
FDA Regulatory Review
Paper Auditing
Presented by Jay P. Siegel, M.D.
Director, Office of Therapeutics Research and Review,
Center for Biologics Evaluation and Research, Food and Drug Administration
The Food and Drug Administration (FDA) clinical reviewer audit focuses on whether a study report and related documents are an accurate reflection of the methods and outcomes of a clinical trial. Several factors determine the extent of an audit. One key factor is the importance of the data, that is, their impact on decision making and labeling. Those aspects of the conduct of the study which are deemed most critical to key conclusions (e.g., subject eligibility, level of drug compliance, and use of concomitant medications) will receive the most attention. During the audit, primary endpoints generally receive greater attention than secondary endpoints. The importance of the clinical trial to the overall Biologics License Application (BLA) or New Drug Application (NDA) is also considered; for example, is it a pivotal study or the principal source of safety data?
The extent of the FDA reviewers' audit of the data is also influenced by aspects of the study design, especially blinding, the objectivity of the endpoints, and whether the trial is designed to demonstrate equivalence or superiority. Lack of blinding increases concern about many aspects of study treatments and assessments. Equivalence studies are far more likely to have errors whereby an investigator will incorrectly accept the hypothesis that the drug has the desired effect (type I error). The general quality of the data report and the amount of missing data can also influence the intensity of the audit; it behooves the sponsor to include an open discussion of data deficiencies.
FDA's experience with a sponsor or investigator may also affect the nature and extent of the audit, particularly when there has been trouble with prior submissions or previous warning letters. FDA clinical reviewers have, to date, not usually considered the extent of monitoring and auditing conducted by the sponsor as a key factor in determining the extent and nature of their own audits. However, studying and attempting to validate the sponsor's quality assurance
(QA) efforts can be powerful and effective. Of note, there is substantial variation in the approach to data quality auditing within FDA, and factors such as deadlines, workloads, and competing priorities can significantly affect the nature and extent of auditing.
FDA clinical reviewers use four basic auditing tools: (1) checks for compliance with the protocol, (2) checks for data consistency, (3) checks of clinical judgment calls, and (4) interactions with field inspectors. Compliance with the protocol is a central part of both medical and statistical reviews and focuses on inclusion and exclusion criteria, blinding, randomization, treatment, assessment, and analysis. Consistency checks include comparisons among centers in multicenter trials, comparisons of data over time, especially in studies with multiyear accrual of subjects, and checks of the consistency of data in various formats (e.g., tables, summaries, listings, and labeling). Assessments that require clinical judgment—such as cause of death, the cause of an adverse event, or success versus failure—are often critically evaluated by clinical reviewers. The clinical reviewer interacts with the field auditor to help decide which sites will be visited, which data will receive the closest scrutiny on-site, and which documents, if any, will be retrieved from study sites for further scrutiny.
Specific elements that are checked include randomization, blinding and unblinding, inclusion and exclusion criteria, treatment of more sensitive populations, the level of drug compliance, and the manner in which efficacy and safety data are reported. Other data points examined on a patient-by-patient basis include death, adverse events that lead to withdrawal from the study, and other serious adverse events. Specific approaches are used for audits of the various trial elements and data types.
Paper audits represent a substantial investment of FDA resources. Frequently, half or more of the time spent by clinical and statistical reviewers reviewing a marketing application is spent on assessing data validity in the broad sense.
Clinical Site Review and Institutional Review Board Audits
Presented by David Lepay, M.D., Ph.D.
Director, Division of Scientific Investigations
Center for Drug Evaluation and Research, Food and Drug Administration
On-site inspections complement paper audits in the Food and Drug Administration's (FDA's) efforts to ensure data quality and integrity in clinical trials. The Bioresearch Monitoring Program, which was established in the late 1970s, seeks to detect sloppiness or misconduct that might affect human subject protection, data integrity, and sound decision making on applications. It also seeks to prevent data quality and integrity problems before they occur. Inspections are conducted in accordance with published standard operating procedures that are updated every 3
years and that focus on five groups: Institutional Review Boards (IRBs), clinical investigators, sponsors, contract research organizations, and monitors.
The primary purpose of an IRB review is to ensure the protection of the rights and welfare of human subjects. FDA's Bioresearch Monitoring Program conducts almost 200 IRB inspections per year, with routine inspections of historically compliant IRBs occurring once every 5 years, on average. However, the Bioresearch Monitoring Program focuses particular attention on new IRBs as well as those that oversee large numbers of studies or studies with large numbers of subjects, those that oversee higher-risk Phase 2 and 3 trials, and IRBs with a history of poor compliance.
Clinical investigator inspections concentrate on individual sites, validating data in the marketing application against original source data. This approach provides the opportunity to interact with clinical investigators and site managers and inquire firsthand about potential data integrity issues. Planning and evaluation of inspections requires communication and coordination not only with the study site, but also across FDA divisions, with sponsors, and with international regulatory authorities.
Review times have decreased since the passage of the Prescription Drug User Fee Act of 1992, but now FDA must meet even stricter time lines under PDUFA-2. This raises several issues:
- Will the Bioresearch Monitoring Program be able to appropriately expand the number of inspections, to address systemic problems that are discovered at one site and that are generalized across a multisite trial, and to have a positive impact on assessments of the study findings?
- Should the sponsor rather than FDA be responsible for performing validity assessments across the entire study when problems are disclosed at a single site?
- Is there sufficient flexibility in the PDUFA timeline to allow for contingencies?
The number of applications to FDA has increased, and with it the inspection workload has also increased. FDA's Center for Drug Evaluation and Research (CDER), for example, experienced a 40 percent increase in New Drug Application (NDA) filings between 1992 and 1997, from 73 in 1992 to 104 in 1997. The number of clinical investigator inspections has increased accordingly, to about 350 per year, but many applications involve scores of sites and investigators. As a result, it is not clear that FDA is inspecting enough sites or investigators per application to ensure public confidence in data quality and integrity. Table 1 presents the average time and costs required to conduct a clinical investigator or an IRB inspection.
TABLE 1 Time and Costs of Each Clinical Investigator or IRB Inspection
|
Type of Inspection |
Time (hours)* |
Expense ($) |
|
Domestic clinical investigator |
70 |
7,350 |
|
Foreign clinical investigator |
91 |
9,400 |
|
IRB |
51 |
|
|
* Includes preparation, conduct of inspection, and report write-up. |
||
In deciding which sites to inspect, FDA tries to select those sites that have the greatest impact in terms of their contribution either to treatment effect or to the statistical significance of that effect (i.e., because the site contributes the greatest number of subjects). All reports of scientific misconduct received by FDA are investigated and may lead to inspections. Still, the selection of investigators for inspection is not by random sampling, and this precludes generalization of the inspection findings to the population of clinical investigators as a whole. Notwithstanding this caveat, results for the 302 domestic clinical investigator inspections conducted in 1997 indicated the following (Figure 2):
- 40.3 percent of domestic inspections exhibited no deviation from regulations and were classified as no action indicated (NAI);
- 56.3 percent of domestic inspections revealed objectionable conditions that were deemed correctable by action or reply by the investigator and were classified as voluntary action indicated (VAI);
- 3.3 percent of domestic inspections revealed major deviations from regulations or official action indicated (OAI).
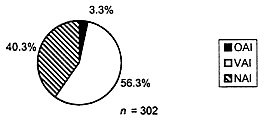
FIGURE 2. A total of 302 domestic clinical investigator inspections were conducted in 1997. A slight majority of inspections revealed at least minor deficiencies and in most cases these deficiencies were of a nature that should be detectable, correctable, and preventable with effective monitoring.
More than 50 percent of domestic clinical investigator inspections revealed at least minor deficiencies. In most cases, these deficiencies were of a nature that should have been detectable, correctable, and preventable with effective monitoring. The nature of these deficiencies has tended to remain constant over time: failure to follow the protocol (25 percent), problems with the consent form or process (21 percent), failure to maintain adequate source records (20 percent), failure to maintain accountability for investigational drugs (13 percent), and problems with adverse event reporting (5 percent).
Scientific misconduct is a rarity, accounting for no more than 1 to 3 percent of all inspections per year, but these cases receive a great deal of attention and can negatively affect public confidence in the clinical trial process. Moreover, seriously noncompliant investigators may work on multiple trials for multiple sponsors and may therefore affect numerous applications submitted to the agency. Failure analysis of six recent cases of serious scientific misconduct revealed that one clinical investigator was working on 91 Investigational New Drugs (INDs) or NDAs for 47 different sponsors (Table 2). At least 13 different sponsors had used two or more of these investigators, and 1 sponsor had used all six. Although most violations should have been detected by adequate monitoring, none were reported to FDA by the study sponsors. Disclosure of involvement with multiple trial or multiple sponsors may help identify and prevent noncompliance among investigators.
The number of clinical investigators is estimated to exceed 30,000 and is increasing at a rate of 8 to 10 percent per year. Clinical trial experience varies among clinical investigators. Other variables may also lead to a variable quality of work among clinical investigators, for example, differences in training, financial pressures, resistance to correction, inclination to delegate, and degree of personal involvement in the study. Qualifications, training, and experience also vary among monitors. This may be reflected in the degree of detail reviewed during inspection and the manner in which monitors respond to and report problems. The quality of monitoring may be a function of the monitoring visits themselves: their number, timing, and choice of sites. The degree to which monitors interact with investigators and sponsors is also important. Many monitors work closely with investigators, providing frank discussion of problems and recommendations for correction. When regulatory compliance cannot be achieved promptly, it is the sponsor's responsibility to terminate a seriously violative clinical investigator. Questions as to whether sponsors are meeting this particular responsibility have surfaced recently.
In addition to its domestic inspection program, CDER conducts international inspections when the data from international sites are pivotal to the regulatory decision-making process. The number of non-U.S. inspections increased from 5 in 1991 to 36 in 1997, reflecting the globalization of clinical trials. FDA has now conducted inspections of clinical investigators in 30 foreign countries. Serious data quality and integrity problems are more common in foreign inspections than in domestic inspections: in 1997, 17 percent of international inspections were classified by FDA as requiring additional regulatory actions, whereas
only 4 percent of domestic sites received this classification (see Figure 2). Some small improvements have been noted among nations that participate in the International Conference on Harmonization (ICH). However, it remains an open question whether the broader adoption of ICH standards will lead to an improvement in data quality and integrity unless the adoption of such standards is accompanied by a mechanism for inspecting and enforcing these ICH standards. The latest revision of FDA's compliance program guidance manual, which includes reference to ICH good clinical practices and which provides guidance on computer systems, is in final review. The FDA's clinical investigator compliance program suggests that investigators keep copies of all source data and documents submitted to a sponsor to ensure FDA's ability to reconstruct the study while on-site.
TABLE 2 Failure Analysis of Recent Cases of Serious Scientific Misconduct
|
Clinical Investigator |
No. of Applications |
No. of Sponsors |
|
A |
91 |
47 |
|
B |
49 |
25 |
|
C |
43 |
21 |
|
D |
21 |
17 |
|
E |
12 |
6 |
|
F |
6 |
6 |
FDA's review divisions and Bioresearch Monitoring Program are available to assist sponsors during the design and execution of clinical trials. This may include straightforward dialogue on the specific data to be collected and the importance of such data in supporting a safety or efficacy determination. In this context, CDER is also willing to meet with sponsors to develop a forthright understanding of the approaches and adequacy of trial monitoring and auditing proposals.
Sanctions
Presented by Stan W. Woollen
Deputy Director, Division of Scientific Investigations
Center for Drug Evaluation and Research, Food and Drug Administration
A major goal of Food and Drug Administration (FDA) sanctions is not to punish wrongdoers but, rather, to protect the integrity of the approval process and the rights and welfare of human subjects. Sanctions accomplish this by notifying affected parties that corrective action is required and excluding the data or the parties that have corrupted the process. Sanctions can be imposed against
(1) clinical investigators, (2) sponsors, and (3) Institutional Review Boards. However, sanctions are rarely imposed because few people or organizations deliberately fail to comply with FDA requirements.
Sanctions available for use against clinical investigators include, in order of increasing severity, warning letters, consent agreements, formal disqualification, debarment, and prosecution under criminal statutes. Warning letters communicate the need for corrective action, and FDA follows up to see that corrective actions are taken. Warning letters are not sent to foreign investigators unless they are working under an Investigational New Drug application. Repeated or deliberate noncompliance or submission of false information leads to formal disqualification, barring the investigator from receiving investigational products. A formal disqualification requires a regulatory hearing and can be a time-consuming process that takes 2 to 4 years, during which time the investigator may continue to conduct studies.
An alternative is a consent agreement, which results in voluntary disqualification or negotiated restrictions on the investigators' activities, such as the number of studies an investigator may perform oversight by another investigator, or third-party verification of data. Consent agreements reduce legal and administrative costs and give FDA the ability to tailor the sanctions imposed.
Debarment under the Generic Drug Debarment Act effectively prevents an individual from working in the drug industry. FDA will not accept or review applications from individuals or companies who have been debarred. Furthermore, prosecution under criminal statutes, for example, for fraud against the government, will also result in debarment, but these most extreme sanctions are rare.
Sanctions may be imposed against sponsors (and against contract research organizations that assume the responsibility of a sponsor) for problems in their FDA submissions and for problems that arise at study sites. Problems with submissions to FDA, such as false statements of material fact or patterns of error that result in widespread problems with data integrity, may be handled under the application integrity policy. Under its application integrity policy, FDA will defer substantive scientific review until a validity assessment is completed and may refuse to approve or may withdraw approval of an application.
Regulations related to monitoring at the clinical site and dealing with non-compliant clinical investigators are vague, and the appropriate sanctions are not defined. Frequently, sponsors fail to report the problems or the corrective actions that have been taken. For example, none of the sponsors who used six egregiously noncompliant investigators reported the investigators to FDA. In these cases, sponsors excluded the data but did not terminate activities at the site and were not required by regulation to report the investigators. Issues concerning how FDA can ensure proper monitoring of clinical sites and correction and reporting of problems in the face of existing regulatory requirements remain to be addressed.
Assessment of Drugs
Presented by Murray Lumpkin, M.D.
Deputy Director for Review Management
Center for Drug Evaluation and Research, Food and Drug Administration
When the results of paper audit and on-site review are completed, two fundamental questions may determine their impact on decision making:
|
1. |
Are the problems with data integrity of such a magnitude that they raise questions regarding the integrity of the entire submission or the complete study? |
|
2. |
If the problems are limited to a single site, will the integrity of the overall study be maintained if the data from the problem site are removed from the analysis? |
Actual cases can illustrate the range of deficiencies that the Food and Drug Administration (FDA) encounters and how they influence the agency's assessment of New Drug Applications (NDAS) for molecular agents. In calendar year 1997, the Center for Drug Evaluation and Research (CDER) initiated actions on 235 NDAs, 121 of which were approved. Some 39 of the approved NDAs were for new molecular entities, and 37 of these were applications whose clinical data had been inspected. (The other two were orphan drug applications with nontraditional clinical data that were not amenable to conventional validation.) Those 37 approved NDAs involved inspections of 180 domestic sites, ranging from a low of 2 sites to a high of 13 sites per application. Of 180 sites, 65 were rated no action indicated and 112 were rated voluntary action indicated. Among the latter, problems at five of the sites were serious enough that FDA requested a formal response from the investigator. Only 3 of the 180 sites had a rating of official action indicated and are described below:
|
1. |
One investigator failed to follow the protocol for women of childbearing age, failed to notify the Institutional Review Board and sponsor of the death of subjects, enrolled 25 percent of his subjects from undocumented sites, and failed to retain source data at his principal site. |
|
2. |
Another investigator failed to conduct required pregnancy tests, enrolled patients who were clearly ineligible, failed to collect the required samples, broke the blinding in the middle of the study, and had numerous discrepancies in the patient records, including treatment records that were dated before the individual's employment at the study site. |
|
3. |
The third investigator failed to collect both baseline and study laboratory data, failed to report on prior or concomitant medications and adverse drug reactions, and had numerous discrepancies between source documents and case report forms. |
Of the five applications with a more serious rating of voluntary action indicated, the number and nature of the discrepancies were less egregious than those at sites with a rating of official action indicated, such as admission of a subject before collection of a signed consent, no record of institutional review board approval for protocol amendments, failure to randomize all subjects, and inability to produce original documents. These problems appeared to occur randomly and were deemed administrative rather than analytic. In such cases it is appropriate to ask if there is a reasonable explanation. If no explanation is forthcoming, however, the agency must determine whether there is a pattern of similar errors at other study sites and whether these errors affect the overall outcome.
In a recent case study, after an efficacy supplement for a cancer drug was approved, regulators discovered that 1 of 157 major sites involved in the trial had falsified clinical data. The original results were quite robust, and the results remained robust when data from the site with discrepant data were removed, but it raised questions about a drug on which many patients' lives depended. To restore public confidence in the overall study, it was necessary to show that this site with discrepant data was an isolated case. It was clearly impractical to audit all 157 sites in the United States and Canada. Alternatively, the agency developed a statistical model based on the impact of data from each site on the overall results of the study. Sensitivity analysis showed it would be necessary to eliminate data from all of the top 15 sites to reach a statistical value not significantly in favor of the drug.
On the basis of this model, FDA conducted full audits of 41 of the 157 sites. Ten were inspected for cause: three were previously inspected as part of the Bio-research Monitoring Program review, two others (the site with discrepant data and the headquarters site) were inspected when the discrepancies emerged, and five other sites were inspected in response to questions that had been raised about data from the site. In addition, based on the statistical model that identified the top 15 sites, FDA inspected all of the top 4 sites, two-thirds of the next 11 sites, one-third of the remaining sites with more than 50 patients, and 15 percent of the 129 remaining sites that had fewer than 50 patients. The records of all subjects at each inspected site were reviewed.
In a large audit, examination of every data point in each source record and case report form is not practical. Accordingly, CDER selected 13 primary data points for examination of efficacy and developed standardized forms and instructions for field auditors. The resulting data were range checked and double entered into independent databases reconciled to identify missing and inconsistent data. Analysis showed that the overwhelming majority of source data were in agreement with the data in the NDA, and no pattern of discrepancy was discernible on either the treatment or placebo side of the study. A further stratified log-rank analysis revealed that even if all data from all sites that had any discrepancies were excluded, there would still be a significantly positive finding in favor of the drug.
This uprecedented audit, which occurred 10 years after the events transpired, was extremely resource-intensive and could not be applied to every ap-
plication. These findings suggest that the majority of investigators are conducting clinical trials correctly. Most of the errors that do occur appear to be through carelessness or misconduct. An audit aims to seek a balance between the detection and prevention of errors without burdening conscientious investigators. The audit described above concluded that the methodologies developed for the design of clinical trials, including randomization, blinding, and monitoring, are the best defense against scientific fraud.
FDA has a long history of working with companies during the planning phase of their clinical trials. In the past 5 to 10 years more companies have been consulting with FDA because of the rising cost of drug development programs. Whether more intensive monitoring procedures produce a corresponding reduction in data problems remains to be determined.
Assessment of Devices
Presented by Susan Alpert, M.D., Ph.D.
Director, Device Evaluation Office
Center for Devices and Radiological Health, Food and Drug Administration
About 12,000 manufacturers of medical devices have products on the market in the United States, and 65 percent of these manufacturers have fewer than 50 employees. Of the 5,000 products that go to market each year, 95 percent do so without any new data from clinical trials. When clinical trials are conducted, they are very different from those conducted for drug safety and efficacy. Clinical trials for medical devices are not always concurrently controlled and they are not always randomized, and masking or blinding of the prescriber is impractical or frequently not possible. The average clinical trial of a medical device involves 3 to 15 sites and about 200 patients, whereas hundreds of sites and tens of thousands of subjects are involved in drug trials.
Nevertheless, the Center for Devices and Radiological Health (CDRH) expects clinical trials of medical devices to be valid, well-designed, controlled evaluations of the safety and effectiveness of these new products. In 1992, CDRH enacted a more rigorous and organized monitoring program for the conduct of its clinical trials. Limited resources, however, restrict the number of site visits or extensive audits, even for smaller trials. The current goal is to monitor the sponsor site and up to three subsidiary sites for each premarket approval application (PMA).
In 1997, 28 of 46 sites with successful PMAs were audited. In some cases the company or the site had been visited recently and did not warrant an audit. In others the nature of the data did not warrant a traditional audit or there were too few subjects to make an audit meaningful. Of 28 audits, 3 (11 percent) required no action, 21 (75 percent) had minor discrepancies, and 4 (14 percent) had significant discrepancies:
- In one case, informed-consent documents for some of the subjects at several sites were absent, and failure to follow the protocol was determined at another site. These discrepancies, however, did not have a significant negative impact on data quality.
- In a second case, the sponsor failed to monitor the trial, resulting in a number of inconsistencies in the data. These occurred primarily as a result of failure to report device failures, patient injuries, and other adverse events. CDRH required the sponsor to change the labeling and conduct a postmarketing safety study as a condition for approval.
- In a third case, one site failed to report adverse events, resulting in inadequate accountability for the experimental devices. These failures did not appear to be systemic, however, and a laborious reevaluation of the data from the remaining sites did not change the outcome of the PMA.
- In a fourth case, inadequate accountability, incomplete or inaccurate case report forms, and lack of Institutional Review Board approval at one site were observed. Because this was a critical product, CDRH worked with the investigators to reconstruct the entire study, an extremely intense audit that took more than a year. The reconstruction required the company to incur substantial financial costs to ensure that the product was indeed safe and would perform as expected.
The distribution and accountability of products, such as artificial hips or defibrillators, are critical areas to be monitored during clinical studies of medical devices. Often, far more devices than the number actually used—sometimes 100 or 200 percent more—are distributed to investigators because investigators will need a variety of sizes to fit the incoming population. When investigators fail to return the extra units, the PMA will lack information on the missing product and CDRH is unable to reconstruct the final disposition of a device.
In another case, the agency worked with a clinical practice group to conduct a retrospective study of pedicel screw implants, devices used to fuse and stabilize vertebrae in back surgery. There was no prior protocol, no case report form, and no consistent control for patients or treatment at the time that the study was undertaken. CDRH worked with investigators to construct retrospectively an entire data set. In addition, an independent monitor visited the major sites and conducted an audit of all sites. Problems with missing data and variations among physicians' decisions in terms of subject inclusion, monitoring, and measurements of outcome were widespread. The lessons learned from this audit demonstrated that having in place a protocol, a good case report form, and a good monitoring program has a positive impact on the quality of the data and the agency's ability to make a regulatory decision.
Sound regulatory decisions do not require perfect data. Instead, they require reliable data that accurately reflect the methods and procedures used and subject outcome. Critical errors are rare, fraud is more rare, and not every error has the same impact. It is the number of errors and where they occur that determine their impact on the analysis and the decision to exclude subjects or study sites or
terminate the study. The case studies provided here demonstrate that audits increase the quality of the data across the board. In addition, audits strengthen the quality of investigators and clinical sites and improve data accuracy. Audits may also improve the ways in which companies conduct clinical trials and therefore help contain costs. Audits also provide the Food and Drug Administration the confidence needed to make an informed decision about marketing applications.
Panel Discussion
During the workshop's second panel discussion, one Food and Drug Administration (FDA) manager reported that, according to his informal survey, FDA statisticians have no established standard procedures or formal criteria for evaluating the quality of the data in the submissions that they receive. Instead, a neutral position is ensured as exploratory data analysis begins, with evaluation of the quality of the data occurring during the process. Both hypothetical questions and data are posed at this stage. In addition, an analysis and data verification are conducted with the sponsor's results. A disagreement in results is an indication of possible data error and poor data quality. In one case, for example, the protocol called for certain analyses, but the results of those analyses were not included in the submission. FDA personnel ran the analysis and obtained a significant result that would have been adverse to the product; they also found that summary statistics in one key table were not derived from the same analysis. The sponsor was unable to adequately state the reasons for these inconsistencies in data quality.
When FDA statisticians find unexpected gaps or errors in the data, an ''untitled letter'' rather than a warning letter is issued. Although clinical investigators and sponsors are not required to respond, they often do, explaining that regulatory requirements were not understood completely and expressing gratitude for having the opportunity to correct and learn from the mistakes that they made. These types of situations reveal the need for additional training of investigators by sponsors, given the fact that FDA personnel are spending a considerable amount of time finding errors and discrepancies that should have been revealed during the monitoring process. Many problems and questions are also being identified during Institutional Review Board (IRB) audits, thereby increasing the pressures on IRBs. Thus, this may be an area that is ripe for collective, cooperative action by sponsors and FDA.
Workshop participants identified the need for collaborative systematic improvement in the area of data quality as more prudent than trying to find data inconsistencies after the fact when auditing is conducted. This is an important consideration because both the number of independent sites for clinical research and the number of clinical investigators will continue to grow. Most of the physicians trained by U.S. medical schools, however, are not specifically trained in clinical investigation, which may contribute to a low level of prestige for clinical investigators within the academic environment. This raises questions about the
need to modify medical education and to certify clinical investigators, as is already done for other subspecialties. Although certification of clinical investigators may also be desirable for research coordinators, there is a need to explore alternative incentives (and reimbursement patterns) that reward quality rather than volume.
These ideas, which were well received by the panel members, further support the findings that 50 percent of physicians who participate in clinical trials are first-time investigators, which confirms that there is still much naivete among the investigators at the clinical sites. The Association of Clinical Research Professionals, which represents 7,600 members, recently spent $500,000 on programs to certify 800 clinical research associates and 3,500 clinical research coordinators in 37 cities. There is now an interest in the development of a similar program for certification of clinical investigators and institution of a code of ethics. These topics were addressed at an April 1998 meeting of the Drug Information Association which was conducted after this workshop (see the box on the Drug Information Association).
|
Drug Information Association The Drug Information Association (DIA), founded in 1964, is a nonprofit, multidisciplinary, member-driven scientific association with more than 20,000 members. Its membership consists primarily of individuals from regulatory agencies; academia; contract support organizations; pharmaceutical, biological and device industries; and from other health care organizations. DIA provides a neutral global forum for the exchange and dissemination of information on the discovery, development, evaluation, and utilization of medical products and related health care technologies. The meetings, workshops, and training courses sponsored by DIA are responsive to the rapidly evolving, multidisciplinary needs of its international membership. |
Participants expressed concern about the possible absence of an informed consumer's perspective in discussions on clinical trials and research. For example, if a goal of clinical research is to provide patients with access to continually improved quality of care, then progress toward risk-free therapy needs to be based on the best possible information and needs to include opinions from informed consumer groups. Moreover, patient participation was recognized as paramount to clinical research, and this requires an informed and willing consumer population. Lack of informed patient participation could undermine public confidence and trust in the regulatory process.
The National Breast Cancer Coalition (NBCC) was noted as one example of a consumer group that has played a vital role in outreach for participation in and accrual in clinical trials. NBCC trains community activists and works with com-
panies in designing programs that permit expanded access to clinical trials (see the box on The National Breast Cancer Coalition). Two important issues for this group include (1) a lack of coordination between FDA and industry and (2) the failure of FDA to join with the National Cancer Institute (NCI) and other agencies and organizations to lobby insurance agencies for reimbursement for the costs associated with patient participation in FDA and NCI clinical trials.
Ensuing discussions requested that panelists focus on the related issues of hierarchical data sets and two-tiered monitoring. They were asked to consider whether both sponsors and FDA are spending proportionately (1) too much effort on traditional audits and inspections, which focus on record keeping, or (2) not enough effort on proactive questions such as definition of error, sensitivity analyses, tolerance of error, and how to design a simpler and more efficient data system. One panelist characterized the traditional audits and inspections as "mindless monitoring" as opposed to "looking at process." Another panelist cited the extreme example of investigators who create a separate set of source documents for clinical trials that are totally divorced from the patient's main medical records because they believe that FDA requires them. Although this practice was rejected as a waste of time and a source of new inconsistencies, it does reinforce the need for FDA to communicate its expectations more clearly. Efforts to build quality into clinical trials are generally not part of the pretrial discussions between FDA and sponsors.
Most panel members agreed that there is a need to define data standards and to distinguish between primary and secondary data. Several suggested that International Committee on Harmonization guidelines (which describe a quality assurance program without specifically requiring it) were a first step in that direction. Others suggested that safety data would always be important, but that a commitment to postmarketing surveillance is also needed, especially if premarketing testing is streamlined. However, a panelist expressed concern that once the patient leaves the clinical trial setting, it becomes almost impossible to distinguish between the therapeutic effect of a drug or a device and the natural course of a disease. The long-term effects of Fen-Phen on cardiac valves, for example, were not detected through systematic surveillance but were identified by astute observations by medical specialists. Managed care organizations could make a considerable contribution to systemic long-term surveillance because of the wealth of data on drug use and patient health over time that they harbor. However, thus far they have expressed little interest in making such a contribution.
Several participants suggested that the best way to simplify databases was not to collect too many data in the first place. Such a protocol may require prior agreement to determine which data should be collected and which data should be excluded. Should this become a viable alternative, the secondary questions on the case report form need to be disregarded so that data are not collected. There is a pressing need for sponsors and FDA to work together to decide which data are not as important and to agree on the areas on which monitoring and auditing should focus. Several panelists also suggested that the investigators' meeting with FDA should be conducted earlier and that the Institutional Review Board
be invited to address data sets and data quality measures as early as possible in the process.
Other panelists expressed reservations about having FDA work closely with sponsors in planning the design of a trial. They expressed concern whether it would be prudent for a sponsor to conduct an objective assessment of the outcome of a clinical trial after working with an FDA reviewer in defining data sets or monitoring schemes. Although some panelists indicated that an objective assessment was a management issue that needed to be handled by FDA, others rejected the idea. The latter group argued that by clarifying expectations before-hand, FDA would in no way compromise scientific integrity. FDA reviewers explained that by helping sponsors correctly conduct the study, it would not mean that the results would be favorable or that the product would be approved. The alternative—knowingly allowing inadequate trials to go forward—would simply be irresponsible in terms of both getting drugs approved and protecting patients.
|
National Breast Cancer Coalition The mission of the National Breast Cancer Coalition (NBCC) is to help eradicate breast cancer through action and advocacy. From its inception, this nonprofit, grassroots organization has acted as an advocate not just within the government, but also within industry and the scientific community. The coalition is a powerful voice that speaks on behalf of breast cancer patients, activists, and others concerned with the breast cancer epidemic. NBCC has been successful in bringing together breast cancer activists from across the country. The sponsored conferences by the coalition are designed to educate and train breast cancer advocates; they have provided beginner and advanced advocacy training as well as information on breast cancer research and public policy. Through its network of activists—consisting of more than 450 organizations and 58,000 individuals—NBCC has initiated fundamental changes over a 7-year period. Some of its accomplishments during this period have included the following:
|
Among the many reasons why NBCC has been successful in achieving its mission and has realized many accomplishments since its inception has been its persistent focus on three goals:
|
An industry representative found it encouraging that FDA reviewers were discussing the probability of collecting fewer data and asked when it would be most appropriate to hold such discussions in the review process. FDA personnel suggested that such discussion should be integrated into meetings on protocol design, before the clinical trial is actually launched. Although a regulatory agency may never have a concrete answer to what is considered sufficient data or sufficient quality, it is best for sponsors to discuss such questions early in the development process.
















