
their applications to nuclear chemistry have been reviewed [Mey80]. Techniques used in radio-chemical separations were reviewed by Irving [Irv76]. Trautmann and coworkers reviewed rapid, continuous methods for the study of nuclei far from stability [Tra81]. The study of short-lived fission products by rapid separation methods was reviewed by Trautmann [Tra85], and the utilization of gas-jet systems for the radiochemical study of fission products has been reviewed [Ren85a]. A short review of more recent fast radiochemical separations in the study of nuclei off the line of stability has been presented by Kratz, Trautmann, and Herrmann [Kra86], and a summary of parts of this work has been presented [Ren86a]. A number of articles reviewing autobatch processes [Mey90], continuous SISAK techniques [Ska90], continuous, gas-phase separation techniques [Ren90a], fast chemical separations for the study of short-lived nuclides [Ren90b], and rapid radiochemical procedures used for the study of neutron-deficient nuclides [Ham90] have appeared recently.
In this monograph, our aim is to review the techniques used in radiochemical separation procedures, that approach the limits of chemical reaction rates; hence, the term “ultrafast.” The main thrust of this monograph is to provide a collection of ultrafast procedures available in the literature for various elements. An arbitrary limit of approximately less than 1 min was set as the separation time for most elements. If a procedure requiring less than 1 min is not available for an element, then procedures with longer separation times have been included. The first part of this report provides the general review of techniques, while the second part is a compilation of published procedures in summary form.
We have divided our review into several sections. Section 2, entitled Batch Processes describes techniques used in batch processes such as precipitation, ion exchange, volatilization, and solvent extraction. Next, Sec. 3, entitled Automated Processes, compares the relative merits of the automated batch processes with those of the continuous chemical techniques. Section 4, entitled Autobatch Techniques, describes automated batch processes, with examples. Various aspects of continuous production and delivery of nuclear reaction products to chemistry systems are discussed in Sec. 5, Delivery Systems for Continuous Techniques. Continuous chemical separation techniques are discussed in Sec. 6 and Sec. 7, which are entitled Continuous Liquid-Phase Chemical Separations and Continuous Gas-Phase Chemistry, respectively. A brief note on the Future of Ultrafast Chemistry appears as Sec. 8. In Sec. 9, References, we list all the references we have accumulated concerning ultrafast chemical separations. For convenience of identification, we have adopted a system wherein the reference is described by the first three letters of the first-mentioned author's last name followed by the last two digits of the year of publication.
The second part of this book is arranged alphabetically according to the name of the element. Under each element, a separate entry is given for each published technique. Each entry summarizes the main technique used, the type of procedure (batch, autobatch, or continuous), the time required for the separation, a brief outline of the procedure, and the specific reference.
2. Batch Processes
The techniques used in batch processes include many standard methods utilized in analytical chemistry: precipitation, solvent extraction, ion exchange, distillation, volatilization, electrolysis, and electrophoresis. Isotopic exchange, absorption, and thermochromatography are examples of other techniques specially used in fast separation procedures. In this section, the various techniques are briefly discussed, and whenever possible reference is given to other review articles discussing the specific method.
2.1 Precipitation
Precipitation, in the context of our review, can be considered as the formation of a crystalline solid phase from a liquid phase containing electrolytes. Taking precipitation of BaSO4 as an example, when the ionic product, Kss, is greater than the solubility product, Ksp, precipitation may occur; i.e., Kss = [Ba2+][SO4−2] > Ksp. There is an incubation period for precipitation, defined as the time between the formation of the supersaturated solution and the occurrence of visible turbidity. The incubation time is related to the concentration of the supersaturated solution by the following equation:
t = Const. × C−n.
(1)
The value of n was found to be 3.3 for CaC2O4, 4.7 for Ag2CrO4, and 9 for CaF2 [Nie55]. The kinetics of precipitation is discussed in depth by Nielsen in a monograph [Nie64] and in an article by O'Rourke and Johnson [Oro55].
The size of the particles formed is influenced by the degree of supersaturation when precipitation occurs. The less supersaturation, the bigger the particle size will be. Bigger particle size is preferable for fast and easy filtration. However, under low supersaturation conditions, the incubation time will be long.
The factors that affect the purity of the precipitate need to be considered along with the kinetics and particle size mentioned above. Contamination of the precipitate may occur even when the ionic product of the contaminant does not exceed its solubility product. This phenomenon, known as coprecipitation, may be classified into four major categories:
-
Formation of mixed crystals: Chemically similar electrolytes known to form isomorphous crystals will form mixed crystals. For example, BaSO4 and PbSO4 form mixed crystals.
-
Occlusions: During the rapid growth of crystal, the adsorbed ions may not be desorbed before the next larger crystal grows. Thus, the adsorbed impurity is trapped inside the crystal. This phenomenon is known as occlusion.
-
Mechanical entrapment: Several small crystals physically lying close together during growth may form a crystalline mass; during this process, part of the solution may be entrapped.
-
Adsorption: On the surface of the crystal, depending on the conditions of the precipitation, various ions could be adsorbed. Most of the adsorbed contaminant can be removed by proper washing of the precipitate. Unfortunately, this is not the case with coprecipitation due to the other three phenomena mentioned above.
The faster the growth, the larger the chance of contamination by occlusion and mechanical entrapment. Coprecipitation can be minimized by slow precipitation from homogeneous solution. For example, the distribution coefficient for coprecipitation of americium with lanthanum oxalate was 6.3 when the precipitation rate was slow, but decreased to 1 at fast rates [Wal67a]. Precipitation from a homogeneous solution has been reviewed by Gordon [Gor55] and by Cartwright and coworkers [Car67]. The theory of coprecipitation is discussed by Kolthoff [Kol32] and Nielsen [Nie83]. Information on adsorption is presented by Kolthoff [Kol36] and Nielsen [Nie83]. The various factors affecting the formation of precipitates have been discussed in detail by Walton [Wal67b].
From the point of view of ultrafast separation, it is imperative that the required radioactive material be precipitated rapidly from the solution and filtered quickly. These conditions are contradictory to the requirement for obtaining a pure solid phase [Gor59]; also, these conditions will lead to crystals of small size that will not have the required filtration characteristics. Because of these limitations, precipitation has not been used extensively in fast radiochemical separations. Figure 1 shows the elements for which a fast precipitation procedure has been reported in the
literature. Table 1 lists the elements, the chemical form of the precipitate, the separation time, and the procedure number given in Part II. Many of the procedures either are for the separation of parent from daughter activities (or vice versa) or are for the separation of a specific element from activities of neighboring elements that could be produced in charged-particle reactions. Technetium was separated from a molybdenum parent by precipitation with tetraphenylarsonium chloride in the presence of tartaric acid. The separation was achieved in 5 to 6 s [Fle56]. Livingood and Seaborg [Liv38] used precipitation of MnO2 to separate manganese activities from deuteron-irradiated iron and chromium and from 4He-bombarded vanadium. None of the procedures, for example, provided the kind of separation needed to isolate a specific element from a complex mixture such as fission products. For example, Lin and Wahl [Lin73] used Sn(II) sulfide precipitation to quickly separate Sn(II) and Sn(IV) formed in fission. Tellurium was later separated from Sn(II) and Sn(IV), purified, and the activity of daughter products measured. Frequently, precipitation is used as the final step to obtain the activities in a convenient form for counting. Nuh and coworkers [Nuh75] volatilized molecular iodine from a fission-product solution and collected it in a bisulfite solution; they precipitated AgI from this solution and used it for counting.
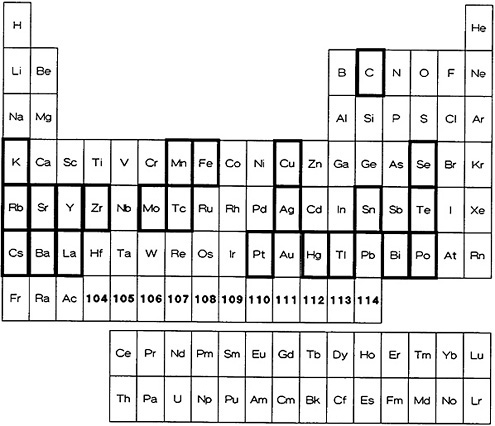
Figure 1. Elements with fast procedures based on precipitation (shown with bold rectangles).
The phenomenon of ion exchange with previously formed precipitate has frequently been used to obtain a fast separation of an element of interest. Silver, bromine, and iodine have been separated using this type of interaction with silver halide precipitate. Discussion of this type of separation is included in Sec. 2.2.
As pointed out earlier, coprecipitation causes difficulty by contaminating the precipitate with elements forming isomorphous crystals. This phenomenon is used to carry trace levels of certain elements on precipitate. For example, insoluble rhenium compounds are routinely used to carry technetium activities. Trautmann and coworkers [Tra72] coprecipitated technetium with tetraphenylarsonium rhenate to prepare a counting sample for technetium. Zychor and coworkers [Zyc83] separated hafnium from lutetium and coprecipitated it with Fe(II) hydroxide. Also, coprecipitation with Fe(III) hydroxide is used to scavenge unwanted activities from solution.
Table 1. Use of precipitation in radiochemical separation procedures.
|
Element |
Reagent |
Timea |
Procedure # |
|
Barium |
Nitrate/sulfate |
Few min |
Ba-3 |
|
Barium |
Nitrate |
Few min |
Ba-4 |
|
Barium (from La) |
Lanthanum hydroxide |
3 – 4 s |
Ba-6 |
|
Bismuth (+ Po) |
Bismuth thionalide |
4 min |
Bi-2 |
|
Carbon |
Barium carbonate |
20 s |
C-1,2 |
|
Cesium |
Chloroplatinate |
3 – 5 min |
Cs-2 |
|
Cesium |
Cobaltinitrite |
<3 min |
Cs-3 |
|
Cesium |
Perchlorate |
~30 s |
Cs-4 |
|
Copper |
Cuprous thiocyanate |
~1 min |
Cu-3 |
|
Iron |
Ferric hydroxide |
3 – 5 min |
Fe-1 |
|
Lanthanum |
Fluoride |
1.7 min |
La-7,8 |
|
Manganese |
Manganese dioxide |
Few min |
Mn-1,2 |
|
Mercury |
Elemental mercury |
Few min |
Hg-3 |
|
Molybdenum |
Amm. phosphomolybdate |
~2 min |
Mo-3 |
|
Molybdenum |
Hydroxide |
2 min |
Mo-4 |
|
Platinum |
Chloroplatinate |
2 – 3 min |
Pt-1 |
|
Polonium (+ Bi) |
Bismuth thionalide |
4 min |
Po-3 |
|
Potassium |
Cobaltinitrite |
Few min |
K-1 |
|
Potassium |
Perchlorate |
Few min |
K-2 |
|
Rubidium |
Perchlorate |
Few s |
Rb-2,3,4 |
|
Selenium |
Elemental selenium |
45 – 50 s |
Se-7 |
|
Silver |
Chloride |
<1 min |
Ag-10 |
|
Strontium |
Sulfate |
3 min |
Sr-2 |
|
Strontium |
Nitrate |
~15 s |
Sr-4,5 |
|
Technetium |
[(C6H5)4As]TcO4 |
5 – 6 s |
Tc-9 |
|
Tellurium |
Elemental tellurium |
Few min |
Te-6 |
|
Thallium |
Thallium iodide |
5 min |
Tl-4 |
|
Tin |
Tin(II) sulfide |
~45 s |
Sn-5 |
|
Yttrium |
Fluoride |
40 s |
Y-5,6 |
|
Zirconium |
Iodate |
Few min |
Zr-5 |
|
a Separation time is for the fastest procedure reported. |
|||
Another troublesome aspect of precipitation, contamination due to adsorption of ions, has been used advantageously either to remove unwanted radioactive ions from a solution or to selectively remove the element of interest. Details are dealt with in Sec. 2.2.
2.2 Adsorption and Isotopic Exchange
The adsorption of species from solution onto a solid surface has been known for quite some time. As pointed out in Sec. 2.1, analytical chemists have been aware of the effects of adsorption on precipitates used for quantitative measurement. The adsorbate (the species adsorbed) can be a simple cation or anion, a complex ion, or a polar or nonpolar molecule. The adsorption may take place at a liquid-solid interface or at a gas-solid interface. The typical adsorbents used in radiochemical procedures are solids such as silver halides, manganese dioxide, tin dioxide, hydrated antimony pentoxide, alumina, and activated charcoal. Silver halides and hydrated antimony pentoxide adsorb specific ions, whereas adsorbents like alumina and charcoal are nonspecific.
Rapid exchange also takes place between ions on the surface of a precipitate and the ions in the solution. This property has frequently been used in fast radiochemical separations to remove the activity of interest or contaminants from a solution. In the following subsections, we briefly discuss the applications of adsorption and exchange in fast radiochemical separation.
2.2.1 Adsorption
Weak intermolecular forces such as dispersion (London) forces, dipole forces, hydrogen bonds, and weak covalent bonding are responsible for adsorption [Sny75]. A variety of materials have been used as adsorbents. The main requirement that determines whether a solid can act as adsorbent is its surface area. Silica gel used for the separation of aromatic hydrocarbons from petroleum fractions has a surface area of about 80 acres/lb (714 m2/g). Coconut-shell carbon has a surface area of nearly 1700 m2/g.
Other important physical characteristics are pore diameter and particle size. Mair [Mai61] has given a table listing the physical properties of some common adsorbents. The adsorbents can be classified as polar and nonpolar adsorbents. Polar adsorbents, such as inorganic oxides, tend to retain polar compounds, whereas nonpolar adsorbents such as carbon retain polarizable compounds. Carbon can also be prepared in polar forms [Sny75]. For information on adsorbents, the articles by Motl and Novotny [Mot79], Snyder [Sny75], and Mair [Mai61] can be consulted. The physical chemistry of adsorption is discussed by Oscik [Osc82] in his monograph.
Gas- and liquid-chromatographic separations based on adsorption have improved significantly in the last quarter of a century. Even closely related, structurally similar compounds can be separated from each other based on the slight differences in their adsorption on a particular adsorbent. However, in fast radiochemical separations, such finer aspects of adsorption chromatography are seldom used; because of the time constrictions, separations are performed under nonequilibrium conditions. The gross difference in adsorption is used for certain separation.
Table 2 lists elements for which fast separation procedures are available where adsorption was used either to selectively retain the elements or to selectively remove the contaminant from the product mixture. Activated-carbon, quartz-wool, and fiberglass filters have been used as adsorbents. Dropesky and Schardt [Dro56] used an activated-carbon trap cooled in dry ice to remove impurities from irradiated neon in their study of 24Ne. Quartz wool was used to adsorb selenium [Zen80, Ren82a] and bromine [Ren82b], while carbon was used to adsorb tellurium [Zen78] from volatile products formed by interaction of ethylene with fission fragments in a gas-jet system. In the selenium procedure [Ren82a] for example, the main separation from fission-products elements is achieved by the interaction with ethylene. The target of uranium was
covered by aluminum foil so that only the lighter fission fragments passed through to interact with ethylene. Selenium and bromine are the high-yield elements that interacted with ethylene to form volatile compounds. Selenium was selectively retained by quartz wool. Weis and Denschlag [Wei81] used a fiberglass filter to selectively retain niobium in their fission-yield study.
Table 2. Fast chemical separation procedures based on adsorption.
|
Element |
Adsorption medium |
Retention/decontamination |
Time |
Procedure # |
|
Arsenic |
Charcoal |
Decontamination from Ge |
Few s |
As-4 |
|
Bromine |
Charcoal |
Retention of Br |
~1 s |
Br-4 |
|
Germanium |
Charcoal |
Retention of Ge |
Few s |
Ge-3 |
|
Neon |
Charcoal |
Decontamination |
Few s |
Ne-1 |
|
Niobium |
Fiberglass filters |
Retention of Nb |
2.2 s |
Nb-1,3,8 |
|
Phosphorus |
Hydrous Zr oxide |
Retention of P |
2 min |
P-1 |
|
Selenium |
Quartz wool |
Retention of Se |
~1 s |
Se-5 |
2.2.2 Adsorption of Ions and Exchanges
Ions in solution may be adsorbed on the surface of a precipitate. Ions in solution may also exchange with the ions on the surface of the precipitate. When a solution is left in contact with a freshly formed precipitate, two processes occur. There is a rapid exchange or adsorption of ions onto the surface of the precipitate. As the precipitate ages, the surface area decreases due to recrystallizations, and the adsorbed or exchanged ions may be incorporated in the crystal lattice (if compatible); also, entrapment of solution may occur. Radioactive tracer studies showed that 95% of 212Pb ions is taken up by PbSO4 and PbCrO4 within a minute [Wal67c]. The effect of aging of precipitates was studied by Kolthoff and O'Brien [Kol39a, Kol39b]. They have shown that the rate of aging depends on the medium with which the precipitate had been in contact during aging. The rate of aging decreased with decreasing solubility of the precipitate; however, there is a certain minimum solubility value beyond which the solubility effect is not present [Kol39a, Kol39b]. Aging also occurs when a precipitate such as silver bromide was air-dried at 25°C. This type of thermal aging was more pronounced at higher temperature [Kol39c].
Langer [Lan42] showed that silver ions in solution exchanged constantly with silver chloride, and that both silver and bromide ions exchanged with silver bromide [Lan43]. Langer later applied this isotopic exchange for the determination of silver [Lan50]. Meinke and Sunderman [Mei55] used this technique for the preparation of beta-ray sources. A nonisotopic technique was used by Arnott and Wells-Cole [Arn53] for the determination of radioiodine in urine samples. They passed the urine through a silver chloride column. The amount of radioactive iodine retained varied with certain conditions, and was in the range of 75 to 98%. Sunderman and Meinke [Sun57] evaluated the application of isotopic exchange for radiochemical separations. Their evaluation showed that radioactive silver can be determined using exchange with silver chloride deposited on a platinum gauze. In less than 15 min, quantitative separation of radioactive silver was achieved. They have determined decontamination factors for a large number of ions. Earlier [Sun55], they showed that 50% of the silver activity present in 4-year-old fission-product solution can be exchanged with the silver chloride on a platinum gauze in about 20 s at 95°C.
The information obtained from the studies referred to in the last few paragraphs was used in the incorporation of exchange techniques in fast radiochemical separations. Most of these procedures involve exchange of silver, bromide, or iodine ions with a freshly formed silver halide precipitate. In an automated procedure, Brüchle and Herrmann [Brü82] used isotopic exchange
with freshly precipitated silver chloride to achieve separation of silver from fission-product solution. The silver chloride was washed, dissolved in ammonium hydroxide, and reprecipitated. The entire procedure was completed in about 4 s. Vine and Wahl [Vin81] separated technetium from molybdenum by exchange/adsorption of TcO4− ions on preformed [(C6H5)4As]ClO4 precipitate.
Exchange between a gas and a solution was used by Tomlinson and coworkers to separate selenium and tellurium from fission products [Tom71, Tom73]. Uranyl chloride solution containing selenium, bromine, tellurium, and iodine carrier was irradiated; by bubbling H2Se or H2Te gas through the solution, they were able to carry the selenium or tellurium activity with the gas.
Adsorption has been used for the separation of niobium from fission products. Ahrens and coworkers [Ahr76] passed a nitric acid solution containing fission products and holdback carriers through silver chloride to remove bromine, iodine, and silver. The nitric acid concentration of the solution was increased and then passed through fiberglass filters. The filters retained niobium. Weis and coworkers [Wei87] investigated the adsorption of niobium on fiberglass filters as a function of sorption medium. Nitric acid was found to be the best medium. The solution, 10M in HNO3 containing zirconium and niobium carriers, was passed through five consecutive filters. The filters were washed with HNO3. The retention of niobium was found to be 94%; 2% of zirconium was present as a contaminant.
Table 3 lists the elements for which an exchange reaction was used as a major step in separation from other associated activities. The exchange medium and the separation time are also included.
Table 3. Use of exchange in fast chemical procedures.
|
Element |
Exchange medium |
Timea |
Procedure # |
|
Iodine |
Pre-formed AgI |
52 s |
I-6 |
|
Iodine |
Pre-formed AgCl |
2 s |
I-7,9 |
|
Selenium |
Hydrogen selenide |
1.0 – 2.3 s |
Se-2 |
|
Silver |
Pre-formed AgCl |
4.1 s |
Ag-1,2 |
|
Silver |
AgCl coated on Pt |
~20 s |
Ag-4,5 |
|
Strontium |
Fine powder of SrSO4 |
Few min |
Sr-3 |
|
Technetium |
[(C6H5)4As]ClO4 |
5 s |
Tc-1 |
|
Tellurium |
Hydrogen telluride |
<1 min |
Te-2 |
|
Tin |
CdS/cellulose |
2 min |
Sn-9 |
|
a Separation time is for the fastest procedure reported. |
|||
In many of the procedures, the exchange reaction has been used advantageously to remove contaminants. In separation from fission products, filtration through freshly precipitated silver chloride has been used to remove bromine, iodine, and silver activities produced in fission. The procedure used for zirconium by Trautmann and coworkers [Tra72] and by Weis and Denschlag [Wei81] used such a step. Similar exchange with silver chloride was used in technetium procedures used by Trautmann and coworkers [Tra72, Tra76b]. Franz and Herrmann [Fra78] utilized isotopic exchange with freshly precipitated barium sulfate to remove barium activity in their procedure for the separation of ruthenium from fission products.
2.3 Ion-Exchange Techniques
According to Rieman and Walton [Rie70a], the first recorded ion-exchange separation may have been that performed by Moses around 3000 BC, when he made the saline water from the spring of Marah drinkable [Bib-1870]. The “bitter” Epsom salt was probably removed by an ion-exchange reaction with the carboxyl groups of the oxidized cellulose (tree), and the sulfuric acid produced reacted with limestone deposits. The first research observations on the ion-exchange properties of soil were made by two British agricultural chemists, Thompson and Way, in 1850. They observed that soil can remove potassium or ammonium salts from aqueous solution and release calcium salts [Tho50, Way50]. The synthesis of organic ion-exchange resin by Adams and Holmes in 1935 [Ada35] rekindled interest in ion-exchange processes. In the last half-century, a variety of organic and inorganic ion exchangers have been produced; their applications for innumerable types of separations, both in laboratory use and on an industrial scale, have been increasing steadily with time. It was the development of ion-exchange technology that made possible separation of pure, individual lanthanides [Ket47].
Ion-exchange techniques have been used extensively in radiochemical separations. Separations of a number of fission products, in both trace amounts and macro amounts, were reported by a number of radiochemists in the 1940s [Tom47, Spe47a, Mar47, Spe47b, Har47, Ket47]. Massart [Mas71] has compiled the radiochemical procedures which use cation-exchange techniques. Mitchell [Mit85] has discussed the use of ion exchange in radiochemistry. Inorganic exchangers have also found a variety of applications. Shukla and coworkers [ShuT1, Shu72] have used manganese dioxide to achieve separation of zirconium from niobium and promethium from Ce(IV). Girardi and coworkers [Gir70] have established the use of hydrated antimony pentoxide for the efficient removal of alkali metal activity from neutron-irradiated samples.
In rapid radiochemical procedures, mostly organic ion exchangers have been utilized. Hence, in the following subsections, use of the organic ion exchangers in radiochemical separations is discussed. The term “ion exchanger” is used specifically to refer to organic ion exchangers. The books by Amphlett [Amp64] and Paterson [Pat70a] and the article by Clearfield, Nancollas, and Blessing [Cle74] provide general information on inorganic ion exchangers.
2.3.1 Principle
Ion-exchange resins can be broadly categorized into two groups: cation exchangers and anion exchangers. The cation exchangers are further classified into strong-, moderate-, and weak-acid types. The anion exchangers are typically classified into weak- and stronger-base types. Apart from cation and anion exchangers, there are other special types of ion-exchange resins, such as the chelating resin and bifunctional resin. All ion exchangers have a functional group attached to a polymer matrix. For example, the strong-acid-type, cation-exchange resin has a sulfonic acid group attached to a cross-linked polystyrene matrix. The exchange reaction involves an ionic equilibrium which can be written as follows:
R–SO3− H+ + M+X− ⇄ R–SO3− M+ + H+X−.
(2)
In Table 4, we list functional groups of common types of ion exchangers. A table of the types of ion exchangers, the structure of the polymer matrix, the functional groups along with their exchange capacity, and trade names is given by Rieman and Walter [Rie70b]. Detailed tables containing the international trade names of ion exchangers can be found in the appendices of the book on ion exchangers by Dorfner [Dor72] and in Wilson and Wilson's comprehensive analytical chemistry volume on ion exchangers by Marhol [Mar82a].
Table 4. Functional groups of common types of ion exchangers.
|
Ion exchanger |
||
|
Category |
Type |
Functional group |
|
Cation |
Strong acid Moderately strong acid Weak acid |
–SO3−H+ –PO(OH)2 –COO−H+ |
|
Anion |
Strong base Weak base |
–(CH2NR3)+Cl− –(CH2NH2R)+Cl− –(CH2NHR2)+Cl− |
For the exchange equation given above, the selectivity coefficient can be written as
(3)
The selectivity coefficient depends on the specific ion exchanger, the number of charges on the ion exchanged, and the conditions of the solution. For strongly acidic cation exchangers, the selectivity for the alkali metal ions is as follows:
Li+ < Na+ < NH4+ < K+ < Rb+ < Cs+.
(4)
For the trivalent lanthanides, it is
Lu3+ < Yb3+ < . . . < Ce3+ < La3+
(5)
For strongly basic anion exchangers, the selectivity for halide ions is as follows:
F− < Cl− < Br− < I−.
(6)
Marhol [Mar82b] has given selectivity lists for a number of ion exchangers, and ion-exchange selectivity has been discussed by Reichenberg [Rei66]. In typical radiochemical applications, as in many analytical applications, it is convenient to use the distribution coefficient rather than the selectivity coefficient. The distribution coefficient D is given by the equation
(7)
Use of radioactive tracers is a convenient method for determining D. The batch and column methods for determining D are discussed by Marhol [Mar82c]. Comparison of the values of D for different elements for a given resin under a given set of conditions will help determine whether the element of interest can be separated from other elements in the mixture. Values of distribution coefficients for different systems are tabulated in several monographs [Mar82d, Str73, Mas71].
The exchange process can be thought of as happening in several stages. The ion in the solution must diffuse through the film adhering to the surface of the resin; the ion then must diffuse to the site of the exchange, which may or may not be on the surface of the resin bead. Once the exchange has taken place, the exchanged ion must diffuse from the exchange site into the solution. The overall rate of the process is controlled by the kinetics of all these steps. For dilute
solutions, often the slowest step is the diffusion of the ion through the film. Ion-exchange kinetics are discussed in detail by Helfferich [Hel66] and by Rieman and Walton [Rie70c]. A number of other monographs and books provide useful information on ion exchangers, separation processes, and other related topics [Nac56, Sam63, Pat70b, Rot73].
2.3.2 Application to Fast Radiochemical Separations
Ion exchange is one of the most frequently used techniques, next to solvent extraction, in fast radiochemical procedures. The time constraint necessitates the use of flow rates and other conditions that are far from optimum. In spite of these facts, ion exchangers have been successfully used to achieve separation of parent and daughter, or the separation of a specific element along with other elements of low yields that will not interfere in terms of their radioactivity. Separation of individual actinides has been accomplished using minicolumns.
The separation of a daughter growing in from a parent activity adsorbed on an ion-exchange column is easy to achieve if the adsorption characteristics of the parent and daughter are very different. Campbell and Nelson used a Dowex-1 anion exchanger to retain parent bismuth activities from O.5M HCl solutions while allowing the daughter lead activities to flow through; they used the resin in the form of a column [Cam53] and also as layers on a sintered glass fiber. Similarly, they were able to separate short-lived iridium activities from parent osmium using the strong sorption of OsCl62− on Dowex-1 [Cam56]. These separations were achieved in a few seconds to allow study of 0.8-s 207Pbm and 49-s 191Irm. Another such example is the separation of short-lived thallium activity from lead reported by Andersson and coworkers [And57] using a Dowex-2 column.
The separation of individual lanthanides and actinides has been a challenge to analytical chemists ever since the discovery of lanthanide elements. As pointed out in Sec. 2.3, it was the development of ion-exchange technology that led to the separation of pure, individual rare earths. Identification of a number of complexing agents led to improved, faster separation of these elements using cation-exchange resins. Citric acid, lactic acid, glycolic acid, and α-hydroxyisobutyric acid have all been used as complexing agents to achieve separation and elution of lanthanides from cation-exchange resins. The separation factors between adjacent lanthanides and some actinides for lactic acid, glycolic acid, and α-hydroxyisobutyric acid at 20°C and 87°C (90°C for lactic acid) are listed by Stevenson and Nervik [Ste61b]. They point out that, based on a consensus of articles in the literature, α-hydroxyisobutyric acid is the best for small-scale, cation-exchange separation of lanthanides followed by lactic acid, glycolic acid, and citric acid [Ste61c].
Ghiorso and coworkers [Ghi55] used α-hydroxyisobutyric acid in their separation and identification of mendelevium. The applicability of this complexing agent for the separation of actinides [Cho56a] and lanthanides [Cho56b] on a tracer scale was shown by Choppin and coworkers. They used a 5-cm by 0.2-cm Dowex-50 X-12 column operated at 87°C. Figure 2 shows the elution curve obtained by them for a mixture containing several lanthanides. This is probably the first separation of lanthanides (with good separation of adjacent elements) within 2 hours! They also indicated that the separation factors do not decrease by more than 10% for elution at 25°C. Smith and Hoffman [Smi56] showed that effective separation can be performed at 25°C, and the usefulness of α-hydroxyisobutyric acid to achieve fast separation of any specific lanthanide element has been shown by Rengan and Meinke [Ren64]. The latter used a small column of Dowex-50 X-12 (0.2-cm diameter by 2.8-cm height, -400 mesh); at room temperature, they were able to separate any specific rare earth from its neighbor in about 16 min. The combination of α-hydroxyisobutyric acid and Dowex-50 has been frequently used for the separation, identification, and study of chemical properties of many of the man-made elements. Silva and coworkers [Sil70b] used such a procedure to separate element 104 within 1 min after
irradiation. This procedure used the fact that the resin will not hold element 104 as strongly as the actinides. Similarly, short-lived mendelevium [Ghi55] has also been separated in a few minutes after irradiation from the recoil products collected from the target.
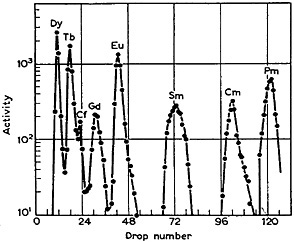
Figure 2. Elution of lanthanides from a Dowex-50 X-12 column (5 cm × 0.2 cm) with α-hydroxyisobutyric acid at 87°C. [Cho56b; reprinted with permission from J. Inorg. Nucl. Chem.]
The separation of individual lanthanides formed in fission is more complicated because of the large number of lanthanide nuclides formed and the variations in fission yields. A fast, autobatch procedure was used by Klein and coworkers in their study of 9.6-s 96Y [Kle75]. The procedure does not achieve separation of yttrium from lanthanides in the dysprosium-terbium region [Her82], but the fact that the low-energy fission yields of those nuclides are small compared to those of yttrium allowed a reasonably pure yttrium sample to be obtained 10 s after the end of irradiation. Table 5 gives a list of procedures that use ion-exchange techniques.
Recent developments in high-pressure liquid chromatography (HPLC) have an impact on fast radiochemical separation of lanthanides and actinides. The application of HPLC for the ion-exchange separation of fission-product lanthanides and actinides is discussed in Sec. 2.5.
2.4 Liquid-Liquid Extraction
Liquid-liquid extraction is one of the most common techniques used in all branches of chemistry. A few examples are its use
-
In analytical chemistry for separation and analysis of mixtures.
-
In organic chemistry for purification of reaction products.
-
For the separation of aromatics from aliphatics in the petroleum industry.
-
In the nuclear industry for the separation of uranium and for separation of plutonium from most of the fission products.
The technique has been frequently used in radiochemical separation procedures.
The first application of liquid-liquid extraction is credited to Peligot, who observed that uranyl nitrate can be recrystallized from diethyl ether [Pel42]. This extraction technique has been used from the time of the Manhattan Project to achieve rapid radiochemical separation. Levinger and coworkers [Lev51] separated iodine within 28 s after irradiation by extraction of molecular I2 with CCl4. A number of ultrafast procedures based on liquid-liquid extraction exist. The following subsections give information on the applications of this technique to ultrafast radiochemical procedures.
Table 5. Fast chemical procedures utilizing ion exchangers.
|
Element |
Ion exchanger used |
Eluting agent |
Timea |
Procedure # |
|
Barium (from Cs) |
Thallium phosphotungstate |
HCl (0.5M) |
Few s |
Ba-5 |
|
Californium |
Dowex-50 X-12 (cation) |
α-Hydroxyisobutyrate |
Few min |
Cf-1 |
|
Hafnium |
Wolfatit KPS (cation) |
HF (0.5M) |
10 – 20 s |
Hf-3 |
|
Iridium |
Dowex-1 (anion) |
HCl (6M) |
Few s |
Ir-3 |
|
Iron |
Dowex-1 (anion) |
HCl (8M) |
150 s |
Fe-3 |
|
Lawrencium |
Dionex (cation) |
α-Hydroxyisobutyrate |
6 min |
Lr-3 |
|
Lead |
Dowex-1 (anion) |
HCl (0.3/0.5M) |
Few s |
Pb-1,2 |
|
Mendelevium |
Dowex-50 (cation) |
α-Hydroxyisobutyrate |
~5 min |
Md-1 |
|
Nobelium |
Dowex-50 (cation) |
α-Hydroxyisobutyrate |
Few min |
No-1 |
|
Silver |
Dowex-1 (anion) |
HCl (10M) |
Few min |
Ag-8 |
|
Thallium |
Dowex-2 (anion) |
Chloride/H2O |
~2 s |
Tl-2 |
|
Tungsten |
Wolfatit KPS (cation) |
(–) complexes in NH3 |
10 – 20 s |
W-2 |
|
Yttrium |
Dowex-50 X-12 (cation) |
α-Hydroxyisobutyrate |
10 s |
Y-2 |
|
Yttrium |
Dowex-1 (anion) |
Concentrated HCl |
Few min |
Y-7 |
|
Z = 104 |
Dowex-50 X-12 (cation) |
α-Hydroxyisobutyrate |
1 min |
104-4 |
|
Zinc |
Dowex-1 (anion) |
HCl (1M) |
~1 min |
Zn-2,4 |
|
a Separation time is for the fastest procedure reported. |
||||
2.4.1 Systems in Use
The simple system involves partitioning of molecular species between an aqueous phase and an organic solvent immiscible with the aqueous phase. Molecules such as Br2, I2, and halides of germanium, arsenic, and antimony can be extracted by a number of solvents. An uncharged chelate complex can be prepared and extracted. Cupferron, dithizone, α-benzoinoxime, and 2-thenoyltrifluoroacetone (TTA), structural formulae of which are shown in Fig. 3, are some examples of reagents that can react with a number of metal ions to form uncharged chelate complexes. TTA, production of which was first reported by Reid and Calvin [Rei50], has found a large number of radiochemical applications because of its ability to form complexes even from fairly acidic solutions.
Poskanzer and Foreman [Pos61] have summarized the TTA extraction coefficients available in the literature. Freiser and Morrison [Fre59] have listed the optimum pH for the complete extraction of a number of elements, while De, Khopkar, and Chalmers [De70a] and Minczewski and coauthors [Min82a] have given tables summarizing the data available in the literature for the optimum extraction conditions for various elements. Available literature information on the chelates of cupferron, dithizone, and α-benzoinoxime as well as other, similar uncharged chelate systems have been summarized by Freiser and Morrison [Fre59], by De, Khopkar, and Chalmers [De70b], and by Minczewski and coworkers [Min82b]. The two systems referred to above (simple partitioning of molecules and extraction of uncharged chelate complexes) are grouped together as extraction of uncharged covalent species.
A broad second group of extraction systems involves ion-association complexes. A simple system of this type involves extraction of halometallic acids such as HAuCl4 and HFeCl4. Similar extractions of thiocyanates and nitrates are also possible. The extractions are typically carried out using diethyl ether or methyl isobutyl ketone as solvent. Freiser and Morrison [Fre59], De et al. [De70c], and Minczewski et al. [Min82b] have provided summaries of such systems.
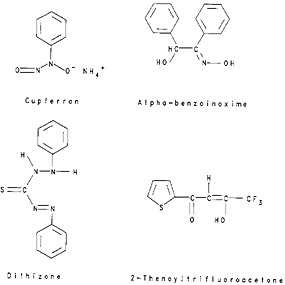
Figure 3. Structural formulae of some complexing agents.
A class of compounds known as liquid-ion exchangers fall under this second group. High-molecular-weight amines dissolved in an organic solvent can extract acid from an aqueous solution to form an amine salt:
(8)
The anion A− can be exchanged for another anion or for an anionic metal complex:
(R3NH+A−)org + MXn− → (R3NH+MXn−)org + A−aq ,
(9)
where MXn− is the anion metal complex. These types of amines are referred to as liquid-anion exchangers. The most common amines used are tri-n-octylamine (TOA) and triisooctylamine (TIOA). General reviews of the uses of liquid-anion exchangers in inorganic analysis are provided by Green [Gre73] and by Hogfeldt [Hog66]. Extraction conditions for various elements are summarized in a table by De, Khopkar, and Chalmers [De70d] and by Minczewski et al. [Min82c]. Moore [Moo60] has discussed the liquid-liquid extraction with high-molecular-weight amines and has provided a collection of selected procedures.
Several organophosphorus compounds are used as extracting agents. Figure 4 shows the structural formulae of some of these compounds. The organophosphoric acids with replaceable hydrogen (e.g., H2MEHP, HDEHP) exchange hydrogen with metal ions and are known as liquid-cation exchangers. The extractability increases with increasing charge of the ion. Peppard and coworkers [Pep57a] showed the feasibility of using HDEHP for the fractionation of lanthanides. They also showed that berkelium can be separated and purified from lanthanides and lower actinides by using the extraction of Bk(IV) by HDEHP [Pep57b].
The most common extractant of this group used in rapid radiochemical separations is HDEHP. The extraction characteristics of HDEHP have been studied extensively, and a large volume of data is available in the literature. Green [Gre73] has reviewed the use of HDEHP in inorganic analysis. Minczewski and coauthors [Min82d], as well as De and coauthors [De70e], have provided a summary table giving extraction data for different metals. The available extraction data are given in the form of a periodic table by Qureshi and coworkers [Qur69]. The
organophosphorus compounds with no exchangeable hydrogen (e.g., TBP, TOPO) form addition compounds such as La(NO3)3·3TBP, UO2(NO3)2·2TOPO, and ZrCl4·2TOPO. Among this group of extractants, TBP and TOPO are the two used most frequently in radiochemical separations. TBP is used in the Purex process for the extraction and separation of uranium and plutonium from irradiated uranium and in the Thorex process for the extraction and separation of thorium and uranium from irradiated thorium [Ben81]. Extraction data summaries are available in the form of tables for TBP, TOPO, and a number of other organophosphoric compounds [De70f, Min82b]. The radiochemistry monograph by White and Ross [Whi61] reviews the extraction characteristics of TOPO.
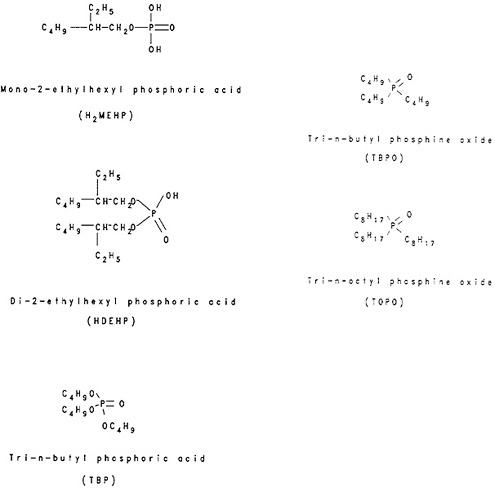
Figure 4. Structural formulae of organophosphorus extractants.
A number of books, monographs, and review series dealing with solvent extraction are available. The book by De, Khopkar, and Chalmers [De70b] reviews solvent extraction of metals. The book discusses each type of solvent-extraction system and gives a summary table of extraction data for each extraction discussed; it also gives selected extraction procedures for metals. The earlier book by Morrison and Freiser [Mor57] also gives a brief procedure for most elements. The book by Stary [Sta64] deals with solvent extraction of metal chelates. Marcus and Kertes discuss solvent extraction and ion-exchange systems in their book [Mar69]. The book by Zolotov [Zol70] discusses various aspects of extraction of chelate compounds. The review series entitled Ion Exchange and Solvent Extraction has several articles related to solvent extraction. Interactions of organophosphorus extractants [Kol71] and the kinetics of metal extraction by organophosphorus extractants [Col71] are discussed in two articles. Separation of lanthanides and actinides by solvent extraction has been reviewed by Weaver [Wea74]. Irving's article [Irv82] provides a general review of liquid-liquid extraction, while Peppard has specifically reviewed the extraction of actinides [Pep71].
2.4.2 Application to Ultrafast Separations
The liquid-liquid extraction technique has been applied in a wide variety of ways for a number of elements to achieve ultrafast separation. Figure 5 shows the elements for which one or more ultrafast separation procedures based on extraction are available in the literature. Many of the procedures were developed for batch-mode operations; some have been adopted for autobatch operations, while a number of continuous-mode procedures exist. Table 6 shows the wide variety of elements for which liquid-liquid extraction procedures are available. Separation times vary from 2 s to several minutes, depending on the complexity of the separation. As can be expected, the slowest step in this technique is the separation of the two phases. Judicious selection of diluents for the extractants aids in the faster separation of phases and reduces the overall time required for the separation. In the remainder of this section, we present some examples of procedures using this technique.
Liquid-liquid extraction procedures were applied even in the early period of fission studies for the investigation of short-lived bromine and iodine isotopes. Sugarman [Sug47] extracted bromine within 30 s after irradiation. Irradiated uranyl nitrate solution containing bromide and iodide carriers was mixed with Na2CO3 solution containing Br− and BrO3− and treated with NaOCl to oxidize I− to IO3− under basic conditions. The solution was then acidified with HNO 3, and the liberated Br2 was extracted with CCl4. The procedure was used to identify the 4.5-s delayed neutron emitter of bromine.
Hulet and coworkers [Hul80] used solvent extraction with a high-molecular-weight quaternary amine to study the chloride complexation of element 104. The element produced by bombardment of 248Cm with 18O was transported by NaCl aerosol and deposited on the surface of a polypropylene rabbit. The deposit was dissolved in 12M HCl and passed through an extraction column containing 0.25M trioctylmethylammonium chloride (Aliquat-336) diluted with o-xylene and supported on fluorocarbon powder. The column was first eluted with five column volumes (150 µL) of 12M HCl, and then the eluent was changed to 6M HCl. Since element 104 is a homolog of zirconium and hafnium, it should form strong chloride complexes and should be retained by the extractant. The actinides can be eluted by 12M HCl. Tracer experiments with hafnium indicated that element 104 should be eluted by 6M HCl. The experiments showed that the actinides were present in the 12M HCl eluent; the 6M HCl eluent showed evidence for the presence of 65-s 261104. The separation of element 104 from actinides was completed in less than 3 min. The entire operation was automated so that the experiment could be repeated every 3 min.
A total of 44 experiments were performed, and six alpha-decays corresponding to 261104 were observed.
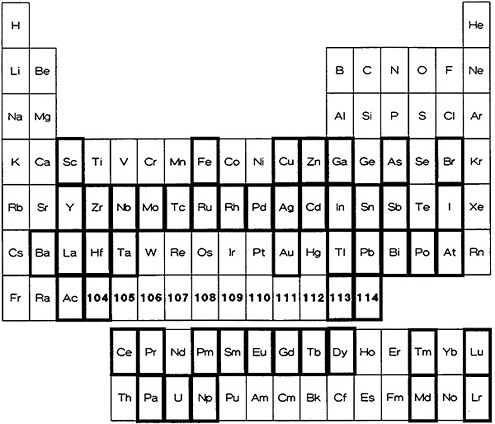
Figure 5. Elements for which a fast procedure based on solvent extraction is available (shown with bold rectangles).
Ultrafast separation procedures have been developed for some lanthanides using oxidation states other than +3 for the specific element or its parent. Hoffman and coworkers [Hof66] used an HDEHP extraction of Ce(IV) to separate cerium from other lanthanides, and the difference in the extractabilities of Ce(IV) and Pr(III) was used to obtain a sample of 2.1-min 148Pr [Ren77]. Figure 6 shows the procedure they used to separate praseodymium from fission products, in the form of a flow chart. The praseodymium chemistry was completed within 5 min after the milking of praseodymium from cerium.
Manual extractions are slow. Denig and coworkers have used solvent-impregnated material to achieve extraction of many fission-product activities [Den66]. The various steps involved in the procedure were automated so that the entire procedure can be carried out in several seconds. Examples of this type of procedure are described in Sec. 4.
Individual separation of lanthanides and actinides can be performed within 20 min using high-pressure liquid-chromatographic techniques. In this case, the extractant HDEHP, which was supported on inert materials, was used as a chromatographic column. Details of this technique are discussed in Sec. 2.5.
Table 6. Fast chemistry procedures based on solvent extraction.
|
Element |
Mode |
Reagenta |
Timeb |
Procedure # |
|
Actinium |
Batch |
HDEHP and TTA |
3 min |
Ac-1 |
|
Actinium |
Batch |
HDEHP,ion exchange |
100 s |
Ac-2 |
|
Antimony |
Batch |
Diisopropyl ether |
~1 min |
Sb-6 |
|
Arsenic |
Continuous |
CHCl3 |
3 or 4 s |
As-6 |
|
Arsenic |
Continuous |
HDEHP |
1.0 s |
As-11 |
|
Astatine |
Batch |
Diisopropyl ether |
90 s |
At-2,4 |
|
Barium |
Continuous/batch |
Crown ethers |
<1 min |
Ba-2 |
|
Bismuth |
Batch |
TBP – dibutyl ether |
5 min |
Bi-1 |
|
Bromine |
Batch |
CCl4 |
30 s |
Br-12,14 |
|
Bromine |
Continuous |
CHCl3 |
~5 s |
Br-9 |
|
Cadmium |
Batch |
MIBK – CHO |
5 – 10 s |
Cd-2 |
|
Cadmium |
Continuous |
MIBK – CHO |
~2 s |
Cd-3 |
|
Cadmium |
Batch |
Dithizone |
2 min |
Cd-5 |
|
Cerium |
Batch |
HDEHP |
~5 min |
Ce-1 |
|
Cerium |
Continuous |
HDEHP |
<5 s |
Ce-3,4,5 |
|
Cerium |
Batch |
Hexone (MIBK) |
2 min |
Ce-7 |
|
Copper |
Continuous |
Oxime |
20 s |
Cu-1 |
|
Dysprosium |
Autobatch |
DHDECMP |
<9 min |
Dy-1 |
|
Europium |
Batch |
Na amalgam |
1 min |
Eu-1 |
|
Gadolinium |
Autobatch |
DHDECMP |
3.2 min |
Gd-1 |
|
Gallium |
Batch |
Ethyl ether |
~1 min |
Ga-1,2 |
|
Gold |
Batch |
Ethyl acetate |
~4 min |
Au- 1,2 |
|
Hafnium |
Batch |
TBP – HDEHP5 |
5 min |
Hf-2 |
|
Indium |
Batch |
MIBK – CHO5 |
5 – 10 s |
In-1 |
|
Indium |
Continuous |
MIBK – CHO |
~2 s |
In-2 |
|
Indium |
Batch |
Chloroethyl ether |
Few s |
In-3 |
|
Iodine |
Batch |
CCl4 |
28 s |
I-9,10 |
|
Iodine |
Continuous |
CCl4 |
~5 s |
I-4 |
|
Iron |
Batch |
Ethyl ether |
Few min |
Fe-2 |
|
Lanthanides |
Autobatch |
Cation exchange |
<3 min |
Ln-1 |
|
Lanthanides |
Batch |
HDEHP |
15 – 20 s |
Ln-4 |
|
Lanthanum |
Continuous |
HDEHP |
<5 s |
La-2,3,5 |
|
Lawrencium |
Batch |
TTA – MIBK |
50 s |
Lr-1,2 |
|
Lawrencium (from Md) |
Autobatch |
HDEHP |
1 – 2 min |
Lr-5 |
|
Lead |
Continuous/batch |
Crown ethers |
<1 min |
Pb-3 |
|
Lead |
Batch |
TBP – dibutyl ether |
5 min |
Pb-4 |
|
Lutetium |
Batch |
TBP – HDEHP |
6 min |
Lu-1 |
|
Mendelevium (from Lr) |
Autobatch |
HDEHP |
1 – 2 min |
Md-3 |
|
Molybdenum |
Autobatch |
IAA – NBA |
5 s |
Mo-1 |
|
Neptunium |
Continuous |
HDEHP |
10 s |
Np-1 |
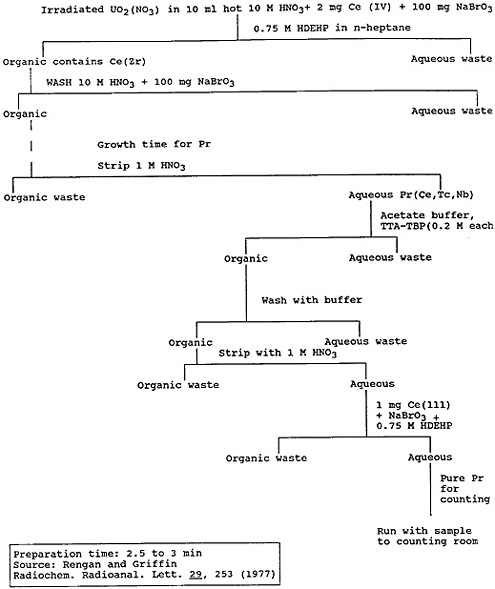
Figure 6. Flow diagram for the solvent-extraction procedure for praseodymium. [Ren77]
2.5 High-Performance Liquid Chromatography
In conventional column chromatography, the solutions flow by gravity, and a typical separation takes hours to days. The theoretical work of Giddings [Gid65] showed that liquid chromatography has several potential advantages over gas chromatography. However, he concluded that column efficiencies and separation time had to be improved. The separation time has been shortened significantly by the development of efficient adsorbent material for chromatographic columns, and by operation of the columns under high pressure. Early investigations into high-performance liquid chromatography were published by a number of researchers in the late 1960s [Hub67, Hor67, Kir69, Fel69, Hub69]. Nearly all the early
developments centered around the separation of organic compounds. Only in the last decade has this powerful technique been applied to inorganic separations.
2.5.1 Principle
High-pressure liquid chromatography (HPLC) equipment has changed rapidly during the last two decades. The availability of microprocessor technology has added new dimensions to HPLC instrument capability. A block diagram of the basic components is shown in Fig. 7. The column, usually made of stainless steel or tantalum, is 2 to 4 mm in diameter and 25 to 50 cm in length. The stationary phase is typically made of particles of 5 to 20 µm and provides high column efficiency. The mobile phase (solvent) is delivered by a high-pressure pump. Different types of pumps are used. In general, the pumps used have the ability to operate at 3000 to 6000 psi and are capable of providing a flow rate of 0 to 10 mL/min. In general, some type of pulse-damping arrangement is used. The sample is injected into the column in small volume (10 to 500 µL). The output of the column can either be fed to a detector location (trap, etc.), or different fractions can be collected using a fraction collector. Details of the various aspects of HPLC are discussed in several books [Raj75, Eng79, Pry79, Sny79, Run81, Ham82].
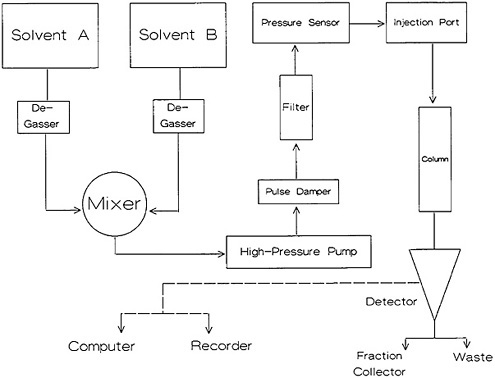
Figure 7. Block diagram of high-performance liquid chromatography equipment.
The radiochemical applications are based on using either ion exchangers or solvent adsorbed on inert material as the stationary phase. The former uses ion-exchange chromatographic separation, whereas the latter separation is based on extraction chromatography. Further details are given in the following subsections.
2.5.2 Separation of Lanthanides
HPLC can be expected to find a variety of applications in radiochemistry. Thompson and coworkers' [Tho54] use of a 2-mm-diameter, 5- to 6-cm-long resin column (200 to 400 mesh) in their separation of einsteinium and fermium from lower actinides can be thought of as the forerunner of HPLC in radiochemical separations. With a flow rate of one drop (35 µL) per 2 min, they were able to elute einsteinium and fermium in about 40 min. Several others (e.g., see [Ghi51] and [Ren64]) also used small ion-exchange columns to achieve fast separations. However, the application of HPLC techniques, as they are presently known, for fast radiochemical separation took place only in the late 1970s. Two types of ion-exchange materials are used: standard styrene-divinylbenzene resins, as small spherical beads (5 to 20 µm), and ion exchangers bonded to silica packings. Both types have been used for the fast separation of lanthanides and actinides. Earlier attempts at separation of lanthanides using HPLC have been reviewed by Cassidy [Cas81a]. The time required for separation ranged from 80 min to 4 h.
Schädel, Trautmann, and Herrmann [Sch77a] investigated three types of column material for the lanthanide separation:
-
Zipax SCX, “Pellicular” material consisting of 30-µm glass beads coated with a 3-µm-thick layer of fluoropolymer containing free sulfonic acid groups.
-
Aminex A5, styrene-divinylbenzene copolymer with sulfonic acid groups, 13-µm particle size.
-
Di(2-ethylhexyl)orthophosphoric acid adsorbed on hydrophobic porous silica-gel particles (HDEHP-SI60).
They used 3-mm i.d., 250-mm-long columns and a flow rate of about 2 mL/min. They compared the efficiency of the three column materials at 25°C and 65°C. They initially characterized the column performance by measuring height equivalent to a theoretical plate (HETP) and found that Aminex A5 and HDEHP-SI60 can be expected to provide better separation for lanthanides; in addition, their data showed that at 25°C Aminex A5 gave better separation of adjacent elements, whereas at 75°C HDEHP-SI60 performed better. Eluents used were α-hydroxyisobutyric acid (α-HIBA) for an Aminex A5 column and nitric acid for an HDEHP-SI60 column. With a gradient elution technique, they were able to obtain separation of seven lanthanide elements and yttrium within 20 min. Elchuk and Cassidy [Elc79] compared the bonded-phase, ion exchanger Nucleosil 105A with Aminex for the separation of lanthanides using α-HIBA as eluent. The best separation was obtained using a 20-cm by 4-mm column of 5-µm bonded-phase resin. Figure 8 shows the elution curve they obtained. The entire separation was completed in 17 min.
The nuclear chemistry group at the Idaho National Engineering Laboratory (U.S.A.) has coupled extraction chromatography and ion-exchange chromatography to separate and study short-lived lanthanide fission products produced in the spontaneous fission of 252Cf [Bak81, Bak82]. The entirety of the lanthanide fission products was separated in the cation exchangers column in less than 8 min, while any specific lanthanide could be separated in less than 3 min from the end of irradiation. Extraction chromatography was used to separate lanthanides as a group from the rest of the fission products. The individual lanthanides were separated using ion-exchange chromatography. Both separations used HPLC systems. The entire system was microprocessor-controlled. Details of the procedure are given in Sec. 4.5. This automated system has been successfully used by the Idaho group to study short-lived isotopes of promethium [Gre82], samarium [Bak80], gadolinium [Geh82a], terbium [Gre83], and dysprosium [Geh82b].
As mentioned earlier, Schädel and coworkers [Sch77a] found extraction chromatography with HDEHP supported on inert material as the stationary phase to be as effective as the cation exchanger. It should be pointed out that the order of elution is reversed from that of a cation exchanger; the elution order follows the atomic number. Horwitz and coworkers [Hor77] have investigated the applicability of extraction chromatography for the separation of lanthanides. They used porous silica microspheres containing 25 to 30 wt% of HDEHP in dodecane. The particles were 46 µm in diameter, the columns used had a 2- to 3-mm i.d., were 2 cm long, and operated at 50°C. They obtained good separation of adjacent lanthanides; four lanthanides were separated in about 30 min.
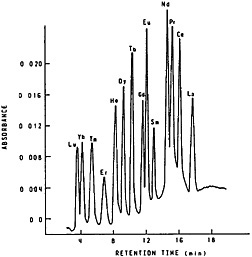
Figure 8. Separation of the lanthanides on 5-µm Nucleosil SCX. Experimental conditions: 10 cm × 4 mm column; 10 µL of a solution containing ~10 µg/mL of each lanthanide; linear program from 0.018 mol/L HIBA to 0.07 mol/L HIBA over a 20-min period at 0.8 mL/min and pH 4.6; detection at 600 nm after post-column reaction with Arsenazo I. [Elc79; reprinted with permission from Anal. Chem., American Chemical Society]
2.5.3 Separation of Other Elements
As pointed out in Sec. 2.5.2, the applications of HPLC for radiochemical separation of lanthanides have been investigated by several workers. Applications for the separation of actinides have been explored by Schädel and coworkers [Sch78] and Horwitz and coworkers [Hor77]. As can be expected, the results are similar to those described for lanthanides in Sec. 2.5.2.
The HPLC technique has not been extensively explored for inorganic ions; radiochemical applications are very few. Cassidy [Cas81b] has written an extensive review of the separation and determination of metal species by modern liquid chromatography. Other short reviews available are those by Schwedt [Sch79] and Fritz [Fri77]. In addition, Analytical Chemistry reviews [Wal80, Maj82, Maj84, Bar86, Bar88, Dor90, Syn90] provide some information on recent articles in this field.
2.6 Distillation and Volatilization
Distillation and volatilization have been used as a technique to isolate a number of elements. Typically, the separation is achieved in two ways:
-
By direct distillation of the volatile component from the sample.
-
By chemically reacting the components of the sample with a specific reagent to produce volatile compounds of element(s) of interest, which are removed.
There are very few elements that are volatile at moderately low temperatures (~500°C); these include elements that are gases at room temperature (noble gases, H2, N2, O2, F2, Cl2), bromine, iodine, astatine, cadmium, indium, mercury, and germanium. A number of elements form volatile chlorides, bromides, or oxides that can be distilled off. Amiel [Ami68] has discussed, with
examples, separation of elements by distillation. Volatilization of chlorides and bromides from HClO4, H3PO4-HClO4, and H2SO4 media were systematically investigated by Hoffman and Lundell [Hof39]. They found that chlorides and/or bromides of As(III), As(V), Cr(III), Ge, Hg, Os, Re, Ru, Sb(III), Sb(V), Sn(II), and Sn(IV) can be distilled off in good yield. They performed their tests using 25 to 100 mg of the elements. Wasowicz and Rutkowski [Was66] carried out distillation of trace amounts of a number of elements from concentrated HCl. Their trace-level data mostly confirmed the macro-level observations of Hoffman and Lundell. Minczewski, Chwastowska, and Dybczynski [Min82e] have reviewed separation by volatilization. They have presented, in tabular form, a list of major constituents separated by distillation along with separation conditions and literature references. Similarly, they have provided a summary table of trace impurities separated by distillation.
2.6.1 Applications
DeVoe [Dev62] has reviewed the application of the distillation technique to radiochemical separations. In addition to a general discussion of the technique, he has provided some detailed procedures and references to procedures in other radiochemistry monographs. Applications of volatilization techniques to rapid radiochemical separations have been reviewed in depth by Amiel [Ami68] and by Herrmann and Trautmann [Her82].
The elements for which there are ultrafast volatilization procedures are identified in Fig. 9. As can be seen, they involve elements that exist as a gas (nitrogen), have a high vapor pressure at room temperature (bromine, iodine), or can be distilled readily in the elemental form at moderate temperature (astatine, mercury), as well as those elements that readily form volatile hydrides (arsenic, selenium, tin, antimony, and tellurium); only ruthenium and osmium are distilled as their tetroxides. Table 7 lists the elements along with the chemical form of the volatilized species, the separation time, and the procedure reference.
The actual procedure used varies from a simple removal of the volatile species by a carrier gas to a series of reactions involving hydride generation and selective removal of contaminants. Dillard and coworkers [Dil51] determined the half-lives of short-lived krypton and xenon fission products by sweeping them in a stream of nitrogen. In their study of short-lived bromine isotopes, Nuh and coworkers [Nuh72] distilled off bromine from the fission product solution, which contained bromide and iodide carrier. They used the heat generated from the addition of concentrated sulfuric acid to this solution after a solution of sodium manganate had just been added. The bromine released was further purified by passing it through a series of traps to remove iodine and ruthenium.
The hydrides of arsenic, selenium, antimony, and tellurium have been produced using a variety of reactions. Greendale and Love [Gre63a] used zinc and H2SO4 to produce AsH3 and SbH3, which was further separated by selective decomposition. They separated tin from fission products using sodium borohydride to generate SnH 4 [Gre63b]. In their study of delayed neutron precursors of arsenic, antimony, and germanium, del Marmol and Néve de Mévergnies utilized selective thermal decomposition of the hydrides [Del67]. Herrmann and coworkers used the hydrides for studying selenium and tellurium nuclides. The volatilized hydrides were passed through sodium hydroxide solution which retained selenium and tellurium. Separation of selenium and tellurium was accomplished by oxidation and column extraction of tellurium [Fol69, Kra70]. Sodium borohydride has been used as the reducing agent to generate AsH3 and SbH3 [Mey80, Hen81]. An electrolytic technique was used by Tomlinson and Hurdus [Tom68] to achieve similar results.
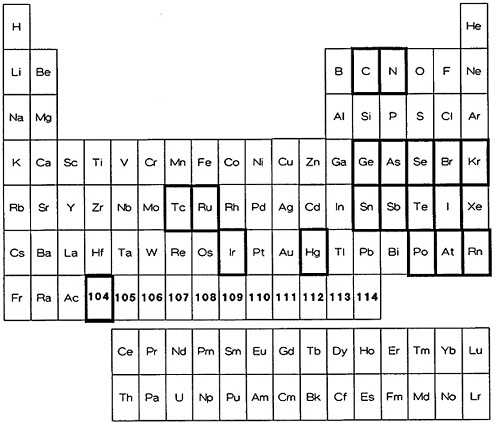
Figure 9. Elements for which a fast procedure based on volatilization is available (shown with bold rectangles).
Ruthenium and osmium can be distilled off as their tetroxide. The common oxidizing agents used to oxidize ruthenium to RuO4 are HClO4 [Ric71], sodium bismuthate [Nai84], and potassium permanganate [Ate55]. Naumann and Gerhart [Nau54] prepared iridium samples by volatilizing osmium from irradiated ammonium perosmate, and it was found that trace iridium accompanied RuO4 during distillation [Ren65]. Addition of milligram amounts of iridium reduced the iridium contamination but did not eliminate it; further steps were necessary to achieve the required decontamination. The behavior of elements in trace level may be different from macro-level behavior; it is essential to be aware of the likely differences between macro- and trace-level behavior.
Table 7. Fast volatilization procedures.
|
Element |
Species volatized |
Timea |
Procedure # |
|
Antimony |
Hydride |
2.7 s |
Sb-1 to 5,7,8,10,11,13 |
|
Arsenic |
Hydride |
1.9 s |
As-1 to 3,5,7,8,10 |
|
Astatine |
Oxide? |
<30 s |
At-1,3 |
|
Bromine |
Element |
~1 s |
Br-1,3,7,11,13 |
|
Carbon |
Oxide |
Few min |
C-3 |
|
Germanium |
Chloride |
~5 s |
Ge-1,2 |
|
Iodine |
Element |
Few s |
I- 1,8,11 |
|
Iridium |
Osmium tetroxide? |
Few s |
Ir-2 |
|
Krypton |
Element |
<1 min |
Kr-2 |
|
Mercury |
Element |
<1 min |
Hg-2,4 |
|
Nitrogen |
Element |
5 – 10 s |
N-1,3 |
|
Polonium |
Oxide? |
<30 s |
Po-4 |
|
Radon |
Element |
40 s |
Rn-1 |
|
Ruthenium |
Oxide |
1 s |
Ru-1,6 |
|
Selenium |
Hydride |
4.0 s |
Se-1,3,8,9 |
|
Technetium |
Chloride |
1 s |
Tc-2,4 |
|
Tellurium |
Hydride |
5.0 s |
Te-1,5 |
|
Tellurium |
Fluoride? |
~20 s |
Te-4 |
|
Tin |
Hydride |
4 s |
Sn-1,2,6 |
|
Z = 104 |
Chlorides |
<1 s |
104-3 |
|
a Separation time is for the fastest procedure reported. |
|||
2.7 Thermochromatography
An entirely different approach from that of volatilization, which we have discussed in Sec. 2.6, is the use of thermochromatography for the separation of different components that are volatilized under a given set of conditions. This technique has found a number of applications in fast radiochemical separations. The principle of the technique is briefly described in the following subsection, while in the subsequent subsection we cite some specific cases.
2.7.1 Principle
Zvara and the Dubna group have done pioneering work in the utilization of the thermochromatographic technique for the study of a number of elements, especially transplutonium elements. The technique uses the differences in the volatility of chlorides, bromides, and oxides/hydroxides of elements. A carrier gas containing thermalized reaction products is mixed with a reactive gas and passed through a reaction chamber kept at a high temperature. The gases coming from the reaction chamber are passed through a long quartz tube (~100 cm); a negative temperature gradient is maintained through the length of the quartz tube. Different volatile products carried by the gas are deposited in different temperature regions of the tube. The least volatile materials are deposited very close to the reaction chamber, while the most volatile compounds are deposited in the cooler regions of the tube. Thionyl chloride, zirconium chloride, niobium chloride, and titanium chloride have been used as the chlorinating agents [Zva70a, Zva71], while Br2 [Tra76a] and a mixture of Br2 and BBr3 [Zva75] have been used as brominating
agents. For the thermochromatography of oxides and hydroxides, usually O2 or air (dry or moist) is used [Dom83]. The deposition temperature depends on the chemical nature of the compound as well as on the nature of the surface on which it is deposited [Gra73]. Gäggeler and coworkers [Gäg86] studied the deposition behavior of a number of elements using vacuum thermochromatography. Based on the results obtained and the theoretical model developed for the evaluation of adsorption enthalpies, they have predicted that vacuum thermochromatography can be used for fast chemical separations. It is important to recognize that the behavior of elements present at trace levels may be different from the behavior of the same element at macro levels.
2.7.2 Specific Cases
Westgaard, Rudstam, and Johnson [Wes69] investigated the effect of various experimental conditions, such as the amount of carrier and pressure, on deposition using zinc chloride. They demonstrated the applicability of this technique for radiochemical separation using AgCl, CdCl2, and InCl3 labeled with 115Inm, 115Cd, and 110Agm. Zvarova and Zvara achieved separation of lanthanides and actinides using the complex with AlCl3 [Zva69, Zva70b]. Zvara and coworkers used this technique to establish that the chloride of element 104 behaves like the chlorides of zirconium and hafnium, and so is most likely a member of group IV B. Using radioactive tracers of hafnium, niobium, and tantalum, Zvara and coworkers [Zva75] showed that bromides can be used for the study of elements 104 and 105. Büchmann and coworkers [Büc76a] established the applicability of this technique for on-line, gas-phase separation. They showed the separation of a number of fission products using a quartz tube coated with BaCl2, NaCl, KCl, and CsCl. They found that BaCl2 improved the deposition of low-volatility chlorides such as those of cerium, lanthanum, and barium. They were able to obtain a decontamination factor of 105 between cerium and zirconium or niobium. Using 252Cf as fission source and an uncoated quartz tube, they were able to separate ruthenium and technetium.
Rudstam and coworkers have developed the use of thermochromatography for the separation of isobaric components in a mass beam obtained from an isotope separator [Rud73]. They studied the deposition temperature for a number of materials in the elemental and compound states; they also investigated deposition on different materials. By combining this technique with the output of an isotope separator, they successfully separated iodine isotopes with half-lives in the range of 1 to 2 s [Gra73]. With an improved apparatus, they used the technique for the study of neutron-rich zinc and cadmium isotopes [Rud81]. Novgorodov and coworkers [Nov80] have investigated the applicability of thermochromatography for the volatilization and separation of ultramicro amounts of spallation products produced in silver and gold. The irradiated silver or gold was placed in a boat kept at 1080°C for silver and 1330°C for gold. The trace amounts of volatilized reaction products (molybdenum, technetium, ruthenium, rhenium, and osmium) were allowed to deposit on quartz and porcelain surfaces. Their experiments showed that the volatilized products can be separated.
Zvara and coworkers have extensively studied the applicability of thermochromatographic techniques for the transition elements, actinides, and transactinides. In one series of investigations, they studied the deposition of metallic elements in titanium chromatographic columns [Hüb80a]. They utilized the thermochromatographic behavior to characterize the properties of heavy elements [Hüb80b, Hüb82]. The technique has also been extensively used for the separation of many actinides and transactinides. Other articles dealing with thermochromatography involve the study of deposition of polonium in copper columns [Eic79], determination of adsorption enthalpies of short-lived nuclides [Rud80], adsorption enthalpies of lanthanide chlorides [Die83], evaluation of the enthalpy of adsorption [Eic82], and Monte Carlo methods of simulation of the thermochromatographic process [Zva85]. Figure 10 shows the elements for which thermochromatographic technique has been applied. A list of elements for which the
thermochromatographic procedure has been reported along with the separation time is given in Table 8. D'Auria [Dau77] has given a brief review of on-line separations using thermochromatography.
Recently, this technique was applied for the continuous separation of fission products in a gas jet. Details of this application are described later (see Sec. 7).
2.8 Electrophoresis
Electromigration techniques have been used from the turn of the century. Kendall and coworkers have applied this technique for the separation of inorganic ions [Ken23, Ken25]. The first radiochemical separation achieved using this technique was the separation of radium from barium [Ken26]. Kendall, Jette, and West used agar gel as the supporting medium. In the late 1940s and 1950s, biochemists discovered the usefulness of this method for the separation of amino acids and proteins. A number of new developments improved the technique and its applicability. Chromatographic paper is now commonly used as the supporting medium.
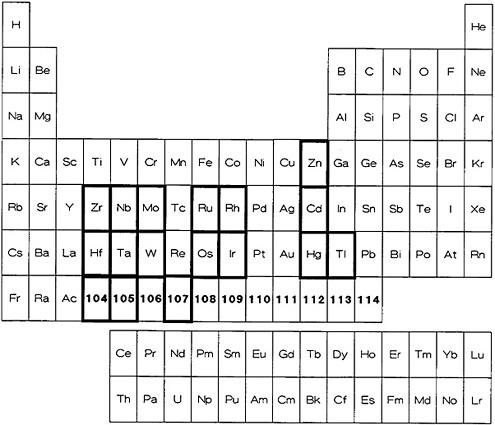
Figure 10. Elements for which a procedure based on thermochromatography is available (shown with bold rectangles).
In general, the solution containing ions to be separated is deposited as a spot on the paper, soaked in supporting electrolyte. Electrodes are placed on both ends of the paper, and a potential difference of 10 to 400 V/cm is applied. Separation of the ions is a direct result of the different migration rates of the ions in the applied electrical field. Because of the faster migration rates at higher applied voltage in most fast radiochemical separation applications, a potential of 100 to 400 V/cm is applied.
Several factors affect the mobility of the ions when a potential is applied. The first and foremost one, as mentioned in the last paragraph, is the magnitude of the applied potential gradient. A higher potential gradient leads to fast migration; efficient heat removal is essential because of the larger heat generation under these conditions. The sign and size of the charge as well as the size of the ions are important factors that affect the migration rate. For a given set of ions, other important factors that affect the relative mobilities are the nature of the supporting electrolyte, the presence of complexing agents, and the ion exchange or adsorption characteristics of the supporting medium. Everaerts and coworkers [Eve83] and Prusik [Pru79] have discussed various aspects of electrophoresis. The theory of electrophoresis has been discussed by Wieme [Wie75a], while Michl [Mic75] has presented the practical aspects of the technique. Information on the general nature of this technique is presented in the book by Smith and Feinberg [Smi72].
Table 8. Fast chemical procedures based on thermochromatography.
|
Element |
Species volatized |
Timea |
Procedure # |
|
Cadmium |
Mass-separated ion |
~1 s |
Cd-1 |
|
Cadmium |
Element |
Few s |
Cd-4 |
|
Hafnium |
Chlorides |
~0.4 s |
Hf-1 |
|
Iridium |
Oxide |
3 – 5 min |
Ir-1 |
|
Mercury |
Element? |
3 – 5 min |
Hg-1 |
|
Molybdenum |
Chloride? |
3 – 5 min |
Mo-2 |
|
Niobium |
Bromides |
0.1 – 0.2 s |
Nb-5 |
|
Niobium |
Chloride |
<3 min |
Nb-6 |
|
Niobium |
Bromides/oxybromides |
12 s |
Nb-9 |
|
Osmium |
Oxide |
3 – 5 min |
Os-1,2 |
|
Rhenium |
Oxide |
<3 s |
Re-1 to 5 |
|
Ruthenium |
Chloride |
3 min |
Ru-9 |
|
Tantalum |
Bromides |
0.1 – 0.2 s |
Ta-1 |
|
Thallium |
Oxide |
2 – 3 min |
Tl-3 |
|
Tungsten |
Oxide |
~5 min |
W-1 |
|
Z = 104 |
Chloride |
~0.4 s |
104-2 |
|
Z = 105 |
Bromides |
0.1 – 0.2 s |
105-1 |
|
Z = 105 |
Bromides/oxybromides |
12 s |
105-2 |
|
Z = 107 |
Oxides |
<3 s |
107-1 |
|
Zinc |
Mass-separated ions |
~1 s |
Zn-1 |
|
Zirconium |
Chloride |
<3 min |
Zr-4 |
|
a Separation time is for the fastest procedure reported. |
|||
During the period 1950 to 1970, various researchers investigated the applicability of paper electrophoresis for the separation of inorganic ions. The separation of lanthanides attracted the most attention. One of the early reports was that of Sato and coworkers [Sat52]. They used various concentrations of lactic acid as well as a mixture of tartaric acid and diammonium tartrate
as supporting electrolytes. A potential of 5 V/cm was applied for 1 to 2 days. They were able to obtain complete separation of parent-daughter mixtures such as strontium-yttrium, barium-lanthanum, and cerium-praseodymium. Schumacher and coworkers have performed extensive research on the application of electrophoresis for the separation of radioactive materials. They reported separation of carrier-free 90Sr-90Y [Sch57] and investigated the effect of a number of chelating agents on the separation of a number of inorganic ions [Sch58]. A number of parent-daughter pairs could be separated in a relatively short time, e.g., yttrium from strontium and lanthanum from barium in 3 min [Sch60]. This technique was used for the analysis of rare-earth mixtures [Fri61]. Pucar and Jakovac achieved electrophoretic separation of rare-earth mixtures using 0.05M lactic acid in 30-min runs [Puc60, Puc62, Jak62]. Use of α-hydroxyisobutyric acid in electrophoresis of lanthanides was reported by Bächmann [Bäc65], where the separation of a mixture of cerium, promethium, and europium was achieved in 30 min. This technique was used by Shukla and Adloff to study the chemical behavior of RaD (210Pb), RaE (210Bi), and RaF(210Po) in different acid media [Shu62a, Shu62b]. Applicability of this technique to achieve separation of actinides has been investigated by several researchers. For example, Kraak and Walz [Kra65] reported electrophoretic separation of americium and curium from a solution containing 0.001M EDTA in a 4-h run, while Bächmann [Bäc66] has reported separation of actinium, americium, and curium from one another, as well as separation of various mixtures of actinides and lanthanides. A summary of electrophoretic radiochemical procedures is available in the radiochemical techniques monograph of Bailey [Bai62].
A number of fast radiochemical procedures using paper electrophoresis are available in the literature; however, such procedures are known only for a few elements, namely, selenium, bromine, strontium, cesium, barium, lanthanum, cerium, and praseodymium. The list of elements and the separation times of procedures are for the separation of a specific lanthanide from fission products. Ohyoshi and coworkers [Ohy72] studied 40-s 144La by separating lanthanum from fission products in about 25 s. They used nitrilotriacetic acid at pH 2.0 as the supporting electrolyte and chromatographic paper as the supporting medium. The irradiated uranium solution, along with lanthanum and cerium carriers, was spotted on the paper wetted with the supporting electrolyte. By applying a potential gradient of 90 V/cm for 70 s, they achieved the separation of lanthanum. Using a similar procedure, but a higher potential gradient (300 to 400 V/cm), Yamamoto and coworkers have achieved separation of lanthanum, cerium, and praseodymium from fission products in 2 to 3 min [Yam80, Yam81].
Table 9. Fast chemical procedures based on electrophoresis.
|
Element |
Reagent |
Timea |
Procedure # |
|
Barium |
NTAb (1 × 10−2M) |
<1 min |
Ba-1 |
|
Bromine |
H2SO3 (1 × 10−2M) |
40 s |
Br-5 |
|
Cerium |
NTA (4 × 110−3M) |
2 – 3 min |
Ce-2,6 |
|
Cesium |
Citric acid (10−2M) |
~40 s |
Cs-1 |
|
Lanthanides |
NTA (4 × 10−3M) |
~5 min |
Ln-2 |
|
Lanthanum |
NTA (4 × 10−3M) |
40 s |
La-1,4,6 |
|
Praseodymium |
NTA (4 × 10−3M) |
3 min |
Pr-1,3,5 |
|
Selenium |
HClO4 (1 × 10−2M) |
<1 min |
Se-7 |
|
Strontium |
NTA (1 × 10−2M) |
<1 min |
Sr-1 |
|
a Separation time is for the fastest procedure reported. b NTA = nitrilotriacetic acid. |
|||





























