
5
Large-Scale Production of Monoclonal Antibodies
About 25,000 mAb are listed in Linscott's Directory (1998–99). Most are produced in small quantities (less than 0.1 g) for bench-related research purposes (de Geus and Hendriksen 1998b). However, some have become commercially successful and so require a scale of production different from that usually experienced in research facilities. Commercial interests consider production scales of 0.1–10 g as small, 10–100 g as medium, and over 100 g as large. Commercial-scale production is generally performed to produce mAb for three purposes: diagnosis, therapy, and research on and development of new therapeutic agents.
Monoclonal Antibody Production for Diagnostic and Therapeutic Purposes
The amount of mAb needed and the importance of such factors as cost, turnaround time, and regulatory compliance depends on the purpose. The very competitive diagnostic industry is concerned with cost, turnaround time, and regulatory requirements. The diagnostic-industry scale of mAb production is usually small to medium and seldom large. The therapeutic industry is considerably less concerned than the diagnostic industry with cost and turn-around time, and its production scale is medium to large. The therapeutic industry is highly regulated and sensitive to regulatory structure and to the very high regulatory cost of any procedural change. The biotechnology industry that develops therapeutic agents produces mAb on a small to medium scale; it is less concerned with cost of production than the diagnostic industry and much more concerned than the therapeutic industry, but turnaround time is very important. Therefore, it requires
rapid turnaround to increase the chance of being first in the marketplace with a product that will probably have a short life span.
Commercial mAb production requires more than the culturing of large batches of cells or their injection into large numbers of mice. It requires considerable preproduction effort to ensure that the cell line is stable, can produce commercially appropriate quantities of a stable antibody, and can produce an uncontaminated product. Commercial production also involves building a high-quality facility for in vivo and in vitro production and for processing of the antibody. There is a need for quality control and quality assurance departments to meet the requirements of good manufacturing practices that are required for commercial products. Product-lot testing is necessary to ensure product reproducibility. Production-process verification and documentation are necessary to protect the consumer and are required by FDA in its regulatory "Points to Consider in the Manufacture and Testing of mAb Products for Human Use" (FDA 1997).
In Vivo and In Vitro Methods for Commercial Production of mAb
Commercial mAb production uses both the mouse ascites method and in vitro methods. Cost is usually the major consideration in determining the method except for marketed therapeutic products.
When all fully-loaded production and pre-production and post-production costs are considered for a commercially viable line, economics usually favor in vivo production. However, as the amount of mAb increases, existing in vitro production technology can become more economical because high, fixed optimization costs (costs associated with selecting a subclone with the best growth and mAb production characteristics and grow in low-serum or serum-free conditions) associated with in vitro production are spread over a larger production amount, making cost per gram competitive with in vivo production, which has a higher and more variable cost structure (figure 2). When production costs are compared for small-scale production, in vitro methods are 1/2 to 6 times higher, depending on the cell line (Hendriksen and de Leeuw 1998; Jackson and others 1996; Peterson and Peavey 1998; Marx 1998; Lipman 1997). However, these costs might not include all factors, such as animal housing costs and technician time. In large-scale production runs, in vitro systems are economically competitive and are usually selected because they reduce animal use and decrease the presence of contaminating foreign antigens if serum-free media can be used. When the time of mAb production is critical and small amounts are required, in vivo production is selected because it takes only 6 weeks. For in vitro systems, time requirements vary considerably. Production time depends on the amount of time required to optimize the hybridoma to the system being used and on the quantity of mAb needed. Commercial-quantity in vitro production of mAb requires more time
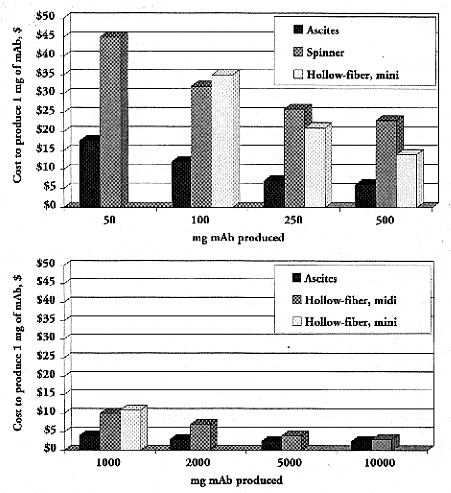
Figure 2.
Cost of producing 1 mg of mAb in mouse ascites, spinner flask (1 to 8L Belco spinners), and mini (spectrum CellMax) and midi (Cellex Accusyst Jr.) hollow-fiber bioreactors (Chandler 1998). Note that absence of a bar does not mean that cost of technique is virtually zero, but rather the in vitro system was not used. Thus, spinner flask is not used above 1,000 mg, mini hollow-reactor is used only in range of 100–1,000 mg, and midi hollow-fiber reactor is used for production needs of 1,000 mg and above.
than in vivo production because of the lengthy optimization process and the increased time for producing a given quantity of mAb (Butler and Huzel 1995; Moro and others 1994; Stoll and others 1996).
The therapeutic industry uses primarily serum-free in vitro technology because of a concern for treatment-related allergic responses caused by repeated foreign-antigen exposure. Immune responses are of concern here because mice are the source of the cell lines used in most mAb production methods. The human immune system tends to reject mouse-derived antibodies, which can lead to allergies or decreased effectiveness of injected mAb. Therefore, techniques that replace most of the mouse's antibody genes with human DNA have been developed. Humanizing antibodies and producing antibody in SCID mice or in an in vitro system have alleviated this problem (Boyd and James 1989; Reuveny and Lazar 1989). In the therapeutic industry, early work to determine whether the mAb will have the desired effect is usually done with in vivo-derived mAb because turnaround time is shorter and production costs are lower. During the same period, the company will develop its final in vitro manufacturing process. When in vitro optimization and product development are completed, the company will develop its final product-effectiveness information and file a final proof-of-process document with FDA.
In diagnostic industry, keen competition leads to overriding cost considerations, whereas the presence of foreign antigens is less important. As a result, in vivo-derived products are commonly used. In vivo procedures are optimized to increase productivity by reducing hybridoma invasiveness and increasing mAb secretion. (Harlow and Lane 1988, p 274–275). This optimization can result in a reduction in animal use by a factor of 2–10 that greatly reduces production costs. Ascites production costs are important because ascites production has a high-variable cost component. However, the research industry—that is, industry concerned with research on and development of new therapeutic agents—is most concerned with production time and binding affinity of the mAb. Therefore, whether in vivo or in vitro methods are used depends on the purpose of the project and on the quality of mAb produced by the cell line in that system. For very-small-scale production, ascites production is often used because it is a much more forgiving procedure than in vitro production and can be done without optimizing cell lines in an in vitro culture.
In Vivo Production
Biologic behavior of a hybridoma cell line is very important in determining whether in vitro culture will be successful or the ascites method must be used. Biologic behavior also affects the concentration of mAb produced and, for the mouse ascites method. the quantity of ascites produced. Researchers, and production facility personnel can optimize production results of both in vitro and in vivo methods by adjusting production variables and selecting appropriate clones.
Experience has shown that when cost is important to a client, system optimization often favors even more the economics of in vivo production for the client's cell line. In vivo optimization is necessary only for cell lines that will go into continuous production, but in vitro optimization is necessary for all cell lines to produce acceptable growth and mAb production (Capiaumont and others 1995; Chua and others 1994a, b; Shacter 1989; Trampler and others 1994). It is an important expense factor for in vitro production because it requires much labor by highly paid, highly trained employees. The optimization process also requires large quantities of disposable supplies, an important factor in the increased costs associated with in vitro production.
The variables affecting in vivo production and optimization (Hendriksen and de Leeuw 1998; Chandler 1987) include age, sex, strain of the host, size of the hybridoma-cell inoculum, number of taps, and type and volume of primer. Those variables can be manipulated to affect ascites yields and mAb concentration. For instance, low-ascites-producing subclones usually form only a few large tumors in the peritoneal cavity, whereas high-producing subclones form numerous colonies of small tumors that grow extensively throughout the mesentery (Cancro and Potter 1976). Therefore, clones that form less-invasive small soft tumors should be selected. Sequential tapping provides the highest yields and greatest mAb concentration from a group of mice (Chandler 1987). Except for very invasive cell lines that allow for only one needle tap, sequential tapping usually reduces the number of mice needed per gram of mAb by a factor of 2-3. The number of needle taps allowed should therefore be based on the clinical condition of the mice, and the maximum, in general, should be three taps (Jackson and others 1999b).
Optimal in vivo production requires reduction of the invasive nature of a cell line so that all of the mice survive completion of a production run. Selecting appropriate clones and altering hybridoma cell concentration injected into the peritoneal cavity of the mice are two ways to optimize production. The volume and concentration of mAb produced depend on the clone selected, and this makes systematic comparisons difficult. Therefore, the best way to achieve maximal in vivo yields is to screen clones in mice and to use the clone that provides the best yield. Cell growth conditions are optimal in vivo, so almost all cell lines will produce antibody, even when they are not optimized (Hendriksen and de Leeuw 1998). That is why injecting into mice usually saves cell lines that are difficult to grow in vitro.
Ascites production is a simple procedure, once proper technique is learned. Daily observation of the mice requires skilled observers to determine the optimal time for tapping the fluid and to determine when the mouse should be euthanized. It is quicker, is more forgiving, is more economical for small-scale and medium-scale production, produces a higher concentration of mAb, and is easier to scale up in production (Chandler 1987). For most cell lines, purification costs are the same as in vitro methods. The major problems associated with in vivo production
are the use of animals, the possibility that the animal could be harmed if technicians are not properly trained and procedures are not followed properly, the presence of endogenous mouse immunoglobulin contamination except when immunodeficient mice are used (Ware and others 1985), and the possibility of contamination with murine pathogens, which requires the use of high-quality animals and a high-quality program for health assurance. High-speed centrifugation of the ascitic fluid brings pristane to the top, where it can be removed easily.
In Vitro Production
Variables affecting in vitro production and optimization are presented in several papers (de Geus and Hendriksen 1998; CAAT 1997; Jackson and others 1996; Beck and others 1987; Seaver 1987; Reuveny and others 1985). Numerous in vitro commercial systems meet the different needs and requirements of users. These systems are of two types: single-compartment systems that allow only low-density cell culture and double-compartment systems that allow high-density cell culture, which results in increased mAb concentration. For very-small—scale production (less than 10 g), the simple low-density cell-culture systems-such as culture flasks, roller bottles, gas permeable bags, and hollow-fiber bioreactors—are used. For small-scale and medium-scale production (10-100 g), double-compartment, high-density cell-culture systems, such as hollow-fiber systems, are used, as well as spinner flasks and roller bottles. High-scale production (over 100 g) is performed in large capital-intensive systems, such as homogeneous suspension culture in deep-tank stirred fermentors, perfusion-tank systems, airlift reactors, and continuous-culture systems.
An antigen-free product can be obtained by adapting the cell line to low-serum or serum-free media, with generally minor inhibitory effects on the cell line (Kurkela and others 1993). Benefits of in vitro production are the absence of live-animal use, although some products in the culture media come from animals; the possibility of low-serum or serum-free media production (Klerx and others 1988); and the absence of host-contributed immunoglobulin or antigens. As the cost of disposable materials decreases further and technologic changes increase production efficiency and decrease equipment costs, the cost of in vitro production should decrease further, so it should become the preferred method of commercial production.
Problems associated with in vitro systems today are as follows (note that the items in the following list do not necessarily apply to all the numerous in vitro systems mentioned above): material, labor, and equipment costs are higher than for the in vivo method (Jackson and others 1996; Peterson and Peavey 1998; Brodeur and Tsang 1986; Lipman 1997); characteristics of the hybridoma are more critical than in vivo; about 3–5% of all clones cannot be maintained in existing in vitro systems (de Geus and Hendriksen 1998; Hendriksen and de Leeuw 1998); the great potential for microbial contamination, poor growth, and
mechanical failure of the system or supporting systems requires constant monitoring and attention every day (Lebherz 1987); production of large quantities of mAb is slower because of low mAb concentration, compared with the ascites method; the increased employee technical capabilities and educational background required by increased training time and system manipulations increase labor expense; the design of downstream processing is emphasized because large volumes of media are required to obtain large quantities of mAb and to ensure product economy and purity (Stang and others 1998); and residual endotoxin, residual DNA from cell death, and bovine IgG contamination with cell lines that require some serum all complicate the process.
It is difficult for a user to choose a particular in vitro system on the basis of manufacturers' claims because of how costs are calculated and because the amount of antibody secreted by different hybridoma lines in identical medium and culture conditions can vary by a factor of as much as 100 (Seaver 1987). Therefore, it is important to compare the productivity of several systems by using several cell lines and to include optimization costs of each system in calculating the overall cost per gram. Numerous commercial-volume systems are available, and none is inexpensive. In the near future, as new technologies (recombinant, transgenic, and so on) are developed and in vitro systems become more economical and efficient, most commercial mAb production will undoubtedly use in vitro systems.
One of the most common causes of failure of in vitro methods is poor adherence to basic tissue-culture techniques, such as sterilization of cultureware, equipment, and media and humidity and temperature control in the system. In large-scale and medium-scale production, it is important to have tight procedural and environmental controls to minimize losses due to system microbial contamination. To help avoid a major economic effect of such losses in commercial production, expensive facilities and tightly controlled procedures are implemented, all of which add to the high fixed cost of in vitro mAb production.
Regulatory Requirements
Regulatory requirements in the United States for in vitro and in vivo manufacturing of mAb have not changed considerably in the last two revisions of the “Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use" (FDA 1997). The required differences for the two methods of production focus on the testing and monitoring of mice used for ascites and the testing of the final product for the 16 adventitious viruses that can come from the animals used in production. Specific guidelines in the "Points to Consider" (FDA 1997) recommend an intense health-monitoring program for the animals, including complete, routine health monitoring of the animal stock. Monitoring also covers mouse antibody production, mycoplasma testing, and complete physiologic and physical examination of the animals. There should
also be a surveillance system that uses animal sentinels for health and serologic screening. These programs need to be continually updated as other adventitious viruses are identified by the FDA.
Protocols for ascites production require specifics on the animals used for manufacturing—such information as sex, age, and species. There are requirements for volume of pristane, cell concentration of the inoculum, and timing for priming, inoculation, and harvesting of ascites. Other requirements are strictly related to the well-being of the animals, such as bedding, feeding schedules, and general housing conditions of the facility involved in manufacturing.
FDA has approved 13 mAb for clinical use, two of which must be produced by the ascites method. Most new-drug applications to FDA are for mAb that are produced in vitro. It is likely that large-scale manufacturing of mAb will use in vitro methods as systems and technology are optimized to reduce the final cost per unit. FDA is encouraging using in vitro methods for producing mAb. In Europe, Germany, the Netherlands, Sweden, Switzerland, and the UK have restricted or prohibited the use of mice for production of ascites and more countries will probably join them.
More important than regulatory differences between the two modes of manufacturing of mAb are requirements that must be met when the mode of manufacturing is changed during product development before licensing. Changes in manufacturing often occur in clinical development of a product. The FDA requires a plan for demonstrating that the products made in different ways are similar. The requirement also applies if there is a scaleup without substantial changes in the manufacturing process during or after completion of phase 3 trials. As presented by the FDA Center for Biologics Evaluation and Research Division (Stein 1998), it could take 3–8 years to obtain data needed to approve a product formerly produced by the ascites method and later produced in vitro. There have been cases in which the two methods of production have yielded similar antibodies that are not comparable. As stated in Stein's presentation, the earlier in the development of the process the changes are made, the better the success of the product. She stated that investigators should adapt the hybridoma to in vitro conditions early when developing mAb for clinical applications.








