
2
Pathophysiology of Acute Hemorrhagic Shock
Definitions of Hemorrhagic Shock
A variety of definitions of hemorrhagic shock have arisen as more understanding of the mechanisms involved have been developed. Several definitions could be considered to be archaic but in general remain accurate (see Box 2-1). A modern definition of shock would acknowledge first that shock is inadequate tissue perfusion and inadequate removal of cellular waste products and second that shock is a failure of oxidative metabolism that can involve defects of oxygen (1) delivery, (2) transport, or (3) utilization, or combinations of all three. The diagnoses of clinical signs of shock are primarily related to organ failure, but organ failure is secondary to failure of the cells.
Many authors have described the "vicious cycles" in shock (see Figure 2-1). They may cascade in a variety of ways such as decreased cardiac output, which leads to a decreased blood pressure, which in turn leads to decreased tissue per-
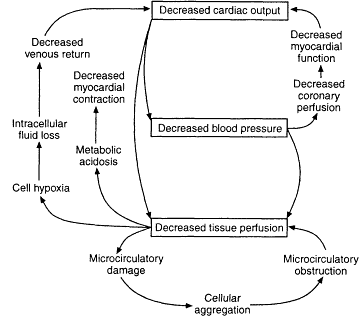
Figure 2-1
Vicious circles in shock. Initiation of shock can occur at any point, but the endpoint is often the same. Source: Reprinted, with permission, from Davis et al., (1995, p. 145). Copyright 1995 by Mosby-Year Book, Inc.
fusion. Increased cardiac work may lead to failing myocardial function and decreased coronary perfusion. Decreased tissue perfusion at the cellular level leads to microcirculatory damage, cellular aggregation, and microcirculatory obstruction, followed by cell hypoxia, transfer of salts and fluid into the cells, and decreased venous return. These events lead to metabolic acidosis, which, if it becomes deep, can result in decreased myocardial contraction. There are time-honored classifications of shock, many of which were initiated by definitions described by Alfred Blalock in the late 1930s. They are:
- Hypovolemic shock: shock secondary to inadequate circulating volume.
- Traumatic shock: shock secondary to inadequate circulatory volume plus soft-tissue injury.
- Cardiogenic shock: failure of the heart to provide circulation.
- Neurogenic shock: failure of the nervous system to provide peripheral vascular resistance.
- Septic shock: hemodynamic instability that may arise as a result of septicemia.
Organ Involvement in Shock
Shock can be evaluated from a number of standpoints (see Table 2-1) but is frequently described in terms of organ failure in the following systems:
- circulatory,
- endocrine,
- metabolic, and
- cellular.
Examples of such alterations are seen in the dynamics of human shock syndromes. Septic shock is manifested by high-output cardiac failure in which there is increased cardiac output but decreased systemic vascular resistance and decreased myocardial contractility. Low-output cardiac failure can also develop in sepsis and reflects a loss of response to catecholamines. Pulmonary changes in shock may include increased lung water levels, increased pulmonary vascular resistance, and increased alveolar-capillary permeability. Hyperpnea may compensate for metabolic acidosis, but the phenomena described above may also result in impaired gas exchange.
The next organ sequentially affected in the organ failure induced by shock is the kidney. Renal failure may ensue as a consequence of shock and, depending on the state of volume resuscitation and other factors, may have the following characteristics:
- initial high level of urine output,
- low pressure in the renal tubules producing sodium retention,
- renal parenchymal damage, and
- renal dysfunction and failure.
Renal failure as a consequence of shock has been a factor in most wars. In the 1973 Yom Kippur War in Israel, the mortality rate among patients who developed renal failure after injury was approximately 60 percent. In the Vietnam conflict in 1972, the mortality rate from renal failure after injury was approximately 70 percent, in the Korean War in 1955, it was about 60 percent, and in World War II, it was about 65 percent. Good civilian series reveal rates of morbidity from acute renal failure after injury that exceed 50 percent. In short, renal failure is a complication of severe shock and, historically in wars and more recently in civilian practice, is associated with a mortality rate of more than 50 percent. Vigorous fluid resuscitation has improved the situation by reducing the incidence of renal failure; early and adequate resuscitation can avoid this dreaded consequence of shock.
TABLE 2-1
Hemodynamic Responses to Different Types of Shock
|
|
Type of Shock |
|
|
|
|
Indicator |
Hypovolemic |
Septic |
Cardiogenic |
Neurogenic |
|
Cardiac index |
|
|
|
|
|
Peripheral resistance |
|
|
|
|
|
Venous capacitance |
|
|
|
|
|
Blood volume |
|
|
|
|
|
Core temperature |
|
|
|
|
|
Metabolic effects |
Effect |
Cause |
Effect |
Effect |
|
Cellular effects |
Effect |
Cause |
Effect |
Effect |
|
NOTE: The hemodynamic response to different types of shock is indicated by arrows to show an increase |
||||
The gastrointestinal consequences of shock include increased acid production and increased permeability of the gastric mucosa. The increased permeability allows tissue penetration by acids, bacteria, and endotoxins. In the past, these complications resulted in the late morbidity from hemorrhagic gastritis, which has a high mortality rate. As understanding of the physiology of this problem has improved, the treatment of patients with shock with serotonin, H2 blockers, and ant-acids has resulted in a marked decrease in this complication's rate of occurrence.
The liver, like all other organs, responds to shock. The effect on the liver is not well delineated but does result in major changes in bilirubin, isoenzymes, protein synthesis, and, perhaps most importantly, the reticuloendothelial system. Decreased consciousness and changes in neural control mechanisms are the responses of the central nervous system to shock.
The metabolic effects of shock manifest as changes in homeostasis or hyperventilation, respiratory alkalosis, metabolic acidosis, and an excretion of nitrogen, phosphate, potassium, magnesium, zinc, and sulfate. Energy depletion of the cell in shock leads to failure of the sodium and potassium adenosin triphosphatase (ATPase) and a drop in resting membrane potential from ˜90mV to ˜60 mV associated with the loss of intracellular potassium and the translocation of sodium and water into the cytoplasm (Chiao et al., 1990; Fantini et al., 1987). Energy depletion is a consequence of hypoxia-induced failure of the Krebs cycle associated with increased lactate levels. Other biochemical effects of shock include hyperglycemia, gluconeogenesis, glycogenolysis, and the synthesis of triglycerides, free fatty acids, lipoprotein, and acute-phase proteins. The hormonal effects of shock result in glycogenolysis, lipolysis, gluconeogenesis, and insulin resistance. These are the results of an adrenergic stimulus and present hemodynamically as tachycardia and a hyperdynamic state. Immunologically
there is an impaired specific immunity at both the cellular and the hormonal levels; there is also an impaired nonspecific immunity of macrophages and opsonins. Hematologic changes include impaired hematopoiesis, altered hemostasis, and in some case, disseminated intervascular coagulation.
Physiologic Responses to Hemorrhage
Acute hemorrhage produces a decrease in arterial systolic, diastolic, and pulse pressures along with an increase in the pulse rate and a decrease in the cardiac stroke volume. The cutaneous veins are generally collapsed and fill slowly when compressed centrally. The skin is pale, moist, and slightly cyanotic. Usually, the respiration is rapid and shallow (Berne, 1983). However, the early stages of hemorrhage result in the initiation of a number of feedback mechanisms that tend to maintain arterial blood pressure in the presence of a decrease in circulating blood volume and a modest decrease in cardiac output. These regulatory mechanisms include (1) central circulatory reflexes originating from atrial stretch receptors and receptors located in the ventricles of the heart, (2) high-pressure baroreceptor reflexes from the carotid sinus and aortic arch, (3) chemoreceptor reflexes, (4) cerebral ischemia responses, (5) reabsorption of tissue fluids at the level of capillaries, (6) release of endogenous vasoconstrictor substances such as vasopressin, and (7) renal conservation of salt and water.
Early loss of less than 10 percent of the circulating blood volume may be associated with no change in arterial pressure because of compensatory increases in sympathetic nervous system activity and both arterial and venous constrictions initiated from low-pressure cardiac receptors (Chien, 1967). A further reduction in circulating blood volume results in decreased arterial pressure and diminished stimulation of aortic arch and carotid sinus baroreceptors and further decreased stimulation of intracardiac receptors. These alterations cause reduced vagal tone to the heart and increased sympathetic nervous system activity to the heart, arterial vessels, and venous capacitance vessels. The changes in peripheral resistance do not occur uniformly in all vascular beds. The cerebral and coronary circulations, which are not primarily regulated by direct sympathetic innervation, participate less in this response. Although the tachycardia and the increased sympathetic tone to arterioles have been studied extensively, the role of the carotid and aortic arch baroreceptors in regulating the sympathetic mediated tone of the regional venous beds has not been studied as extensively. Regulation of splanchnic capacitance vessels appears to involve conventional carotid and aortic arch baroreceptors and a significant contribution by cardiac receptors. Deep limb vessels are similarly regulated, but cutaneous vessels are less influenced by baroreceptors (Rothe, 1963). The activation of sympathetic innervation to splanchnic and deep limb veins provides a short-term autotransfusion of blood from venous reservoirs. Although more pronounced in some species such as the dog, this reflex mechanism also exists in the human (Guyton, 1986). In the early stages of moderate hemorrhage, the changes in total renal vascular resistance are slight because intrinsic autoregulatory mechanisms
within the kidney tend to maintain renal blood flow. The intense splanchnic and renal vasoconstriction may protect the heart and brain but can eventually lead to ischemic injury of the kidney and bowel resulting in kidney failure and further vascular injury and loss of fluids from the vascular compartment into the interstitial space.
When the arterial pressure falls below 60 millimeters of mercury (mm Hg), hypoxia of the peripheral chemoreceptors in the carotid body (because of decreased perfusion) results in activation of chemoreceptor reflexes. This augments peripheral sympathetic nervous system activity and also provides respiratory stimulation, resulting in increased breathing frequency and often an increase in minute ventilation. At very low levels of arterial pressure, below 40 mm Hg, inadequate cerebral blood flow produces an extremely strong activation of the sympathetic nervous system and intense vasoconstriction in response to cerebral ischemia.
A number of endogenous vasoconstrictors are released during hemorrhage. As a direct response to sympathetic nervous system activation, the release of epinephrine and norepinephrine from the adrenal medulla reinforces the actions of direct sympathetic nervous system innervation of the heart and peripheral circulation. Vasopressin, which is a potent vasoconstrictor, is actively secreted by the posterior pituitary gland in response to hemorrhage. Vasopressin release is activated by both the baroreflexes and receptors located in the left atrium. Diminished renal perfusion results in the secretion of renin from the juxtaglomerular apparatus and the subsequent conversion of angiotensinogen to angiotensin, which is also a powerful vasoconstrictor.
Shock Decompensation
Loss of the ability of the compensatory mechanisms described above to maintain arterial blood pressure and cardiac output in the presence of prolonged hemorrhage is usually the result of decreased cardiac function and failure to maintain sympathetically induced arterial and venous vasoconstriction. The decline in cardiac function and vasoconstriction may be the result of toxic peptides (Lefer, 1978, 1985) released from ischemic tissues in combination with metabolic acidosis. These changes are accompanied by alterations of immune function, aberrations in blood clotting, reticuloendothelial system dysfunction, and an inability to regenerate high-energy phosphate reserves at the cellular level. Tissue injury as a result of prolonged ischemia results in the loss of cell membrane integrity with respect to the maintenance of essential ionic gradients, widespread inhibition of metabolic activity because of mitochondrial dysfunction, and the activation of cellular hydrolases that further contribute to cellular injury.
The goal of early volume replacement in fact is to delay or prevent the chain of events that lead to irreversibility in severe prolonged shock, and restoration of blood volume, treatment of acidosis, and management of the metabolic
derangements can provide temporary restoration of blood pressure and cardiac output in severe prolonged shock. Delay in treatment usually results in death (Guyton, 1986). The question that has been posed repeatedly in shock research is what factor or factors lead to the eventual total deterioration of circulatory compensation. The answer to this question seems to be that beyond a certain point so much tissue injury has resulted in the release of toxic mediators, the destruction of metabolic machinery of the cell by lytic enzymes, and so much acidosis and failure of regulatory mechanisms aimed at preservation of homeostasis that even the most vigorous resuscitative measures are not capable of maintaining life (Hannon et al., 1990). The multifactorial nature of the processes leading to irreversibility in severe shock make it seem unlikely that simple therapeutic measures that occur late in shock will have any significant effect on outcome. It is more likely that early interventions that prevent the late extensive tissue injury phenomena are more likely to succeed in changing the outcome.
Cellular Responses to Shock
Reduced tissue perfusion and the pathologic events reviewed above initiate a syndrome characterized by alterations in:
|
1. |
energy metabolism, ion compartmentalization, lipid metabolism, and radical production and metabolism; |
|
2. |
macrophage function; |
|
3. |
transcription and translation that may lead to apoptosis; and |
|
4. |
the secretion of and cellular responsiveness to growth factors. |
Although during the last 30 years there has been considerable controversy regarding the ionic content and concentration of intravenous fluids used for resuscitation of patients in hemorrhagic shock, this controversy has not led to further consistent reductions in mortality. This suggests the possibility that the barrier to substantially improved care resides not in minutiae of volume resuscitation protocols but rather in an inadequate understanding of the molecular and cellular responses triggered by the shock syndrome. Therefore particular attention is paid to these phenomena and the causal mechanisms associated with them.
Altered Energy Metabolism, Ion Compartmentalization, Lipid Metabolism, and Radical Production and Metabolism
The physiologic cardiovascular and neurohumoral responses described above are able to provide considerable compensation for loss of up to 30 to 40 percent of the blood volume. However, when shock persists or volume loss continues to increase, hypoperfusion of first the bowel and then other vital organs impinges on the requirement for the delivery of oxygen (O2) and metabolic
substrates to support adequate cellular levels of high-energy phosphate compounds such as adenosine triphosphate (ATP). The general consequences of this have been studied in a variety of organs, especially in the brain, which is extraordinarily sensitive to damage from ischemia and reperfusion.
ATP can be generated by anaerobic metabolism, which produces a net 2 moles of ATP for each mole of glucose metabolized. The end product of this pathway is pymvate, which is then normally transferred to further oxidative metabolism in the mitochondria. Mitochondrial aerobic metabolism is much more efficient than anaerobic metabolism in the production of ATP; in the aerobic system 36 moles of ATP are generated for each mole of glucose oxidized ultimately to carbon dioxide (CO2) and water (H2O). When aerobic metabolism fails because of inadequate O2 delivery, persistent anaerobic metabolism and the conversion of accumulated pyruvate to lactate by pyruvate dehydrogenase lead to cellular accumulation of unoxidized reducing equivalents and decreased pH because of lactic acidosis.
The mitochondrial metabolism of the substrate generates electrons, which are added to O2 one electron at a time. That is, the reduction of O2 is stepwise, and the reaction can be written as follows:
where e- is an electron, *O2- is superoxide, H2O2 is hydrogen peroxide, *OH is the hydroxyl radical, and OH- is the hydroxide ion. Mitochondria utilize the energy derived from the reduction of O2 to drive the pumping of protons out of the inner mitochondrial volume into the space between the inner and outer mitochondrial membranes. The phosphorylation of adenosine diphosphate (ADP) to generate ATP is then driven from the energy stored in the hydrogen ion gradient across the inner mitochondrial membrane. Two aspects of this system that are very important to understanding the role of mitochondria in postischemic reperfusion injury are the single-electron reduction of O2 and the hydrogen ion gradient across the inner mitochondrial membrane.
Depletion of ATP occurs during ischemia; this develops most rapidly in the brain, where the concentration of ATP is reduced to near zero within approximately 5 minutes of complete ischemia (reviewed by O'Neil et al., 1996). This ATP depletion degrades the energy-dependent maintenance of ionic gradients across the plasmalemma, and sodium ions (Na+) and calcium ions (Ca2+) enter the cell, and potassium ions (K+) exits the cell down their respective concentration gradients. The Ca2+ concentration in the extracellular fluid and within the endoplasmic reticulum (ER) is about 104 greater than the cytosolic Ca2+ concentration, and early massive overload of the cytosol with Ca2+ is a major consequence of tissue ischemia.
Unlike Na+ and K+, Ca2+ is both a signaling molecule (Clapham, 1995) and a cofactor for a number of important enzymes. Important enzymatic consequences of ischemia-induced cytosolic Ca2+ overload and ER Ca2+ depletion are activation of (1) phospholipases, (2) the proteolytic enzyme µ-calpain, (3) the
phosphatase calcineurin, and (4) a kinase responsible for inhibition of the mechanism by which the first amino acid is introduced during initiation of protein synthesis. The consequences of all of these events will be addressed below. Another important consequence of cytosolic Ca2+ overloading during early re-perfusion is mitochondrial diversion of the energy derived from O2 reduction to the sequestration of Ca2+ instead of ATP production. In the situation of high cytosolic Ca2+ concentrations, mitochondria utilize the proton gradient across the inner mitochondrial membrane to drive the influx of Ca2+ rather than to drive the production of ATP (Carafoli and Crompton, 1978).
Free fatty acids, and in particular arachidonate, are released from membrane lipids during brain ischemia as a consequence of the activity of both phospholipase C (Abe et al., 1987) and phospholipase A2 (Drenth et al., 1976; Moskowitz et al., 1984). The concentration of free arachidonate can reach 180 µM during ischemia and remains elevated during early reperfusion (Bazan, 1970; Katsuki and Okuda, 1995; Rehncrona et al., 1982; Umemura, 1990; Yasuda et al., 1985; Yoshida et al., 1980). Oxidative metabolism of arachidonate occurs during re-perfusion and is an important source of superoxide (Bakhle, 1983), which is also generated by reperfused mitochondria. Superoxide is not a potent oxidizer, but it does readily reduce insoluble iron ion (Fe3+) in storage proteins to soluble ferrous iron (Thomas et al., 1985), which catalyzes the formation of strong oxidants that initiate the lipid peroxidation chain reactions (Aust and White, 1986). During brain reperfusion both the release of iron from high-molecular-weight stores (Krause et al., 1987) and lipid peroxidation in selectively vulnerable neurons (White et al., 1993) occur, and similar evidence exists for peroxidative damage to other vital organs. This lipid peroxidation can generate substantial ultrastructural damage to the plasmalemma such that it is no longer able to partition ions (Kumar et al., 1987).
Although the role of ionic iron in catalyzing radical-mediated damage to tissue macromolecules is well established, the identity of the oxidizing chemical species that initiates the injury has been elusive. Recent observations suggest that during early reperfusion there is a burst of cellular nitric oxide (NO) synthesis and that NO can react directly with superoxide to generate the potent oxidizer peroxynitrite. In addition to initiating radical damage to tissue macromolecules, peroxynitrite can nitrosylate amino acid side chains (Alvarez et al., 1999) and modify the structure and activity of enzymes. Phosphorylation of NO synthase inhibits the activity of the enzyme, which is activated by dephosphorylation by calcineurin, and agents that inhibit calcineurin-mediated activation of NO synthase have neuroprotective effects (Dawson et al., 1993)
Radical damage is normally opposed by superoxide dismutase and catalase; transcription for both of the enzymes is regulated by the SP1 housekeeping promoter sequence. The transcription of another set of important antioxidant enzymes comprising the antielectrophile response is regulated by the AP1 promoter (reviewed by O'Neil et al., 1996). Activator Protein 1 (AP1) is a heterodimer comprising c-Fos and c-Jun; messenger RNAs (mRNAs) for both these transcription factors are generated in response to brain ischemia and reperfusion
(reviewed by O'Neil et al., 1996) but are not efficiently translated because of inhibited protein synthesis, which is discussed below.
Alterations in Macrophage Function
In the 1880s Ilya Metchnikoff demonstrated in the larva of marine starfish that puncture injury induced a massive response by macrophages, the only type of immunologic cell in this simple creature (Meltzer and Nacy, 1987). Macro-phage ligand receptors for cytokines, their secretion of interleukin-1 (IL-1) and tumor necrosis factor α (TNF-α) in response to cell injury and antigen-antibody complexes, and their phagocytic activity indicate the central roles that they play in inflammation. Macrophages present antigen to T lymphocytes, and this together with IL-1 causes T lymphocytes to produce IL-2, an obligate growth factor for T-cell proliferation. IL-1 also acts as a required signaling factor for B-lymphocyte maturation and antibody production (Meltzer and Nacy, 1987).
Circulating IL-1α and IL-1β are not detectable before or after hemorrhagic shock, but elevated levels of IL-1α occur in tissue in response to hemorrhagic shock and are not reduced by resuscitation (Molina et al., 1997). This evidence implicates activation of tissue macrophages. IL-1 signal transduction involves binding to plasmalemmal receptors followed by intracellular phosphorylation cascades that lead to activation of nuclear factor κB (NFκB) (O'Neill, 1995), which is essential for inducible TNF-α transcription (Im et al., 1997). Hemorrhagic shock induces increased levels of macrophage mRNA for TNF-α and IL-6 and increased levels of mesenteric TNF-α and IL-6, and these effects are exacerbated by fluid resuscitation (Tamion et al., 1997). The levels of TNF-α in other tissues are also elevated by hemorrhagic shock and are not reduced in tissue by resuscitation or during the early postresuscitation period (Molina et al., 1997). Circulating TNF-α is normally undetectable but its levels are markedly elevated in hemorrhagic shock (Molina et al., 1997). Endotoxin shock induces similar alterations in the cytokines, but in this case volume resuscitation does not enhance the effect (Tamion et al, 1997). Kupffer cells also release IL-6 in response to trauma-induced hemorrhage (Wichmann et al., 1997). The initial macrophage-mediated cytokine response is followed by reduced immunocompetence of the macrophages, and it has been shown that the shift to cytokine release by macrophages following hemorrhagic shock is associated with a reduced capacity for antigen presentation by macrophages (Ayala et al, 1996).
Pulmonary infection following hemorrhagic shock can greatly augment leukocyte sequestration in the lung. This effect is associated with alveolar macrophage expression of cytokine-induced chemoattractant (CINC) mRNA and protein but not with expression of macrophage-inflammatory protein 2, and it is inhibited by treatment with an antioxidant or antibody against CINC (Fan et al., 1998). Leukocyte adhesion and penetration into the endothelium also depend on expression of the β-integrins CD11b/CD18 (Sun et al., 1996), which in patients are induced by hemorrhagic shock associated with lactic acidosis
(Botha et al., 1997). Finally, focal injury to some organs can induce a generalized response throughout the body. Brain compression as a primary insult upregulates mRNA expression of lymphocyte and macrophage products in a wide variety of peripheral tissues, and this is a major concern in the use of organs from brain-dead donors (Takada et al., 1998).
A much more detailed knowledge of the signal transduction and of the transcriptional and translational regulation of these phenomena of the inflammatory response is highly desirable. For example, ligand activation of the TNF-α receptor has both pro- and antiapoptotic consequences, and thus, macrophage activation may have a significant impact on the survival of cells in many organs. The development of such detailed knowledge of the regulation of fundamental cellular processes by cytokines is very likely to provide specific molecular approaches to patient management.
Surgical Bleeding Disorders
Hemostasis is the physiologic cessation of bleeding. The ability to achieve adequate hemostasis is critical to the success of surgical operations and recovery from injury. Under normal circumstances, blood maintains its fluidity because of the balance of various procoagulant and anticoagulant influences, including interactions at the blood-endothelium interface and a variety of circulating factors (Pearson, 1994). Hemostatic and coagulation mechanisms allow the prompt repair of a local injury in the microcirculation without progression to a systemic reaction. Injured blood vessels can therefore be repaired and hemorrhage can be controlled locally while blood continues to flow normally in other uninjured areas. Once the injury to the blood vessel has been repaired, the lysis of clots that had formed in the area begins and the vessel may ultimately regain patency. Localized injury to the vascular system thus activates hemostatic mechanisms at the injury site and circulating coagulation mechanisms. Once healing has occurred, a system of fibrinolysis reestablishes patency of the blood vessel and degrades the products of coagulation.
Fundamental Alterations in Transcription and Translation: Apoptosis
During the public presentations made to the committee as background for the preparation of this report, apoptosis was repeatedly implicated in the progression of tissue injury associated with hemorrhagic shock and volume resuscitation. Apoptosis is a complex mechanism of cell suicide and is triggered by intracellular molecular signals that may originate from a variety of subcellular organelles including the plasmalemma, mitochondria, endoplasmic reticulum, and nucleus (Bredesen, 1996; Hale et al., 1995). Apoptosis in the context of tissue ischemia and reperfusion must be viewed in a way that sorts the molecu-
lar mechanisms into those involved in (1) actual execution of cell death and (2) signaling that activates the execution program.
In early work, the generation of DNA fragments with lengths that were multiples of the interhistone distance (approximately 180 nucleotides)—reflecting double-strand breaks generated by dioxyribonuclease (DNase) action in internucleosomal regions—was thought to be the hallmark of apoptosis (Arends et al., 1990; Hale et al., 1995). Increased intracellular Ca2+ levels were found to be involved in stimulation of the DNase(s) activity (Arends et al., 1990), which is inhibited by zinc. Subsequently, it was recognized that additional morphologic alterations are characteristic of apoptosis; these include chromatin condensation, extensive alterations in the microtubular array, fragmentation of the cell into ''apoptotic bodies,'' and detachment of the cell from its neighbors or the culture substrate (Hale et al., 1995). The alterations in a specific instance of apoptosis are somewhat variable, and it has now become clear that cytoplasmic apoptosis can occur without DNA fragmentation (Jacobson et al., 1994).
During development, the human fetus generates many more cells than are finally needed (Hale et al., 1995), and developmental apoptosis has been extensively studied in Caenorhabditis elegans, which generates exactly 1,090 cells, of which 131 undergo apoptosis (Ellis et al., 1991). Particularly important genes of the apoptosis system in this organism include ced-3 (C. elegans death gene), ced-4, and ced-9 (Hale et al., 1995). The peptide products of these genes are homologous to mammalian proteins: CED-3 is homologous to cysteine-dependent aspartate proteases (caspases) (Hale et al., 1995), CED-4 is homologous to apoptosis-activating factor 1 (APAF-1) (Zou et al., 1997), and CED-9 is homologous to the antiapoptotic protein Bcl-2 (Hale et al., 1995).
Mammalian plasma membrane receptors for the activation of caspases include FAS and the tumor necrosis factor receptor (TNFR) (Nagata and Golstein, 1995). Both FAS and TNFR include a cytoplasmic domain of approximately 70 amino acids called the "death domain" (Hale et al., 1995), which is a protein-protein interaction zone also present in the molecules FADD (MORT1) and CRADD (Hale et al., 1995). Ligand binding to FAS or TNFR leads to recruitment of FADD or CRADD by association of death domains (Ahmad et al., 1997). FADD and CRADD are adapter linkers (Ahmad et al., 1997) and recruit Machl (FLICE, caspase 8), which then activates other caspases by cleaving zymogen procaspases into the active proteases (Ahmad et al., 1997). Several viruses encode death domain-containing proteins that inhibit receptor-induced apoptosis (Bertin et al., 1997; Muzio et al., 1997; Zhou et al., 1997), indicating that a key function of the receptor-mediated caspase activation system is to provide a viral defense in which infected cells are induced to die.
The level of the FAS protein is increased approximately 130-fold in post-ischemic myocytes at 2 hours reperfusion following myocardial ischemia (Kajstura et al., 1996), and it is clear that apoptosis is involved in the post-ischemic death of cardiac myocytes (Brömme and Holtz, 1996). The situation with regard to plasmalemma receptor-induced apoptosis during brain reperfusion is different. There is little FAS mRNA in normal brains, and even after 30
minutes of ischemia and 6 hours of reperfusion FAS mRNA is not found in vulnerable hippocampal or cortical neurons (Matsuyama et al., 1994). Similarly, Chopp's group (Zhang et al., 1997) showed appearance of TNFR as quickly as 1 hour after middle cerebral artery occlusion in mice, but the protein was localized to microvessels. Furthermore, TNFR-knockout mice showed exacerbated brain damage in response to focal ischemia (Brace et al., 1996), possibly because TNFR also leads to activation of the transcription factor NFκB (Der et al., 1997), which counterregulates apoptotic mechanisms.
Although a plasmalemma receptor-activated caspase response is not a primary death mechanism in the vulnerable neurons, the apoptotic mechanisms are more complex than the FAS, FADD/CRADD, and Machl system. For example, although the traditional view has been that caspase expression is constitutive (Muzio et al., 1997), ischemia and reperfusion induce in vulnerable neurons the transcription of CPP32 (Gillardon et al., 1997) (caspase 3, apopain, Yama), which is required for normal fetal brain development (Kuida et al., 1996) but which is essentially absent from the adult neurons (Ni et al., 1997). The relatively specific CPP32 inhibitor N-benzyloxycarbonyl-Asp-Glu-Val-Asp-flouromethylketone (where Asp is aspartic acid; Glu is glutamic acid; and Val is valine) decreased the amounts of caspase cleavage products, reduced the level of tissue damage, and improved functional outcome in a stroke model in rats and mice (Hara et al., 1997).
In various model systems CPP32 is activated before several other caspases (Martins et al., 1997) and is the predominant enzyme involved in the very rapid cleavage of the DNA repair enzymes poly(ADP ribose) polymerase (PARP) (Margolin et al., 1997; Van de Craen et al., 1997) and DNA-dependent protein kinase (Le Romancer et al., 1996). PARP is substantially conserved from Drosophila to humans (Uchida et al., 1993), recognizes strand nicks in DNA, and marks damaged DNA for repair by generating poly(ADP ribose) chains on adjacent nuclear proteins (Rosenthal et al., 1997). PARP is required for cellular recovery after radical-mediated DNA damage (Shah et al., 1996), upregulates the tumor suppressor p53 (Whitacre et al., 1995), and is required for insertion of retrovirus-derived DNA into the genome (Gaken et al., 1996). CPP32 also cleaves actin to produce a characteristic 15-kilodalton (kDa) peptide recognized by a specific antibody (Mashima et al., 1997), which represents a potentially important assay for apoptosis.
The details of non-receptor-mediated caspase mechanisms involved in an apoptosis occurrence can reflect the initiating event. Nedd2 is another caspase that, like CPP32, is expressed in neural precursor cells, is developmentally downregulated (Allet et al., 1996), and efficiently cleaves PARP (Gu et al., 1995). The processing of Nedd2 from the proenzyme to the active form is inhibited in a neural cell line by Bcl-2 (Srinivasan et al., 1996). Reduction of Nedd2 synthesis by antisense mRNA rescues both sympathetic neurons and PC12 cells from apoptosis caused by growth factor withdrawal (Troy et al., 1997). However, in these same cells apoptosis induced by superoxide dismutase
deficiency is inhibited by fluoromethylketone caspase inhibitors but not by the reduction of Nedd2 (Troy et al., 1997).
The prototypic caspase CED-3 can be activated by direct association with CED-4 (Irmler et al., 1997). Transgenic expression of CED-4 in apoptosis-naive Schizosaccharomyces pombe induces chromatin condensation and death associated with immunolocalization of CED-4 to the nucleus (James et al., 1997). In this model system mutation of the nucleotide-binding motif of CED-4 eliminates the lethal effects, and coexpression of CED-4 and CED-9 blocks the nuclear localization of CED-4 and its lethal effects (James et al., 1997) through a direct molecular interaction (Spector et al., 1997). The analogous mammalian system appears to involve activation of CPP-32 by a complex of APAF-1 and cytochrome c (Zou et al., 1997) released from mitochondria after the formation of a nonspecific large pore (approximately 2 nm) in the mitochondrial membrane under the simultaneous influence of matrix Ca2+ overload and radical stress (Andreeva et al., 1995). It is interesting that the iron chelator deferoxamine blocks radical-associated mitochondrial permeability transition and cell death (Nieminen et al., 1997). The human genes encoding CED-9-related proteins include Bcl-2, Bax, Bad, Bcl-x, mcl-2, Bak, and Bak-2 (Hale et al., 1995). Bcl-2 inhibits the processing of caspase proenzymes to active proteases (Srinivasan et al., 1996) and the release of Ca2+ from ER (Lam et al., 1994), an event that, as shown below, is crucial to the activation of intracellular apoptosis mechanisms. Several mammalian viruses carry genes encoding Bcl-2 homologues that inhibit apoptosis (Henderson et al., 1993).
Bax forms ion channels in mitochondrial and ER membranes and promotes apoptosis; Bcl-2 binds to Bax and blocks these channels (Antonsson et al., 1997). The tumor suppressor p53 both upregulates Bax and downregulates Bcl-2 transcription (Hale et al., 1995). These observations suggest that the ratio of Bcl-2 to Bax is important in the regulation of apoptosis.
It is becoming clear that major changes in the translation initiation system for protein synthesis are crucial in apoptosis. During translation initiation, mRNA selection and delivery of the first amino acid (methionine) for the new peptide are two critical steps that are affected by the mechanisms of apoptosis. mRNA is delivered to the translation initiation complex by eukaryotic initiation factor-4 (eIF4). Most mammalian mRNAs include an m7GTP (where GTP is guanosine triphosphate) cap that is bound by the subunit eIF4E. The eIF4 complex also includes eIF4A and eIF4B, which facilitate unwinding of secondary structure in the 5' leader of the mRNA, and eIF4G, which provides docking sites for the other eIF4 subunits and for attachment via eIF3 to the small ribosomal subunit. Methionine is delivered to the small ribosomal subunit by a ternary complex that comprises eukaryotic initiation factor-2 (eIF2), GTP, and methionine-transfer RNA (met-tRNA) (Merrick, 1992). Translation initiation leads to hydrolysis of the eIF2-associated GTP and generation of an eIF2-GDP (where GDP is guanosine diphosphate) complex. The enzyme eIF2B must then exchange that GDP for another GTP on eIF2 before the next round of translation initiation. This GTP exchange reaction is competitively inhibited by eIF2 con-
taining a serine at position 51 (ser-51)-phosphorylated α-subunit [eIF2α(P)] (Rowlands et al., 1988). Because the molar ratio of eIF2 to eIF2B is about 5 to 1, when approximately 20 percent of eIF2 is in the eIF2α(P) form, new protein synthesis is substantially inhibited.
Apoptosis is associated with caspase-mediated degradation of eIF4G (Clemens et al., 1998), which is also degraded during brain ischemia by the calcium-activated protease µ-calpain (Neumar, 1998) and by an unknown protease during early brain reperfusion (DeGracia et al., 1996). Several studies have directly implicated activation of calpain in apoptosis (Estaquier et al., 1996; Jordan et al., 1997; Squier and Cohen, 1997). Furthermore, there is a more than 20-fold increase in the brain eIF2α(P) level so that this isoform represents over 23 percent of total eIF2 within the first 10 minutes of post ischemic reperfusion, and eIF2α(P) maps to selectively vulnerable neurons that display chromatin condensation consistent with early apoptosis (DeGracia et al., 1996, 1997). Kaufman's group has shown that expression of a mutant that substitutes alanine (which cannot be phosphorylated) for the Ser-51 phosphorylation site in eIF2α blocked TNF-α-induced apoptosis (Kaufman and Srivastava, 1996) and induced downregulation of Bax expression (Kaufman et al., 1998). Furthermore, expression of a conditional eIF2α mutant imitating the negative charge at Ser-51-P (by substituting aspartate for Set-51) induced immediate apoptosis (Kaufman and Srivastava, 1996) and activation of CPP32 (Kaufman et al., 1998).
The proteolytic degradation of eIF4G and phosphorylation of eIF2α not only will result in general depression of overall protein synthesis, but will also exert a major effect on message selection for residual translation. Although m7GTP cap-dependent translation cannot occur without a fully competent eIF4 complex, eukaryotic mRNAs containing internal ribosome entry site sequences can circumvent this limitation. Furthermore, long 5' mRNA leaders before the AUG start codon are known to favor translation in the presence of elevated eIF2α(P) levels because the long leader prolongs ribosomal scanning time and thus the opportunity to find a good ternary complex. Although little is known about the characteristics of proapoptotic mRNAs, these alterations in the translation initiation system could favor them. Indeed, mRNA leaders with these characteristics are present on the message for APAF-1 (Doldwell et al., 1998) and several stress-response proteins including platelet-derived growth factor-2 (Sella et al., 1998), vascular endothelial growth factor (Stein et al., 1998), fibroblast growth factor-2 (Vagner et al., 1996), and the X-linked inhibitor of apoptosis (Holcik et al., 1998).
The mechanism for phosphorylation of eIF2α in response to ischemia and reperfusion is not understood as well as the degradation of eIF4G by proteases. Phosphoprotein phosphatase 1 is responsible for the dephosphorylation of eIF2α(P) (DeGracia et al., 1996), and ischemia and reperfusion do not appear to cause a loss of this phosphatase activity (DeGracia et al., 1998). Three mammalian eIF2α kinases are known to exist (Samuel, 1993; Sood et al., 1998; Wek, 1994): (1) the hemin-regulated inhibitor (HRI kinase), which is also activated by
transition metals and lipid hydroperoxides, (2) the "double-stranded viral RNA-activated" protein kinase (PKR), and (3) GCN2.
A major function of PKR is to block virus-directed protein synthesis (Proud, 1995). In this regard, a number of viruses, including human immunodeficiency virus (HIV) and members of the smallpox virus family, encode proteins that are PKR inhibitors (Jagus and Gray, 1994). Recent observations have revealed a much broader cellular role for PKR (Petryshyn et al., 1994; Proud, 1995) and have identified a number of endogenous PKR inhibitors and activators (Jagus and Gray, 1994). Transcription of the PKR gene is induced by interferon, and PKR activity is antagonized by intracellular signaling activated by insulin-family growth factor receptors (Bandyopadhyhay and Sen, 1992). It is particularly interesting that activated PKR appears to be required for induced transcription of the gene for the FAS receptor (Der et al., 1997; Takizawa et al., 1995). Cells from PKR-knockout mice are resistant to apoptosis induced by TNF-α, lipopolysachrides, or viral RNA (Der et al., 1997; Kaufman and Srivastava, 1996). However, phosphorylation of eIF2α during early brain reperfusion is not reduced in PKR knockout mice.
Depletion of Ca2+ from the ER leads to a rapid suppression of protein synthesis (Brostrom et al., 1983, 1985, 1989; Chin et al., 1987; Kumar et al., 1989) by phosphorylation of eIF2α (Kimball and Jefferson, 1992; Prostko et al., 1992, 1993); the detailed mechanism of this is unknown. However, ER Ca2+ depletion, eIF2α phosphorylation, and suppression of protein synthesis are induced by free arachidonate concentrations above 10 micromolar (µM) in cell culture models (Fleming and Mellow, 1995; Rotman et al., 1992). On the ER, two Ca2+ channel-regulating receptors are known: the ryanodine and the inositol triphosphate (IP-3) receptors (RyR and IP-3R). Both receptors are present in vulnerable CA1 neurons, with IP-3R being more prominent in these cells and RyR being more prominent in the vulnerable hilar neurons (Sharp et al., 1993). Arachidonate appears to compete at the ryanodine receptor (Uehara et al., 1996) and has not been studied at the IP-3 receptor. However, it has been shown that IP-3R-deficient T cells are resistant to apoptosis induced by ionizing radiation, FAS ligand, and glucocorticoids (Marks, 1997).
Both RyR and IP-3R are formed by a tetramer of their respective primary proteins, and one 12-kDa peptide that binds to the transplant immunosuppressant tacrolimus (or FK506, thus, the FK-binding protein [FKBP12]) is associated with each monomer of both receptors (Cameron et al., 1995; Marks, 1996). Expression of cloned RyR in otherwise RyR- and FKBP12-naive Spodoptera frugiperda generated tetrameric RyR receptors that exhibited only Ca2+ subconductance at four distinct levels, but coexpression of FKBP 12 generated channels that exhibited full ligand-activated conductance (Marks, 1996). FK506 dissociates FKBP12 from both receptors (Cameron et al., 1995; Timermann et al., 1995), resulting in only Ca2+ subconductance (Marks, 1996). Thus, the overall effect of FK506 is to reduce ER Ca2+ conductance in response to these ligands.
FK506 has been shown to salvage vulnerable neurons after brain ischemia (Sharkey and Butcher, 1994) and to allow them to synthesize stress-response
proteins in response to copious transcript formation, which FK506 did not affect during reperfusion (Kamlya et al., 1997; Katayama et al., 1997). FK506 is a macrolide antifungal immunosuppressant (Thomson et al., 1995) structurally related to rapamycin but not to cyclosporin A (Bierer et al., 1991), neither of which are as neuroprotective during reperfusion (Sharkey and Butcher, 1994). Cyclosporin A and FK506 bind to cellular proteins called immunophilins (Thomson et al., 1995), many of which catalyze the cis-trans isomerization of peptidyl-prolyl bonds (by means of peptidyl-prolyl isomerase) (Bierer et al., 1991) and in conjunction with heat shock proteins help peptides fold correctly (Fischer et al., 1993; Galat, 1993). Immunophilins are classified as cyclophilins and FK-binding proteins. Cyclophilins A, B, and C are DNases (Montague et al., 1997). The DNase activities of cyclophilins A and B are stimulated by increased Ca2+ levels and are inhibited by K+ (50 percent inhibitory concentration = 83 millimolar [mM] concentration) (Montague et al., 1997), and NUC 18, thought to be responsible for apoptotic internucleosomal DNA fragmentation (Montague et al., 1994), appears to be identical to cyclophilin A (Wine et al., 1997). Recent evidence suggests that NUC 18 is activated by proteolysis of an approximately 60-kDa precursor (Fraser et al., 1996). An approximately 21-kDa cyclophilin (Tanveer et al., 1996) is an integral part of the pore formed in the mitochondrial membrane under the simultaneous influence of matrix Ca2+ overload and radical stress (Andreeva et al., 1995), and both cyclosporin A and FK506 inhibit Ca2+ transit of this pore (Schweizer et al., 1993). Complexes of immunophilins and cyclosporin A or FK506 also inhibit calcineurin (Liu et al., 1992), which dephosphorylates and activates NO synthetase (Dawson et al., 1993).
In summary, apoptotic signals originate from cellular organelles. At the plasmalemma this starts with FAS or TNF-α binding to receptors; in the case of mitochondrial damage it involves the release of cytochrome c which interacts with APAF-1; at the ER activation of IP-3R or RyR by ligands that may emerge from membrane damage leads to ER Ca2+ depletion and eIF2α phosphorylation; and in the nucleus sufficient DNA damage and poly(ADP) ribosylation activate p53, which leads to enhanced Bax transcription and repressed Bcl-2 transcription, with the consequence of pore formation in the ER and mitochondria by Bax. All multicellular eukaryotic organisms appear to have apoptotic systems, but single-celled eukaryotes (yeast) are naive. Finally, viruses have developed a variety of mechanisms that inhibit eukaryotic apoptosis. These observations indicate that both apoptosis and viral resistance to apoptotic mechanisms emerged after the development of multicelled eukaryotes and suggest that execution mechanisms of inhibition of protein synthesis, proteolytic degradation of a DNA recombination mechanism (PARP), and degradation of polynucleotides are all primarily antiviral strategies. This theoretical approach has the advantage of not requiring more or less simultaneous evolution of apoptosis execution and regulation strategies for a primary role in embryogenesis and suggests that the growth factor signal transduction mechanisms that govern developmental apoptosis were developed later in evolution.
From this basic perspective, the complexity of the system arises from the multiple input pathways and the interconnection of the execution mechanisms. For example, Ca2+ release from the ER can be induced by activation of IP-3R or RyR or by an excess of Bax over Bcl-2. This can lead to PKR activation, the consequent inhibition of protein synthesis, and the stimulus of transcription for FAS, which invites activation of the caspase system followed by degradation of PARP and induction of DNase activity. Similarly, primary DNA damage would induce PARP activity followed by p53 activation, Bax expression, ER Ca2+ depletion, formation of pathologic mitochondrial pores, and so on. Membrane receptor-induced caspase activity results in degradation of eIF4G as well as degradation of PARP and cytoskeletal components.
This suggests that apoptosis induced by acute disease processes involves inappropriate activation of an ancient viral defense system that leads to the death of cells essential to survival of the organism. Therapeutic approaches to this system that are beginning to emerge include the use of inhibitors of calpain and caspases and agents that appear to have diverse antiapoptotic activity such as cyclosporin, FK506, and peptide growth factors.
Alterations in Secretion of and Cellular Responsiveness to Growth Factors
Insulin and other growth factors such as nerve growth factor, insulin-like growth factor-1 (IGF-1) (Zhu and Auer, 1994), and fibroblast growth factor have established neuron-sparing effects in the setting of ischemia and reperfusion (reviewed by O'Neil et al., 1996). In the case of insulin, this effect does not involve reduction of blood sugar (Voll and Auer, 1991a), and on the basis of doses thought to be pharmacologically equivalent, insulin is more effective than IGF-1 (Zhu and Auer, 1994). Other studies have also shown that insulin improves neurologic deficit scores and cognitive function and prevents necrotizing brain damage after transient forebrain ischemia (Fukuoka and Yeh, 1989; Strong et al., 1990; Voll and Auer, 1991b).
The evidence presented above suggests that insulin has a neuron-sparing effect that is mediated by direct interaction with receptors in the central nervous system (Zhu and Auer, 1994), even though insulin does not regulate glucose handling in the brain. Insulin is known to bind to only two receptors: the insulin receptor and the IGF-1 receptor. It does not bind to the IGF-2 receptor (Le Roith et al., 1993) or the insulin receptor-related receptor (Jui et al., 1994; Zhang and Roth, 1992). Insulin binds in the dentate and CA1 regions of the hippocampus (Hill et al., 1986; Kar et al., 1993; Werther et al., 1987), and its receptor is found most prominently on the perikarya and processes of the CA1 and CA2 pyramidal cells and the neurons of the dentate gyms; immunohistochemistry does not identify the insulin receptor on glia (Unger et al., 1989). The insulin receptor comprises two covalently linked homodimers formed by its α and β subunits. Ligand binding activates auto-tyrosine phosphorylation of the β subunit fol-
lowed by tyrosine phosphorylation of other peptide substrates, including insulin-receptor substrate-1 (IRS-1). Srchomology-2 domains on phosphorylated IRS-1 activate intracellular signaling cascades. Both pancreatic insulin secretion and the kinase activity of the insulin receptor are markedly downregulated by adrenergic signaling mediated through cyclic adenosine monophosphate and protein kinase A (PKA), and hypovolemia is clinically well known to be associated with insulin resistance. Thus, insulin-mediated growth factor signaling is inhibited by ischemia and reperfusion.
Sullivan and colleagues (1998) have recently used simultaneous autoradiography of pulse-labeled protein synthesis and immunohistochemical mapping of eIF2α(P) to confirm the colocalization of inhibited protein synthesis and eIF2α(P) during brain reperfusion. That same study found that 20 units of insulin per kilogram of body weight administered intravenously at reperfusion caused restoration of normal protein synthesis in and elimination of eIF2α(P) from vulnerable hippocampal neurons by 90 minutes of reperfusion after a 10-minute cardiac arrest.
There is precedent for insulin-mediated downregulation of eIF2α kinases; activation of Ras, an intermediate in insulin signaling, leads to activation of a 97-kDa inhibitor of PKR (Bandyopadhyhay and Sen, 1992), although as already indicated, this enzyme is not required for phosphorylation of eIF2α during brain reperfusion. The reversal of eIF2α phosphorylation in vulnerable neurons by insulin during early reperfusion also might be due to the activation of an eIF2α(P) phosphatase. PP1, which is activated in response to insulin signaling (Begum, 1995; Srinivasan and Begum, 1994), is the enzyme responsible for the dephosphorylation of eIF2α(P) in vivo (Ernst et al., 1982; Foulkes et al., 1983; Ingebritsen and Cohen, 1983; Redpath and Proud, 1990), and isoforms PP1α and PP1γ are present in the brain and are concentrated in the neocortex and hippocampus (Hubbard and Cohen, 1993; Ouimet et al, 1995; Takizawa et al., 1994).
There are several possible explanations for the high dose of insulin required for neuron sparing during postischemic reperfusion. Adrenergic downregulation of insulin secretion and of the insulin receptor itself has already been mentioned. Glucocorticoids also inhibit insulin transport into the central nervous system (Baura et al., 1996). Alternatively, other signaling mechanisms may also decrease the responsiveness of the insulin receptor. TNF-α levels are elevated in the reperfused brain (Lavine et al., 1998), and TNF-α induces insulin resistance by increasing serine and threonine phosphorylation of the insulin receptor and of the major insulin receptor substrates IRS-1 and IRS-2 (Paz et al., 1997). It is also possible that the neuroprotective effects of insulin occur by activation of the IGF-1 receptor (Gluckman et al., 1993). Although insulin and IGF-1 have significant sequence homology, the affinity of insulin for the IGF-1 receptor is about 100-fold lower than that of IGF-1 (Le Roith et al., 1993). Autoradiographic studies have shown large quantities of [125I]IGF-1 receptors in hippocampal neurons (Bohannon et al., 1988; Kar et al., 1993; Lesniak et al., 1988).
IGF-1 has also been shown to have a neuroprotective effect in transient fore-brain ischemia.
It is noted that insulin resistance and hyperglycemia in patients in shock is a well known phenomenon. The above adrenergic and TNF-α signaling mechanisms that down-regulate insulin secretion, the tyrosine kinase activity of the insulin receptor, and the major intracellular substrates of the insulin receptor are in play in shock and are likely involved in the associated insulin resistance and hyperglycemia.
Hematologic Abnormalities Associated With Shock and Resuscitation
The administration of blood products and the management of bleeding disorders are important therapeutic modalities used by surgeons caring for patients with a wide variety of acute and chronic problems. When used with a thorough understanding of appropriate indications, risks, and benefits, blood transfusion is safe and effective. Because blood transfusion is lifesaving for many patients, a knowledge of the appropriate indications, potential risks, and available alternatives should allow clinicians to exercise judgment in using this important resource.
Although it is now routine, the ability to successfully transfuse blood is relatively recent. Accounts of bloodletting and phlebotomy, but not blood transfusion, appear in many early historical references and were recommended for many ailments, including insanity. The routine, safe administration of blood products required several important scientific advances. The discovery of the A, B, and O blood types by Landsteiner in 1900 and the AB blood type by Von Decastello and Sturli in 1902 began the era of modem blood transfusion. By the 1940s, techniques of cross-matching, anticoagulation, and storage of blood and the establishment of blood banks made routine blood transfusion possible. The ability to replace blood lost intraoperatively is an important prerequisite in modem surgical practice.
Modem blood banking is based on the concept that a donated unit of blood can be divided into its components and the different components can be applied to the specific needs of a patient, such as a heightened need for clotting factors. Specifically, after donation, platelets whose shelf life is much shorter than red blood cells, are removed, making them available for platelet transfusion. Clotting factors are also removed, and the residual fluid contains mostly red blood cells suspended in plasma. In the military, citrate-preserved blood (i.e., without platelets) has been the standard transfusion fluid for over 50 years. On a practical basis, any stored blood should be assumed to be deficient in both clotting factors and platelets. From time to time, in acute situations, donors provide whole blood for very specific but quite unusual situations. In some past military experiences, however, inaccuracy in identification of the blood type made the possibility of a severe or fatal reaction occurring from mismatched blood.
Transfusion of the Patient in Shock
During World War I, it was believed that vascular collapse in injured patients was caused by toxins (MacLean, 1985). Experiments in the 1930s showed that fluid was lost from the circulation into damaged tissues. In World War II, plasma became the resuscitation fluid of choice. Subsequent experimental work indicated that extracellular fluids shifted into the intracellular space after significant hemorrhage with shock (Canizaro, 1973). The provision of resuscitation fluid in a volume in excess of the volume of blood that had been shed then became an acceptable practice for the maintenance of adequate circulation.
During World War II, acute tubular necrosis was a common consequence of hypovolemic shock. Because fluid resuscitation became more prevalent during the Korean and Vietnam conflicts, the incidence of acute tubular necrosis dramatically decreased. Although acute tubular necrosis after hypovolemic shock became less of a problem with better fluid resuscitation, the shock lung syndrome (i.e., adult respiratory distress syndrome) became increasingly common. The lung injury in adult respiratory distress syndrome is a function of the shock state rather than the resuscitation solution used.
The goal of resuscitation from shock is prompt restoration of adequate per-fusion and transport of oxygen. Restoration of circulation allows the cell to clear the products of anaerobic metabolism and restore aerobic metabolism. The American College of Surgeons Committee on Trauma developed a classification of shock that permits useful guidelines for resuscitation (1997). Crystalloid is infused at a 3:1 ratio for every unit of red blood cells administered, and therapy is monitored by hemodynamic response. Because crystalloid solutions are universally available and some delay is required for the preparation of blood products, crystalloid is the proper initial resuscitation fluid. Resuscitation then proceeds with the use of blood products, depending on the patient's response. The choice of a colloid solution (e.g., albumin or plasma) or a crystalloid solution (e.g., lactated Ringer's solution) has been controversial. Both can expand the extracellular space and provide effective resuscitation. However, crystalloid solutions are favored because they are less expensive, need not be cross-matched with the patient, do not transmit disease, and probably create less fluid accumulation in the lungs. No experimental data indicate that colloid solutions are less apt to prevent pulmonary edema than are crystalloid solutions. Some work (Holcroft and Trunkey, 1975; Lewis et al., 1979), based on measurements of pulmonary extravascular water volume or lung water measurements, indicates that albumin, if used as a resuscitation fluid, moves across the cell membrane and draws in extracellular fluid by osmosis, thereby exacerbating the pulmonary edema.
Several crystalloid solutions are available for resuscitation, but isotonic solutions should be used to avoid overload of free water. Lactated Ringer's solution has been recommended as initial therapy. Metabolic alkalosis is common after successful resuscitation with lactated Ringer's solution and blood products, because the lactate in Ringer's solution and the citrate in banked blood are both
converted to bicarbonate if the patient has a functioning, perfused liver. Lactated Ringer's solution contains calcium, and if it is mixed with a unit of blood product, the blood may clot in the bag. Normal saline solution is an acceptable alternative to lactated Ringer's solution, but large volumes can produce a hyperchloremic metabolic acidosis, which may complicate the care of the patient in shock.
Massive Transfusion
Massive transfusion has been defined as replacement of the patient's blood volume with stored red blood cells in 24 hours or as transfusion of more than 10 units of blood over a few hours. Massive transfusion can create significant changes in the patient's metabolic status because of the infusion of large volumes of cold citrate-containing blood that has undergone changes during storage (Canizaro, 1973). If a large volume of stored blood is infused rapidly, significant effects may be seen in the recipient, depending on the recipient's metabolic state, including (1) significant degrees of hypothermia, exacerbated in patients who have an open thoracic or abdominal cavity, which accelerates heat loss, increases the affinity of hemoglobin for oxygen, impairs the function of platelets, and increases the potential for hypocalcemia; (2) production of alkalosis with subsequent undesirable effects on myocardial contractility; and (3) changes due to citrate, such as hypotension, narrowed pulse pressure, and elevated left ventricular, end-diastolic, pulmonary artery, and central venous pressures. Other effects include dilutional thrombocytopenia due to the fact that the number of platelets is almost nil in blood stored for 24 hours.
In summary, although a massively resuscitated and transfused patient may develop bleeding disorders because of shock and resuscitation, the major changes in the massively transfused patient are opposite what one might expect on the basis of the changes that occur in blood during storage. The administration of liquid-preserved blood and crystalloid, the usual routine for the resuscitation of hypovolemic patients, results in profound alterations in the amounts of clotting factors, for example. Additionally, platelet counts fall from the normal value of 250,000 to 300,000 µ1 to approximately 50,000 µ1. This phenomenon, called dilutional thrombocytopenia, occurs in the context of lowering primarily clotting Factors V and VIII and prolongs the clotting time. The clotting time, however, in successful resuscitation will usually not be associated with an excess tendency for bleeding. In other words, although laboratory demonstration of the prolongation of clotting time presents a potential problem, in reality, excess bleeding in groups of patients studied in this situation rarely occurs. Similarly, the use of citrate-preserved blood results in a net effect of alkalosis after one pass through the liver following resuscitation. Transient lowering of the ionized calcium is a theoretical problem, but the resuscitated patient will mobilize enough ionized calcium to balance any transient lowering of ionized calcium. Whole blood usually increases levels of potassium because of the progressive death of red blood cells in liquid preservation. This theoretically could
produce a problem in the face of lowered calcium and potassium load. The realities of this are, however, that the metabolic environment of the resuscitation situation is such where sodium is retained and potassium wasted. The net effect of massive transfusion, then, is a relatively normal level of calcium and a somewhat lowered level of sodium because of the lack of isotonicity of the common resuscitation solution.
Risks of Blood Transfusion
The transfusion of blood products can cause numerous serious complications, even death (Linden et al., 1992). A transfusion of incompatible red blood cells is potentially fatal, but other significant concerns exist when a patient receives blood products, including the transfusion of infectious pathogens and immunologic effects. Because a transfusion exposes the recipient to a complex mixture of donor cells and proteins, it is in many ways a transplant. Blood components contain viable lymphocytes that can provoke a graft-versus-host response in severely immunocompromised recipients. Transfusion can modify the recipient's immune response, as has been demonstrated in patients undergoing renal transplantation.
The most severe acute transfusion reactions involve complement-mediated red blood cell destruction. Because the red blood cells are rapidly destroyed intravascularly, peptides derived from complement are released and produce hypotension, compromise renal blood flow, activate the clotting cascade, and lead to disseminated intravascular coagulation (DIC) (see below). Most reported acute fatalities from transfusion reactions result from ABO-incompatible transfusions (Linden et al., 1992; Sazama, 1990). A hemolytic reaction can occur hours to days after transfusion although delayed hemolysis is rarely serious.
Vital and bacterial diseases may be transmitted by blood transfusion. Viruses include HIV, hepatitis viruses, cytomegalovirus, human T-cell leukemia virus types I and II, and Epstein-Barr virus. Bacterial diseases include infection with Pseudomonas fluorescens, Yersinia enterocolitica, staphylococcus epidermis, staphylococcus aureus, or salmonella choleraesuis. The rate of contamination of units (but not necessarily transmission) is: red cells 0-2 percent and platelets 0-10 percent (Wagner et al., 1994). Advances in the ability to detect the hepatitis C virus and more efficient screening of blood products have made the currently available U.S. supply of blood from volunteer donors extremely safe, and the risk of posttransfusion infection is significantly reduced (Fakhry and Sheldon, 1994).
Blood Substitutes and Alternatives to Transfusion
Red blood cell transfusion is the only acceptable clinical method for acutely increasing the oxygen-carrying capacity. Development of red blood cell substitutes would eliminate the risk of transfer of infectious agents through blood
transfusion and provide a ready source of universally compatible product. Several substances have been considered as red blood cell substitutes and can be divided into two general groups: (1) synthetic molecules, such as the porphyrins and the perfluorocarbon compounds, and (2) molecules that incorporate hemoglobin in their structures, such as the cross-linked and polymerized hemoglobin solutions. Acceptable red blood cell substitutes must be able to carry at least as much oxygen as hemoglobin normally carries (1.34 milliliters of oxygen per gram of hemoglobin). In addition, these molecules should be stable and should have an acceptable half-life. The red blood cell substitute should have properties that allow it to become completely saturated with oxygen at the standard fraction of inspired oxygen, but that allow it to unload substantial portions of its transported oxygen at the partial pressure of oxygen levels found in tissue. In addition, the solutions must be highly purified and free of contaminants and endotoxins (Fakhry and Sheldon, 1994).
Perfluorocarbons efficiently transport significant quantities of oxygen and carbon dioxide and thus have the potential to be an effective red blood cell substitute. Several perfluorocarbon molecules have been tested with humans, with limited success (Seaghell et al., 1990). At present they have no clinical application as red blood cell substitutes.
Early attempts to prepare hemoglobin solutions consisted of pooling outdated blood, breaking the red blood cells open, and extracting the hemoglobin molecules. Because the antigenic properties of red blood cells are associated with the membrane, hemoglobin solutions prepared in this way can be infused into patients with all blood types. This solution is termed stroma-free hemoglobin. Limitations to the use of stroma-free hemoglobin include its very short half-life in the circulation, its relatively low oxygen-carrying capacity, and its clearance through the kidneys, which causes significant side effects. Clinical trials are under way to determine the efficacy of the solution for acute blood loss and perioperative applications.
The salvage of intraoperative blood loss effectively minimizes the need for blood transfusion. This technique has had successful applications in various operative procedures, including cardiac surgery, spine surgery, liver transplantation, trauma procedures, and vascular surgery. Reports of intraoperative cell salvage in trauma patients with enteric contamination have demonstrated that the procedure can be used safely in such situations, provided that the cells are washed before reinfusion (Boudreaux et al., 1983). The use of devices for the collection of blood lost from the thoracic cavity through a chest tube can also decrease the amount needed for transfusion.
Disseminated Intravascular Coagulation
DIC is a syndrome rather than a specific disease. Although DIC is generally considered a hemorrhagic disorder because of the obvious bleeding problems that are encountered, it is important to recognize the very serious sequelae re-
sulting from microvascular (and sometimes large-vessel) thrombosis that always accompanies true DIC and that leads to end-organ failure and death (Bick, 1994). The disorder may have a spectrum of presentations, from low-grade DIC, with minimal symptoms and minor laboratory abnormalities, to fulminant DIC, with life-threatening bleeding and protein coagulation abnormalities producing end-organ dysfunction and death. Various disorders ranging from sepsis to malignancy have been described in association with DIC. Although the diagnosis of DIC is made in patients receiving massive transfusions, the diagnosis of platelet dysfunction due to hypothermia or a specific factor deficiency should be excluded before making a diagnosis of DIC.
Laboratory abnormalities in DIC are variable and are related to the many diseases that are associated with this condition. Common abnormalities include abnormal prothrombin and activated partial thromboplastin times with depressed fibrinogen levels and abnormal platelet counts. Levels of fibrin-degradation products and D-dimer are commonly elevated. Because of the continued activation of coagulation, thrombin-antithrombin complexes will be formed and their levels can be measured. Levels of thrombin-antithrombin and antithrombin III are depressed. In addition, the levels of various fragments from coagulation factor degradation are elevated, including those of F1.2 and FpA.
Low-grade DIC generally responds to management of the underlying disorder, with some patients requiring heparin therapy. The appropriate therapy for fulminant DIC remains controversial, and this is compounded by the lack of objective studies and the many underlying causes. Despite improved diagnostic and therapeutic modalities, the rate of mortality from DIC remains high and is closely related to the underlying disorder.
Conclusions and Recommendations
Traditionally, clinicians have evaluated patients for the presence and progression of cellular injury or death by assay of cellular proteins in serum that are not specific to injury processes. The new understanding of lethal cellular processes suggests that serum should be examined for the products of these processes. Indeed, immunochemical assays now exist for:
- conjugates of lipid peroxidation products and proteins,
- calpain-specific degradation fragments of spectrin,
- caspase-specific degradation products from actin, and
- phosphorylated eIF2α.
These species are all specifically pathologic and, in fact, reveal the presence of pathochemical processes for which emerging therapeutic approaches exist.
Furthermore, the processes reflected by these products (destruction of membranes by lipid peroxidation, degration of the cytoskeleton by calpain, caspase-mediated proteolysis, and apoptosis-associated blockade of normal
protein synthesis by phosphorylated eIF2α) are probably all independently lethal. In this situation, pharmacologic interference with only a single injury mechanism is unlikely to achieve any dramatic or even clinically noticeable improvement in outcome. The development of polypharmaceutical treatment approaches is a difficult problem, but current cancer chemotherapy shows that this approach can be fruitful. Morever, the availability of assays for individual injury mechanisms indicates that rigorous evaluation of this probably essential therapeutic approach is now possible.
The committee found that traumatic shock is a complex metabolic and cellular process and not just a hemodynamic event. Significant advances in the treatment of traumatic shock are unlikely to result from any simple management protocol alterations directed only at hemodynamic abnormalities. Rather, the major therapeutic advances will result from approaches that address the metabolic and cellular consequences of shock. Therefore, the committee recommends the following:
Recommendation 2.1 Develop and validate diagnostic assays for substances in serum that indicate the specific mechanisms involved in the molecular processes of cellular injury and cell death induced by shock and resuscitation.
Recommendation 2.2 Expand the use of transgenic experimental animals to further evaluate the role of specific proteins and enzymes in cellular injury and death induced by shock and resuscitation.
Recommendation 2.3 Study and rigorously evaluate polypharmaceutical approaches directed against the multiple and independent mechanisms of cellular injury and death induced by shock and resuscitation.
| This page in the original is blank. |





























