
Introduction
The Topic
The development of a new drug is a challenging and time-consuming process. If preclinical testing suggests that a promising compound might be well tolerated in humans, it is tested for safety and pharmacokinetics in healthy volunteers (Phase 1). If the results of Phase 1 trials warrant further investigation, a limited number of patients with the target disease are challenged with the drug under carefully controlled conditions to evaluate its efficacy and further establish safety and proper dosages (Phase 2). If these trials are successful, the drug enters large-scale trials to better characterize its safety and efficacy in patients (Phase 3). Typically, clinical trials are coordinated by either contract research organizations (CROs) or academic medical centers that are sponsored by the pharmaceutical manufacturer. Physicians at these institutions conduct the clinical trials and care for the patients. The Food and Drug Administration (FDA) is the regulatory body involved during the development, preclinical, and clinical trial phases of new drug discovery and testing in humans.
A significant proportion of the time and expense of conducting clinical trials arises from the need to assure that the resulting data are accurate. Patients are selected, treated, and evaluated by a meticulous protocol, and results are recorded on standardized forms that are collected and analyzed by the sponsor or its designee. To ensure the validity and accuracy of the data, the pharmaceutical company periodically sends monitors to study sites to verify that patients are treated according to the study protocol and that the information is reported according to the study protocol. Monitoring alone can represent up to 30 percent of the costs of a clinical trial. Most pharmaceutical companies also have separate quality assurance departments to review forms and audit data and safety departments to monitor and prepare reports on adverse events.
From the pharmaceutical manufacturer's perspective, the key issue in data quality and integrity is how to collect only the information that is necessary to
assess the safety and effectiveness of the experimental therapy, as well as how to ensure the quality and integrity of that information, while controlling costs and reducing the time consumed by the clinical trial process. From FDA's perspective, the key issue is ensuring that data submitted in support of an application are a valid representation of the clinical trial, especially as the data pertain to drug safety, pharmacokinetics, and efficacy.
Background
Under the Federal Food, Drug, and Cosmetic Act, pharmaceutical manufacturers must obtain a research or marketing permit before beginning studies on certain commodities such as new human drugs, medical devices, veterinary drugs, and food additives and bringing them into interstate commerce. FDA approves these permits, and also regulates biomedical research whose results are to be submitted in support of an application for such a permit. FDA has two principal objectives in regulating this research: (1) to protect the rights and welfare of human research subjects and (2) to assure the quality and integrity of the biomedical research data used to support the initiation or expansion of clinical trials, the approval of new products and new indications, and the labeling of these products. The second objective is the subject of this report.
Organizationally, FDA works to protect human subjects and assure data quality and integrity through an intramural review process, conducted at its headquarters in Rockville, Maryland, and through the Bioresearch Monitoring Program, whose field agents inspect clinical research sites. Figure 1 depicts the review process for a New Drug Application (NDA), which is reviewed by the Center for Drug Evaluation and Research (CDER); the processes for medical devices and biologics are different in their details but follow the same general steps. The components of a marketing application are relatively uniform, and although the exact requirements are a function of the nature of the specific product or device, the application must provide all relevant data and information that a sponsor has collected during the research and development of the product.
In this system, the purpose of a clinical trial is to collect the information that will allow FDA to make regulatory decisions about the safety and efficacy of the product. The clinical trial protocol represents the agreement between the sponsor, investigator, and FDA as to how the clinical trial will be conducted. Consequently, an important focus of FDA reviews, both intramural and on-site, is data auditing to ensure that the study was conducted and analyzed as specified and that deviations from the protocol in conducting the trial or handling the data are adequately addressed. The purpose of such reviews is not only to rule out fraud, but also to ensure that the quality and integrity of the data are not compromised by sloppiness or poor compliance with the protocol. FDA does not have a set of standardized practices for its reviewers to follow in conducting these audits, but it is developing such good review practices.
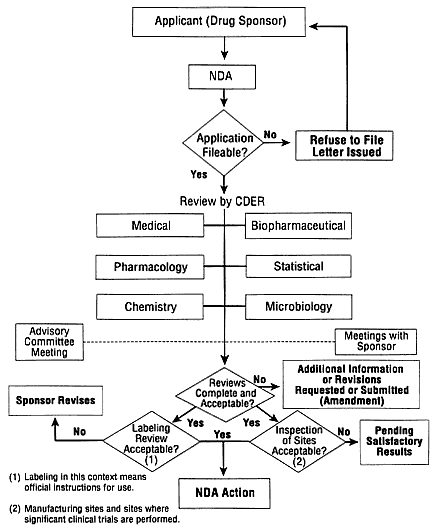
FIGURE 1.
The Food and Drug Administration (FDA) works to protect human subjects and assure data quality and integrity through an intramural review process. The review process for a New Drug Application, which is reviewed by the Center for Drug Evaluation and Research, is depicted. The diagram does not accurately represent the review of a medical device and biologic application; however, it adequately depicts the steps related to such an action. SOURCE: FDA.
Overview of Issues
Presented by Janet Woodcock, M.D.
Director, Center for Drug Evaluation and Research,
Food and Drug Administration
The goal of improving health through the use of new medicines and medical devices cannot be achieved without public confidence in the clinical trials process. The purpose of clinical trials during drug development is to generate data on the safety and efficacy of a new product, data that will become part of the marketing application. The importance of informed consent and human subjects protection to the integrity of clinical trials has been widely discussed. Assuring the quality and therefore the usefulness of data from human clinical trials has received less attention, but it is also vital. Subjects should not be needlessly exposed to risks in trials that fail to yield valid data. In addition, it is essential that these data be reliable, because they form the basis for regulatory and medical decision making. For all these reasons, there is widespread agreement that such trial data should be of high quality.
Despite this fundamental agreement, many issues remain. The current processes for assuring data quality were developed individually in response to various problems or crises, rather than in a comprehensive quality management framework. Although the current system is successful, it is relatively expensive and time-intensive, may limit the overall investment in clinical trials, and may not provide the best-attainable quality for the degree of investment. Additionally, there is no consensus definition for "quality" as it applies to data from clinical trials. Finally, many changes that have the potential to affect data quality are occurring in the area of clinical practice and clinical trials. Widespread computerization of data entry and handling, use of CROs for performance of trials, the increased frequency of multinational trials, and the changes in the health care delivery system all may have impacts. The workshop described here explored these challenges.
The Workshop
Industry's role is to assure data quality and validity as the data are generated and processed. It does this by developing standard operating procedures and quality assurance checks at each stage of the trial, including the design of forms, investigator training, clinical site monitoring, and data cleanup. There is relatively little government guidance on the conduct of this phase of investigation. The International Conference on Harmonization recently developed a comprehensive governing document entitled Good Clinical Practice . This document has had a major impact on the conduct of clinical trials in Europe and Japan. In the United States, the Food and Drug Administration (FDA) has issued guidance on the monitoring of clinical trials and the maintenance of electronic records.
The FDA role includes oversight of the Institutional Review Boards for the protection of human subjects, and the Investigational New Drug process for trials that are being performed in the United States. Once a marketing application is filed, FDA will inspect some of the clinical sites and compare the data in the application with those from among the sites. FDA also conducts data verification, a process that is highly varied because of the complexity and variety of the applications themselves. FDA is currently drafting ''good review practices'' internally.
In the past 10 to 20 years in the United States the quality of data from clinical trials has improved remarkably. Historically, most data problems have been the result of poorly trained investigators who either enrolled the wrong patients, did not follow the clinical trial protocol, did not record the required information, or had poorly designed protocols. Problems resulting from missing data or missing source records have also surfaced, as have problems resulting from faulty data entry, transposition, or analysis. Cases of outright fraud are rare. Fraud is a serious threat to public confidence, however, the prevention of fraud has been a major force in shaping the current system.
The current system for assuring data quality and validity during drug development and testing evolved over time, with parts of it enacted as problems emerged or in response to various crises. Consequently, the current system does not provide the harmonization and close integration of a prospectively designed system. During the workshop participants explored opportunities for improving quality by taking a systems approach.
Another issue explored at the workshop is the lack of a consensus standard for defining the quality of clinical trials data. Much of the industry quality control focuses on the detection of and elimination of missing data, transcription errors, or similar data problems. However, other aspects of trial conduct, such as how well investigators followed the protocol, are also extremely important to the validity of the trial results. Some of the discussions addressed how data quality in clinical trials should be assessed.
Several forces that will influence the future functioning of this system are at work. These include (1) new scientific discoveries, particularly in the area of pharmacogenetics; (2) the increasing automation of handling of clinical trials data, including remote data entry; (3) the potential role of managed care, which thus far has shown little interest in participating in clinical trials; and (4) the growth of outsourcing and the emergence of CROs. Each of these forces holds considerable promise for improving data quality but also has the potential to engender new problems for industry, government, private organizations, and consumers, and participants considered these during the workshop.
The members of the Roundtable identified three additional issues for speakers to address during the workshop. One issue was the question of how good is good enough. Although data quality is a continuum, there can be no "perfect" data set; instead, there may be a decreasing marginal benefit from pursuing such a goal. Quality data would therefore be defined as data that sufficiently support conclusions and interpretations equivalent to those derived from error-free data.
A second issue concerned streamlining, which addressed whether the existing system is effective and efficient, and produces the highest-quality data for the lowest possible investment. If the answer was negative, the Roundtable members asked the speakers to identify how the system could be improved. Finally, Roundtable members asked the speakers what could be done to improve the quality of data generated from trials performed in some foreign countries.
The workshop was successful in broadening the dialog among FDA, industry, and the public on the subject of data quality and validity in clinical trials for the regulatory decision-making process. Although the participants presented and discussed many important issues such as the identification of opportunities for significant improvement in the overall process, the nature of any workshop is that it cannot serve as an exhaustive exploration of the subject matter being addressed. Therefore, the proceedings in this report contain only the information that emerged from the workshop itself.






