
4
The Role of Computational Biology in the Genomics Revolution
Jeffrey Skolnick,
Jacqueline Fetrow,
Angel R. Ortiz,
and
Andrzej Kolinski
Scripps Research Institute
Abstract
The various genome sequencing projects are providing a plethora of protein sequence information, but with no information about protein structure or function. The most effective method for sifting out useful proteins from these genomic databases is the computer prediction of protein function. However, current methods, which are mainly sequence-based, are limited by the extent of similarity between sequences of unknown and known function; they increasingly fail as the sequence identity diverges into and beyond the twilight zone of sequence identity. In practice, between 30 and 60 percent of all proteins can be functionally identified using current sequence-based software. To extend the level of molecular function annotation to a broader class of protein sequences, methods for identification of protein function based directly on the sequence-to-structure-to-function paradigm will need to be developed. One such approach is presented. The idea is to predict the native structure first by using ab initio folding or threading techniques and then to identify its molecular or biochemical function by matching the active site in the predicted protein structure to that in a protein of known function. Application of this approach to genomic screening is then described. Based on these preliminary results, the next 5 to 10 years are likely to see the development of computational tools that will allow for the medium-resolution prediction of the tertiary structure of single domain proteins, the more robust identification of protein ligands, techniques to predict proteins having specific quaternary interactions, and the beginnings of a bottom-up approach to identify important proteins in metabolic and signal transduction pathways.
Introduction
The various genome sequencing projects are providing a vast quantity of protein sequence data,1 but what is needed is information about protein function (Rastan and Beeley, 1997). To enhance the efficiency of the drug design process, one must identify the sequences of functionally important proteins
|
1 |
See the GenBank index at <http://www.ncbi.nlm.nih.gov/Web/Genbank/index.html>. |
that are hidden in these large databases. For example, microbial genomes contain potential protein targets that can be utilized to kill pathogens or that can be developed into commercially useful enzymes to produce or degrade various substances. By far the most effective method for sifting out useful proteins from these genomic databases relies on the computer-based prediction of protein function (Rastan and Beeley, 1997). However, most current methods, being mainly sequence-based, are limited by the extent of sequence similarity between sequences of unknown and known function (Pearson and Lipman, 1988; Henikoff and Henikoff, 1991; Attwood and Beck, 1994; Bairoch, Bucher et al., 1995; Altschul, Madden et al., 1997; Attwood, Beck et al., 1997). They increasingly fail as the sequence identity between two proteins crosses into and beyond the twilight zone of sequence identity, which is about 30 percent (Fetrow and Skolnick, 1998). In practice, current sequence-based software can identify the molecular or biochemical function of roughly 30 to 60 percent of all proteins in a given genome (Bult, White et al., 1996; Casari, Ouzounis et al., 1996). The full annotation of entire genomes is likely to be a major computational and experimental challenge over the next 5 to 10 years, but one which, when successfully addressed, will provide a revolution in disease diagnosis and treatment as well as in our conceptual understanding of biology. To be fully successful, this will require a multidisciplinary approach involving biology, chemistry, physics, and computer science.
Here, we describe one promising means of extending the ability to annotate the remaining orphan sequences based on the sequence-to-structure-to-function paradigm (Fetrow, Godzik et al., 1998; Fetrow and Skolnick, 1998). Logically, this process can be divided into two parts. First, one employs techniques to determine protein structure from sequence (Godzik, Skolnick et al., 1992; Ortiz, Kolinski et al., 1998 a,b,c). Secondly, one employs tools for function prediction based on the identification of active sites in the predicted or experimental structure. The ability to determine function from structure will be very important given the emerging structural genomics initiatives where the goal is to determine all possible protein folds. This reverses the more traditional approach where one first identifies the function of the protein of interest and then subsequently determines its structure.
Prediction of Protein Structure from Sequence
Currently, there exist two basic theoretical approaches for the prediction of protein structure from sequence when homology modeling (which requires significant sequence identity between the probe sequence and its template structure) (Sali and Blundell, 1993) cannot be applied: threading (Bryant and Lawrence, 1993; Miller, Jones et al., 1996), and ab initio folding (Skolnick, Kolinski et al., 1997; Ortiz, Kolinski et al., 1998 a,b,c). In threading, the idea is to match the sequence of interest to a template structure in a library of known structures (Godzik, Kolinski et al., 1993); thus, this approach is conceptually similar to standard homology modeling, except that now the goal is to match probe sequences to template structures when there is no apparent sequence relationship between the two. In ab initio folding, one attempts to fold a protein starting from a random conformation (Kolinski and Skolnick, 1996). The advantage of threading is its speed and the fact that it can be applied to large proteins. In contrast, ab initio folding is computationally more demanding and is, in practice, currently limited to proteins smaller than 100 residues (Ortiz, Kolinski et al., 1998 a,b,c). However, ab initio folding does not demand that an example of a native structure be already solved. Thus, it can be used to identify proteins having a novel native structure. Recent results indicate that for small proteins (those less than 100 residues), ab initio folding approaches can predict structures at a level of quality (4- to 6-Å coordinate root mean square deviation for the backbone atoms) comparable to that provided by threading (Ortiz, Kolinski et al., 1998a,b).
Description of Ab Initio Protein Folding Methodology
In what follows, we describe a newly developed method for structure prediction, MONSSTER, which attempts to address the aforementioned problems. As depicted in Figure 4.1, prediction of protein structure can be conceptually divided into four stages: (1) restraint derivation; (2) structure assembly; and (3) selection of the native conformation. In addition, for those sequences whose structures are known either before or after the prediction is made, following the structure selection process, (4) objective, rigorous validation criteria are applied to judge the success of the prediction.
For (1), restraint derivation, a multiple sequence alignment with the sequence of interest is generated (Sander and Schneider, 1991). Then, predicted secondary structure restraints are obtained from a standard secondary structure prediction scheme (Rost and Sander, 1993; Rost, Schneider et al., 1993) supplemented by our LINKER algorithm (Kolinski, Skolnick et al., 1997)—a quite accurate technique for predicting where the chain reverses global direction. We term such regions "U-turns" (Kolinski, Skolnick et al., 1997). The predicted secondary structural elements between these U-turns define the predicted core regions of the molecule. Tertiary contacts (restraints), termed "seeds," between these core elements are then predicted from multiple sequence alignments. Multiple sequence information is used to derive such seed side-chain contacts based on patterns of residue conservation (Aszodi, Gradwell et al., 1995; Mumenthaler and Braun, 1995) or residue covariation in a set of homologous sequences
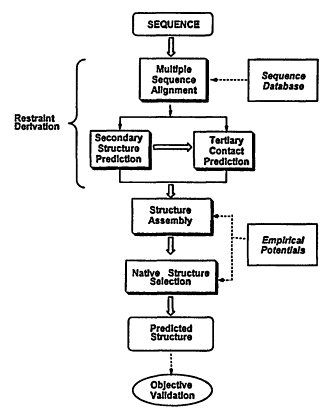
Figure 4.1
Schematic overview of the procedure for tertiary structure prediction.
(Göbel, Sander et al., 1994; Thomas, Cesari et al., 1996; Olmea and Valencia, 1997). Both might be combined for increased sensitivity (Olmea and Valencia, 1997). Here, for the sake of simplicity, we slightly modify the approach of Göbel and coworkers (Göbel, Sander et al., 1994) and calculate the covariation between all residues predicted to be in the putative core of the molecule (Olmea and Valencia, 1997; Ortiz, Kolinski et al., 1998a,b). Unfortunately, there are too few of these seed contacts to assemble a protein from the unfolded state using MONSSTER. Thus, these seed contacts between predicted topological elements (i.e., α-helices and β-strands between U-turns) are enriched by an inverse folding approach that typically produces about N/4 contacts—the number required for successful topology assembly (Olmea and Valencia, 1997; Skolnick, Kolinski et al., 1997; Ortiz, Kolinski et al., 1998a,b).
In (2), the structure assembly step, the set of predicted restraints is used in the MONSSTER method (Skolnick, Kolinski et al., 1997) to drive the conformational search. This uses a reduced-protein-lattice model to assemble the global fold. First, a series of up to 1,000 independent, simulated annealing structure-assembly runs are performed, and the resulting structures are clustered on the basis of their pairwise coordinate root mean square deviation (cRMSD). If the resulting structures do not cluster into several topologies, then no structural prediction is made. If at least a subset of the structures cluster, then we proceed to the structure selection step.
The native structure selection stage (3) consists of long isothermal runs from which the putative native topology is chosen on the basis that it has the lowest average energy. If the differing topologies cannot be selected on this basis, then the prediction consists of several lowest-average-energy representatives of the various generated topologies. In all cases, we report the average cRMSD values corresponding to the lowest-average-energy structures and not the best cRMSD values because in a blind prediction, we would have no means of selecting such structures.
Once the native conformation of the protein of interest is known, we judge the success of the prediction (4) as follows: First, we calculate the global C-α cRMSD between the predicted (lowest average energy) and experimental structures. Since our approach often results in structures whose C-α cRMSD is in the range of 6 Å, there may be substantial topological errors between the native and predicted structure; therefore, a more rigorous assessment of success is necessary. Thus, the predicted fold is subjected to a structural similarity search over a representative database using the DALI (Holm and Sander, 1997) structural superimposition program. We note that a very similar approach has been used to assess the quality of structures predicted by threading techniques, where a sequence is matched to a fold in a library of known structures (Wodak and Rooman, 1993). When a known homologue of the native structure is chosen (or the native structure itself), then the tertiary structural prediction protocol is considered to be successful. If two or more topologies are isoenergetic, both would be subjected to this protocol; if one matches the native topology, we consider this to be a partial success. If the next lowest average energy topology (as predicted by MONSSTER) matches the native fold rather than the lowest average energy structure, this is also considered to be a partial success. Otherwise, by this rigorous criterion, the prediction is unsuccessful.
Validation on Proteins of Known Structure
The above protocol was applied to the set of 19 proteins listed in Table 4.1. On average, for the set of proteins whose native conformation was known in advance, the predicted secondary structure is 69 percent correct; this is slightly less than the reported average for this technique, which is 72 ± 9 percent (Rost and Sander, 1993; Rost, Schneider et al., 1993). Such a large test set is necessary to demonstrate that the current approach can handle a wide variety of folds and different secondary structure types. All
TABLE 4.1 List of Proteins of Known Structure That Constitute the Validation Set
|
Protein |
Nres |
Class |
Fold Description |
Name |
|
3cti |
29 |
small |
disulfide-bound fold, beta hairpin with adjacent disulfide |
Trypsin inhibitor from squash (Cucurbita maxima) |
|
1ixa |
39 |
small |
EGF-like (disulfide-rich fold; nearly all beta) |
Factor IX from human (Homo sapiens) |
|
protA |
47 |
α |
Three-helix bundle |
Protein A |
|
1gpt |
47 |
small |
disulfide-bound fold, beta hairpin with adjacent disulfide |
Gamma-thionine from barley (Hordeum vulgare) |
|
1tfi |
50 |
small |
Rubredoxin-like (metal-bound fold, with 2 CXXC motifs) |
Transcriptional factor SII from human (Homo sapiens) |
|
6pti |
58 |
small |
BPTI-like (disulfide-rich α+β fold) |
Pancreatic trypsin inhibitor from bovine (Bos taurus) |
|
1fas |
61 |
small |
Snake toxin-like (disulfide rich; nearly all beta) |
Fasciculin from green mamba (Dendroaspis angusticeps) |
|
1shg |
62 |
β |
SH3-1ike barrel (partly opened; n* = 4, S* = 8; meander) |
alpha-Spectrin, SH3 domain from chicken (Gallus gallus) |
|
1cis |
66 |
α+β |
CI-2 family (α+β sandwich; loop across free side of β) |
Hybrid protein from barley (Hordeum vulgare) hiproly strain |
|
1ftz |
70 |
α |
DNA-binding 3-helix bundle (right-handed twist; up-down) |
Fushi Tarazu protein from fruit fly (Drosophila melanogaster) |
|
1pou |
71 |
α |
DNA-binding domain (4 helices, folded leaf, closed) |
Oct-I POU-specific domain from human (Homo sapiens) |
|
1c5a |
73 |
α |
Anaphylotoxins (4 helices; irreg. array, disulfide linked) |
C5a anaphylotoxin from pig (Sus scrofa domestica) |
|
3icb |
75 |
α |
EF-hand (2 EF-hand connected with Ca bind loop) |
Calbindin D9K from bovine (Bos taurus) |
|
1ubi |
76 |
α+β |
β-grasp (single-helix packs against β-sheet) |
Ubiquitin from human (Homo sapiens) |
|
1lea |
84 |
α |
DNA-binding 3-helix bundle (right-handed twist; up-down) |
LexA repressor, DNA-binding domain (Escherichia coli) |
|
1ego |
85 |
α/β |
Thioredoxin-like (3 α/β/α layers; β-sheet order 4312) |
Glutaredoxin from bacteriophage t4 |
|
1hmd |
85 |
α |
Four helical up-and-down bundle(left-handed twist) |
Hemerythrin from sipunculid worm (Themiste dyscrita) |
|
1poh |
85 |
α+β |
α+β sandwich |
Histidine-containing phosphocarrier proteins (Escherichia coli) |
|
life |
100 |
α+β |
IF3-1ike (β-α-β—α-β(2); 2 layers; mixed sheet 1243) |
Translation initiation factor IF3 from Escherichia coli |
are outside the set of proteins employed in the derivation of the empirical potentials. It is very important to emphasize that all predictions use the identical parameter set and folding protocol. Table 4.2 shows the accuracy of the predicted secondary structure and tertiary contacts, as well as the results from the folding simulations. Only about 78 percent of the native contacts are correct within ±2 residues; these are typical of results seen on an even larger class of proteins. Often, there are also a number of grossly incorrect restraints that can lead to non-native topologies. Using this information, in about 10 to 30 percent of the assembly runs, native-like topologies, as subsequently assessed by their global cRMSD
and DALI (Holm and Sander, 1997), are recovered for all classes of proteins. But on average, helical proteins are predicted better than alpha/beta proteins, which are predicted better than beta proteins.
In 14 of 19 cases, success or partial success was obtained, with the lowest average C-α cRMSD values ranging from 3.5 to 6.7 Å. For one partial success whose lowest average energy structure has a higher cRMSD from native life, the lowest-energy fold basically adopts the native topology despite its unsatisfactory cRMSD. Here, a strand region is found at the back of the protein rather than at the edge of the fold. The topology with the next higher energy, i.e., the first excited state, is the native one. For the five unsuccessful cases—3cti, 1tfi, 6pti, 1lea, and 1poh—DALI fails to find any structure that is significantly related to the lattice model; thus the prediction is labeled as being unsuccessful. This is true in spite of the fact that the topology of 6pti is native, and for 1poh, a slightly misfolded state is recovered, but by the DALI selection criterion, this simulation is unsuccessful. Furthermore, for mainly helical proteins such as lpou, the alternative low-energy fold is the topological mirror image (where the helices are right-handed, but the chirality of the turns is reversed from the native conformation). In some situations, e.g., for life and 1poh, the alternative topology differs in the placement of one or two topological elements. In other cases, the alternative and native topology do not have much in common.
Blind Predictions
We next present a representative prediction of the tertiary structure of the 81-residue KIX domain of the CREB binding protein, which is involved in gene expression as mediated by AMPc (Brindle and Montminy, 1992; Radhakrishnan, Perez-Alvarado et al., 1997).
As shown in Figure 4.2, the secondary structure prediction scheme suggests that KIX should adopt a three-helix bundle fold. Correlated mutation analysis provides four seed contacts (22-35, 22-73, 35-73, 17-72) that yielded 38 predicted tertiary contacts when enriched; this is a rather large number as compared to other entries in Table 4.2. A series of 10 independent fold assembly simulations were done; all yielded either a left- or right-handed three-helix bundle. As indicated in Table 4.2, on the basis of their average energies, the two topologies are essentially isoenergetic. Decomposing the energy into its constituent contributions (Ortiz, Kolinski et al., 1998c), the pair interactions, secondary-structure preferences, and hydrogen-bond terms favor the right-handed bundle, whereas the burial energy and terms designed to generate protein-like densities favor the left-handed bundle. The difficulty in distinguishing topological mirror images is a problem that this method often experiences with helical proteins, and indicates that improvements in the empirical potential are necessary. When subsequent predictions were done using the subset of restraints that satisfy each of the two topologies, then the native topology was found to be substantially lower in energy than the incorrect alternative.
Structure to Function
In the prediction of protein function from sequence, there are a number of key questions that must be answered. In particular, does one need a protein structure to predict protein function or is sequence information sufficient? If a protein's tertiary structure is needed, how close does it have to be to the native state to permit the protein's function to be identified? Is there a one-to-one relationship between protein structure and protein function? If not, can one construct a library of active sites so that one can search structures for appropriate active sites? In what follows, we address each of these questions in turn.
TABLE 4.2 Summary of Prediction Results
|
Proteina |
Type |
Nb |
Q3c |
Ncd |
Npe |
Nwf |
d=0g |
d=2g |
|
Proteins with Known Structure in Advance of Prediction |
||||||||
|
3cti |
small |
29 |
50.5 |
39 |
6 |
0 |
83.3 |
100.0 |
|
1ixa |
small |
39 |
70.5 |
48 |
5 |
0 |
100.0 |
100.0 |
|
1gpt |
small |
47 |
70.6 |
70 |
13 |
0 |
46.1 |
100.0 |
|
1tfi |
small |
50 |
60.0 |
84 |
37 |
0 |
21.6 |
88.8 |
|
protAo |
α |
47 |
77.6 |
91 |
17 |
0 |
0 |
70.5 |
|
1ftz |
α |
56 |
63.5 |
149 |
12 |
1 |
25.0 |
58.3 |
|
1c5a |
α |
66 |
85.8 |
105 |
43 |
1 |
24.4 |
73.3 |
|
1pou |
α |
71 |
78.8 |
122 |
49 |
0 |
28.6 |
89.8 |
|
3icb |
α |
75 |
82.3 |
154 |
25 |
0 |
28.0 |
68.0 |
|
1hmd |
α |
85 |
90.6 |
157 |
20 |
2 |
10.0 |
65.0 |
|
1shg |
β |
57 |
67.1 |
109 |
39 |
0 |
28.2 |
100.0 |
|
1fas |
β |
61 |
67.1 |
98 |
25 |
1 |
26.3 |
78.9 |
|
6pti |
αβ |
56 |
58.8 |
92 |
19 |
0 |
68.4 |
100.0 |
|
1cis |
αβ |
66 |
64.7 |
144 |
23 |
0 |
8.6 |
78.2 |
|
1lea |
αβ |
73 |
63.5 |
131 |
41 |
2 |
9.7 |
75.6 |
|
1ubi |
αβ |
76 |
62.3 |
153 |
17 |
0 |
23.5 |
94.1 |
|
1poh |
αβ |
85 |
65.9 |
162 |
36 |
3 |
8.3 |
55.5 |
|
1ego |
αβ |
85 |
60.0 |
223 |
33 |
0 |
15.1 |
93.9 |
|
1ife |
αβ |
100 |
75.3 |
148 |
21 |
3 |
14.2 |
38.0 |
|
Result from Blind Prediction |
||||||||
|
KIX |
α |
81 |
87.7 |
320 |
37 |
11 |
26.3 |
57.9 |
|
NOTES: a Protein refers to the Brookhaven National Laboratory's Protein Database (PDB) access number for the protein studied. b N is the number of residues in the protein in the PDB file. c Q3 is the percent of correctly predicted secondary structure. All proteins have a Q3 within one standard deviation of the average. d Nc is the number of contacts in the native structure. e Np is the number of predicted contacts. f Nw is the number of contacts that are incorrect when no native contact is found within ±5 residues of a predicted contact. g Percent of predicted contacts within d residues of a native contact. h rmsn is the cRMSD deviation in angstroms from the native structure. i En is the lowest average energy (in kT) after refinement for the nativelike topology j rsn is the number of restraints satisfied in the nativelike topology. k rmsw is the cRMSD deviation from native in angstroms of the alternative topology of lowest energy. l Ew is the lowest average energy (in kT) in the alternative topology after refinement runs. m rsw is the number of restraints satisfied in the alternative topology. n Relationship of lowest average energy structure to the native conformation if known. S indicates that the full structural selection criterion as assessed by the energy and DALI are "successful," PS indicates that the tertiary structure prediction is "partially successful," and U indicates that the tertiary structure prediction is "unsuccessful." o The B domain of protein A. |
||||||||
|
Native Topology |
Lowest Energy, Nonnative Topology |
|||||
|
rmsnh |
Eni |
rsnj |
rmswk |
Ewl |
rswm |
Final Scoren |
|
3.8 |
-107 |
6 |
6.7 |
-103 |
6 |
U |
|
5.6 |
-130 |
5 |
7.7 |
-131 |
5 |
S |
|
5.9 |
-276 |
9 |
6.6 |
-142 |
10 |
S |
|
5.9 |
-202 |
28 |
7.0 |
-191 |
31 |
U |
|
3.1 |
-246 |
2 |
9.4 |
-240 |
10 |
S |
|
5.1 |
-277 |
11 |
10.1 |
-270 |
15 |
S |
|
4.2 |
-194 |
20 |
9.8 |
-182 |
26 |
S |
|
3.5 |
-418 |
18 |
11.9 |
-364 |
22 |
S |
|
4.5 |
-406 |
21 |
12.6 |
-342 |
11 |
S |
|
4.6 |
-458 |
3 |
9.3 |
460 |
13 |
PS |
|
4.5 |
-420 |
19 |
6.7 |
-397 |
18 |
S |
|
6.2 |
-330 |
19 |
9.37 |
-284 |
20 |
S |
|
4.7 |
-410 |
19 |
9.7 |
-397 |
18 |
U |
|
6.4 |
-240 |
7 |
7.6 |
-232 |
7 |
S |
|
6.1 |
-136 |
26 |
9.4 |
-115 |
27 |
U |
|
6.1 |
-238 |
9 |
11.5 |
-203 |
8 |
S |
|
6.5 |
-336 |
42 |
11.7 |
-299 |
23 |
U |
|
5.7 |
-417 |
20 |
9.0 |
-396 |
16 |
S |
|
6.7 |
-419 |
15 |
8.2 |
-482 |
16 |
PS |
|
5.8 |
-477 |
19 |
10.7 |
-479 |
26 |
PS |

Figure 4.2
For KIX, the primary sequence and a comparison of the predicted and observed secondary structure. Here, H denotes a helix, U a U-turn; PRDSEC (OBSEC) is the predicted (observed) secondary structure from PHD and LINKER.
Limitations of Sequence-based Methods
As residue identity falls into the twilight zone, standard sequence-alignment algorithms will pick up false positive sequences as well as miss false negative sequences. Similarly, as sequence diversity increases, the local sequence signatures found in the Prosite (Bairoch, 1990; Bairoch, Bucher et al., 1995), Blocks (Henikoff and Henikoff, 1991), and Prints (Attwood and Beck, 1994; Attwood, Beck et al., 1997) databases will no longer be strong enough to recognize protein sequences as belonging to a functional family, even though the specific active site residues might be strictly conserved. (See Table 4.3.) To illustrate this inability to recognize local sequence signatures as the sequences diverge, we performed an analysis of the Prosite database (Release 13.0, November 1995). Of 1,152 patterns in this release of Prosite, 908 (79 percent) of the patterns were absolutely specific for their sequences (using the set of true and false positives and negatives as identified by the Prosite developers). However, as the number of instances of a local pattern increases, the number of false positives also tends to increase. For 10.5 percent of the patterns, 90 to 99 percent of the selected sequences were true positives, while for the remaining 10.5 percent of the patterns, fewer than 90 percent of the selected sequences were true positives. To overcome this deficiency, the developers of the Prosite database have begun to use weight matrices or profiles for detection of domains. Unlike the typical Prosite, Blocks, and Prints methods, they create profiles of sequence information such as residue type and solvent accessibility (Gribskov, McLachlan et al., 1987) based on the complete protein sequence, not just a small segment. As with domain-matching methods, problems inherent in matching highly divergent parts of the sequence, as well as the highly conserved functional regions, still reassert themselves.
Similarity of Global Tertiary Structure Does Not Always Imply Similarity of Function
In principle, additional information might be provided by comparing the complete tertiary structures of proteins; however, comparison of overall structure is also not enough to classify protein function unequivocally. The structural databases such as SCOP (Murzin, Brenner et al., 1995), CATH, and DALI (Holm and Sander, 1997) show significant redundancy in domain structures. Proteins such as the barrels and the sandwiches can exhibit very similar structures even though they have very different functions. Valuable information can be obtained from overall tertiary structure comparison (Murzin, 1996), but two proteins with the same global tertiary structure do not necessarily have the same function.
Proteins with Similar Function Conserve the Local Structure Around the Active Site, Even If the Global Fold Is Dissimilar
As the families become more diverse, the sequence similarity among many proteins in the family falls into and below the twilight zone. Then, standard sequence alignments have difficulty establishing a significant relationship between sequences even though one might exist. For example, the mammalian and bacterial serine proteases demonstrate that proteins with very similar functions can have very different three-dimensional structures (Branden and Tooze, 1991). The geometry of the active site would not be recognized by local sequence signatures or by overall comparison of global tertiary structures, but only from an analysis of the structure of the functional residues around the active site.
TABLE 4.3 Data for Classification of Possible Thioredoxin Sequences by the Prosite, Prints, and Blocks Algorithms
|
|
|||||||||
|
|
TP |
FP |
FN |
TP |
FP |
FN |
TP |
FP |
FN |
|
Possible Thioredoxins |
|||||||||
|
DSBC_HAEIN |
|
|
X |
|
|
X |
X |
|
|
|
THIO_CHLLT |
|
|
X |
X(2)e |
|
|
X |
|
|
|
THIO_CHRVI |
|
|
X |
X |
|
|
X |
|
|
|
THIO_RHORU |
|
|
X |
X |
|
|
|
|
X |
|
YX09_MYCTU |
|
|
X |
X |
|
|
|
|
X |
|
Y039_MYCTU |
|
|
X |
|
|
X |
|
|
X |
|
YB59_HAEIN |
|
|
X |
|
|
X |
X |
|
|
|
May Be Thioredoxins (treated here as if they are thioredoxins) |
|||||||||
|
BS2_TRYBB |
X |
|
|
X |
|
|
X |
|
|
|
FIXW_RHILE |
X |
|
|
|
|
X |
X |
|
|
|
GSBP_CHICK |
X |
|
|
X |
|
|
X |
|
|
|
RESA_BACSU |
X |
|
|
X(2)e |
|
|
X |
|
|
|
YME3_THIFF f |
|
|
X |
|
|
X |
X |
|
|
|
Probably Not Thioredoxins |
|||||||||
|
YNC4_CAEEL |
|
X |
|
|
|
|
|
|
|
|
POLG_PVYC |
|
X |
|
|
|
|
|
|
|
|
POLG_PVYN |
|
X |
|
|
|
|
|
|
|
|
POLG_PVYHU |
|
X |
|
|
|
|
|
|
|
|
POLG_PVYO |
|
X |
|
|
|
|
|
|
|
|
a Prosite: recent Prosite database online (thioredoxin examples updated 9/10/97). b TP = true positives; FP = false positives; FN = false negatives. c Prints: search of OWL26.0 database. d Blocks: search of SwissProt32. e Prints uses three different sequence signatures to recognize the thioredoxins. “2” means that this sequence was recognized by only two of the three signatures. f A plasmid in E. coli expressing this gene product complements a thioredoxin mutant, providing experimental evidence that this protein may be a glutaredoxin or thioredoxin. |
|||||||||
Development of a Three-dimensional Library of Functional Motifs
What these examples suggest is that one might be able to excise the local structure around the active site and use this local conformational signature to identify function. In fact, proteins function because of the arrangement of specific residues in three-dimensional space, The residues involved in protein function, particularly those at enzyme active sites, will be highly conserved throughout evolution. This statement seems obvious and it was clearly demonstrated experimentally by the serine protease presented above. The problem with recognizing these residues by sequence alignment is that they are likely to be distant along the sequence, even if they are close together in three-dimensional space. This makes recognition by multiple-sequence-alignment methods problematic. If protein function relies on the
specific tertiary placement of residues, then one should use that geometric information to describe functional families. We term these geometric (e.g., distances and angles) and conformational (e.g., a residue must be in a helix) descriptors "fuzzy functional forms" (FFFs). These methods do not rely on evolutionary conservation of local sequence as do the local sequence signature methods, but instead involve the construction of three-dimensional descriptors of protein function.
There are several distinct advantages to using geometric and conformational descriptors rather than local sequence signatures to describe protein function. It permits classification of proteins into families, even if there is little or no sequence identity to other proteins in the database. Thus, proteins that fall below the twilight zone of sequence identity will still be amenable to analysis. Nor does it rely on matching of the overall protein structure. Thus, proteins with similar structures but different functions will be classified differently by this method. Note that the term "function," as used here, is defined very narrowly; what is meant is the biochemical activity of the protein of interest.
The one major disadvantage of this method is that the structure of the protein must be known. However, as described below, FFFs are specific and unique enough that the structure does not have to be known to high resolution. Low- to moderate-resolution structures are sufficient for functional recognition, and current state-of-the-art prediction algorithms can often predict protein structure at sufficient resolution to allow identification of function using the FFFs. Finally, these prediction algorithms can be scaled up to analyze complete genomes.
Representative Case: The Glutaredoxin/Thioredoxin Family
Overview
In what follows, we consider the glutaredoxin/thioredoxin protein family. These proteins were selected because members of these families have tertiary structures that have been predicted by ab initio methods (e.g., in Table 4.2, 1ego is a glutaredoxin). This family also satisfies the requirement that the functional motif is not simply local in sequence, which could mean that difficulties might be expected in identifying all members of the family from sequence based-methods. Members of the glutaredoxin/ thioredoxin protein family are small proteins that catalyze thiol-disulfide exchange reactions via a redox-active pair of cysteines in the active site. While glutaredoxins and thioredoxins catalyze similar reactions, they are distinguished by their differential reactivity. Glutaredoxins contain a glutathione binding site, are reduced by glutathione (which is itself reduced by glutathione reductase), and are essential for the glutathione-dependent synthesis of deoxyribonucleotides by ribonucleotide reductase. Thioredoxins are reduced directly by the specific flavoprotein thioredoxin reductase and act as more general disulfide reductases. Ultimately, however, reducing equivalents for both proteins come from NADPH. Protein disulfide isomerases (PDIs) have been found to contain a thioredoxin-like domain and thus also have a similar activity.
The active site of the redoxin family contains three invariant residues: two cysteines and a cis-proline. Mutagenesis experiments have shown that the two cysteines separated by two residues are essential for significant protein function. The side chains of these two residues are oxidized and reduced during the reaction (Yang and Wells, 1991; Bushweller, Aslund et al., 1992). However, this local sequence signature is not sufficient to specifically select the members of the family. These two cysteines are also located at the N-terminus of an a-helix. Peptide studies suggest that the positive pole of the helix macrodipole affects the ionization of the cysteines and is important for protein function (Kortemme and Creighton, 1995, 1996). Another unique feature of the redoxin family is the presence of a cis-proline located close to the two cysteines in structure, but not in sequence. While this proline is
structurally conserved in all glutaredoxin and thioredoxin structures (Katti, Robbins et al., 1995) and is invariant in aligned sequences of known glutaredoxins and thioredoxins, its functional importance is unknown. Other residues, particularly charged residues, are also important for the Specific thiol ionization characteristics of the cysteines, but are not essential and can vary within the family (Dyson, Jeng et al., 1997).
The FFF for the glutaredoxin/thioredoxin family is based on the three-dimensional structural comparison of bacteriophage T4 glutaredoxin, 1aaz (Eklund, Ingelman et al., 1992), human thioredoxin, 4trx (Kay, Clore et al., 1990), and proline disulfide isomerase, ldsb (Martin, Bardwell et al., 1993), as well as on literature searches to find residues and structures shown to be functionally important. It consists of two cysteines separated by two residues at the N-terminus of a helix and close to a proline residue. The exact distances are described elsewhere (Fetrow and Skolnick, 1998).
Ability of the FFF to Identify the Active Site in Experimentally Determined Structures
The FFF is sufficient to distinguish proteins belonging to the redoxin family uniquely from a data set of 364 non-redundant proteins from the Brookhaven database. For this set of 364 proteins, 13 have the sequence signature -C-X-X-C-. Of these, three have a proline within the requisite distances. Of these three, only 1thx (a thioredoxin) and 1dsb (chain A, a disulfide binding protein) have the cysteines at or near the N-terminus of a helix. These two proteins are the only two true positives in the test data set, showing that this simple FFF is quite specific for the redoxin protein family. Thus, the FFF can be applied to experimental structures to identify active sites.
Application of the FFF to Predicted Structures
Is this FFF sufficient to identify the function of an inexact model of a protein, or is a high-resolution crystal or solution structure required? The structure of glutaredoxin, 1ego, was predicted with a 5.7-Å cRMSD by MONSSTER (Ortiz, Kolinski et al., 1998a,b). The sequence of this glutaredoxin exhibits less than 30 percent sequence identity to any of the three structures used to create the FFF. The redoxin FFF was applied to 25 correct structures and 56 incorrect or misfolded structures generated by MONSSTER on the 1ego sequence during the isothermal runs. It specifically selects all 25 ego-like structures as belonging to the redoxin family and rejected all 56 misfolded structures. A set of 267 correctly and incorrectly predicted structures produced by the MONSSTER algorithm for five different proteins was then created. The glutaredoxin/thioredoxin FFF was specific for the correctly folded ego structures and did not recognize any of the other correctly or incorrectly folded structures.
Screening of Entire Genomes
This sequence-to-structure-to-function concept has been applied to the analysis of the complete E. coli genome; i.e., all E. coli open reading frames (ORFs) are screened for the thiol-disulfide oxidoreductase activity of the glutaredoxin/thioredoxin protein family. The method can identify the active site residues in 10 sequences that are known to or proposed to exhibit this activity. Furthermore, oxidoreductase activity is predicted in two other sequences that have not been previously identified. These results are summarized in Table 4.4. The method distinguishes protein pairs with similar active sites from protein pairs that are just topological cousins, i.e., those having similar global folds, but not necessarily similar active sites.
TABLE 4.4 Glutaredoxins and Thioredoxins Identified in E. coli Strain K-12
|
|
Functional Motifa |
|||||||
|
Database Nameb |
Thrd/FFFc |
Blst/FFFd |
ps |
pps |
pb |
b |
Database Description |
|
|
GLR1_ECOLI |
x |
x |
x |
x |
x |
x |
glutaredoxin 1 |
|
|
GLR2_ECOLI |
x |
|
x |
|
xe |
x |
glutaredoxin 2 |
|
|
GLR3_ECOLI |
x |
x |
x |
x |
x |
x |
glutaredoxin 3 |
|
|
THIO_ECOLI |
x |
x |
x |
x |
x |
x |
thioredoxin |
|
|
DSBA_ECOLI |
x |
x |
x |
|
xf |
x |
thiol-disulfide interchange protein |
|
|
DSBC_ECOLI |
x |
|
x |
|
xe |
x |
thiol-disulfide interchange protein |
|
|
DSBD_ECOLI |
x |
x |
x |
x |
x |
x |
c-type cytochrome biogenesis protein (inner-membrane Cu tolerance protein) |
|
|
DSBE_ECOLI |
x |
|
x |
xe |
x |
x |
thiol-disulfide interchange protein; (cyto c biogenesis protein CCMG) |
|
|
YFIG_ECOLI |
x |
x |
x |
x |
x |
x |
hypothetical thioredoxin-like protein |
|
|
NRDH_ECOLI |
x |
|
|
|
xf |
x |
glutaredoxin-like NRDH protein |
|
|
NRDG_ECOLI |
x |
|
|
|
|
|
anaerobic ribonucleoside triphosphate inactivating protein |
|
|
B0853 |
x |
|
|
|
|
|
ORF; putative regulatory protein |
|
|
YIEJ_ECOLI |
|
x |
|
|
|
|
hypothetical protein in tnaB-bglB intergenic region |
|
|
a Functional motif: Search of each sequence found by either BLAST/FFF or Thread/FFF protocols against the local signature databases Prosite, Prints using the Prosite scoring method, Prints using the Blocks scoring method, or Blocks. Each motif database was searched with the given sequence, and the returned scores were analyzed to see if the thioredoxin or glutaredoxin families were identified. b Database name: This is the database identifier for each sequence. All sequences come from the SwissProt database, except B0853, which is the label given by the E. coli genome database. This sequence can also be accessed by the GenBank accession number ECAE000187. c Thrd/FFF: Alignment of E. coil open reading frame (ORF) to the sequences of 1ego, 1dsb (chain A), or 2trx (chain A) using a threading algorithm, followed by analysis of the resulting sequence-sequence alignment for the active site residues specified by the fuzzy functional form (FFF) for the thiol-disulfide oxidoreductase activity of the glutaredoxin/ thioredoxin family. Threading results are for a combination of three different scoring methods, sq, br, and tt, as described by Godzik and coworkers (Jaroszewksi et al. 1998). d Blst/FFF: Alignment of each E. coli ORF to the sequences of the lego, ldsb, chain A, and 2trx, chain A proteins using the BLAST search protocol, followed by analysis of the resulting sequence-sequence alignment for the active site residues specified by the thiol-disulfide oxidoreductase activity of the glutaredoxin/thioredoxin family. Results reported here are for a combination of the gapped-BLAST protocol and the PSI-BLAST alignment protocols. All sequences marked are found by both gapped- and PSI-BLAST, except YIFJ-ECOLI, which is found only by gapped-BLAST. e Prints has three patterns for glutaredoxin/thioredoxin activity. This sequence hits only one of the patterns. f Prints has three patterns for glutaredoxin/thioredoxin activity. This sequence hits only two of the patterns. |
||||||||
Computational Requirements for Genome Scale Structure/Function Prediction
The computational requirements of this type of genomic screening analysis are quite substantial. For example, contemporary ab initio protein-folding methods are applicable to single domain proteins—up to about 150 or so residues in length—and can identify possible novel protein folds. Threading is
TABLE 4.5 CPU Requirements for Protein Structure Prediction on the Genomic Scale
|
Genome |
Number of ORFS |
Number of ORFS <150 Residues |
Ab Initio Folding CPU Timea |
CPU Timeb |
|
M. genitalium |
408 |
82 |
4,920 |
2 |
|
H. influenzae |
1,680 |
369 |
22,410 |
8.4 |
|
M. jannaschii |
1,735 |
425 |
25,500 |
8.9 |
|
E. coli |
4,290 |
879 |
52,740 |
21.5 |
|
S. cerevisiae |
5,885 |
1,433 |
85,980 |
29.4 |
|
a Assumes an average of 60 CPU days to perform 1,000 folding simulations per sequence on a single processor of an SGI ORIGIN 200 running at 180 megahertz. b 200 sequences threaded through 1,000 structures takes 1 CPU day. |
||||
significantly less expensive. Table 4.5 gives a summary of the CPU requirements for protein structure prediction on the genomic scale. Thus, given the extensive CPU requirements and the large number of genomic sequences, this type of sequence-to-structure-to-function paradigm would greatly benefit from the availability of teraflops-class machines. This would allow for the construction of low- to moderate-resolution predicted structures of a substantial fraction of proteins in the genome, as well as the prediction of their molecular function. Since these calculations are basically data parallel, they should be done on a machine composed of a large number of loosely coupled processors; e.g., farms of PCs are one means of achieving this. This is typical of many but not all types of calculations at the interface of chemistry and biology.
Outlook for the Future
While low- to moderate-resolution models can be used to predict protein biochemical activity, they are too crude to be used in drug ligand design. Techniques that allow for refinement of these low-resolution to higher-resolution models must be developed. One can imagine a hierarchical approach where the overall topology of the protein is predicted using a reduced protein model, and then atomic detail is added. Such simulations being done at atomic detail will be very CPU-intensive and can profitably exploit the parallelism of current molecular dynamics codes such as AMBER (Pearlman, Case et al., 1991) or CHARMM (Brooks, Bruccoleri et al., 1983). Recently there has been encouraging progress along this direction both for folding of small proteins at atomic detail (Duan, Wang et al., 1998) and for the refinement of protein structures starting from a reduced protein model and finishing with molecules at atomic detail (Simmerling, Lee et al., 1998). To accomplish this goal, in general, will require the development of more efficient conformational sampling algorithms as well as better potentials that can discriminate the native conformation from the myriad of alternative structures.
In the area of structural genomics, where the objective is to determine the structure of all possible types of protein folds (Holm and Sander, 1996), computation will also play a key role. This will happen in sequence selection where the goal is to identify sequences likely to adopt novel folds and where ab initio techniques may prove to be particularly useful, as well as in the development of techniques that will allow for more rapid structure determination. Here, approaches that combine a limited amount of experimental data with structure prediction may prove to be particularly powerful (Monge, Friesner et al., 1994; Aszodi, Gradwell et al., 1995; Monge, Lathrop et al., 1995; Mumenthaler and Braun, 1995; Dandekar and Argos, 1996; Skolnick, Kolinski et al., 1997; Kolinski and Skolnick, 1998). Such
experimental data may come from nuclear magnetic resonance, from electron microscopy, and from low-resolution X-ray crystal structures.
Another promising area of investigation will be in the prediction of protein binding regions. This will be the first step toward identifying multidomain interactions, both in the sense of predicting which proteins interact as well as where they interact. Then, the simulation of more complex interactions involving the components of various signaling pathways and metabolic cascades will have to be addressed. The very elegant studies of Schulten and coworkers on the light harvesting complex are an excellent example of the power of such approaches (Hu and Schulten, 1998). More generally, the simulation of membrane proteins and the prediction of their structure and function will also be a very important, computer-intensive area of investigation (Milik and Skolnick, 1992, 1993; Heijne, 1994, 1995; Stowell and Rees, 1995; Casadio, Fariselli et al., 1996) and will be the active focus of future research in the next 5 to 10 years. In addition to studies at full atomic detail, hierarchical approaches that represent the system at different levels of detail will be developed. In this regard, an interesting preliminary study is found in the simulation of virus coat protein assembly (Rapaport, Johnson et al., 1998).
Another very important area of investigation that touches on the areas of computer science, biology, and chemistry will be in the development and presentation of large databases containing all that is known about a given protein, its structure, and molecular and physiological function. Basically, since so much information is and will be available, means must be developed to make it usable and understandable to both the specialist and the nonspecialist alike. This is a very outstanding unsolved problem, but it is a reasonable guess that Web-based tools are going to be very important.
Summary
These studies demonstrate that protein function prediction based on the sequence-to-structure-to-function paradigm can successfully compete with more standard sequence-based approaches and may well identify the function of additional proteins in the twilight zone of sequence identity. What is very encouraging is that low-resolution structures as provided by state-of-the-art tertiary structure predictions can identify active sites by using appropriate three-dimensional conformational descriptors, the fuzzy functional forms. Future methodological developments may allow for the prediction of protein structures at the resolution required for automated drug design. This will enable the sequence-to-structure-to-function paradigm to realize its full potential. More generally, large-scale simulations that describe the interactions of large protein (and/or membrane) aggregates will be undertaken in the near future. Such simulations will not only provide fundamental insights into how various cellular processes work at the microscopic and mesoscopic level, but may also suggest therapeutic approaches at the molecular level for the treatment of numerous diseases. These advances in algorithms and techniques at the interface of biology and chemistry will rely on the use of large numbers of inexpensive computers. Often, these can be loosely coupled, but other problems demand closely coupled, parallel machines. Whatever the mode of parallelism, advances in computational biology will, depending on the specific problem, require the availability of 1 to 100 teraflops-class machines. Given the advances in raw CPU power as well as theoretical understanding, there is every reason to believe computational biology and chemistry will play a major role in the genomics revolution.
Literature Cited
Altschul, S., T. Madden, et al. (1997). Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402.
Aszodi, A., M.J. Gradwell, et al. (1995). Global fold determination from a small number of distance restraints. J. Mol. Biol 248: 308-326.
Attwood, T. and M. Beck (1994). PRINTS—A protein motif fingerprint database. Protein Eng. 7: 841-848.
Attwood, T., M. Beck, et al. (1997). Novel developments with the PRINTS protein fingerprint database. Nucleic Acids Res. 25: 212-216.
Bairoch, A. (1990). Prosite: A Dictionary of Protein Sites and Patterns . Department de Biochimie Medicale, Universite de Geneva, Geneva.
Bairoch, A., P. Bucher, et al. (1995). The PROSITE database, its status in 1995. Nucleic Acids Res. 241: 189-196.
Branden, C. and J. Tooze (1991). Introduction to Protein Structure . New York and London, Garland Publishing, Inc.
Brindle, P., K. and M.R. Montminy (1992). The CREB family of transcription factors. Curr. Opin. Genet. Develop. 2: 199-204.
Brooks, B.R., R. Bruccoleri, et al. (1983). CHARMM: A program for macromolecular energy minimization, and molecular dynamics. J. Comp. Chem. 4: 187-217.
Bryant, S.H. and C.E. Lawrence (1993). An empirical energy function for threading protein sequence through folding motif. Proteins 16:92-112.
Bult, C.J., O. White, et al. (1996). Complete genome sequence of the methanogenic archaeon Methanococcus jannaschii. Science 273: 1058-1073.
Bushweller, J.H., F. Aslund, et al. (1992). Structural and functional characterization of the mutant Escherichia coli glutaredoxin (C14-S) and its mixed disulfide with glutathione. Biochemistry 31: 9288-9293.
Casadio, R., P. Fariselli, et al. (1996). A predictor of transmembrane a-helix domains of proteins based on neural networks. Eur. Biophys. J. 24: 165-178.
Casari, G., C. Ouzounis, et al. (1996). GeneQuiz II: Automatic function assignment for genome sequence analysis. The First Annual Pacific Symposium on Biocomputing. World Scientific, pp. 708-709.
Dandekar, T. and P. Argos (1996). Identifying the tertiary fold of small proteins with different topologies from sequence and secondary structure using the genetic algorithm and extended criteria specific for strand regions. J. Mol. Biol. 256: 645-660.
Duan, Y., L. Wang, et al. (1998). The early stage of folding of villin headpiece subdomain observed in a 200 nanosecond fully solvated molecular dynamics simulation. Proc. Natl. Acad. Sci. U.S.A. 95: 9897-9902.
Dyson, H.J., M.F. Jeng, et al. (1997). Effects of buried charged groups on cysteine thiol ionization and reactivity in Escherichia coli thioredoxin: Structural and functional characterization of mutants of Asp 26 and Lys 57. Biochemistry 36: 2622-2636.
Eklund, H., M. Ingelman, et al. (1992). Structure of oxidized bacteriophage T4 glutaredoxin (thioredoxin). Refinement of native and mutant proteins. J. Mol. Biol. 228: 596-618.
Fetrow, J., A. Godzik, et al. (1998). Functional analysis of the Escherichia coli genome using the sequence-to-structure-to-function paradigm: Identification of proteins exhibiting the glutaredoxin/thioredoxin disulfide oxidoreductase activity. J. Mol. Biol. 282: 703-711.
Fetrow, J. and J. Skolnick (1998). Method for prediction of protein function from sequence using the sequence-to-structure-to-function paradigm with application to glutaredoxins/thioredoxins and T1 ribonucleases. J. Mol. Biol. 281: 949-968.
Göbel, U., C. Sander, et al. (1994). Correlated mutations and residue contacts in proteins. Proteins 18: 309-317.
Godzik, A., A. Kolinski, et al. (1993). De novo and inverse folding predictions of protein structure and dynamics. J. Comp. Aided Mol. Design 7: 397-438.
Godzik, A., J. Skolnick, et al. (1992). A topology fingerprint approach to the inverse folding problem. J. Mol. Biol. 227: 227-238.
Gribskov, M., A.D. McLachlan, et al. (1987). Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. U.S.A. 84: 4355-4358.
Heijne, G.v. (1994). Membrane proteins: From sequence to structure. Annu. Rev. Biophys. Biomol. Struct. 23: 167-192.
Heijne, G. v. (1995). Membrane protein assembly: Rules of the game. Bioessays 17(1): 25-30.
Henikoff, S. and J. Henikoff (1991). Automated assembly of protein blocks for database searching. Nucleic Acids Res. 19: 6565-6572.
Holm, L. and C. Sander (1996). Mapping the protein universe. Science 273: 595-602.
Holm, L. and C. Sander (1997). Dali/FSSP classification of three dimensional protein folds. Nucleic Acids Res. 25: 231-234.
Hu, X. and K. Schulten (1998). Model for the light harvesting complex I (B875) of Rhodobacter spheroides. Biophys. J. 75: 683-694.
Jaroszewski, L,Rychlewski, L., et al. (1998). Fold prediction by a hierarchy of sequence, threading, and modeling methods. Protein Sci. 7: 1431-1440.
Katti, S.K., A.H. Robbins, et al. (1995). Crystal structure of thioltransferase at 2.2 Å resolution. Protein Sci. 4: 1998-2005.
Kay, J.D.F., G.M. Clore, et al. (1990). Studies on the solution conformation of human thioredoxin using heteronuclear15N-1H nuclear magnetic resonance spectroscopy. Biochemistry 29: 1566-1572.
Kolinski, A. and J. Skolnick (1998). Assembly of protein structure from sparse experimental data: An efficient Monte Carlo model. Proteins 32: 475-494.
Kolinski, A., J. Skolnick, et al. (1997). A method for the prediction of surface U-turns and transglobular connections in small proteins. Proteins 27: 290-308.
Kolinski, A.K. and J. Skolnick (1996). Lattice Models of Protein Folding, Dynamics and Thermodynamics. Austin, Tex., R.G. Landes Company.
Kortemme, T. and T.E. Creighton (1995). Ionisation of cysteine residues at the termini of model alpha-helical peptides. Relevance to unusual thiol pKa values in proteins of the thioredoxin family. J. Mol. Biol. 253: 799-812.
Kortemme, T. and T.E. Creighton (1996). Electrostatic interactions in the active site of the N-terminal thioredoxin-like domain of protein disulfide isomerase. Biochemistry 35: 14503-14511.
Martin, J.L., J.C. Bardwell, et al. (1993). Crystal structure of the DsbA protein required for disulphide bond formation in vivo. Nature 365: 464-468.
Milik, M. and J. Skolnick (1992). Spontaneous insertion of polypeptide chains into membranes: A Monte Carlo model. Proc. Natl. Acad. Sci. U.S.A. 89: 9391-9395.
Milik, M. and J. Skolnick (1993). Insertion of peptide chains into lipid membranes. An off-lattice Monte Carlo dynamics models. Proteins 15: 10-25.
Miller, R.T., D.T. Jones, et al. (1996). Protein fold recognition by sequence threading: Tools and assessment techniques. Federation of American Societies for Experimental Biology (FASEB) Journal 10: 171-178.
Monge, A., R.A. Friesner, et el. (1994). An algorithm to generate low-resolution protein tertiary structures from knowledge of secondary structure. Proc. Natl. Acad, Sci. U.S.A. 91: 5027-5029.
Monge, A., E.J.P. Lathrop, et al. (1995). Computer modeling of protein folding: Conformational and energetic analysis of reduced and detailed protein models. J. Mol. Biol. 247: 995-1012.
Mumenthaler, C. and W. Braun (1995). Predicting the helix packing of globular proteins by self-correcting distance geometry. Prot. Sci. 4: 863-871.
Murzin, A.G. (1996). Structural classification of proteins: New superfamilies. Curr. Opin. Struct. Biol. 6: 386-394.
Murzin, A.G., S.E. Brenner, et al. (1995). Scop: A structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 247: 536-540.
Olmea, O. and A. Valencia (1997). Improving contact predictions by the combination of correlated mutations and other sources of sequence information. Folding & Design 2: S25-S32.
Orengo, C.A., A.D. Michie, et al. (1997). CATH—A hierarchic classification of protein domain structures. Structure 5: 1093-1108.
Ortiz, A., A. Kolinski, et al. (1998a). Fold assembly of small proteins using Monte Carlo simulations driven by restraints derived from multiple sequence alignments . J. Mol. Biol. 277: 419-448.
Ortiz, A., A. Kolinski, et al. (1998b). Nativelike topology assembly of small proteins using predicted restraints in Monte Carlo simulations. Proc. Natl. Acad. Sci. U.S.A. 95: 1020-1025.
Ortiz, A., A. Kolinski, et al. (1998c). Tertiary structure prediction of the KiX domain of CBP using Monte Carlo simulations driven by restraints derived from multiple sequence alignments. Proteins 30: 287-294.
Pearlman, D.A., D.A. Case, et al. (1991). Assisted Model Building with Energy Refinement (AMBER) code. University of California, San Francisco.
Pearson, W. and D. Lipman (1988). Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. U.S.A. 85: 2444-2448.
Radhakrishnan, I., G.C. Perez-Alvarado, et al. (1997). Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator:coactivator interactions. Cell 91: 741-752.
Rapaport, D.C., J.E. Johnson, et al. (1998). Supramolecular self-assembly: Molecular dynamics modeling of polyhedral shell formation. Comput. Phys. Commun., submitted.
Rastan, S. and L. Beeley (1997). Functional genomics: Going forwards from the databases. Curr. Opin. Genet Devel. 7: 777-783.
Rost, B. and C. Sander (1993). Prediction of secondary structure at better than 70% accuracy. J. Mol. Biol. 232: 584-599.
Rost, B., R. Schneider, et al. (1993). Progress in protein structure prediction? TIBS 18: 120-123.
Sali, A. and T. Blundell (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234: 779-815.
Sander, C. and R. Schneider (1991). Database of homology derived protein structures and the structural meaning of sequence alignment. Proteins 9: 56-68.
Simmerling, C., M. Lee, et al. (1998). Combining MONSSTER and LES/PME to predict protein structure from amino acid sequence: Application to the small protein CMTI-1 J. Am. Chem. Soc., submitted.
Skolnick, J., A. Kolinski, et al. (1997). MONSSTER: A method for folding globular proteins with a small number of distance restraints. J. Mol. Biol. 265: 217-241.
Stowell, M.H.B. and D.C. Rees (1995). Structure and stability of membrane proteins. Adv. Protein Chem. 46: 279-311.
Thomas, D.J., G. Cesari, et al. (1996). The prediction of protein contacts from multiple sequence alignment. Protein Eng. 11: 941-948.
Wodak, S.J. and M.J. Rooman (1993). Generating and testing protein folds. Curr. Opin. Struct. Biol. 3: 247-259.
Yang, Y.F. and W.W. Wells (1991). Identification and characterization of the functional amino acids at the active center of pig liver thioltransferase by site-directed mutagenesis. J. Biol. Chem. 266: 12759-12765.
Discussion
William Winter, SUNY-ESF, Syracuse: Glycosylation has to play a major role in the final selection of a particular protein conformation in many proteins where it does occur. Are you doing anything at all to use that kind of information to make further selections once you have determined a family of possible structures?
Jeffrey Skolnick: Not yet, but we are aware of the problem. So far we have picked molecular functions that are basically self-contained by design because we did not pick the hardest case first. But you are absolutely right, glycosylation is extremely important. The problem there is that not a lot is known. Even the potentials that you should put in to describe the conformational spectrum are not well established. People are still developing these, so that field is very much in its infancy. Our view has been, yes, we recognize it is important, and especially in a biological context it is very, very important; it protects the proteins and keeps them from being chewed up, but we quite frankly wanted to consider the simplest cases first to see if the basic approaches could work—choose molecular functions or biochemical functions where it is apparently not believed to be important and then work our way up. But, yes, you are absolutely right. One day we or someone else will have to deal with that problem, but I think it is premature at this stage of the game.
David Dixon, Pacific Northwest National Laboratory: Jeff, have you looked at or have you started thinking about the fact that there is also spatial resolution within a cell, and have you looked at how you connect your proteins up into cell signaling pathways?
Jeffrey Skolnick: Yes, we have already started, at least on a very schematic level, simulating peptide insertion and protein insertion into membranes, treating the system, you know, with spatial anisotropy. You have a membrane region that could be treated at various levels of detail in the interfacial regions, bulk regions, but only on a very, very schematic level at this point. As it is, these kinds of calculations really tax any resources that we can get hold of, and we are not sure about adding additional details other than on a very simplified level. And then we are not even sure that the descriptives are sufficiently good that it would be worthwhile. I mean, we are trying to proceed on a very building-block basis: establish something that works, validate it, move on, make it more complicated, move on. My guess is the next thing we are going to do is membrane protein tertiary structure prediction, and there there are some encouraging results.


















