem. Radioanal. Lett. 19, 43–53 ( 1974).
PROCEDURE: See procedure Re-2 under “Rhenium,” Bayar, B., et al., Radiochem. Radioanal. Lett. 19, 43–53 (1974). Also see Bayar, B., et al., Soviet Radiochemistry (Eng. Tr.) 16, 333–338 (1974).
Os-2
Osmium
SEPARATION TIME: <5 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Adilbish, M., Zaitseva, N. G., Kovach, Z., Novgorodov, A. F., Sergeev, Yu. Ya., and Tikhonov, V. I., “Volatilization from molten gold and thermochromatographic separation of ultramicroquantities of rhenium and osmium oxides at low pressures of oxygen-containing gases,” Soviet Radiochemistry (Eng. Tr.) 20, 652–662 ( 1978).
PROCEDURE: Irradiated gold was kept in a crucible and placed in a furnace maintained at 1330°C. Dry oxygen was maintained at a pressure of 1 × 10−3 mm. Osmium deposited in the thermochromatographic column near the temperature region of 200 to 250°C. Good separation was achieved from rhenium (separation factor of 800 to 1200; higher value is for separation time >5 min).
Os-3
Osmium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Distillation
PRODUCTION MODE: Batch
REFERENCE: Hofstetter, K. J., and Daly, P. J., “Decay properties of neutron-deficient osmium and rhenium isotopes. I. Decay modes of 179Re, 180Re, 180Os, and 181Os,” Phys. Rev. 152, 1050–1055 ( 1966).
PROCEDURE: The irradiated tungsten targets were dissolved in a mixture of HF – HNO3 (1:5); osmium and rhenium carriers were added to the solution. The OsO4 was distilled and collected in cold, 6M NaOH solution. The distillate contains 18F impurity and can be separated by precipitating osmium sulfide and using the sulfide precipitate for counting.
O-1
Oxygen
SEPARATION TIME: ~1 min
SEPARATION TECHNIQUE: Absorption
PRODUCTION MODE: Batch
REFERENCE: Livingston, M. S., and McMillan, E., “The production of radioactive oxygen,” Phys. Rev. 46, 437–438 ( 1934).
PROCEDURE: Nitrogen gas was irradiated with deuterons, mixed with oxygen and an excess of hydrogen, and passed over heated, platinized asbestos. The active oxygen, probably present as NO2, N2O, or O2, was reduced to H2O and collected in a CaCl2 drying tube.
O-2
Oxygen
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Sorption
PRODUCTION MODE: Batch
REFERENCE: McMillan, E., and Livingston, M. S., “Artificial radioactivity produced by the deuteron bombardment of nitrogen,” Phys. Rev. 47, 452–457 ( 1935).
PROCEDURE: Irradiated air was mixed with hydrogen and passed through a tube containing platinized asbestos kept at 500°C. The mixture was carried by nitrogen through a CaCl2 tube. Oxygen activity produced, along with the oxygen in the irradiated air, was converted to H2O and adsorbed by CaCl2.
Pd-1
Palladium
SEPARATION TIME: 135 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Meikrantz, D. H., Gehrke, R. J., McIsaac, L. D., Baker, J. D., and Greenwood, R. C., “An automated system for selective fission-product separation: decays of 113–115Pd,” Radiochim. Acta 29, 93–101 ( 1981).
PROCEDURE: Fission products from a 252Cf source were transported by a NaCl aerosol and deposited on a Mylar film. The deposit was dissolved in hot (70°C) HNO3 (8M) – HCl (0.1M) solution containing palladium, ruthenium, rhenium, and tellurium carriers. The pH of the solution was adjusted to 1 with NH4OH. Dimethylglyoxime (1% in methanol) and dichloromethane were added, and the mixture was transferred to the first centrifugal contactor. The palladium extract from the first centrifuge was washed with HNO 3 (1M) in the next centrifuge; in the third centrifuge, palladium was stripped with NH4OH (3M). The ammonia solution was passed through a column of Zn (30 mesh). Palladium deposited in the zinc column was counted.
Pd-2
Palladium
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Aronsson, P. O., Ehn, E., and Rydberg, J., “New isotope of palladium, 116Pd,” Phys. Rev. Lett. 25, 590–593 ( 1970).
PROCEDURE: This procedure was used to identify short-lived palladium nuclide through its daughter product. A HNO3 solution (1M) containing uranyl nitrate (2M) and acetylacetone (0.1M) was passed through an irradiation cell. The irradiated solution passed through a delay line before entering the first mixer centrifuge. The solution was extracted with toluene, and the aqueous phase containing silver and other fission products was discarded. The organic phase containing palladium passed through a second delay line before entering the second mixer centrifuge. The silver daughter activities were stripped with 0.001M HNO3, exchanged with preformed AgI, and counted.
Pd-3
Palladium
SEPARATION TIME: ~2 s
SEPARATION TECHNIQUE: Plating, chemical
PRODUCTION MODE: Batch
REFERENCE: Weiss, H. V., Ballou, N. E., Elzie, J. L., and Fresco, J. M., “Nuclear charge distribution in symmetric fission of 235U with thermal neutrons: yields of 117Ag, 118Ag, and 118Pd,” Phys. Rev. 188, 1893–1896 ( 1969).
PROCEDURE: This procedure was used for the determination of the half-life of 118Pd through the isolation of cadmium activities. The irradiated uranium solution in concentrated HBr, containing ruthenium, rhodium, palladium, and silver carriers, was passed through a column containing copper powder (2 g); the copper powder was washed with concentrated HBr. Under these conditions, palladium was selectively retained by the copper column. The copper was dissolved in concentrated HNO3, and cadmium was isolated and purified after the required delay. The half-life determination was achieved by isolating palladium at different times after irradiation. Also see Weiss, H. V., et al, Phys. Rev. 172, 1269–1271 (1968).
P-1
Phosphorus
SEPARATION TIME: 2 min
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Apt, K. E., and Knight, J. D., “Decay of P-35,” Phys. Rev. C 6, 842–846 ( 1972).
PROCEDURE: Triton-irradiated LiCl or NaCl was dissolved in HCl solution (pH 1) containing Al(III) holdback carrier, bromine water, and hydrous zirconium oxide (HZO). The bromine oxidized lower-oxidation-state phosphorus to phosphate; the phosphate was sorbed by the HZO. The mixture was stirred well, filtered, and washed twice with HCl (pH 1). The HZO was then washed with acetone, dried, and mounted for counting.
Pt-1
Platinum
SEPARATION TIME: 2 to 3 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Facetti, J., Trabal, E., McClin, R., and Torres, S., “A new isotope, 201Pt,” Phys. Rev. 127, 1690–1692 ( 1962).
PROCEDURE: The irradiated HgO sample was dissolved in 6M HCl. Platinum was precipitated as K2PtCl6 or (NH4)2PtCl6 by the addition of KCl or NH4Cl. Thg precipitate was filtered, washed, and mounted for counting. If Hg(NO3)2·H2O was used for irradiation, the sample was dissolved in 3M HNO3; using the method of Noyes and Bray, a gold-mercury fraction was removed (see “Qualitative analysis for rare elements,” A. Noyes and W. Bray, The McMillan Company, New York, 1952, pp. 114–116). Potassium chloroplatinate was precipitated after the removal of the gold-mercury fraction. The time required was about 4 min.
Po-1
Polonium
SEPARATION TIME: ~1 min
SEPARATION TECHNIQUE: Plating, chemical
PRODUCTION MODE: Batch
REFERENCE: Spiess, F. N., “Alpha-emitting isomer: 211Po,” Phys. Rev. 94, 1292–1299 ( 1954).
PROCEDURE: A solution of irradiated lead carbonate was prepared in hot HCl (6M) containing bismuth holdback carrier. A silver foil was kept in the solution, and the solution was stirred for about 1 min. The silver foil was removed, washed with distilled water, and counted. Polonium was chemically plated on the foil.
Po-2
Polonium
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Pappas, A. C., and Wiles, D. R., “New short-lived isotopes of tin found in fission,” J. Inorg. Nucl. Chem. 2, 69–78 ( 1956).
PROCEDURE: A solution containing lead and polonium was adjusted to a pH of 8.5 and extracted with 0.01% dithizone in CCl4. The organic phase was washed with a pH 8.5 solution. Polonium was stripped with 1M HCl. This procedure achieved a fast and clean separation of polonium from lead. Also see procedure Sn-4 under “Tin,” Pappas, A. C., and Wiles, D. R., J. Inorg. Nucl. Chem. 2, 69–78 (1956).
Po-3
Polonium (+Bi)
SEPARATION TIME: 4 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Spiess, F. N., “Alpha-emitting isomer: 211Po,” Phys. Rev. 94, 1292–1299 ( 1954).
PROCEDURE: Irradiated lead carbonate was dissolved in hot HNO3 (5 mL) containing bismuth carrier. The volume of the solution was reduced to precipitate most of the lead as lead nitrate. The supernatant solution was transferred to a tube containing NaOH. Two drops of 1% solution of thionalide (thioglycollic acid beta-amino naphthalide) was added to the tube. Bismuth was precipitated as an organic complex; polonium was carried by the precipitate. The precipitate was centrifuged, washed, and then dissolved in acetone. The solution was evaporated on a hot platinum disk, flamed, and used for counting.
Po-4
Polonium
SEPARATION TIME: <30 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Perlman, I., Asaro, F., Ghiorso, A., Larsh, A., and Latimer, R., “Isomeric state of 212Po,” Phys. Rev. 127, 917–922 ( 1962).
PROCEDURE: The bismuth or lead oxide targets were heated with a gas torch, and the volatile polonium and astatine were deposited on a platinum plate. The separation of polonium from astatine was achieved by selective volatilization of astatine at 450°C. The polonium fraction remaining on the platinum plate was used for counting. Also see procedure At-3 under “Astatine,” Thoresen, P. E., Asaro, F., and Perlman, I., J. Inorg. Nucl. Chem. 26, 1341–1347 (1964).
K-1
Potassium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Henderson, W. J., Ridenour, L. N., White, M. G., and Henderson, M. D., “The radioactivity of K-38,” Phys. Rev. 51, 1107 ( 1937).
PROCEDURE: NaCl, irradiated with alpha particles, was dissolved in an aqueous solution containing potassium carrier. Potassium cobaltinitrate was precipitated by the addition of a solution of sodium cobaltinitrite. The precipitate was filtered, washed, dried, and mounted for counting. See also procedure K-2 under “Potassium,” Hurst, D. G., and Walke, H., Phys. Rev. 51, 1033–1037 (1937).
K-2
Potassium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Hurst, D. G., and Walke, H., “The induced radioactivity of potassium,” Phys. Rev. 51, 1033–1037 ( 1937).
PROCEDURE: The deuteron-irradiated calcium metal was dissolved in HCl solution; potassium chloride carrier was added, and KClO4 was precipitated by the addition of HClO4 and ethanol. The precipitate had scandium contamination.
Pr-1
Praseodymium
SEPARATION TIME: 3 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Yamamoto, H., Ikeda, Y., Kawade, K., Katoh, T., and Nagahara, T., “Decay studies of 143La and 147Pr,” J. Inorg. Nucl. Chem. 43, 855–865 ( 1981).
PROCEDURE: See procedure La-1 under “Lanthanum,” Yamamoto, H., et al., J. Inorg. Nucl. Chem. 43, 855–865 (1981).
Pr-2
Praseodymium
SEPARATION TIME: 10 to 20 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Aronsson, P. O., Skarnemark, G., and Skarestad, M., “Short-lived isotopes of lanthanum, cerium, and praseodymium studied by the SISAK technique,” J. Inorg. Nucl. Chem. 36, 1689–1696 ( 1974).
PROCEDURE: Fission products formed in a target of uranyl sulfate complex adsorbed on Dowex-1 X8 were continuously eluted with (NH 4)2SO4 solution (pH 5) at 85°C. Cerium was oxidized with a mixture of HNO3 – H2SO4 – K2Cr2O7 and transferred to a SISAK system. In the first centrifuge, cerium was extracted with HDEHP (0.3M) in kerosene. In the second centrifuge, the organic phase was contacted with an oxidizing solution to remove any praseodymium daughter that had grown in. The solution was passed through a Dowex-50W X8 column, which retained praseodymium and its daughters. Also see Aronsson, P. O., et al., Inorg. Nucl. Chem. Lett. 10, 753–762 (1974), and Skarnemark, G., et al., Radiochim. Acta 23, 98–103 (1976).
Pr-3
Praseodymium
SEPARATION TIME: 3 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Ikeda, Y., Yamamoto, H., Kawade, K., Katoh, T., and Nagahara, T., “Level properties of 146Nd in the decay of 146Pr,” J. Phys. Soc. Japan 45, 725–732 ( 1978).
PROCEDURE: The parent isotope, 146Ce, was separated using the procedure Ce-2 reported under “Cerium,” Yamamoto, H., et al., J. Inorg. Nucl. Chem. 42, 1539–1546 (1980). The daughter 146Pr was milked from 146Ce.
Pr-4
Praseodymium
SEPARATION TIME: ~5 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Rengan, K., and Griffin, H. C., “Rapid radiochemical isolation of praseodymium from fission products, ” Radiochem. Radioanal. Chem. 29, 253–260 ( 1977).
PROCEDURE: Irradiated uranium solution was mixed with hot HNO3 (10M) containing Ce(IV) carrier and NaBrO3; cerium was extracted with HDEHP (0.75M) in n-heptane. The cerium extract was washed with HNO3 (10M) – NaBrO3 solution and kept aside for praseodymium growth. After an appropriate growth time, praseodymium was stripped with HNO3 (1M), mixed with ammonium acetate solution, and extracted with TTA–TBP (0.2M each) in cyclohexane. The praseodymium was stripped with HNO3 (1M). Further purification from cerium was achieved by addition of cerium carrier, oxidation of cerium to (IV), and extraction with HDEHP. The aqueous phase was counted.
Pr-5
Praseodymium
SEPARATION TIME: 6.5 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Ohyoshi, A., Ohyoshi, E., Tamai, T., and Shinagawa, M., “Short-lived isotopes of lanthanum, cerium, and praseodymium in neutron-irradiated uranium,” J. Inorg. Nucl. Chem. 34, 3293–3302 ( 1972).
PROCEDURE: See procedure La-6 under “Lanthanum,” Ohyoshi, A., et al., J. Inorg. Nucl. Chem. 34, 3292–3302 (1972). For praseodymium separation, 86 to 93 V/cm was applied for 180 s. The praseodymium zone was located by color reaction with Arsenazo III. Also see Ohyoshi, E., et al., J. Nucl. Sci. Technol. 10, 101–105 (1973).
Pm-1
Promethium
SEPARATION TIME: <3 min
SEPARATION TECHNIQUE: Extraction, ion exchange
PRODUCTION MODE: Autobatch
REFERENCE: Greenwood, R. C., Gehrke, R. J., Baker, J. D., and Meikrantz, D. H., “Identification of a new isotope, 155Pm, produced in 252Cf fission,” Radiochim. Acta 30, 57–60 ( 1982).
PROCEDURE: See procedure Ln-1 under “Lanthanides,” Baker, J. D., et al., J. Radioanal. Chem. 74, 117–124 (1982).
Pa-1
Protactinium
SEPARATION TIME: ~30 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., “Rapid separations of protactinium and uranium radioisotopes from cyclotron-bombarded thorium nitrate,” J. Chem. Phys. 20, 754 ( 1952).
PROCEDURE: Irradiated thorium nitrate was dissolved in HNO3 (4M) and extracted with a solution of 2-thenoyltrifluoroacetone (TTA) in benzene (0.25M). The organic layer was washed with HNO3 (4M) and used for counting. In TTA extraction, zirconium, niobium, and hafnium will also follow protactinium. Also see Meinke, W. W., et al., Phys. Rev. 81, 782–798 (1951), and Meinke, W. W., U.S. Atomic Energy Commission report AECD-2750 (1949), pp. 9–10.
Pa-2
Protactinium
SEPARATION TIME: ~2 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Carswell, D. J., and Lawrance, J. J., “Radiochemical experiments: a 234Th “cow” for 234Pa,” J. Chem. Ed. 36, 499–501 ( 1959).
PROCEDURE: The thorium was obtained by periodic milking of uranium adsorbed on an anion-exchange column. The thorium was purified from traces of uranium and kept in an HCl (6M) medium. Protactinium was extracted from the HCl solution using diisopropyl ether as a solvent. The ether phase was washed with HCl (6M) and used for counting. (Forty percent tri-n-octylamine in xylene can also be used as a solvent to extract protactinium; diisopropyl ether gave a fast-phase separation.)
Pa-3
Protactinium
SEPARATION TIME: 2 to 3 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Bas-May, A., Kratz, J. V., and Trautmann, N., “Absence of delayed fission in the beta-decay of 2.3-min 238Pa,” Z. Phys. A 322, 457–462 ( 1985).
PROCEDURE: The irradiated uranyl nitrate powder was transferred automatically and dissolved in a hot solution of HNO3 (0.5M) containing SO2. The solution was passed through a layer of AgCl to remove halogen activities. The effluent was made 9M in HCl by the addition of HCl (11M) containing H2C2O4 (0.1M). The solution was passed through a filter bed of KEL F powder coated with diisobutylcarbinol (DIBC), which extracted Pa(V); the filter bed was washed with HCl (12M) – H2C2O4 (0.1M) solution. A solution of HCl (12M) – HF (0.25M) was used to back-extract protactinium. The final sample was prepared by coprecipitation of protactinium with iron as hydroxide, after complexation of fluoride with boric acid.
Rn-1
Radon
SEPARATION TIME: 40 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Valli, K., Nurmia, M. J., and Hyde, E. K., “Alpha-decay properties of neutron-deficient isotopes of emanation, ” Phys. Rev. 159, 1013–1021 ( 1967).
PROCEDURE: The irradiated metallic targets were transferred to a quartz tube connected to a counting chamber, and the system was evacuated. The quartz tube was heated rapidly to melt the target and release the radon. The released radon was counted.
Rn-2
Radon
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Condensation
PRODUCTION MODE: Continuous
REFERENCE: Hildebrand, N., Frink, C., Greulich, N., Hickmann, U., Kratz, J. V., Trautmann, N. Herrmann, G., Brügger, M., Gäggeler, H., Summerer, K., and Wirth, G., “A cryosystem for the detection of alpha and spontaneous-fission activities in volatile species,” Nucl. Instrum. Meth. Phys. Res. A 260, 407–412 ( 1987).
PROCEDURE: Recoiling nuclear reaction products were thermalized and carried by argon gas. The gas stream was passed through a series of traps to remove nonvolatile products and volatile impurities like H2O, CO2, and O2. Finally, the gas was led into the cryogenic unit maintained at 40 K. Radon condensed on the surface of the solar cell. Surface barrier detectors recorded the alpha spectra. The system was tested with 219Rn (T1/2 = 3.96 s) recoiling from 227Ac, with 220Rn (T1/2 = 55.6 s) recoiling from 228Th, and with 222Rn (T1/2 = 3.8 d) recoiling from 226Ra. For additional details see procedure 118-1 under “Z = 118,” Hildebrand, N., et al., Nucl. Instrum. Meth. Phys. Res. A 260, 407–412 (1987).
Re-1
Rhenium
SEPARATION TIME: <3 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayar, B., Novgorodov, A. F., and Zaitseva, N. G., “Rapid gas-thermochromatographic separation of radioactive elements. I. Production of radioactive rhenium isotopes,” Radiochem. Radioanal. Lett. 15, 231–242 ( 1973).
PROCEDURE: Irradiated NH4ReO4 or osmium metal was heated in a stream of oxygen. A temperature of 800°C was used for NH4ReO4, while osmium was heated at 850°C. The gases flowed through a thermochromatographic column, where the temperature dropped from 800°C to room temperature over ~40 cm. Macro rhenium was deposited in the region near 200°C, while carrier-free rhenium was deposited in the region near 600°C. Continued heating moved the carrier-free rhenium to cooler parts.
Re-2
Rhenium
SEPARATION TIME: 3 to 5 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayar, B., Novgorodov, A. F., Vocilka, I., and Zaitseva, N. G., “Rapid gas-thermochromatographic separation of radioactive elements. II. Gold as a universal target for the rapid producing of radioactive rhenium, osmium, iridium, and mercury isotopes,” Radiochem. Radioanal. Lett. 19, 43–53 ( 1974).
PROCEDURE: Irradiated gold metal was heated at 1160°C in a stream of carrier gas (moisture-free air, O2, or helium). The gas flowed through a quartz tube with a negative temperature gradient and then through a PVC tube. The oxides of rhenium and iridium deposited in the 500°C to 300°C and 180°C to 80°C regions, respectively, while mercury deposited in the 80°C to 25°C region. Osmium deposited in the PVC tube. Iridium was not volatilized in helium. Volatilization of rhenium was faster in O2. Also see Bayar, B., et al., Soviet Radiochemistry (Eng. Tr.) 16, 333–338 (1974).
Re-3
Rhenium
SEPARATION TIME: ~3 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Adilbish, M., Zaitseva, N. G., Kovach, Z., Novgorodov, A. F., Sergeev, Yu. Ya., and Tikhonov, V. I., “Volatilization from molten gold and thermochromatographic separation of ultramicroquantities of rhenium and osmium oxides at low pressures of oxygen-containing gases,” Soviet Radiochemistry (Eng. Tr.) 20, 652–662 ( 1978).
PROCEDURE: Irradiated gold was kept in a crucible and placed in a furnace maintained at 1330°C. It was heated in a crucible at reduced H2O vapor pressure of 1 × 10−2 mm. Rhenium deposited in the thermochromatographic column in the temperature region of ~500°C. Under these conditions, good separation was achieved from osmium (separation factors of 850 to 1300 were obtained, depending on the time of separation; a higher value resulted if time was >3 min).
Re-4
Rhenium
SEPARATION TIME: ~5 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayar, B., Vocilka, I., Zaitseva, N. G., and Novgorodov, A. F., “Fast gas-thermochromatographic separation of radioactive elements. V. Production of volatile rhenium oxides and hydroxides in rhenium-tungsten system and their gas-thermochromatographic behaviour,” Radiochem. Radioanal. Lett. 34, 75–88 ( 1978).
PROCEDURE: Irradiated tungsten was maintained at 1160°C in a stream of oxygen containing water vapor. The gases passed through a quartz thermochromatographic column. Carrier-free rhenium was deposited in the 100 ± 20°C temperature zone. Also see Bayar, B., et al., Soviet Radiochemistry (Eng. Tr.) 16, 871–876 (1974).
Re-5
Rhenium
SEPARATION TIME: <3 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Domanov, V. P., Khyubener, Z., Shalaevskii, M. R., Timokhin, S. N., Petrov, D. V., and Zvara, I., “Experimental approach to the identification of element 107 as ekarhenium. I. Continuous gas-thermochromatographic isolation of radiorhenium, ” Soviet Radiochemistry (Eng. Tr.) 25, 23–28 ( 1983).
PROCEDURE: See procedure 107-1 under “Z = 107,” Domanov, V. P., et al., Soviet Radiochemistry (Eng. Tr.) 25, 23–28 (1983). Also see Zvara, I., et al., Soviet Radiochemistry (Eng. Tr.) 26, 72–76 (1984).
Re-6
Rhenium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Hofstetter, K. J., and Daly, P. J., “Decay properties of neutron-deficient osmium and rhenium isotopes. I. Decay modes of 179Re, 180Re, 180Os, and 181Os,” Phys. Rev. 152, 1050–1055 ( 1966).
PROCEDURE: The irradiated tantalum targets were dissolved in HF containing a small amount of HNO3, in the presence of rhenium carrier. The pH of the solution was adjusted to 9, and rhenium was extracted as tetraphenyl arsonium perrhenate into CHCl3. The rhenium was precipitated as rhenium sulfide after back-extraction into aqueous solution.
Rh-1
Rhodium
SEPARATION TIME: 140 s
SEPARATION TECHNIQUE: Distillation
PRODUCTION MODE: Batch
REFERENCE: Franz, G., “The decay of 79.8-s 109Rh into levels of 109gPd,” J. Inorg. Nucl. Chem. 40, 1467–1472 ( 1978).
PROCEDURE: Irradiated plutonium solution containing Ru(III) carrier was mixed with concentrated H2SO4 and HClO4 and heated to 140°C after a delay of 20 s. The distilled RuO4 was absorbed in NaOH (1M). After waiting 30 s for the ruthenium to decay, the solution was acidified with HCl, rhodium carrier was added, and the solution was passed through a layer of fine-grained plastic support coated with TBP. Rhodium was precipitated from the filtrate by zinc powder, filtered, washed with HCl, and counted.
Rb-1
Rubidium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Recoil
PRODUCTION MODE: Batch
REFERENCE: O'Kelley, G. D., Lazar, N. H., and Eichler, E., “Decay of 89Rb,” Phys. Rev. 102, 223–227 ( 1956).
PROCEDURE: A target of enriched uranium (0.3-mg/cm2 thick) was covered with a 3.12-mg/cm2 aluminum foil and enclosed in a specially designed rabbit. A cover foil absorbed the heavy fission fragments. The krypton gas, collected in the rabbit, was flushed with an air stream and drawn through a glass-wool trap cooled in a mixture of acetone and dry ice. The gas mixture was collected in a vessel containing a negatively charged aluminum foil; the rubidium daughter deposited in the foil and contained a slight contamination of cesium.
Rb-2
Rubidium
SEPARATION TIME: 3 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REPERENCE: Arlt, R, Beyer, G. J., Herrmann, E., Habenicht, W., and Tyroff, H., “On the identification of 78Rb/T = 19 min,” Radiochem. Radioanal. Lett. 10, 173–175 ( 1972).
PROCEDURE: The following procedure was used after initial mass separation. A Zr-Nb alloy target irradiated with the external beam of a synchrocyclotron was used in the ion source of a mass separator. The surface ionization source produced only rubidium and strontium ions. After mass separation, the ions were collected on an aluminum-coated polyester tape. The deposited rubidium and strontium were dissolved in a few drops of aqua regia. The solution was mixed with 60 mg each rubidium and strontium carriers; strontium was precipitated with H2SO4 and filtered. From the filtrate, rubidium was precipitated as perchlorate by the addition of perchloric acid.
Rb-3
Rubidium
SEPARATION TIME: ~1 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Litz, L. M., Ring, S. A., and Balkwell, W. R., “The 1.25-min 82Rbm daughter of 27-day 82Sr,” Phys. Rev. 92, 288–290 ( 1953).
PROCEDURE: This procedure was used for separation of rubidium from purified strontium parent. After the establishment of the parent-daughter equilibrium in the initially purified strontium solution, perchloric acid, rubidium, and strontium carriers were added, and the mixture was boiled to fuming. The solution was cooled in ice, and rubidium was precipitated as perchlorate by the addition of cooled absolute alcohol. The precipitate was filtered, washed with alcohol, and Used for counting.
Rb-4
Rubidium
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Fritze, K., and Kennett, T. J., “The identification and half-lives of fission-product 92Rb and 93Rb,” Can. J. Phys. 38, 1614–1622 ( 1960).
PROCEDURE: This procedure was used to isolate rubidium from fission products and to analyze for rubidium activity by means of yttrium daughter products. The irradiated uranium solution was mixed with 2M HCl containing rubidium carrier. The solution was poured into a millipore filtration apparatus containing HClO4 (20%), and the precipitated rubidium perchlorate was filtered, washed with 10% HClO4, dissolved, and used for further processing.
Ru-1
Ruthenium
SEPARATION TIME: 1 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Continuous
REFERENCE: Matschoss, V., and Bächmann, K., “Selective, on-line, gas-phase separation methods for technetium and ruthenium,” J. Inorg. Nucl. Chem. 41, 141–147 ( 1979).
PROCEDURE: Recoiling fission fragments from a 252Cf source were thermalized in a stream of N2, mixed with O2, and passed through a heated quartz tube. Ruthenium, technetium, and iodine were transferred to a cooled aluminum disc. Flushing the aluminum disc with a stream of HCl at 150°C removed technetium and iodine. The remaining fraction on the aluminum disc showed ruthenium and rhodium gamma rays. The aluminum disc was changed every 0.8 s.
Ru-2
Ruthenium
SEPARATION TIME: 40 to 50 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Franz, G., and Herrmann, G., “Identification of short-lived ruthenium and rhodium isotopes in fission by rapid chemical separations,” J. Inorg. Nucl. Chem. 40, 945–955 ( 1978).
PROCEDURE: An irradiated uranium or plutonium solution containing I− and Ru(III) carriers was mixed with a suspension of Ce(IV) iodate in HClO4 (2M). The resulting solution was filtered through a layer of Voltalef coated with TBP and HDEHP and then through a layer of freshly prepared BaSO4. The filtrate was mixed with HIO4 solution (3%) and passed through a layer of Chromosorb 102 saturated with petroleum ether or CCl4. Ruthenium was retained by this layer. The Chromosorb layer was first washed with HIO4 – HClO4 solution, then with HCl – HF (10:1) solution, and finally with water. The Chromosorb layer was counted.
Ru-3
Ruthenium
SEPARATION TIME: 8.3 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Franz, G., and Herrmann, G., “Identification of short-lived ruthenium and rhodium isotopes by rapid chemical separations,” J. Inorg. Nucl. Chem. 40, 945–955 ( 1978).
PROCEDURE: Irradiated uranium or plutonium solution containing Ru(III) carrier and IO3− was filtered through a fiberglass filter, and the filtrate was collected in an aqueous suspension of Ce(IV) iodate. The filter was washed with HClO4 containing Ru(III) and collected with the filtrate. The solution was passed through a layer of Voltalef coated with TBP and HDEHP and then through a layer of freshly formed BaSO4. The filtrate was collected in HIO4 solution. The solution was then passed through a layer of Chromosorb 102 saturated with petroleum ether. The Chromosorb layer was washed with HIO4 – HClO4, then with concentrated HCl – HF (10:1), and finally with NaOH (5M). The Chromosorb layer was counted.
Ru-4
Ruthenium
SEPARATION TIME: ~60 s
SEPARATION TECHNIQUE: Distillation
PRODUCTION MODE: Batch
REFERENCE: Franz, G., “The decay of 79.8-s 109Rh into levels of 109Pd,” J. Inorg. Nucl. Chem. 40, 1467–1472 ( 1978).
PROCEDURE: See procedure Rh-1 under “Rhodium,” Franz, G., J. Inorg. Nucl. Chem. 40, 1467–1472 (1978).
Ru-5
Ruthenium
SEPARATION TIME: ~5 s
SEPARATION TECHNIQUE: Distillation
PRODUCTION MODE: Batch
REFERENCE: Riccato, M. T., “Uber die schnelle trennung des rutheniums von den spaltprodukten des urans,” Radiochim. Acta 15, 3 ( 1971).
PROCEDURE: Ruthenium was distilled from a solution of uranium in HClO4 containing sodium chlorate. The distillation was carried out in a stream of air at a temperature of 150°C. The distillate was counted. The chemical yield was about 50%.
Ru-6
Ruthenium
SEPARATION TIME: ~5 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Fettweis, P., and del Marmol, P., “Study of short-lived ruthenium isotopes produced in thermal-neutron fission of 235U,” Z. Physik A 275, 359–367 ( 1975).
PROCEDURE: An irradiated uranium solution was mixed with hot (80°C) NaOH solution (1M) containing Ru(III) carrier. Cl2 gas was passed through the solution, oxidizing ruthenium to RuO4. The RuO4 was carried by the gas stream and was party deposited on polyethylene pellets heated to 85°C. The time for the whole chemical process was 5 s, and the yield was about 5%. The sample had bromine, iodine, and noble-gas contamination. For additional details about the chemistry, see under “Ruthenium,” Jenson, D. K., Health Phys. 12, 923 (1966).
Ru-7
Ruthenium
SEPARATION TIME: 30 to 40 s
SEPARATION TECHNIQUE: Distillation
PRODUCTION MODE: Batch
REFERENCE: Nair, A. G. C., Srivastava, A., Srivastava, B. K., Prakash, S., and Ramaniah, M. V., “Cumulative yields of the short-lived ruthenium isotopes in the spontaneous fission of 252Cf,” J. Radioanal. Nucl. Chem. 82, 263–267 ( 1984).
PROCEDURE: The fission products from 252Cf were collected on an ammonium nitrate pellet for the required time. The pellet was dissolved in H2SO4 (3M) containing ruthenium carrier. Ruthenium was distilled as RuO4 using sodium bismuthate as the oxidizing agent and collected in HCl (1M).
Ru-8
Ruthenium
SEPARATION TIME: 5 to 6 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Skarnemark, G., Brodén, K., Yun, M., Kaffrell, N., and Trautmann, N., “Rapid, continuous-separation procedures for arsenic and ruthenium from complex reaction-product mixtures,” Radiochim. Acta 33, 97–100 ( 1983).
PROCEDURE: Recoiling fission products, which were stopped and transported by a KCl – N2 gas jet, were dissolved in a solution of H2SO4 (0.2M) containing RuCl3 (10−4M) and degassed at 70°C. The solution was then mixed with H2SO4 (0.2M) – Ce(SO4)2 (0.015M) at 70°C and passed on to a SISAK system. In the first centrifuge, RuO4 was extracted by CCl4, along with small amounts of Br2 and I2. In the second centrifuge, ruthenium was stripped using HCl (0.1 M) – Na2SO3 (0.05M) – KI (10−3M); part of the bromine accompanied the ruthenium. The aqueous phase was mixed with HNO3 (4M) containing KBrO3 (0.1M), and in the third centrifuge, bromine was removed by extraction with CCl4. The aqueous phase was counted for ruthenium.
Ru-9
Ruthenium
SEPARATION TIME: 3 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayar, B., Votsilka, I., Zaitseva, N. G., and Novgorodov, A. F., “Rapid gas-thermochromatographic isolation of neutron-deficient isotopes of ruthenium from silver chloride,” Soviet Radiochemistry (Eng. Tr.) 20, 642–652 ( 1978).
PROCEDURE: Irradiated silver chloride was heated at 870°C in a stream of a mixture of chlorine and oxygen. The gases passed through a thermochromatographic column (quartz) with a temperature gradient from 400°C to 20°C over ~50 cm. Ruthenium was deposited in the temperature zone around 190°C and was well separated from other elements such as zirconium, niobium, molybdenum, and technetium.
Ru-10
Ruthenium
SEPARATION TIME: ~1 min
SEPARATION TECHNIQUE: Distillation
PRODUCTION MODE: Batch
REFERENCE: Aten, Jr., A. H. W., and De Vries-Hamerling, T., “A short-lived ruthenium isotope,” Physica 21, 544 ( 1955).
PROCEDURE: Irradiated molybdenum metal was dissolved in a mixture of HNO3 and HF. Potassium permanganate and H2SO4 were added to the solution, and ruthenium was distilled off and collected.
Ru-11
Ruthenium
SEPARATION TIME: ~3 min
SEPARATION TECHNIQUE: Distillation
PRODUCTION MODE: Batch
REFERENCE: Glendenin, L. E., “Short-lived ruthenium-rhodium decay chains,” National Nuclear Energy Series, Vol. 9, “Radiochemical Studies: The Fission Products,” Coryell, C. D., and Sugarman, N. (Eds.), Book 2, p. 849 ( 1951).
PROCEDURE: Ruthenium in a fission product solution was oxidized with NaBiO3 in acid solution. The ruthenium was absorbed in NaOH solution and precipitated by boiling with alcohol. The ruthenium oxide precipitate was centrifuged and used for counting.
Sm-1
Samarium
SEPARATION TIME: ~11 min
SEPARATION TECHNIQUE: Extraction, ion exchange
PRODUCTION MODE: Autobatch
REFERENCE: Baker, J. D., Gehrke, R. J., Greenwood, R. C., Meikrantz, D. H., and Novich, V. J., “A new isotope, 158Sm; comments on the decay of 157Sm,” J. Inorg. Nucl. Chem. 42, 1547–1553 ( 1980).
PROCEDURE: See procedure Ln-1 under “Lanthanides,” Baker, J. D., et al., J. Radioanal. Chem. 74, 117–124 (1982).
Sc-1
Scandium
SEPARATION TIME: ~1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Rogers, P. C., and Gordon, G. E., “Decay of Sc-42m and the levels of Ca-42,” Phys. Rev. 129, 2653–2659 ( 1963).
PROCEDURE: Irradiated KCl (or calcium salt) was dissolved in HCl solution (0.1 to 5M) and extracted with 0.5M 2-thenoyltrifluoroacetone (TTA) in benzene. Note that titanium will also be extracted under these conditions.
Se-1
Selenium
SEPARATION TIME: 5 s
SEPARATION TECHNIQUE: Volatilization, extraction
PRODUCTION MODE: Autobatch
REFERFNCE: Kratz, J. V., and Herrmann, G., “Half-lives, fission yields, and neutron-emissionv probabilities of 87Se and 88Se and evidence for 87As,” J. Inorg. Nucl. Chem. 32, 3713–3723 ( 1970).
PROCEDURE: The irradiated uranium solution was mixed with HCl (12 M) containing arsenic, selenium, antimony, and tellurium carriers; the hydrides of the carrier elements were generated by the production of nascent hydrogen (by the addition of zinc powder). The gases produced were passed through NaOH solution (0.5M), which retained selenium and tellurium. The solution was made 5 M in HCl, oxidized with Br2, and poured through a layer of plastic grains coated with TBP; tellurium and Br2 were extracted by TBP. The filtrate containing selenium was counted. Also see Kratz, J. V., and Herrmann, G., Proceedings of the 3rd Symp. Physics and Chemistry of Fission, IAEA publication, Vol. 2 (Rochester, 1970), p. 95. Also see Folger, H., et al., Radiochem. Radioanal. Lett. 1, 185–190 (1969).
Se-2
Selenium
SEPARATION TIME: 1.0 to 2.3 s
SEPARATION TECHNIQUE: Exchange
PRODUCTION MODE: Batch
REFERENCE: Tomlinson, L., and Hurdus, M. H., “Delayed neutron precursors. IV. 87Se, 88Se, and 89Se; half-lives, neutron-emission probabilities, and fission yields, ” J. Inorg. Nucl. Chem. 33, 3609–3620 ( 1971).
PROCEDURE: Uranyl chloride in HCl (0.5M) containing selenium, bromine, and iodine carriers and a reducing agent was irradiated in a vessel. H2Se gas was bubbled through the solution and flushed with helium. The gas was allowed to flow through soda-lime traps. Selenium was retained mostly in the first trap. Also see Tomlinson, L., and Hurdus, M. H., J. Inorg. Nucl. Chem. 33, 3609–3620 (1971).
Se-3
Selenium
SEPARATION TIME: 4 to 8 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: del Marmol, P., and Van Tigchelt, H., “A rapid radiochemical separation method for selenium,” Radiochim. Acta 12, 57–59 ( 1969).
PROCEDURE: An irradiated uranium solution in H2SO4 (30%) containing selenium carrier was transferred to a vessel containing zinc powder. The gaseous hydrides generated were bubbled through water containing selenium carrier and NaHSO3. The resulting solution was filtered through an AgCl precipitate, and the filtrate was counted for selenium (nota bene: the arsenic and antimony hydrides do not dissolve in water, and the tellurium hydride was not formed to an appreciable extent). Also see del Marmol, P., and Perricos, D. C., J. Inorg. Nucl. Chem. 32, 705–712 (1970).
Se-4
Selenium
SEPARATION TIME: 1 to 2 s
SEPARATION TECHNIQUE: Thermal decomposition
PRODUCTION MODE: Continuous
REFERENCE: Zendel, M., Stender, E., Trautmann, N., and Herrmann, G., “Chemical reactions in a gas-jet recoil-transport system: continuous separation procedure for selenium and tellurium from fission products, ” Nucl. Instrum. Methods 153, 149–156 ( 1978).
PROCEDURE: Recoiling fission fragments were thermalized and carried by a C2H4 – N2 stream. The nonvolatile fission products carried by C2H4 clusters were removed by passing the stream through two filter papers. The gas was then allowed to enter a quartz spiral heated to 860° C and then passed through a quartz-wool trap coated with silver. The halogens remained in the heated chamber or in the capillary connecting the chamber and the trap. Selenium was retained by the silver-coated quartz wool. The noble gases flowed through. Also see Zendel, M., et al., J. Inorg. Nucl. Chem. 42, 1378–1395 (1980).
Se-5
Selenium
SEPARATION TIME: ~1 s
SEPARATION TECHNIQUE: Adsorption
PRODUCTION MODE: Continuous
REFERENCE: Rengan, K., Lin, J., Massey, T. N., Zendel, M., and Meyer, R. A., “Use of organometallic reactions for the isolation and study of short-lived selenium fission products and simultaneous suppression of daughter bromine activity,” Radiochem. Radioanal. Lett. 50, 385–391 ( 1982).
PROCEDURE: Recoiling fission fragments were thermalized in a stream of C2H4 – N2 mixture and carried to a reaction chamber. The gas was passed through a quartz-wool trap, which retained the nonvolatile fission products carried by clusters. Br2 gas was introduced at this stage, and the volatile fission products in the stream were allowed to react with it. The stream was then passed through a quartz-wool trap, which retained selenium. The noble gases and halogens passed through. The elemental Br2 vapor continuously removed the active bromine daughter products generated in the quartz-wool trap.
Se-6
Selenium
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Tamai, T., Matsushita, R., Takada, J., and Kiso, Y., “Gamma-my energies of 85Se and 86Se,” Inorg. Nucl. Chem. Lett. 9, 1145–1152 ( 1973).
PROCEDURE: An irradiated uranium solution and selenium carrier were spotted on a chromatographic paper wetted with the supporting electrolyte (1 × 10−2M HClO4, pH 2.2). KMnO4 solution was spotted at a position 3 mm from the sample position, toward the cathode side. A potential gradient of 500 V/cm was applied for 20 s. The selenium position, previously determined by tracer run, was cut and counted.
Se-7
Selenium
SEPARATION TIME: 45 to 50 s
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Sugarman, N., “Determination of the ranges of the fission fragments emitting delayed neutrons. Chemical identification of the 4.51-s delayed neutron activity, ” J. Chem. Phys. 15, 544–551 ( 1947).
PROCEDURE: Uranyl chloride in HCl (4M), containing SeO32− carrier, was irradiated. The solution was mixed with concentrated HCl, and elemental selenium was precipitated by the addition of NaHSO 3. The selenium precipitate was centrifuged and used for counting.
Se-8
Selenium (Te)
SEPARATION TIME: <10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Naeumann, R., Folger, H., and Denschlag, H. O., “Determination of the nuclear charge distribution in the chain 132 from thermal neutron fission of 235U and 233U,” J. Inorg. Nucl. Chem. 34, 1785–1797 ( 1972).
PROCEDURE: See procedure Te-5 under “Tellurium,” Naeumann, R., et al., J. Inorg. Nucl. Chem. 34, 1785–1797 (1972).
Se-9
Selenium
SEPARATION TIME: ~5 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Tomlinson, L., and Hurdus, M. H., “Delayed neutron precursors—III. 87Se,” J. Inorg. Nucl. Chem. 30, 1995–2002 ( 1968).
PROCEDURE: Uranium solution containing Ge, As, Se, Br, Sn, Sb, Te, and I carriers and thiourea was irradiated. The irradiation vessel was maintained at 1000°C. The hydrides were generated by the addition of zinc to the irradiation solution. A flow of helium carried the hydrides generated; selenium was selectively retained by a large calcium sulfate trap (6.3 g). For additional details, see Tomlinson, L., and Hurdus, M. H., J. Inorg. Nucl. Chem. 30, 1649–1661 (1968).
Si-1
Silicon
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Hot-atom reaction
PRODUCTION MODE: Continuous
REFERENCE: Hardy, J. C., Schmeing, H., Geiger, J. S., and Graham, R. L., “The superallowed beta decay of 18Ne, 22Mg, and 26Si,” Nucl. Phys. A246, 61–75 ( 1975).
PROCEDURE: The irradiation cell contained a stack of enriched 24Mg foils. H2 gas at atmospheric pressure flowed through the cell. The recoiling atoms were thermalized by the H2 gas; the silicon atoms presumably combined with hydrogen, forming a gaseous compound related to SiH4. The gas was passed through a glass-wool filter cooled by dry ice in alcohol.
Ag-1
Silver
SEPARATION TIME: 14 to 29 s
SEPARATION TECHNIQUE: Exchange
PRODUCTION MODE: Batch
REFERENCE: Brüchle, W., and Herrmann, G., “Decay properties of neutron-rich silver isotopes,” Radiochim. Acta 30, 1–10 ( 1982).
PROCEDURE: Irradiated uranium solution was mixed with H2SO4 (1M) saturated with K2Cr2O7 and containing Ce(IV) and palladium carriers. The solution was filtered through a layer of freshly formed AgCl [precipitated by mixing hot solutions of HCl (0.05M, 6 mL) and AgNO3 (0.05M, 5 mL)]. The AgCl layer was washed first with H2SO4 (4M) – K2Cr2O7 solution and then with water. The AgCl was then dissolved in NH4OH (6M) and used for counting. The procedure takes 29 s; it can be finished in 14 s if two persons work together and use smaller volumes of solutions.
Ag-2
Silver
SEPARATION TIME: 4.1 s
SEPARATION TECHNIQUE: Exchange
PRODUCTION MODE: Autobatch
REFERENCE: Brüchle, W., and Herrmann, G., “Decay properties of neutron-rich silver isotopes,” Radiochim. Acta 30, 1–10 ( 1982).
PROCEDURE: The recoiling fission fragments collected in NH4NO3 were transported and dissolved in H2SO4 (1M) containing K2Cr2O7 and Ce(IV) carrier. The solution was filtered through a preformed AgCl layer and washed first with H2SO4 – K2Cr2O7 solution and then with water. The AgCl was dissolved in NH4OH (6M) and collected in HCl (6M). The AgCl was filtered on a movable fiberglass filter and moved to counting position.
Ag-3
Silver
SEPARATION TIME: ~3 s
SEPARATION TECHNIQUE: Plating, chemical
PRODUCTION MODE: Batch
REFERENCE: Björnstad, T., and Alstad, J., “Decay of 116gAg and 116mAg,” J. Inorg. Nucl. Chem. 36, 2159–2166 ( 1974).
PROCEDURE: CdO dissolved in acetic acid was irradiated. The irradiated solution was mixed with hexamethylenetetramine and passed through a column of copper powder mixed with rockwool. Silver was reduced and deposited on copper. The detector was positioned to measure the activity of the copper column. For further details, see Greendale, A. E., and Love, D. L., Nucl. Instr. Methods 23, 209 (1963).
Ag-4
Silver
SEPARATION TIME: ~20 s
SEPARATION TECHNIQUE: Exchange
PRODUCTION MODE: Batch
REFERENCE: Sunderman, D. N., and Meinke, W. W., “Radiochemical separations by isotopic exchange: a rapid, high-decontamination method for silver,” Science 121, 777 ( 1955).
PROCEDURE: Silver chloride-coated platinum gauze was immersed in a solution containing AgCl. A decontamination factor of 1 × 104 was obtained from 4-year-old fission products. silver activity at 95°C for 20 s. Nearly 50% of the silver activity was found to exchange with the
Ag-5
Silver
SEPARATION TIME: ~20 s
SEPARATION TECHNIQUE: Exchange
PRODUCTION MODE: Batch
REFERENCE: Alexander, J. M., Schindewolf, U., and Coryell, C. D., “Short-lived isotopes of palladium and silver of masses 113–117,” Phys. Rev. 111, 228–236 ( 1958).
PROCEDURE: Palladium was initially separated from irradiated uranium. Silver was separated from palladium using an exchange technique. The procedure was used to search for silver activities with half-life <20 s. For exchange-procedure details, see procedure Ag-4 under “Silver,” Sunderman, D. N., and Meinke, W. W., Science 121, 777 (1955).
Ag-6
Silver
SEPARATION TIME: 5 to 10 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Semkow, T., and Wahl, A. C., “Extraction of Ag(I), Cd(II), In(III), Sn(II), Sn(IV), Sb(III), and U(VI) from aqueous solutions by ketone solutions using single-step batch and continuous SISAK methods,” J. Radioanal. Chem. 79, 93–101 ( 1983).
PROCEDURE: See procedure In-1 under “Indium,” Semkow, T., and Wahl, A. C., J. Radioanal. Chem. 79, 93–101 (1983). Silver was extracted with indium from the aqueous solution using the solvent mixture methyl isobutyl ketone – cyclohexanone.
Ag-7
Silver
SEPARATION TIME: ~2 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Semkow, T., and Wahl, A. C., “Extraction of Ag(I), Cd(II), In(III), Sn(II), Sn(IV), Sb(III), and U(VI) from aqueous solutions by ketone solutions using single-step batch and continuous SISAK methods,” J. Radioanal. Chem. 79, 93–101 ( 1983).
PROCEDURE: See procedure In-2 under “Indium,” Semkow, T., and Wahl, A. C., J. Radioanal. Chem. 79, 93–101 (1983). The organic solvents extracted 98 to 99% of the silver.
Ag-8
Silver
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Schindewolf, U. I., Winchester, J. W., and Coryell, C. D., “Decay properties of 74-s 111mAg,” Phys. Rev. 105, 1763–1765 ( 1957).
PROCEDURE: This procedure was developed for fast milking of silver activities from palladium. Palladium was oxidized to Pd(IV) using Cl2; the solution was adjusted to 10M in HCl. The solution was passed through a Dowex-1 column which retained palladium. Silver daughter activities can be eluted using 10M HCl.
Ag-9
Silver
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Recoil
PRODUCTION MODE: Batch
REFERENCE: Miskel, J., “The genetic relationship in 110Ag,” Phys. Rev. 79, 403–404 ( 1950).
PROCEDURE: This procedure was used for the separation of the short-lived, isomeric daughter product of 110Ag. The long-lived silver activity was prepared as silver tetraphenylporphin in ether. Two copper electrodes were immersed in the ether solution, separated by 3 mm, and kept at a potential difference of 600 V. After an appropriate time, the electrodes were removed, washed with ether, and counted.
Ag-10
Silver
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Hicks, H. G., and Gilbert, R. S., “A new isotope of palladium, 1.5-min 113Pd,” Phys. Rev. 94, 371 ( 1954).
PROCEDURE: This procedure was used for milking silver daughter activities from palladium. The fission-product solution containing palladium holdback carrier was scavenged twice with silver chloride. The supernatant solution from the second silver chloride scavenge was maintained at 900°C; a known amount of silver carrier was added, and the precipitate was centrifuged. Addition of silver carrier and separation of AgCl were repeated at known intervals.
Ag-11
Silver
SEPARATION TIME: ~2 s
SEPARATION TECHNIQUE: Plating, chemical
PRODUCTION MODE: Batch
REFERENCE: Weiss, H. V., Ballou, N. E., Elzie, J. L., and Fresco, J. M.. “Nuclear charge distribution in symmetric fission of 235U with thermal neutrons: yields of 117Ag, 118Ag, and 118Pd,” Phys. Rev. 188, 1893–1896 ( 1969).
PROCEDURE: This procedure was utilized for fast isolation of silver from fission products with later assay by means of the daughter activities. The irradiated uranium solution containing ruthenium, rhodium, palladium and silver carriers, sodium citrate, and hexamethylenetetramine was passed through a column of copper powder (0.5 g, previously washed with 4M HNO3). The column was washed with 0.1M HNO3 (16 mL); silver was selectively retained by the column. The cadmium daughter activities were stripped from the column by periodic washing with 0.1M HNO3 (8 mL); the wash solution containing the cadmium was purified radiochemically.
Sr-1
Strontium
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Electrophoresis
PRODUCTION MODE: Batch
REFERENCE: Tamai, T., Takada, J., Matsushita, R., and Kiso, Y., “Gamma-ray energies of 95Sr and 94Sr,” Inorg. Nucl. Chem. Lett. 9, 245–251 ( 1973).
PROCEDURE: Irradiated uranium solution and the carrier solution containing barium and strontium were spotted on the chromatographic paper wetted with the supporting electrolyte (1 × 10−2M nitrilotriacetic acid, pH 4.1). K2CrO4 solution was spotted 1 cm from the sample position, toward the cathodic side. An electrical potential of 5000 V/15 cm was applied for 20 s, starting 20 s after the end of irradiation. The strontium position in the paper, previously determined by a 90Sr tracer run, was cut and counted.
Sr-2
Strontium
SEPARATION TIME: 3 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Arlt, R., Beyer, G. J., Herrmann, E., Habenicht, W., and Tyroff, H., “On the identification of 78Rb/T = 19 min,” Radiochem. Radioanal. Lett. 10, 173–175 ( 1972).
PROCEDURE: After initial mass separation, strontium was separated from rubidium by precipitation as SrSO4. For details, see procedure Rb-2 under “Rubidium,” Arlt, R., et al., Radiochem. Radioanal. Lett. 10, 173–175 (1972).
Sr-3
Strontium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Exchange
PRODUCTION MODE: Batch
REFERENCE: Bakhru, H., and Mukherjee, S. K., “The decay of 93Sr and the gamma spectrum of 93Y,” Nucl. Phys. 61, 56–64 ( 1965).
PROCEDURE: This procedure was used for fast separation of strontium from yttrium and zirconium. The irradiated zirconium powder was dissolved in HF; yttrium holdback carrier and NH4F were added to the solution. Finely powdered SrSO4 was then added to the solution, mixed for a few minutes, and centrifuged. The SrSO4 was washed and used for counting.
Sr-4
Strontium
SEPARATION TIME: ~15 s
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Fritze, K., Kennett, T. J., and Prestwich, W. V., “Half-lives of 94Rb, 94Sr, 94Y, 95Rb, 95Sr, 95Y,” Can. J. Chem. 39, 675–680 ( 1961).
PROCEDURE: This procedure was used for fast separation of strontium from fission products and later determination using yttrium daughter activities. The irradiated uranium solution was mixed with strontium carrier, and Sr(NO3)2 was precipitated by the addition of cold, fuming HNO3. The nitrate precipitate was filtered using a glass-filter crucible of medium porosity. The precipitate was dissolved and used for further processing.
Sr-5
Strontium
SEPARATION TIME: ~1.7 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Fritze, K., Kennett, T. J., and Prestwich, W. V., “Half-lives of 94Rb, 94Sr, 94Y, 95Rb, 95Sr, 95Y,” Can. J. Chem. 39, 675–680 ( 1961).
PROCEDURE: The irradiated uranium solution was mixed with strontium and barium carriers, and the two elements were precipitated as nitrates. The precipitate was dissolved, and barium precipitated as chromate. The filtrate containing strontium was used for gamma-ray counting.
Ta-1
Tantalum
SEPARATION TIME: 0.1 to 0.2 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Zvara, I., Belov, V. Z., Domanov, V. P., and Shalaevskii, M. R., “Chemical isolation of nilsbohrium as ekatantalum in the form of the anhydrous bromide. II. Experiments with a spontaneously fissioning isotope of nilsbohrium,” Soviet Radiochemistry (Eng. Tr.) 18, 328–334 ( 1976).
PROCEDURE: See procedure 105-1 under “Z = 105,” Zvara, I., et al., Soviet Radiochemistry (Eng. Tr.) 18, 328–334 (1976). Also see Belov, V. Z., et al., Soviet Radiochemistry (Eng. Tr.) 17, 87–92 (1975).
Ta-2
Tantalum
SEPARATION TIME: 3 to 5 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Rezanka, I., Ladenbauer-Bellis, I. M., Rasmussen, J. O., Ribbe, W., and der Mateosian, E., “High-spin rotational states in 169Hf from the 159Tb (14N,4nγ) reaction and decay of 169Ta,” Phys. Rev. C 11, 1767–1785 ( 1975).
PROCEDURE: The irradiated terbium metal was dissolved in hot 6M HCl, and a few drops of concentrated HF were added to precipitate lanthanide fluorides. The mixture was then centrifuged, and the supernatant solution was transferred to a separatory funnel. Tantalum was extracted with diisopropylketone. For additional details see Felber, Jr., F. F., University of California report UCRL-3618 (1957).
Ta-3
Tantalum
SEPARATION TIME: 10 to 20 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Bruchertseifer, H., Eichler, B., Estevez, J., and Zvara, I., “Fast, continuous, radiochemical isolation of the short-lived isotopes of hafnium, tantalum and tungsten produced by heavy-ion-induced reactions, ” Radiochim. Acta 47, 41–46 ( 1989).
PROCEDURE: The nitrogen gas containing KCl aerosol thermalized and carried the recoiling nuclear-reaction products. The stream was passed through a nuclear filter that retained the KCl particles. The flow was diverted to another identical filter unit while the KCl carrying the reaction products was dissolved in a mixture of 0.5M HF + 1.0M HCl + methyl isobutyl ketone (MIBK). The mixture was transferred to a separatory funnel, the aqueous phase was discarded, and the organic phase was counted. The MIBK extracted tantalum; hafnium and lanthanides remained in the aqueous phase.
Tc-1
Technetium
SEPARATION TIME: 5 s
SEPARATION TECHNIQUE: Adsorption
PRODUCTION MODE: Batch
REFERENCE: Vine, E. N., and Wahl, A. C., “Fractional independent yields of 104Tc and 105Tc from thermal neutron induced fission of 235U and of 239Pu,” J. Inorg. Nucl. Chem. 43, 877–883 ( 1981).
PROCEDURE: Technetium was separated from molybdenum by adsorption of TcO4− on preformed [(C6H5)4As]ClO4 precipitate. Irradiated uranium or plutonium solution in H2SO4 containing Ce(IV) and [(C6H5)4As]Cl was filtered through a layer of freshly formed precipitate and washed with water.
Tc-2
Technetium
SEPARATION TIME: 20 to 30 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Neidhart, B., Bächmann, K., Kramer, S., and Link, I., “Selective separation of fission technetium using solid chlorides, ” Radiochem. Radioanal. Lett. 12, 59–69 ( 1972).
PROCEDURE: UCl4 granules (40 µm) were mixed with KCl (or SrCl2) containing ZrCl4 and irradiated. After irradiation, the mixture was heated in a stream of N2 gas to a temperature of 700 to 750°C. Technetium was preferentially volatilized.
Tc-3
Technetium
SEPARATION TIME: 2.5 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Trautmann, N., Kaffrell, N., Ahrens, H., and Dittner, P. F., “Identification of 109Tc and 110Tc in fission of 249Cf,” Phys. Rev. C 13, 872–874 ( 1976).
PROCEDURE: Recoiling fission fragments collected in NH4NO3 catcher were dissolved in HNO3 (0.1M) containing SO2 and tartaric acid. The solution was passed through two preformed layers of AgCl to remove halogens. The filtrate was passed through a layer of Voltalef 300 LD-PL micropowder coated with [(C6H5)4]AsCl in CHCl3 (0.025M). Technetium extracted by this column was washed with HNO3 containing tartaric acid and transported for counting.
Tc-4
Technetium
SEPARATION TIME: 1 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Autobatch
REFERENCE: Matschoss, V., and Bächmann, K., “Selective on-line, gas-phase separation methods for technetium and ruthenium,” J. Inorg. Nucl. Chem. 41, 141–147 ( 1979).
PROCEDURE: Recoiling fission fragments were thermalized and carried by a stream of N2 which was then mixed with H2O vapor and O2 and passed through a quartz tube heated to 900°C. The gas flowed through a segment of the tube coated with silver, which retained iodine. HCl was then introduced, and the last segment of the tube was maintained at 150°C. Ruthenium remained in the last region, while technetium passed through and was collected in a trap at room temperature.
Tc-5
Technetium
SEPARATION TIME: 7 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Brodén, K., Skarnemark, G., Björnstad, T., Eriksen, D., Haldorsen, I., Kaffrell, N., Stender, E., and Trautmann, N., “Rapid continuous separation procedures for zirconium, niobium, technetium, bromine, and iodine from complex reaction product mixtures,” J. Inorg. Nucl. Chem. 43, 765–771 ( 1981).
PROCEDURE: Recoiling fission fragments from a 239Pu target were thermalized and carried by a KCl aerosol. The fission products were dissolved in a HNO3 (0.1M) – KBrO3 (0.1M) solution, degassed at 80°C, and transferred to a SISAK-2 system. Technetium was extracted in the first centrifuge with Alamine-336 (0.05M) in CHCl3. In the next centrifuge, technetium was stripped with HNO3 (2M). The small contaminants were removed in the next step by an extraction with di-(2-ethylhexyl)phosphoric acid (0.5M) in CHCl3. Technetium in the aqueous phase was counted. Also see Summerer, K., et al., Nucl. Phys. A339, 74–88 (1980) and Stachel, J., et al., Radiochim. Acta 26, 127–133 (1979).
Tc-6
Technetium
SEPARATION TIME: 7.5 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Trautmann, N., Kaffrell, N., Behlich, H. W., Folger, H., Herrmann, G., Hubscher, D., and Ahrens, H., “Identification of short-lived isotopes of zirconium, niobium, molybdenum, and technetium in fission by rapid solvent extraction technique, ” Radiochim. Acta 18, 86–101 ( 1972).
PROCEDURE: Irradiated uranium or plutonium solution in HNO3 (0.2M) containing SO2 and tartaric acid was passed through two layers of AgCl. The AgCl layers were washed with HNO3 (0.1M) containing tartaric acid and antimony and niobium holdback carriers. The filtrate and the washings were collected in (NH4)2S2O8 (0.2M) containing AgNO3. The solution was filtered through a layer of Chromosorb coated with Ph4AsCl in CHCl3. The solvent layer was washed with a HNO3 – tartaric acid – antimony – niobium solution, and technetium was eluted with HNO3 (2M) containing NH4ReO4. The eluate was collected in Ph4AsCl solution; technetium was precipitated with Ph4AsReO4. The precipitate was filtered on a fiberglass filter, washed with HNO 3 (0.1M), and transferred for counting. Also see Kaffrell, N., et al., Phys. Rev. C8, 320–322 (1973).
Tc-7
Technetium
SEPARATION TIME: 7 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Stachel, J., Kaffrell, N., Trautmann, N., Brodén, K., Skarnemark, G., and Eriksen, D., “The collective structure of 106,108Ru,” Z. Phys. A316, 105–119 ( 1984).
PROCEDURE: For extraction details, see procedure Tc-5 under “Technetium,” Brodén, K., et al., J. Inorg. Nucl. Chem. 43, 765–771 (1981). The aqueous phase containing technetium was passed through a polyethylene chamber (1 cm × 4 cm) containing Dowex-1 X4. Technetium reached this chamber in ~7 s and was retained for an average of 30 s.
Tc-8
Technetium
SEPARATION TIME: 5 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Stachel, J., Kaffrell, N., Trautmann, N., Brodén, K., Skarnemark, G., and Eriksen, D., “The collective structure of 106,108Ru,” Z. Phys. A316, 105–119 ( 1984).
PROCEDURE: For extraction details, see procedure Tc-5 under “Technetium,” Brodén, K., et al., J. Inorg. Nucl. Chem. 43, 765–771 (1981). The third step of the extraction of contaminants with HDEHP was dropped. The HNO3 strip of technetium was passed through a polyethylene chamber (2 cm × 4 cm) in front of the detector.
Tc-9
Technetium
SEPARATION TIME: 5 to 6 s
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Flegenheimer, J., and Seelman-Eggebert, W., “Determination of the half-life of 102Tc,” Proc. Internat. Conf. on the Peaceful Uses of Atomic Energy 7, 152–153 ( 1956).
PROCEDURE: This procedure was used for the separation of short-lived technetium from parent molybdenum. Molybdenum separated from fission products was complexed with tartaric acid in an HCl medium. An excess of tetraphenylarsonium chloride was added to the solution. By the addition of a few drops of perrhenate ion to the solution, technetium was precipitated.
Te-1
Tellurium
SEPARATION TIME: 5.0 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Autobatch
REFERENCE: Folger, H., Kratz, J. V., and Herrmann, G., “Rapid volatilization of arsenic, selenium, antimony, and tellurium in the form of their hydrides,” Radiochem. Radioanal. Lett. 1, 185–190 ( 1969).
PROCEDURE: See procedure Se-1 under “Selenium,” Kratz, J. V., and Herrmann, G., J. Inorg. Nucl. Chem. 32, 3713–3723 (1970). The tellurium retained by the plastic grains coated with tributyl phosphate (TBP) was washed with HCl – KClO3 solution, and the TBP layer was counted.
Te-2
Tellurium
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Exchange
PRODUCTION MODE: Batch
REFERENCE: Tomlinson, L., and Hurdus, M. H., “Exchange and reducing reactions of hydrogen telluride: rapid radiochemical separation of tellurium,” Radiochim. Acta 17, 199–202 ( 1973).
PROCEDURE: Uranyl chloride in HC1 (0.03M) containing arsenic, selenium, tellurium, bromine, and iodine carriers and a reducing agent was irradiated. H2Te was passed through the solution, and the system was flushed with helium. The gas flowed through a furnace kept at 350°C and then through soda lime and charcoal traps, respectively. Tellurium, which was deposited in the furnace, was dissolved in HCl (10M) and counted.
Te-3
Tellurium
SEPARATION TIME: 1 to 2 s
SEPARATION TECHNIQUE: Adsorption
PRODUCTION MODE: Continuous
REFERENCE: Zendel, M., Stender, E., Trautmaan, N., and Herrmann, G., “Chemical reactions in gas-jet recoil-transport system: continuous-separation procedure for selenium and tellurium from fission products,” Nucl. Instr. Methods 153, 149–156 ( 1978).
PROCEDURE: Recoiling fission products were thermalized and carried by a C2H4 – N2 stream. The gas was passed through a column of charcoal (0.5 cm × 10 cm) which adsorbed selenium, bromine, and iodine and allowed the fission products carried on clusters to go through. The stream was then allowed to enter a quartz spiral heated to 860°C. The clusters decomposed, and the volatile compounds of tellurium formed, along with noble gases, were transported by the stream to a charcoal trap. Tellurium was retained in the trap.
Te-4
Tellurium
SEPARATION TIME: ~20 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch or continuous
REFERENCE: Weber, M., Trautmann, N., and Herrmann, G., “Volatilization of fission products from solid uranium tetrafluoride, ” Radiochem. Radioanal. Lett. 6, 73–80 ( 1971).
PROCEDURE: Irradiated UF4, spread in a boat (~40 mg/cm2), was kept in a thermochromatographic apparatus and heated to 800 °C in a stream of nitrogen. The other end of the apparatus was cooled by water. Experiments showed that tellurium can be volatilized at 800°C and deposited in the cooler part of the apparatus with a half-time of 20 s.
Te-5
Tellurium (Se)
SEPARATION TIME: <10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Naeumann, R., Folger, H., and Denschlag, H. O., “Determination of the nuclear distribution in the chain 132 from thermal neutron fission of 235U and 233U,” J. Inorg. Nucl. Chem. 34, 1785–1797 ( 1972).
PROCEDURE: An irradiated uranium solution was mixed with concentrated HCl containing antimony and tellurium carriers, and then zinc powder was added. The nascent hydrogen produced reduced arsenic, antimony, selenium, and tellurium to hydrides and swept them through a series of traps. The first trap, quartz wool impregnated with NaOH (0.5 M), retained the hydrides of selenium and tellurium. The second trap, quartz wool impregnated with a saturated solution of KOH in ethanol, retained arsenic and antimony. Daughter products were separated from the traps, purified, and used for fission-yield determinations.
Te-6
Tellurium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Reising, R., and Pate, B. D., “The decay of 6-min 115Te,” Nucl. Phys. 61, 529–541 ( 1965).
PROCEDURE: This procedure was utilized to isolate and purify tellurium from irradiated tin. The tin foil was dissolved in a mixture of HCl and H2O2 in the presence of tellurium carrier. The excess of H2O2 was decomposed by boiling with HBr. Tellurium was precipitated with SO2.
Tb-1
Terbium
SEPARATION TIME: <3 min
SEPARATION TECHNIQUE: Extraction, ion exchange
PRODUCTION MODE: Autobatch
REFERENCE: Greenwood, R. C., Gehrke, R. J., Baker, J. D., Meikrantz, D. H., and Reich, C. W., “Identification of a new isotope, 165Tb,” Phys. Rev. C27, 1266–1270 ( 1983).
PROCEDURE: See procedure Ln-1 under “Lanthanides,” Baker, J. D., et al., J. Radioanal. Chem. 74, 117–124 (1982).
Tl-1
Thallium
SEPARATION TIME: 5 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Andersson, G., Tove, P. A., Jung, B., and Svensson, I. B., “The isomeric transitions in 197Tl and 195Tl,” Nucl. Phys. 3, 493–505 ( 1957).
PROCEDURE: This procedure was developed to separate short-lived thallium daughter products from a lead parent. The lead activity was kept in HCl (6M) solution. Thallium, which may be present as Tl(I), was oxidized to Tl(III) by the addition of a drop of KMnO4. TlCl3 was extracted with ether from the HCl solution. The ether phase was separated and counted.
Tl-2
Thallium
SEPARATION TIME: ~2 s
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Andersson, G., Tove, P. A., Jung, B., and Svensson, I. B., “The isomeric transitions in 197Tl and 195Tl,” Nucl. Phys. 3, 493–505 ( 1957).
PROCEDURE: This procedure was developed for the separation of short-lived thallium daughter products from lead. A column of Dowex-2 was loaded with phosphate ions. The active lead solution in a chloride medium was passed through the column. The thallium daughter product was eluted with water. (The total resin used was 30 mg.)
Tl-3
Thallium
SEPARATION TIME: 2 to 3 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayar, B., Zaitseva, N. G., and Novgorodov, A. F., “Fast gas-thermochromatographic separation of radioactive elements. VI. Extraction of thallium isotopes from irradiated lead oxides, ” Radiochem. Radioanal. Lett. 34, 89–98 ( 1978).
PROCEDURE: Irradiated lead oxide, in a platinum boat, was introduced into a furnace kept at 830°C. Dry oxygen at a flow rate of 20 mL/min was used as the carrier gas. The gas was passed through a thermochromatographic column. Thallium deposited in the temperature zone 220 ± 20 °C. Also see Bayar, B., et al., Soviet Radiochemistry (Eng. Tr.) 16, 877–881 (1974).
Tl-4
Thallium
SEPARATION TIME: 5 min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., Chemical Procedures Used in Bombardment Work at Berkeley, U.S. Atomic Energy Commission report AECD-2738, p. 234 ( 1949).
PROCEDURE: This procedure was developed by Brooks for the separation of thallium from irradiated HgNO3. The irradiated target was dissolved in hot, saturated KI solution (20 mL); TlNO3 carrier solution (20 mg of thallium) was added. The precipitate of Tl(I) was separated and counted.
Tm-1
Thulium
SEPARATION TIME: 11 min
SEPARATION TECHNIQUE: Extraction chromatography
PRODUCTION MODE: Batch
REFERENCE: Latuszynski, A., Mikulski, J., Penev, I., Potempa, A. W., Zielinski, A., Zuber, K., and Zuber, J., “The new 157Tm isotope, T1/2 = 3.6 ± 0.3 min.,” J. Inorg. Nucl. Chem. 38, 585–587 ( 1976).
PROCEDURE: The chemical separation was performed after an isobaric separation with a mass separator. The ions with A = 157 were focused on a glass plate coated with NH4Cl. The NH4Cl layer was dissolved in 1M HCl, and the solution was transferred to a silica-gel column containing di-(2-ethylhexyl)phosphoric acid (HDEHP). The lanthanides were eluted with 8M HCl saturated with HDEHP. For the column and flow rate used, thulium appeared after 9 min.
Sn-1
Tin
SEPARATION TIME: ∼10 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Greendale, A. E., and Love, D. L., “A rapid radiochemical procedure for tin,” Anal. Chem. 35, 1712–1715 ( 1963).
PROCEDURE: The fission-product solution containing Sn(IV) carrier was mixed with HCl (0.6M) containing Sb(III) carrier and added to NaBH4 solution. The hydrides generated, carried by a stream of N2 gas, were passed through a Drierite tube and then through a heated quartz tube. SnH4 was decomposed and deposited as metal.
Sn-2
Tin
SEPARATION TIME: 45 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Izak, T., and Amiel, S., “Half-lives and gamma rays of tin isotopes of masses 129, 130, 131, and 132,” J. Inorg. Nucl. Chem. 34, 1469–1477 ( 1972).
PROCEDURE: Tin and antimony were volatilized as hydrides by reduction of the fission product solution containing tin and antimony carriers with NaBH4. See procedure Sn-1 under “Tin,” Greendale, A. E., and Love, D. L., Anal. Chem. 35, 1712–1715 (1963). SnH4 was selectively adsorbed on activated charcoal (column or filter). SbH3 was partially removed by ascarite.
Sn-3
Tin
SEPARATION TIME: ∼20 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Semkow, T., and Wahl, A. C., “Extraction of Ag(I), Cd(II), In(III), Sn(II), Sn(IV), Sb(III), and U(VI) from aqueous solutions by ketone solutions using single-step batch and continuous SISAK methods,” J. Radioanal. Chem. 79, 93–101 ( 1983).
PROCEDURE: See procedure In-1 under “Indium,” Semkow, T., and Wahl, A. C., J. Radioanal. Chem. 79, 93–101 (1983).
Sn-4
Tin
SEPARATION TIME: 45 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Pappas, A. C., and Wiles, D. R., “New, short-lived isotopes of tin found in fission,” J. Inorg. Nucl. Chem. 2, 69–78 ( 1956).
PROCEDURE: Irradiated (PyH)2UCl6 was dissolved in 10% ammonium acetate solution containing Sn(II) carrier. Potassium sodium tartarate (10%) was added to the solution, and the pH was adjusted to 8.5 by the addition of NH4OH (4M). Tin was extracted by shaking the solution with 0.01% dithizone in CCl4. The organic phase was then washed with ammonium acetate (5%) – potassium sodium tartarate (5%) solution of pH 8.5. Palladium, cadmium, and indium were also extracted along with tin under these conditions, but because of their low fission yield, they did not interfere. Thallium, lead, bismuth, and polonium also would extract under these conditions. The antimony daughter can be back-extracted with pH 8.5 solution.
Sn-5
Tin
SEPARATION TIME: ∼45 s
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Lin, C .C., and Wahl, A. C., “Chemical behavior of short-lived tin fission products,” J. Inorg. Nucl. Chem. 35, 1–9 ( 1973).
PROCEDURE: Uranium solution containing Sb(III), Sn(II), and Sn(IV) carriers and HI (0.15M) and HCl(1M) was irradiated. After irradiation, the solution was mixed with 10 mL of HF (0.68M) containing Sn(II). Hydrogen sulfide gas was passed through the solution for 20 to 30 s to precipitate Sn(II). The precipitate was filtered. The filtrate contained Sn(IV). This procedure achieved a fast separation of Sn(II) from Sn(IV).
Sn-6
Tin
SEPARATION TIME: 4 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Batch
REFERENCE: Naeumann, R., Folger, H., and Denschlag, H. O., “Determination of the nuclear charge distribution in the chain 132 from thermal neutron fission of 235U and 233U,” J. Inorg. Nucl. Chem. 34, 1785–1797 ( 1972).
PROCEDURE: Irradiated uranium solution was mixed with HCl (0.8M) containing lead, antimony, and tellurium carriers. Sodium borohydride (5M) in NaOH (0.2M) was added to the uranium solution, and the hydrides produced were swept through a series of traps by the H2 gas evolved. The first trap contained quartz wool soaked in NaOH (0.5M) or KOH (1M) in ethanol and retained selenium and tellurium. The gases bubbled through a second trap containing HCl (2M), Br2, and Sb(V); the trap retained antimony. The last trap contained quartz wool impregnated with AgNO3 (1M) or HCl (6M) saturated with Br2; tin hydride was absorbed in this trap.
Sn-7
Tin
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Morris, D. F. C., and Reed, G. L., “The short-lived 127Sn isomer,” J. Inorg. Nucl. Chem. 27, 1713–1715 ( 1965).
PROCEDURE: This procedure was utilized for the separation of tin in (n,α) reaction on tellurium. The irradiated tellurium metal was dissolved in 3M HCl. Tin carrier was added to the solution along with bromine water. The solution was cooled in ice, and NH4CNS (3M) was added to it. Tin was extracted with diethyl ether. The organic phase was washed twice with dilute HCl containing NH4CNS and then used for counting.
Sn-8
Tin
SEPARATION TIME: 3 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Chu, P., and Marinsky, J. A., “Isomers of 129Sn,” J. Inorg. Nucl. Chem. 28, 1339–1342 ( 1966).
PROCEDURE: This procedure was used for the fast separation of tin from fission products; the antimony daughter activities were periodically milked, purified, and assayed. The irradiated uranium solution was mixed with Sn(IV) carrier, sufficient NaI and HClO4 were added to make the solution 2.5M in both I− and ClO4−. The solution was extracted with benzene and washed twice with NaI (2.5M) – HClO4 (2.5M) solution containing Sb(III) carrier. Antimony activities were milked from the organic phase by shaking it periodically with the NaI – HClO4 - Sb(III) solution.
Sn-9
Tin
SEPARATION TIME: 2 min
SEPARATION TECHNIQUE: Exchange
PRODUCTION MODE: Batch
REFERENCE: Fowler, M. M., Goth, G. W., Lin, C. C., and Wahl, A. C., “Half-lives of tin and antimony fission products with A = 128 to 133, ” J. Inorg. Nucl. Chem. 36, 1191–1198 ( 1974).
PROCEDURE: Uranium solution in HCl (0.3M), saturated with H2S, was irradiated; the solution was passed through a CdS-cellulose filter bed. Using 9M HCl containing tin carrier, the tin and antimony retained by the bed were eluted. The eluent was mixed with HCl solution (6M) containing Sb(III), I−, and I2. After the addition of HF and dilution, Sb2S3 was precipitated from the solution and filtered. The filtrate was used for counting.
W-1
Tungsten
SEPARATION TIME: ∼5 min
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Batch
REFERENCE: Bayar, B., Novgorodov, A. F., Vocilka, I., and Zaitseva, N. G., “Fast gas-thermochromatographic separation of radioactive elements. III. Production and thermochromatographic behavior of volatile oxides and hydroxides of radioactive tungsten,” Radiochem. Radioanal. Lett. 22, 53–62 ( 1975).
PROCEDURE: Irradiated gold or tantalum was heated to 1160°C in a stream of O2 containing H2O vapor. The gas passed through a thermochromatographic column with a negative temperature gradient. The volatilized, carrier-free tungsten was deposited in the region with temperature near 560°C. Also see Bayar, B., et al., Soviet Radiochemistry (Eng. Tr.) 16, 339–344 (1974).
W-2
Tungsten
SEPARATION TIME: 10 to 20 s
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Autobatch
REFERENCE: Bruchertseifer, H., Eichler, B., Estevez, J., and Zvara, I., “Fast, continuous, radiochemical isolation of the short-lived isotopes of hafnium, tantalum and tungsten by heavy-ion-induced reactions, ” Radiochim. Acta 47, 41–46 ( 1989).
PROCEDURE: The nitrogen gas containing KCl aerosol thermalized and carried the recoiling nuclear-reaction products. The stream was passed through a porous glass filter along with a solution of NH3. The filter partly retained tantalum, hafnium, and the lanthanides. The solution was then passed through a column of Wolfatit KPS resin (cation exchanger). Tantalum, hafnium, and the lanthanides were retained by the ion-exchange resin; tungsten was present in the effluent. The effluent was counted for short-lived tungsten activities.
U-1
Uranium
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Ahmad, S., and Skarnemark, G., “Extraction of thorium, protactinium, uranium, and neptunium by PMBP from aqueous solutions at short phase-contact times,” J. Radioanal. Nucl. Chem. Lett. 85, 181–192 ( 1984).
PROCEDURE: Nitric acid solution (2M) containing short-lived uranium isotopes produced by heavy-ion reactions was passed to a high-speed centrifuge. Thorium, protactinium, and neptunium were extracted with 1-phenyl-3-methyl-4-benzopyrazolone-5 (PMBP) dissolved in toluene (0.2M). Aqueous phase was passed to the next centrifuge, where uranium was extracted with Alamine-336 or di-(2-ethylhexyl)phosphoric acid. In the third centrifuge, uranium was stripped from the organic phase with water.
U-2
Uranium
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Ahmad, S., Thoren, A., and Skarnemark, G., “Separation of uranium from complex nuclear reaction-product mixtures, ” J. Radioanal. Nucl. Chem. Lett. 86, 247–254 ( 1984).
PROCEDURE: The reaction products carried by a gas jet were dissolved in a mixture of HNO3 (2M) – NH4NO3 (8M) and passed on to a high-speed centrifuge, after degassing. Elements such as thorium, protactinium, Np(IV), Pu(IV), zirconium, indium, and cadmium were extracted by 0.2M 1-phenyl-3-methyl-4-benzoylpyrazolone-5 (PMBP) in toluene. The aqueous solution was passed on to a second centrifuge, where uranium was extracted by Alamine-336 (0.2M) – Dodecanol (0.5%) in cyclohexane. Small amounts of zirconium, niobium, technetium, and iodine were also extracted, along with uranium. The organic phase was passed on to a third centrifuge, where uranium was stripped with H2O. Also see procedure U-1 under “Uranium,” Ahmad, S., and Skarnemark, G, J. Radioanal. Nucl. Chem. Lett. 85, 181–192 (1984).
U-3
Uranium
SEPARATION TIME: 1.4 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., Ghiorso, A., and Seaborg, G. T., “Further work on heavy collateral radioactive chains,” Phys. Rev. 85, 429–431 ( 1952).
PROCEDURE: Irradiated thorium nitrate was dissolved in saturated ammonium nitrate solution and extracted with diethyl ether. The ether layer was washed with ammonium nitrate solution and then transferred to a platinum plate. The ether was removed by evaporation and the plate flamed.
U-4
Uranium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Huizenga, J. R., Rao, C. L., and Engelkemeir, D. W., ”27-min isomer of 235U,“ Phys. Rev. 107, 319–320 ( 1957).
PROCEDURE: Plutonium in aqueous solution was reduced with Fe(II), and the solution was saturated with ammonium nitrate. Uranium was extracted with diethyl ether. The organic phase was washed with saturated ammonium nitrate solution containing Fe(II). The ether solution was evaporated on a platinum plate.
U-5
Uranium
SEPARATION TIME: ∼30 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Meinke, W. W., “Rapid separations of protactinium and uranium radioisotopes from cyclotron-bombarded thorium nitrate,” J. Chem. Phys. 20, 754 ( 1952).
PROCEDURE: Irradiated thorium nitrate was dissolved in a solution of magnesium nitrate (3M) – nitric acid (0.1M) and extracted with diethyl ether. The ether layer was washed with magnesium nitrate – nitric acid solution and finally with ammonium nitrate solution. The ether layer was used for preparing counting samples.
Xe-1
Xenon
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Emanation
PRODUCTION MODE: Batch
REFERENCE: Ford, G. P., Wolfsberg, K., and Erdal, B. R., “Independent yields of the isomers of 133Xe and 135Xe for neutron-induced fission of 233U, 235U, 238U, 239Pu, and 242mAm,” Phys. Rev. C 30, 195–213 ( 1984).
PROCEDURE: The fissile materials were used in the form of thin layers of oxides deposited on platinum or as stearates. The target was covered with praseodymium stearate and kept in an aluminum tube with rubber stoppers. The tube was evacuated and then irradiated. After irradiation, the tube was connected to a set of two charcoal traps through one of the rubber stoppers. The trap closest to the tube was maintained at 150°C, and the second one was kept at liquid-nitrogen temperature. Inactive xenon was admitted into the tube through the other stopper, allowed to mix with active xenon, and passed on to the charcoal traps. The operation was repeated, and the cold trap was sealed.
Xe-2
Xenon
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Emanation
PRODUCTION MODE: Batch
REFERENCE: Wolfsberg, K., “Nuclear charge distribution in fission: fractional yields of krypton and xenon isotopes from thermal neutron fission of 233U and 239Pu and from 14-MeV neutron fission of 235U and 238U,” Phys. Rev. 137, B929–B935 ( 1965).
PROCEDURE: See procedure Kr-1 under “Krypton,” Wolfsberg, K., Phys. Rev. 137, B929–B935 (1965).
Y-1
Yttrium
SEPARATION TIME: 100 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Rengan, K., and Griffin, H. C., “Fast radiochemical separation of yttrium from fission products,” Radiochem. Radioanal. Lett. 24, 1–8 ( 1976).
PROCEDURE: The irradiated uranium solution was mixed with HCl (0.5 M) containing Ce(III) carrier and H2O2. Yttrium was extracted with di-(2-ethylhexyl)phosphoric acid (0.75 M) in petroleum ether. The solvent phase was washed first with HCl (1M) containing H2O2 and then with HCl (1M). Finally, yttrium was back-extracted with HCl (8M). The HCl phase with yttrium was counted.
Y-2
Yttrium
SEPARATION TIME: 10 s
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Autobatch
REFERENCE: Klein, G., Kaffrell, N., Trautmann, N., and Herrmann, G., “Identification of 96Y in fission by a rapid chemical-separation procedure,” Inorg. Nucl. Chem. Lett. 11, 511–518 ( 1975).
PROCEDURE: Irradiated uranium solution in HCl, containing EDTA and SnCl2, was passed through a layer of Dowex-50 X12 in H+ form and washed with an HCl solution of EDTA and SnCl2. The Dowex layer was washed with NH4Cl (to remove alkali metals) and then with α-hydroxyisobutyric acid (AHIB, pH 3.0) to remove alkaline earths and heavy lanthanides. A second washing with AHIB (pH 3.0) removed yttrium and the light lanthanides; this solution was passed through another layer of Dowex-50 X12 in NH4+ form. The resin retained light lanthanides, while yttrium passed through. The yttrium was collected in a YCl3 solution (0.1M), precipitated as oxalate, filtered, and transferred for counting.
Y-3
Yttrium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Maxia, V., Kelly, W. H., and Horen, D. J., “The neutron-deficient yttrium isotopes 82Y, 83Y, and 84Y,” J. Inorg. Nucl. Chem. 24, 1175–1179 ( 1962).
PROCEDURE: The irradiated arsenic metal was dissolved in hot aqua regia and boiled to dryness. The residue was dissolved in 0.1M HCl, and yttrium was extracted with 1.5M di-(2-ethylhexyl)phosphoric acid in toluene. The organic phase was washed with 0.1M HCl. The half-lives of yttrium isotopes were determined by timed milking of strontium and rubidium daughter activities.
Y-4
Yttrium
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Goldhaber, M., Der Mateosian, E., Schraff-Goldhaber, G., and Sunyar, A. W., “Isomeric state of 89Y and the decay of 89Zr,” Phys. Rev. 83, 661–662 ( 1951).
PROCEDURE: The deuteron-irradiated Y2O3 was dissolved, and the zirconium activities were extracted with 2-thenoyltrifluoroacetone solution. The yttrium daughter activity was stripped from the organic phase with 2M HClO4.
Y-5
Yttrium
SEPARATION TIME: 40 s
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Vallis, D. G., and Perkin, J. L., “Yttrium-96 and strontium-93: new nuclear data,” J. Inorg. Nucl. Chem. 22, 1–5 ( 1961).
PROCEDURE: The irradiated zirconium solution was mixed with yttrium (2 mg) and strontium (50 mg) carriers in 0.5 mL of 6M HNO3 kept in the centrifuge cup of the automatic chemistry apparatus. YF3 was precipitated by the addition of a solution of 12% NH4F (1 mL); centrifugation removed the supernatant liquid automatically. The precipitate was washed once with 0.5% NH4F (2 mL) and thrice with water (2 mL). The washes removed SrF2. The YF3 was dissolved in 6M HCl saturated with boric acid and transferred for counting.
Y-6
Yttrium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Yu, Y. W., and Caretto, Jr., A. A., “The half-lives of 84Zr and 85Zr,” J. Inorg. Nucl. Chem. 33, 3223–3225 ( 1971).
PROCEDURE: This procedure was used to milk yttrium daughter product from the zirconium parent. The irradiated ZrO2 was dissolved in hot 17M HF. To the solution containing zirconium, yttrium solution (5 mg in 0.5 mL) was added; the YF3 precipitate was centrifuged and removed. The YF3 scavenging was repeated three times. Periodically, the yttrium daughter growing in the zirconium solution was removed as YF3. The YF3 precipitate was centrifuged, dissolved in 8M HCl containing a few drops of HNO3, passed through a column of Dowex-1 X10, and the yttrium was eluted with 8M HCl. The yttrium was finally precipitated as oxalate, filtered, and used for counting.
Y-7
Yttrium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Butement, F. D. S., and Briscoe, G. B., “Neutron-deficient isotopes of yttrium and zirconium,” J. Inorg. Nucl. Chem. 25, 151–157 ( 1963).
PROCEDURE: This procedure was used for milking the yttrium daughter activities from the parent zirconium. The irradiated Y2O3 sample was dissolved, and the zirconium activities were extracted using 0.5M 2-thenoyltrifluoroacetone in xylene (procedure of F. L. Moore, Anal. Chem. 28, 997, 1956 ). The zirconium was back-extracted with 2 mL of concentrated HCl; the solution was passed through a Dowex-1 column, which retained the zirconium. Yttrium daughter activities were removed periodically by washing the column with concentrated HCl.
104-1
Z = 104
SEPARATION TIME: 2 to 3 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Hulet, E. K., Lougheed, R. W., Wild, J. F., Landrum, J. H., Nitschke, J. M., and Ghiorso, A., “Chloride complexation of element 104,” J. Inorg. Nucl. Chem. 42, 79–82 ( 1980).
PROCEDURE: Recoil products produced in the reaction 248Cm(18O,5n) were transported by a NaCl – helium gas jet and deposited on the end surface of a rabbit. The products were dissolved in the chemistry apparatus and passed through a column of fluorocarbon coated with trisoctylmethylammonium chloride dissolved in o-xylene (0.25M). Element 104 was eluted with HCl (6M).
104-2
Z = 104
SEPARATION TIME: ∼0.4 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Zvara, I., Belov, V. Z., Chelnokov, L. P., Domanov, V. P., Hussonois, M., Korotkin, Yu. S., Schegolev, V. A., and Shalayevsky, M. R., “Chemical separation of kurchatovium,” Inorg. Nucl. Chem. Lett. 7, 1109–1116 ( 1971).
PROCEDURE: Recoiling nuclear reaction products were carried by flowing nitrogen gas and mixed with chlorinating agents SOC12 and TiCl4 vapor. The mixture was passed through a 200-cm-long glass column. The first ~125 cm was maintained at 400 ± 5°C and in the rest of the column (125 to 200 cm), the thermochromatographic section, the temperature was gradually dropped from 400 to 50°C. Experiments with hafnium activity showed that hafnium deposited in this region, and that it takes ~0.4 s for hafnium to reach the deposition zone from the production region. Element 104, ekahafnium, is expected to deposit in this region. Evidence for a spontaneously fissioning isotope of Z = 104 with a mass of 259 was obtained using this procedure. Also see Zvara, I., et al., Soviet Radiochem. (Eng. Tr.) 14, 115–118 (1972).
104-3
Z = 104
SEPARATION TIME: <1 s
SEPARATION TECHNIQUE: Volatilization
PRODUCTION MODE: Continuous
REFERENCE: Zvara, I., Chuburkov, Yu. T., Tsaletka, R., Zvarova, T. S., Shalaevskii, M. R., and Shilov, B. V., “Chemical properties of element 104,” Sov. J. At. Energy 21, 709–710 ( 1966).
PROCEDURE: The recoiling reaction products were chlorinated by reaction with NbCl5 or ZrCl4 in the vapor phase. The chlorides of group III elements were deposited on the walls of the gas-flow tube or adsorbed on the special filters. The activities of element 104 were carried by the gas flow to the detector. Also see procedure 104-2 under “Z = 104,” Zvara, I., et al., Inorg. Nucl. Chem. Lett. 7, 1109–1116 (1971).
104-4
Z = 104
SEPARATION TIME: 1 min
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Silva, R., Harris, J., Nurmia, M., Eskola, K., and Ghiorso, A., “Chemical separation of rutherfordium,” Inorg. Nucl. Chem. Lett. 6, 871–877 ( 1970).
PROCEDURE: The recoiling products were collected on a platinum foil coated with NH4Cl. The products were dissolved in a small volume (~50 µL) of ammonium α-hydroxyisobutyrate (0.1M, pH 4.0). The solution was loaded on to a column of Dowex-50 X12 (2-mm diameter by 2 cm long) kept at ~80°C. The column was eluted with ammonium α-hydroxyisobutyrate. Drops number 3 to 6 were collected, evaporated to dryness, and heated to ~500°C. The sample was transferred to a counter.
105-1
Z = 105
SEPARATION TIME: 0.1 to 0.2 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Zvara, I., Belov, V. Z., Domanov, V. P., and Shalaevskii, M. R., “Chemical isolation of nilsbohrium as ekatantalum in the form of the anhydrous bromide. II. Experiments with a spontaneously fissioning isotope of nilsbohrium,” Soviet Radiochemistry (Eng. Tr.) 18, 328–334 ( 1976).
PROCEDURE: Recoiling nuclear reaction products were slowed in a nickel chamber through which helium carrier gas containing Br2 and BBr3 vapor flowed continuously. The carrier gas containing the reaction products was passed through a thermochromatographic column, where the temperature dropped from 250°C to 25°C over a distance of 150 cm. Nilsbohrium bromide, less volatile than its homologs niobium and tantalum, deposited in a higher-temperature region of the column. The tracks on mica detectors located at the deposition position provided evidence for element 105. The time, taken from the formation to the deposition, was measured with radioactive niobium serving as a fiducial.
105-2
Z = 105
SEPARATION TIME: 12 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Nai-Qi, Ya., Jost, D. T., Baltensperger, U., and Gäggeler, H. W., “The Saphir gas-jet and a first application to an on-line separation of niobium,” Radiochim. Acta 47, 1–7 ( 1989).
PROCEDURE: See procedure Nb-9 under “Niobium,” Nai-Qi, Ya., Jost, D. T., Baltensperger, U., and Gäggeler, H. W., Radiochim. Acta 47, 1–7 (1989).
105-3
Z = 105
SEPARATION TIME: 55 s
SEPARATION TECHNIQUE: Extraction (HPLC)
PRODUCTION MODE: Autobatch
REFERENCE: Kratz, J.W., Zimmermann, H.P., Scherer, U.W., Schädel, M., Brüchle, W., Gregorich, K.E., Gannett, C.M., Hall, H.L., Henderson, R.A., Lee, D.M., Leyba, J.D., Nurmia, M.J., Hoffman, D.C., Gäggeler, H., Jost, D., Baltensperger, U., Ya Nai-Qi, Fürler, A., Lienert, Ch., “Chemical properties of element 105 in aqueous solution: halide complex formation and anion exchange into triisooctyl amine,” Radiochim Acta 48, 121-133 ( 1989).
PROCEDURE: Element 105 (A=262), produced by 249Bk(18O,5n) reaction, was carried by He/KCl gas jet and deposited on a polyethylene frit. After one minute of collection the jet was directed to a second frit; the first frit was moved to a position on top of an extraction column containing Voltalef (inert support) coated with triisooctyl amine (TIOA). The reaction products deposited on the frit were dissolved in 12 M HCl - 0.02 M HF and the solution passed through the column. The amine extracted halide complexes of Nb, Ta, Pa, and other transplutonium elements while other elements passed through. The column was washed with 4M HCl - 0.02 HF for ten seconds; the effluent (Pa, Nb fraction) was collected on a Ta disk and evaporated to dryness. The column was then washed with 6 M HNO3-0.015 M HF for ten seconds and the effluent (Ta fraction) was collected and evaporated to dryness. Counting of the samples started 55 s after the end of collection on the frit. In an 8-hour shift 300 separations were carried out.
107-1
Z = 107
SEPARATION TIME: <3 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Domanov, V. P., Khyubener, Z., Shalaevskii, M. R., Timokhin, S. N., Petrov, D. V., and Zvara, I., “Experimental approach to the identification of element 107 as ekarhenium. I. Continuous gas-thermochromatographic isolation of radiorhenium, ” Soviet Radiochemistry (Eng. Tr.) 25, 23–28 ( 1983).
PROCEDURE: Pure air containing water vapor at a pressure of 600 Pa (corresponding to saturated water vapor pressure at 0°C) was used as carrier gas. The recoiling nuclear reaction products were stopped in a quartz filter kept at 900°C. The carrier gas passed through a quartz thermochromatographic column with an initial temperature of 400°C and a temperature gradient of 3.5°C/cm. Under these conditions, rhenium, the homolog of element 107, was adsorbed in a temperature zone of 100 ± 20°C. Element 107, if formed, should also deposit in this zone. Track detectors kept at this position can provide evidence for spontaneous fission events from element 107. Also see Zvara, I., et al., Soviet Radiochemistry (Eng. Tr.) 26, 72–76 (1984).113,114-1
113,114-1
Z = 113,114
SEPARATION TIME: <1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous/batch
REFERENCE: Langrock, L. J., Bazarkina, T. V., and Czosnowska, W., “Procedures for selective solvent extraction of superheavy elements 113+ and 1142+ by use of crown ethers,” Radiochim. Acta 30, 229–231 ( 1982).
PROCEDURE: A possible rapid-separation procedure has been developed for Z = 113 and Z = 114 based on their predicted ionic radii. The extraction of Pb2+ and Ba2+ was studied since their ionic radii are very close to the predicted values for 113+ and 1142+. The ions were taken in an aqueous solution buffered at a pH of 4.6 and contained sodium picrate (0.02M). The ions Pb2+ and Ba2+ were extracted with ~100% efficiency by 18-crown-6-ether in CHCl 3. Lanthanides and actinides were not extracted. The ions can be stripped with HCl (6M). This procedure can be adapted for continuous on-line separation.
118-1
Z = 118 (112 and 114 )
SEPARATION TIME: Few s
SEPARATION TECHNIQUE: Condensation
PRODUCTION MODE: Continuous
REFERENCE: Hildebrand, N., Frink, C., Greulich, N., Hickmann, U., Kratz, J. V., Trautmann, N., Herrmann, G., Brügger, M., Gäggeler, H., Summerer, K., and Wirth, G., “A cryosystem for the detection of alpha and spontaneous-fission activities in volatile species,” Nucl. Instr. Meth. Phys. Res. A 260, 407–412 ( 1987).
PROCEDURE: Recoiling nuclear-reaction products (48Ca bombardment of 248Cm or 238U bombardment of 238U) were thermalized and carried by argon gas. The gas was allowed to flow through a quartz tube filled with quartz powder and kept at 1250 K. Nonvolatile products were deposited in the tube, and clusters were destroyed. The gas then flowed through a quartz tube filled with tantalum chips maintained at 1000 K; this removed H2O, CO2 and O2 from the gas. The gas was then led into the cryogenic unit kept at 40 K. Volatile species condensed on top of a solar cell. Surface barrier detectors were used to record alpha spectra. The system was tested with short-lived radon isotopes.
Zn-1
Zinc
SEPARATION TIME: ∼1 s
SEPARATION TECHNIQUE: Thermochromatography
PRODUCTION MODE: Continuous
REFERENCE: Rudstam, G., Aagaard, P., Hoff, P., Johansson, B., and Zwicky, H. O., “Chemical separation combined with an ISOL-system,” Nucl. Instr. Methods 186, 365–379 ( 1981).
PROCEDURE: See procedure Cd-1 under “Cadmium,” Rudstam, G., et al., Nucl. Instr. Methods 186, 365–379 (1981).
Zn-2
Zinc
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Cumming, J. B., “A new zinc isotope, 61Zn,” Phys. Rev. 99, 1645A ( 1955).
PROCEDURE: Zinc was separated from gallium, copper, nickel, cobalt, iron, and manganese by anion exchange. The zinc fraction showed the presence of 61Cu, daughter of 1.5-min 61Zn. Conditions of the separation were not given.
Zn-3
Zinc
SEPARATION TIME: ∼1 min
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Batch
REFERENCE: Lindner, L., and Brinkman, G. A., “60Zn and 61Zn,” Physica 21, 747–748 ( 1955).
PROCEDURE: Irradiated nickel was dissolved in HNO3. Zinc di-beta-naphthylthio-carbazone was extracted into CHCl3. The extraction separated zinc from copper, nickel, cobalt and iron.
Zn-4
Zinc
SEPARATION TIME: 1 to 2 min
SEPARATION TECHNIQUE: Ion exchange
PRODUCTION MODE: Batch
REFERENCE: Hoffman, E. J., and Sarantities, D. G., “Decay schemes of 60Zn and 62Zn,” Phys. Rev. 177, 1640–1647 ( 1969).
PROCEDURE: Irradiated nickel foil was dissolved in hot, concentrated HNO3. The solution was heated with concentrated HCl to eliminate HNO3 and was adjusted to be 1M in HCl. Zn2+ and Cu2+ carriers were added (0.05 mg each) to the solution and mixed, then a few mg of Dowex-1 resin was added, stirred, and filtered. The resin was washed with 1M HCl and mounted for counting. Also see Cumming, J. B., Phys. Rev. 114, 1600–1604 (1959).
Zr-1
Zirconium
SEPARATION TIME: 2.2 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Weis, M., and Densclag, H. O., “Fractional independent yields of 99Y, 99Zr, 99mNb, and 99Nb in the thermal neutron induced fission of 235U,” J. Inorg. Nucl. Chem. 43, 437–444 ( 1981).
PROCEDURE: Fission product solution in HNO3 containing molybdenum and antimony carriers and tartaric acid was filtered through a preformed AgCl layer. The filtrate was made 7.5 M in HNO3 and passed through a layer of Voltalef 300 LD-PL micropowder coated with tri-n-butyl phosphoric acid. This layer retained zirconium.
Zr-2
Zirconium
SEPARATION TIME: ∼7 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Continuous
REFERENCE: Brodén, K., Skarnemark, G., Björnstad, T., Eriksen, T., Haldorsen, I., Kaffrell, N., Stender, E., and Trautmann, N., “Rapid continuous separation procedures for zirconium, niobium, technetium, bromine, and iodine from complex reaction-product mixtures,” J. Inorg. Nucl. Chem. 43, 765–771 ( 1981).
PROCEDURE: Recoiling fission products, thermalized and carried by KCl aerosol, were dissolved in H2SO4 (1M), degassed at 70°C, and transferred to the first centrifuge of a SISAK-2 system. The solution was contacted with Alamine-336 (0.1M) in Shellsol-T containing n-dodecanol (5%), which extracted zirconium, niobium, technetium, and some other elements. In the second centrifuge, zirconium and niobium were stripped by HNO3 (0.3M), leaving contaminants in the organic phase. The aqueous phase was made 3M in HNO3 and 1M in H2O2. Then, zirconium was extracted with HDEHP (1M) in Shellsol-T in the third centrifuge. Zirconium was counted in the organic phase. After one more extraction for zirconium, the niobium in the aqueous phase was counted.
Zr-3
Zirconium
SEPARATION TIME: 4.0 s
SEPARATION TECHNIQUE: Extraction
PRODUCTION MODE: Autobatch
REFERENCE: Trautmann, N., Kaffrell, N., Behlich, H. W., Folger, H., Herrmann, G., Hubscher, D., and Ahrens, H., “Identification of short-lived isotopes of zirconium, niobium, molybdenum, and technetium by rapid solvent-extraction technique,” Radiochim. Acta 18, 86–101 ( 1972).
PROCEDURE: Irradiated uranium solution in HNO3 (0.5M), containing SO2 and tartaric acid, was passed through two layers of preformed AgCl. The filtrate was collected in concentrated HNO3 containing KClO3. The resulting solution, which was 7.5M in HNO3, was filtered through a layer of Voltalef 300 LD-PL micropowder coated with tri-n-butyl phosphoric acid. Zirconium retained by this layer was washed with HNO3 (7.5M) containing KClO3 and transported for counting.
Zr-4
Zirconium
SEPARATION TIME: <3 min
SEPARATION TECHNIQUE: Thermocromatography
PRODUCTION MODE: Batch
REFERENCE: Bayer, B., Vocilka, I., Zaitseva, N. G., and Novgorodov, A. F., "Fast gas-thermochromatographic separation of radioactive elements. IV. Extraction of neutron-deficient zirconium and niobium isotopes from irradiated silver chloride melt," Radiochem. Radioanal. Lett. 34, 63-74 (1978).
PROCEDURE: Irradiated AgCl was heated to 800°C to 900°C in a stream of dry HCl gas. The gas passed through a quartz thermochromatographic column wth a negative temperature gradient. Zirconium deposited in the temperature region that corresponded to the boiling point of NbCl5 (246°C). Increasing the evporation time moved the zirconium and niobium regions close to each other. Heating in an additional O2 stream (following HCl) fixed the zirconium and niobium in nonoverlapping regions, zirconium in the temperature zone 420 ± 40°C and niobium in 230 ± 30°C. Also see Bayer, B., et al., Soviet radiochemistry (Eng. Tr.) 16, 345-351 (1974).
Zr-5
Zirconium
SEPARATION TIME: Few min
SEPARATION TECHNIQUE: Precipitation
PRODUCTION MODE: Batch
REFERENCE: Dubridge, L. A., and Marshall, J., "Radioactive isotopes of strontium, yttrium, and zirconium," Phys. Rev. 58, 7-11 (1940).
PROCEDURE: The irradiated YCl3 was dissolved in HNO3, and zirconium carrier was added to the solution. Zirconium was precipitated as the iodate.



































































































































































































